
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
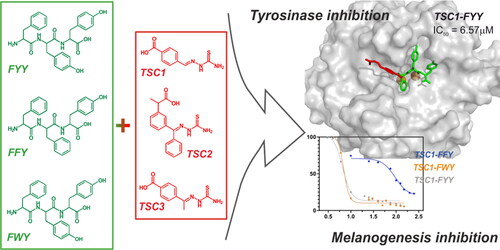
The development of skin-care products is recently growing. Cosmetic formulas containing active ingredients with proven efficacy, namely cosmeceuticals, are based on various compounds, including peptides. Different whitening agents featuring anti-tyrosinase activity have been applied in the cosmeceutical field. Despite their availability, their applicability is often limited due to several drawbacks including toxicity, lack of stability, and other factors. In this work, we present the inhibitory effect on diphenolase activity of thiosemicarbazone (TSC)-peptide conjugates. Tripeptides FFY, FWY, and FYY were conjugated with three TSCs bearing one or two aromatic rings via amide bond formation in a solid phase. Compounds were then examined as tyrosinase and melanogenesis inhibitors in murine melanoma B16F0 cell line, followed by the cytotoxicity assays of these cells. In silico investigations explained the differences in the activity, observed among tested compounds. Mushroom tyrosinase was inhibited by TSC1-conjugates at micromolar level, with IC50 lower than this for kojic acid, a widely used reference compound. Up to now, this is the first report regarding thiosemicarbazones conjugated with tripeptides, synthesised for the purpose of tyrosinase inhibition.
Graphical abstract
Introduction
The interest in the development of skin whitening agents is growing. A report on skin lightening products market published by Grand View Research shows that the global market of skin-lightening products was valued at USD 9.96 billion in 2021 and is expected to expand by 5.5% at a compound annual growth rate (CAGR) from 2022 to 2030.Citation1
Melanin is the pigment responsible for skin and hair colour in mammals. It also absorbs ultraviolet radiation protecting the skin from damage.Citation2 The key enzyme in melanin production is tyrosinase (EC 1.14.18.1) which is able to oxidise both monophenols to o-quinones and o-diphenols to o-quinones in two different steps during melanogenesis.Citation3 Two copper ions in the active site of tyrosinase are important for catalytic activity.Citation4 Overproduction, accumulation, and uneven distribution of melanin in mammalian skin lead to numerous pigmentary disorders, involving freckles, age spots, melasma, sites of actinic damageCitation5 or even malignant melanoma, a type of skin cancer that develops from pigment-producing cells (melanocytes).Citation6 Therefore, one of the most common approaches for skin pigmentation control involves the inhibition of tyrosinase, the key enzyme in melanin production. Various tyrosinase inhibitors of both natural and synthetic sources have been already discussed in some relevant reviews.Citation7–11
Despite the availability of a great number of compounds revealing anti-tyrosinase activity, their application in the cosmetic industry is very limited due to several drawbacks including toxicity, lack of stability, off-odours, off-flavours, and economic sustainability. Hydroquinone and kojic acid,Citation12 known components of anti-pigmentation cosmetics, have been reported to be unsafe in concentrations required for a substantial skin-lightening effect.Citation13,Citation14 Therefore, there is a need to search for novel effective and safe tyrosinase inhibitors.
Peptides are rapidly expanding as cosmeceutical active ingredients.Citation15,Citation16 Due to their safety, tolerability decreased side effects, and low toxicity they are considered attractive ingredients for many skin-related indications.Citation17–20 The latest review on peptides as tyrosinase inhibitors point out that peptides are able to efficiently form hydrogen and hydrophobic bonds, as well as interact with copper in the active site of the enzyme.Citation21 It was proven that the presence of hydrophobic amino acids at the N- and C-termini of the peptide chains are involved in additional non-covalent interactions within copper-containing active sites of tyrosinase.Citation22 Tyrosine located at the C-terminus of the peptide sequences enhanced the overall inhibitory effect and this phenomenon was confirmed by molecular modelling.Citation23 Moreover, only short sequences, i.e. di-, tri-, and tetrapeptides were found to be capable of binding histidine residues in the active site.Citation22
Both short and long peptides can be conjugated with small organic molecules, hence acting in different modes. The peptide moiety can be the active ingredient, but it can also be a carrier or a stabilising agent.Citation16,Citation24,Citation25 Recently, the use of peptide conjugates has increased, with particular attention on pharmaceutical applications, as demonstrated by ConjuPepDB, a database of peptide-drug conjugates, recently published by Balogh et al.Citation26,Citation27 In most cases, the peptide is not mandatory for the activity, but it enhances the overall efficacy, for example, by additional binding specificity.Citation25,Citation28,Citation29 Various chemical linkages are used to perform the conjugation, the most frequently utilised are amide, sulphonamide, disulphide, glucuronide, carbamate, ether, and ester bonds.Citation26,Citation28 It was reported that tripeptides conjugated with a small organic molecule, i.e. kojic acid,Citation30,Citation31 mimosine,Citation32 kaffeic acid,Citation33 are more potent tyrosinase inhibitors than the original organic molecule or the tripeptide alone. For both conjugates, the amino acid sequences in kojic acid-tripeptideCitation30 and mimosine-tripeptideCitation32 of the most effective tripeptides were consistent. Phenylalanine was the first amino acid in the sequence conjugated with the organic molecule, tyrosine was consequently located at the C-terminus, while one of the amino acids containing an aromatic residue (Phe, Tyr, or Trp) was located in the middle of the chain.
Thiosemicarbazones (TSCs) represent a versatile class of ligands. They possess multiple biological, pharmacological, and therapeutic properties that make them effective antibacterial,Citation34 antifungal,Citation35 antiviral,Citation36 and anticancerCitation37 agents. Over the past ten years, numerous aromatic thiosemicarbazone derivatives endowed with potent anti-tyrosinase activity have been reported.Citation38–44 Possibly, the aromatic residue may interact with the hydrophobic cavity of the enzyme via van der Waals interactions, and the N-N-S tridentate coordination scaffold may act as a chelator of catalytic copper ions.Citation45 In our previous papers we reported that para-substituted acetophenone TSC derivatives are more potent tyrosinase inhibitors than their analogues substituted at ortho or meta positions, and in many cases than unsubstituted onesCitation46,.Citation47 Similar observations were made for benzaldehyde TSC.
The main purpose of the present work is to evaluate the inhibitory activity of a series of TSC-peptide conjugates on diphenolase activity of tyrosinase. Nine tripeptides conjugated with TSCs bearing one or two aromatic rings have been synthesised. The effect of these conjugates on the activity of mushroom tyrosinase has been investigated. The nature of tyrosinase-inhibitor interactions was explained with molecular docking. We finally studied the inhibition of melanin production of the new thiosemicarbazone conjugates in murine melanoma B16F0 cell line and their cytotoxicity in the same cells. The above-mentioned results may be useful for designing novel tyrosinase inhibitors with cosmeceutical relevance, possessing skin-whitening properties.
Materials and methods
Synthesis, purification, analysis
Materials
For the synthesis of TSCs, all solvents were of commercial quality and purchased from a local supplier (Avantor). Thiosemicarbazide (catalogue nr T33405); racemic ketoprofen (catalogue nr K1751); 4-acetylbenzoic acid (catalogue nr 177458); 4-formylbenzoic acid (catalogue nr 124915); and p-toluenesulfonic acid monohydrate (catalogue nr T35920) were purchased from Merck (Darmstadt, Germany).
For peptide synthesis, all Fmoc-protected amino acids Fmoc-Phe-OH, Fmoc-Tyr(tBu)-OH, and Fmoc-Trp(Boc)-OH were purchased from Iris Biotech GmbH (Marktredwitz, Germany). Peptide synthesis grade N,N′-Dimethylformamide (DMF) and acetonitrile (ACN) were purchased from Carlo Erba (Milano, Italy). Dichloromethane (DCM), trifluoroacetic acid (TFA), triisopropylsilane (TIS), methanol (MeOH), piperidine, N,N′-Diisopropylethylamine (DIPEA), N-Methylmorpholine (NMM), 1,2-Ethanedithiol (EDT), and acetic anhydride were purchased from Sigma-Aldrich (Milano, Italy). Fmoc-Tyr(tBu)-Wang resin (100–200 mesh, loading: 0.70 mmol/g) was purchased from Novabiochem (Merck, Darmstadt, Germany).
Purification of peptides and TSC-conjugates
Crude peptides and TSC-conjugates were purified by Reverse-Phase Flash Liquid Chromatography (RP-HPLC) on Isolera One Flash Chromatography (Biotage, Uppsala, Sweden) using a SNAP Ultra C18 column (40 g) at 20 ml/min flow. Eluent systems: 0.1% TFA in H2O (A), 0.1% TFA in ACN (B). Due to the poor solubility in water of final compounds, the samples for purification were dissolved in the mixture of solvents A and B at the initial percentage of the purification gradient and treated with ultrasounds. The linear gradient applied for each purification was: 35–65% B in A in 30 min.
LC-MS analysis
HPLC-MS experiments of peptides and TSC-conjugates were performed using Alliance Chromatography model 2695 (Waters, Milford, U.S.A.) with a Phenomenex Kinetex C18 column (2.6 μm, 3.0 × 100 mm), flow 0.6 ml/min, coupled to a single quadrupole ESI-MS (Micromass ZQ) at 6 ml/min of: 0.1% TFA in H2O (A), 0.1% TFA in 84% ACN/H2O (B), λ = 254 nm, gradient: 30–90% B in 5 min (for the conjugates) and 20–70% (for control tripeptides), injection volume: 10 µL. High-resolution Mass Spectra of thiosemicarbazones 1–3 were recorded on an LCT Premier XE Waters apparatus, in ESI + mode.
NMR analysis
The 1H and 13C NMR spectra were recorded on a Jeol ECZ 400S spectrometer in DMSO-d6 as solvent. Chemical shifts (δ) are given in parts per million (ppm) relative to solvent signals (2.50 ppm and 39.52 ppm in 1H NMR and in 13C NMR spectra, respectively). Multiplicity is reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sext = sextet, hept = heptet, m = multiplet, br = broad, bs = broad singlet.
Synthesis of thiosemicarbazones
Thiosemicarbazones 1–3 were prepared according to the procedure described previously in our papers.Citation46–48 Detailed protocol and NMR data of the obtained products are reported in the Supplementary Information.
Solid-phase peptide synthesis
Peptides Ac-FXaaY-OH (where Xaa = F,W,Y) were synthesised manually in solid-phase following the Fmoc/tBu orthogonal protection strategy as described elsewhere.Citation49,Citation50 Detailed protocol, chromatograms, and mass spectra of the obtained products are reported in the Supplementary Information.
Tripeptide conjugates synthesis
Conjugates of peptides and TSCs were synthesised according to the procedure described for the peptides. TSCs were dissolved in DMF and coupled to the free N-terminus peptide chain after Fmoc-deprotection, following the standard coupling protocol (double coupling with 1.5 eq.). TSCs, well soluble in DMF, do not require any particular treatment and the conjugation undergoes easily. Chromatograms and mass spectra of the obtained products are reported in the Supplementary Information.
Tyrosinase inhibition assay
The tyrosinase enzymatic assay was performed according to the protocol described previously in our papersCitation46–48 using L-3,4-dihydroxyphenylalanine (L-DOPA) (Sigma-Aldrich, Germany) as a substrate. L-DOPA was dissolved in an aqueous 0.15 mM phosphoric(V) acid solution, preventing its oxidation. The final L-DOPA concentration for the assay was 5 mM. TSCs were dissolved in DMSO, achieving the concentration of stock solutions of 10 mM. TSC-conjugates and unmodified peptides were dissolved in a solution of DMSO/sodium phosphate buffer (1:9, 0.1 M, pH 6.8; assay buffer) (v/v), achieving the concentration of stock solutions of 1 mM. All compounds were then diluted in the assay buffer to test concentrations (DMSO concentrations in final reaction mixtures did not exceed 1% volume). Tyrosinase powder (Sigma-Aldrich, Germany) was dissolved in the assay buffer to obtain a concentration of 50,000 units/mL. This stock solution was divided into 50 μL aliquots, each one containing 2500 units of tyrosinase, and stored at −80 °C. Shortly before the assay, one of the aliquots was reconstituted in the assay buffer up to 3000 μL. First, 10 μL of the diluted enzyme solution was preincubated with samples at various concentrations for 5 min at 25 °C. Afterwards, 20 μL of L-DOPA solution was added. The reaction was immediately monitored by measuring the change in absorbance of the colour product (dopachrome) for 10 min in kinetic mode with 5 s intervals (λ = 475 nm, 25 °C) using Molecular Devices SpectraMax Plus 384 Microplate Reader. The enzyme control (EC) sample did not contain the inhibitor; the blank sample did not contain either the inhibitor or the substrate. Kojic acid (Sigma-Aldrich, Germany), as a positive control, was treated as the other inhibitors.
Cell proliferation assay
B16F0 murine melanoma cell line was cultured in DMEM (Biowest, Nuaillé, France) medium supplemented with 100 U/ml streptomycin (Sigma-Aldrich, Taufkirchen, Germany), 100 U/ml penicillin (Sigma-Aldrich, Taufkirchen, Germany), and 10% foetal bovine serum (Sigma-Aldrich, Taufkirchen, Germany). Cells were maintained in 5% CO2 at 37 °C. Cells viability was estimated by MTT colorimetric assay as described by Bellei et al.Citation51 B16 cells were seeded into 96-well plate (4 × 103 cells/well) and incubated in the presence of the investigated compounds at various concentrations (1000, 400, 100, 40, 10, 1, 0.1 μM) for 48 h. Kojic acid and DMSO were used as controls. MTT was added into wells (0.625 mg/ml, Sigma-Aldrich, Taufkirchen, Germany) for the last 4 h. Then lysing buffer was added to dissolve insoluble formazan. The absorbance was measured (λ = 570 nm) using Thermo Lab Multiscan RC microplate reader.
Measurement of melanin production
Measurement of melanin production in B16F0 murine melanoma cell line was performed as described by Bellei et al.Citation51 The cells were seeded into 96-well plate (5 × 103 cells/well) and stimulated with α-MSH (100 nM, Sigma-Aldrich, Taufkirchen, Germany). After 24 h cells were treated with the investigated compounds as well as DMSO and kojic acid at a final concentration of 100, 40, 10, 4, and 1 μM. After 48 h of incubation with inhibitors, melanin production was measured by evaluating the absorbance (λ = 405 nm) using a microplate reader.
Molecular modelling
Before the simulation of the docking process, ligand structures were prepared with Discovery Studio Client. The structures of all the ligands were optimised in the program Gaussian16 at the B3LYP/6-311g (d,p).Citation52 During the structure optimisation, solvent model PCM was applied. The crystal structure of tyrosinase from Agaricus bisporus was obtained from the RCSB Protein Data Bank (ID: 2Y9X).Citation53 The ligand, besides copper ions, was removed from the crystal structure of the enzyme. Lacking protons and charges were added to the protein using the H++ serve according to pH value 6.8Citation54.Citation55 The active site sphere was selected based on the location of tropolone with a radius 18Ang. Molecular docking in the defined active site of tyrosinase was carried out using GOLD Algorithm (2021.3.0 version, CCDC, Cambridge, United Kingdom) using the ChemPLP scoring function. The docking using genetic algorithm used default settings (population size: 100, selection pressure: 1.1; the number of operations: 100000; the number of islands: 5; niche size: 2; crossover frequency: 95; mutation frequency: 95; migration frequency: 10). For each inhibitor, ten distances were obtained. Analysis of inhibitor–enzyme interactions of docked molecules was performed with Discovery Studio Visualiser 5 (Dassault Systemes BIOVIA, U.S.A.).
Results and discussion
Design of the new peptide conjugates
According to our previous findings, the sulphur atom in the thiourea moiety interacts with copper ions in the active site of tyrosinase, and the aromatic residue is responsible for the hydrophobic interactions.Citation46,Citation47 It may be assumed that the preferable position for the peptide chain attachment should not be located in the proximity of the thiourea group. Therefore, we synthesised different thiosemicarbazones possessing free -COOH group (). Compounds 1 and 3, with the carboxyl group at position para of the aromatic ring, differed from each other by one additional methyl group. TSC 2, bearing a second branched phenyl ring, was included to evaluate the influence of the presence of two aromatic moieties on the inhibitory properties of the final product. Thus, peptide sequences treated as bulky substituents would have been located in the most convenient position distanced from the thiourea group of each TSC.
Figure 1. Thiosemicarbazones 1–3 with a free and reactive carboxyl group, available for peptide conjugation.

As mentioned in the introduction, the peptide sequences chosen for conjugation were selected from the literature, where it was reported that the preferable amino acids should be aromatic and hydrophobic (Phe, Tyr, Trp), and Phe residue should be located at the N-terminus. Thus, we considered the conjugation with three different tripeptides: Phe-Trp-Tyr (FWY), Phe-Tyr-Tyr (FYY), and Phe-Phe-Tyr (FFY).
Synthesis of thiosemicarbazones
Synthesis of all thiosemicarbazones (TSCs 1–3) was performed according to the procedure described in detail in our previous papers.Citation46–48 As shown in Scheme 1, TSC 1–3 were prepared by the reaction of the appropriate carbonyl precursor with thiosemicarbazide.
Scheme 1. Synthesis of the thiosemicarbazones TSC 1 and TSC 3 (top) and TSC 2 (bottom).

Thiosemicarbazones TSC 1 and TSC 3 were obtained by heating stoichiometric amount of thiosemicarbazide and 4-formylbenzoic acid or 4-acetylbenzoic acid in ethanol without any acid catalyst. The preparation of thiosemicarbazone of ketoprofen, TSC 2, required the use of p-toluenesulfonic acid as an acid catalyst and an excess of thiosemicarbazide. TSC 1 and 3 were obtained as single E isomers, as confirmed on the basis of the chemical shift of the signal of N2H proton (C = N-NH-) in the 1H NMR spectrum. In the case of the thiosemicarbazones of aromatic aldehydes, the mentioned signal usually appears over 11 ppmCitation56,,Citation57 while the thiosemicarbazones of aryl alkyl ketones give this signal at about 10.2 to 10.6 ppm.Citation40,Citation57,Citation58 In the case of TSC 1 and 3, the N2H signals were observed at 11.53 and 10.30 ppm, respectively. Since the starting ketoprofen was a racemic mixture, TSC 2 could theoretically exist as a mixture of two pairs of isomers, i.e. R/S mixture of E isomer and R/S mixture of Z isomer. Thiosemicarbazone TSC 2 gives a 1H NMR spectrum with two sets of signals in the 45:55 ratio (based on the integration of the quartettes of CH groups), confirming that TSC 2 was obtained as a nearly equimolar mixture of a racemic mixture of E and Z isomers.
Synthesis of peptides and conjugates
Peptides were synthesised in the solid phase according to the standard orthogonal protection Fmoc/tBu strategy, following the procedure described in the Materials and methods section. Briefly, all conjugates were obtained by coupling the corresponding TSC to the peptide sequence in N-terminal position via amide bond formation. The conjugation was performed in the solid phase after Fmoc-deprotection of the N-terminal position of the tripeptide. Cleavage of the final products, both unmodified sequences and conjugates, was performed in acidic conditions with trifluoroacetic acid (TFA). The conjugated thiosemicarbazones were resistant to both basic and acidic conditions and displayed good solubility in DMF. Peptides and the corresponding conjugates were purified by HPLC and lyophilised for further enzymatic assays. Analytical data (chromatograms and mass spectra) of the synthesised compounds are reported in the Supporting Information.
Tyrosinase inhibition assay
The screening of tyrosinase activity of the synthetic compounds was performed according to the procedure previously reported in our articles.Citation46–48 As reported in most of the research dealing with tyrosinase inhibition, we employed mushroom tyrosinase, due to the lack of information and the limited availability of human tyrosinase.Citation59,Citation60 All the compounds were evaluated at different concentrations after incubation with tyrosinase in solution for 5 min at room temperature. Afterwards, the substrate L-DOPA was added to start the enzymatic reaction. The absorbance provided by the colourful derivative, dopachrome, was measured every 5 s for 10 min in order to monitor the progress of the reaction over time. The percentage of tyrosinase activity was calculated by the formula (1), where: Sampleslope – is the slope value of linear fragment of the absorbance observed for a sample in the presence of the inhibitor; ECslope – is the slope value of a sample without inhibitor (enzyme control; EC).
(1)
(1)
The effect on the diphenolase activity of the enzyme was expressed as IC50 ( and ) using GraphPad Prism software.Citation61 All TSCs and the tripeptide-conjugates demonstrated dose-dependent inhibitory activity. Significant differences between TSCs 1, 2, and 3 were observed. The bulkiest TSC 2 acted as the worst tyrosinase inhibitor among the three tested thiosemicarbazones. It provided a maximum of 34% of enzyme inhibition at 150 μM concentration. At higher concentrations of TSC 2 no further decrease of the tyrosinase activity was observed. On the other hand, the two structurally similar TSC 1 and 3 demonstrated comparable inhibition at micromolar level (IC50 5.3 and 5.8 μM, respectively). Moving forward to peptides and their conjugates with TSCs, firstly we analysed unmodified peptides 4–6. In the tested range of concentrations from 0 to 150 μM, unmodified peptides did not show any significant inhibitory activity (max. observed 5% of inhibition). In accordance with the results obtained for TSCs, the TSC 2-tripeptide conjugates 10–12 were poor inhibitors of tyrosinase (max. 50% of inhibition for CM=250 μM). In contrast, the conjugates with TSC 1 generally maintained the inhibitory activity of the thiosemicarbazone (IC50=6.57 and 6.66 μM), except for FFY sequence (IC50=95.56 ± 25 μM). The tripeptide conjugates with TSC 3, i.e. 13–15, displayed a decrease in inhibitory activity compared to the unconjugated TSC molecule. Once again, FYY- and FWY-tripeptide derivatives (14 and 12) displayed similar tyrosinase inhibition (IC50=26 and 25 μM, respectively), but that was remarkably worse in the case of the FFY-conjugate 13 (62% of inhibition for 250 μM).
Figure 2. Plots with results of tyrosinase inhibition assay. TSC 1 and 3, and their tripeptide conjugates 7–9 and 13–15 were found to be the most active inhibitors of tyrosinase among the tested group (IC50 representation – relative activity in % versus log[inh] in μM).Citation61
![Figure 2. Plots with results of tyrosinase inhibition assay. TSC 1 and 3, and their tripeptide conjugates 7–9 and 13–15 were found to be the most active inhibitors of tyrosinase among the tested group (IC50 representation – relative activity in % versus log[inh] in μM).Citation61](/cms/asset/96b0e243-a808-4809-a2b6-0e8a73b50ee4/ienz_a_2193676_f0002_c.jpg)
Table 1. Results of tyrosinase inhibition assay.
Melanogenesis inhibition
The effect of the synthetic compounds on melanogenesis inhibition was performed in B16F0 murine melanoma cell line. The mouse model of human skin is often employed in skin biology and cancer research due to numerous similarities.Citation62 B16F0 murine melanoma cell line shares most of the melanogenesis with human melanocytes. Therefore, it is the proper model for screening the effect of potential inhibitors on melanogenesis.Citation63
The results of percentage inhibition of melanogenesis by synthetic compounds 1–15 as well as dose-response measurements (IC50) along with confidence intervals for melanin production and cell proliferation in B16F0 mouse melanoma cells are shown in . Kojic acid was used as a standard control, DMSO was used as a solvent control, unconjugated thiosemicarbazone TSC 1–3 and tripeptide sequences 4–6 were used as controls for TSC-tripeptide conjugates 7–15.
Table 2. Results of inhibition of melanin production and cell proliferation in B16F0 cell line.
The results show that melanogenesis in B16F0 cells was inhibited by all the synthetic compounds 1–15 (except for compound 11), kojic acid, and DMSO in a dose-dependent manner. Almost all compounds (3–14) inhibited 0–3% of melanogenesis at the concentration of 10 µM, and 4–13% at 40 µM, which does not place them among the most potent melanogenesis inhibitors. Only TSC 1–3 and the tripeptide conjugate 15 inhibited melanogenesis more than kojic acid at these two concentrations.
Interestingly, TSC 2 possessing the bulkiest structure turned out to be the most potent melanogenesis inhibitor. It inhibited 77% of melanogenesis activity at 40 µM. This result is in agreement with the results of one of our previous papersCitation48 where thiosemicarbazone of benzophenone and of (E)-2-phenylacetophenone (possessing an additional aromatic bulky substituent) revealed IC50 value for melanogenesis below 2 µM. Interestingly, this phenomenon seemed to be independent of the tyrosinase inhibition, as thiosemicarbazone of benzophenone was an efficient tyrosinase inhibitor, while thiosemicarbazone of (E)-2-phenylacetophenone did not inhibit tyrosinase at all.
All the investigated compounds (except for compound 11) revealed IC50 at the micromolar level. TSC 1 and 2 as well as the tripeptide conjugates 12 and 15 showed stronger potency of melanogenesis inhibition than the positive control kojic acid (lower IC50 values in comparison with kojic acid, which gave IC50 of 121 µM). These results were verified with the effect of tested inhibitors on the proliferation of B16F0 cells (). Low melanin production might have been caused by high toxicity (low IC50 of inhibition of cell proliferation) of the inhibitors which, in turn, can lead to inhibition of melanin production. IC50 of cell proliferation is in the range of 49.8–113.6 µM, and all are lower than kojic acid (162.2 µM). The investigated compounds are not highly toxic to B16F0 cells but at the same time, they inhibit melanogenesis with relatively low efficacy. The ratio between the potency of TSCs to inhibit melanin production vs. cell toxicity is visualised in .
Figure 3. Inhibition of B16F0 cell proliferation versus inhibition of melanin production. The bar graph represents the ratio of IC50 for B16F0 cell proliferation to IC50 referred to melanin production, counted for all investigated compounds (except compound 11) and kojic acid.

The higher bars in indicate the higher ratio of melanogenesis inhibition to toxicity. Low ratio values, especially <1.0, do not let to conclude if melanin production inhibition depends on the direct activity of the inhibitor or on the inhibition of cell proliferation. Therefore, IC50 of melanin production should be higher than IC50 of cell proliferation. The only suitable ratio refers to TSC 2 (ratio: 3.6) and this is the only thiosemicarbazone displaying a better ratio value than kojic acid (ratio: 1.3). TSC 2 inhibits melanogenesis in B16F0 cell line but shows no significant inhibition towards tyrosinase. It appears that TSC 2 inhibits the pathway at different stages than the step catalysed by tyrosinase, for example affecting the activity of other key enzymes in melanin biosynthesis, e.g. dopachrome tautomerase, microphthalmia-associated transcription factor, or tyrosinase c protein 1.Citation64
Molecular docking
Understanding the nature ofguest–host interactions is mandatory for the rational design of inhibitors. Molecular modelling, especially simulation of the docking process, can contribute to understanding the physical nature of those interactions. Performing docking simulations allow us to better understand the results of biological tests. During the preliminary steps, structures of the investigated thiosemicarbazones and their conjugates with the designed tripeptide sequences have been optimised by using Gaussian16 code at the B3LYP/6-311g(d,p) level with a solvent model and water as solvents. In the case of TSC 2, containing an asymmetric carbon atom, both R and S isomers have been considered. Isomer E was docked as the default configuration, however during the docking process there was a possibility to freely change between E and Z. No significant results were found for Z isomer; therefore, all the presented data refer to the preferential E configuration. In the second preliminary step, the structure of tyrosinase was protonated at pH 6.8 using H++ web serverCitation54,.Citation55 Tyrosinase structure has been obtained from Protein Data Bank (PDB ID: 2Y9X).Citation53 Calculated structures of inhibitors were docked to the active site of tyrosinase by using Gold ver. 2021.3 algorithm.Citation65 Ten orientations of each investigated inhibitor were obtained, among which the ones with the best value of the ChemPLP scoring function were selected for further investigations.
As it was shown in our previous results of docking studies, the thiourea moiety of thiosemicarbazone inhibitors usually interacts with copper ions of tyrosinase molecule.Citation46–48 Because of that, according to the previously published procedure, we adopted a docking protocol without any additional inhibitor-enzyme constraints.
Structural analysis of inhibitor-enzyme complexes for TSC 1–3 shows that the alignment of each thiosemicarbazone inhibitor is stabilised by π-π stacking interaction between the inhibitor phenyl moiety and the imidazole ring of His263 (see , top). Additionally, strong interaction between the negatively charged carboxylate moiety of inhibitors and copper ions (see Figure SI 32) firmly anchors them into the active site.
Figure 4. Orientation of the thiosemicarbazone TSC 1–3 in the tyrosinase active site. Top: free docking; bottom: constrained docking. Intermolecular interactions are represented as dash lines – green: H-bond; purple: π-π interaction. Copper ions are shown as red spheres.

These unexpected results deriving from calculations show that inhibitors TSC 1–3 interact with the enzyme not through the thiourea moiety, but through the carboxylate one. To confirm this unexpected preference, we performed docking studies using additional structural constraints, forcing the distance between the sulphur atom of the thiourea group and copper ions in the range of 1.5–5.0Ang. The use of these structural constraints allowed us to obtain distances in which the thiourea group interacted with copper ions (see bottom). However, comparing the scoring function values we observed that these positions are less preferred regardless of the considered inhibitor (see Table SI 2). Similar conclusions can be drawn from comparing free and constrained docking results for peptide conjugates. Nevertheless, in the case of these inhibitors, differences are not significant.
The relationship between inhibitory activity and the sequence of the tripeptide linked in the conjugates can be analysed using the example of thiosemicarbazone TSC 1. As mentioned above, the scores of the scoring function obtained in free and constrained docking are relatively similar, suggesting that interaction via the thiourea group is also possible. Therefore, we analysed the impact of the tripeptide sequence on the ability to interact with the protein surface for both docking variants.
Generally, in the case of free docking, the small cavity binding of both copper ions is occupied mainly by the phenyl moiety of Phe1 side-chain of the tripeptide conjugate. Similar behaviour is also found for the other thiosemicarbazone conjugates. Only a few distances, less than 5% of all obtained orientations, present different ways of impact. Apart from the minimal frequency of their occurrence, they are also characterised by lower values of the scoring function. This observation is a bit surprising in the face of the results summarised in,Citation22 where interaction through the tyrosine side-chain, which is a natural substrate, is suggested.
Comparing the arrangement of the tripeptide conjugates TSC1-FYY (8), TSC1-FWY (9), and TSC1-FFY (7), the main differences can be found in how the peptide side-chains interact with the tyrosinase surface. For the TSC1-FYY (8) and TSC1-FWY (9) inhibitors, the highest number of intermolecular hydrogen bonds has been found. The position of TSC1-FYY (8) in the enzymatic active site is stabilised by four intermolecular hydrogen bonds involving hydroxyl groups of Tyr2 and Tyr3 sidechains. The alignment of TSC1-FWY (9) is stabilised by three intermolecular hydrogen bonds, involving indole amide proton of Trp2 and hydroxyl group of Tyr3 of the inhibitor. The orientation of the TSC1-FFY (7) in the tyrosinase active site is stabilised only by two intermolecular hydrogen bonds, involving carbonyl oxygen (in the backbone) from Phe2 and Tyr3. Detailed information on the formed hydrogen bonds is presented in . Moreover, the alignments of all inhibitors are stabilised by π-π interactions (TSC1-FYY (8): π-π stacking Phe1-His263; π-π edge-to-face Ph(TSC1)-Phe264; Tyr2-His244; TSC1-FFY (7): π-π edge-to-face Ph(TSC1)-Phe264). Additionally, in the case of TSC1-FYY (8) and TSC1-FWY (9) some stabilising interactions of the sulphur atom of the thiourea group with the π-electrons of the Trp227 indole ring can also be observed. Obtained orientations of the investigated inhibitors in the active site are shown in (top). In contrast, in the case of constrained docking, the alignment of each inhibitor is stabilised by the π-electron-sulphur atom interaction. This result seems to be expected due to the fact that such an orientation of the inhibitors towards the active cavity is forced.
Figure 5. Orientation of the thiosemicarbazone TSC 1 tripeptide conjugates in the tyrosinase active site. Top: free docking; bottom: constrained docking. Intermolecular interactions are shown as dash lines – green: H-bond; purple: π-π interaction. Copper ions are shown as red spheres.

Table 3. List of the residues of inhibitors 7–9 and tyrosinase, involved in the intermolecular hydrogen bonds formed during the free and constrained docking.
The sulphur atom of thiourea moiety interacts mainly with the aromatic ring of His61, Phe90, and His296, regardless of the peptide sequence of the conjugates. As in the case of free docking and constraint docking, differences in the strength of the interaction of inhibitors with the enzyme can be explained on the basis of the number of intermolecular hydrogen bonds. Similarly, the arrangement of TSC1-FYY (8) is stabilised by the highest number of hydrogen bonds (). Moreover, in the case of TSC1-FWY (9) the observed interaction formed by Trp2 involves the tryptophane backbone instead of the amino acid side chain. The orientations obtained for the investigated inhibitors in the active site are shown in (bottom).
The presented results allowed us to conclude that introducing an amino acid in the second position without the donor hydrogen bond in the side chain causes a drastic decrease in the ability to form intermolecular hydrogen bonds. This, in turn, significantly limits the inhibitory properties, which is reflected in the values of the scoring function. The inhibitory properties of the tripeptide conjugates containing tyrosine or tryptophan in the second position are comparable. On the other hand, inhibitors containing a phenylalanine residue at this position have a much lower affinity for the tyrosinase active cavity.
Conclusions
Conjugation of peptides with small organic molecules has been recently widely applied, mainly for their application as carriers or stabilising agents.Citation25–27 An interesting example is represented by the use of various peptides and their conjugates as cosmeceutical active ingredients, described in recent reviewsCitation15,.Citation16 In this work, we report the synthesis of nine conjugates of three thiosemicarbazones with designed tripeptides, based on the Phe-Xaa-Tyr motif, where Xaa = Phe, Trp, or Tyr. Our aim was to evaluate the tyrosinase inhibitory potential of such conjugates, following the encouraging results published in the past years when we demonstrated the strong inhibitory properties of various thiosemicarbazones. Furthermore, several examples of peptide conjugates, e.g. with a commercially available inhibitor of tyrosinase, such as kojic acid, resulted in increased inhibitory activity and/or enhanced stability, or decreased cytotoxicity of the final product.
New thiosemicarbazone structures (TSCs) were selected considering our previous results, demonstrating that the thiourea group is responsible for copper coordination in the tyrosinase active cleft. Considering these data, we designed three new thiosemicarbazones with a free carboxyl group, located distantly from the active thiourea moiety. Therefore, three new TSCs possessing free carboxyl moieties necessary for the conjugation were synthesised. Tripeptide sequences were selected on the base of previously published data, suggesting the relevance of N-terminally located Phe residue and the fundamental role of aromatic and hydrophobic residues for tyrosinase inhibition. TSCs were conjugated to the tripeptide chains in the solid phase via standard coupling strategy, by N-terminal modification via amide bond formation. Final conjugates were cleaved from the resin, purified, and characterised. Pure products were then evaluated as inhibitors of tyrosinase, demonstrating a good inhibitory activity of two structurally similar thiosemicarbazones TSC 1 and 3, and poor activity of the bulky TSC 2 (IC50 5.3 and 5.8 μM versus > 150 μM, respectively). At variance, the unmodified peptides 4–6 did not display significant inhibition, in accordance with the results from the literature (e.g. IC50 of Ac-FFY-OH found in the literature: 240 μM).Citation23 Finally, we tested the conjugates, observing that the tripeptide conjugates containing TSC 2 showed weak inhibitory activity, the set of TSC 1-derivatives 7–9 contains the most active compounds 8 and 9 (IC50 value: 6.5 μM for both conjugates), while the conjugates of TSC 3, compounds 14 and 15, feature similar intermediate activity (IC50 around 25 μM). Surprisingly, we found that the derivatives including FFY sequence (compounds 7 and 13) are significantly weaker inhibitors of tyrosinase regarding FWY and FYY conjugates (IC50 >150 μM). Molecular modelling supports the explanation of the influence of the amino acid sequence on the activity of conjugates. In silico results showed that the reason for significant differences in the activity of Phe-containing derivatives should be sought in the lack of a hydrogen bond donor group, caused by introducing a phenylalanine residue in the central position of the peptide.
Tests performed on B16F0 cell line show that all investigated compounds inhibited melanogenesis at a micromolar level. However, only TSC 1 and 2 as well as the tripeptide conjugates 12 and 15 showed stronger potency of melanogenesis inhibition than kojic acid, so they can be considered as medium melanogenesis inhibitors. It should be noted that bulky thiosemicarbazone TSC 2, unexpectedly, revealed the highest inhibitory potency towards melanogenesis showing relatively low toxicity towards the cells. TSC 2 showed no significant inhibition towards tyrosinase, so it probably inhibits melanogenesis at a different stage of the process.
For most compounds, IC50 of cell proliferation inhibition turned out to be lower than IC50 of melanin production. This observation lets raise the question if the inhibited melanin production is the result of inhibitor activity towards melanogenesis process, or of toxic effect on the cells. Further investigations to explain the mechanism of inhibition of tyrosinase in normal cells, and the role of peptide sequence in this mechanism should be performed in further studies.
According to the results of molecular modelling, in the case of inhibitors 1–3, their interaction with the active cavity of the enzyme through free carboxyl groups should be taken into account. This interaction seems so significant that it may force an orientation opposite to that postulated for this class of compounds.Citation48 Our previous studies indicated that introducing an additional methyl group at the R position significantly improved inhibitory activity. This could be explained by a better fit for the active cave and, thus, an increase in van der Waals interactions, which would only occur thanks to the interaction via the thiourea moiety. On the other hand, similar tyrosinase inhibiting abilities of thiosemicarbazones 1 and 3 presented in this work indicate that the presence of this additional methyl group does not have such a significant effect on the activity, which would suggest their different interaction with the enzyme. Moreover, the differences in the number of interactions between TSC 1 and derivatives TSC1-FYY (8) and TSC1-FWY (9) are significant. While TSC 1 interacts with tyrosinase through π-π stacking interaction provided by the TSC 1 phenyl moiety, peptide derivatives 8 and 9 form numerous hydrogen bonds between amino acid residues and the tyrosinase. Peptide conjugates 8 and 9 maintain the in vitro inhibitory properties of TSC 1 alone, but it should be noticed that the interactions observed in silico of the peptide chain and tyrosinase active cleft significantly stabilise the complex.
The above-mentioned results might be helpful in designing and developing novel skin-whitening products based on tyrosinase inhibitors.
Supplemental Material
Download PDF (2.1 MB)Acknowledgements
The authors declare the support from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Science and Technology, from the University of Florence, and from Polish National Agency for Academic Exchange (the Iwanowska Programme Scholarship for KH, agreement no. PPN/IWA/2019/1/00116/U/00001).
PL is the participant of “BioTechNan” project – Interdisciplinary Environmental Doctoral Studies KNOW in the field of Biotechnology and Nanotechnology, co-financed by the European Union. The PhD of PL is performed in the context of a doctorate between the PhD Schools in Drug Research and Innovative Treatments of the University of Florence (XXXV Ciclo) and in Chemical Sciences of the Wroclaw University of Science and Technology (Supervisors: PR and RL).
We kindly acknowledge the Erasmus + Programme for Traineeship at the Faculty of Chemistry of Wroclaw University of Science and Technology (September-December 2022) for MSc thesis of GC, co-tutored by RL and PR.
The molecular modelling was carried out using hardware and software resources of The Supercomputing and Networking Centre in Wroclaw (grant no. 197).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Grand View Research. 2022. https://www.grandviewresearch.com/industry-analysis/skin-lightening-products-market
- d‘Ischia M, Wakamatsu K, Cicoira F, Di Mauro E, Garcia-Borron JC, Commo S, Galván I, Ghanem G, Kenzo K, Meredith P. Melanins and melanogenesis: from pigment cells to human health and technological applications. Pigment Cell Melanoma Res. 2015;28(5):520–544.
- Ramsden CA, Riley PA. Tyrosinase: the four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg Med Chem. 2014;22(8):2388–2395.
- Ismaya WT, Rozeboom HJ, Schurink M, Boeriu CG, Wichers H, Dijkstra BW. Crystallization and preliminary X-ray crystallographic analysis of tyrosinase from the mushroom Agaricus bisporus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67(Pt 5):575–578.
- Parvez S, Kang M, Chung H-S, Bae H. Naturally occurring tyrosinase inhibitors: mechanism and applications in skin health, cosmetics and agriculture industries. Phytother Res. 2007;21(9):805–816.
- Hridya H, Amrita A, Sankari M, George Priya Doss C, Gopalakrishnan M, Gopalakrishnan C, Siva R. Inhibitory effect of brazilein on tyrosinase and melanin synthesis: Kinetics and in silico approach. Int J Biol Macromol. 2015;81:228–234.
- Peng Z, Wang G, Zeng Q-H, Li Y, Liu H, Wang JJ, Zhao Y. A systematic review of synthetic tyrosinase inhibitors and their structure-activity relationship. Crit Rev Food Sci Nutr. 2022;62(15):4053–4094.
- Panzella L, Napolitano A. Natural and Bioinspired Phenolic Compounds as Tyrosinase Inhibitors for the Treatment of Skin Hyperpigmentation: Recent Advances. Cosmetics. 2019;6(4):57.
- Pillaiyar T, Manickam M, Namasivayam V. Skin whitening agents: medicinal chemistry perspective of tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2017;32(1):403–425.
- Pillaiyar T, Namasivayam V, Manickam M, Jung S-H. Inhibitors of Melanogenesis: An Updated Review. J Med Chem. 2018;61(17):7395–7418.
- Lee SY, Baek N, Nam T. Natural, semisynthetic and synthetic tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2016;31(1):1–13.
- Phasha V, Senabe J, Ndzotoyi P, Okole B, Fouche G, Chuturgoon A. Review on the Use of Kojic Acid—A Skin-Lightening Ingredient. Cosmetics. 2022;9(3):64.
- Burnett CL, Bergfeld WF, Belsito DV, Hill RA, Klaassen CD, Liebler DC, Marks JG, Shank RC, Slaga TJ, Snyder PW, et al. Final report of the safety assessment of kojic acid as used in cosmetics. Int J Toxicol. 2010;29(6 Suppl):244S–2473.
- Andersen FA, Bergfeld WF, Belsito DV, Hill RA, Klaassen CD, Liebler DC, Marks JG, Shank RC, Slaga TJ, Snyder PW. Final amended safety assessment of hydroquinone as used in cosmetics. Int J Toxicol. 2010;29(6 Suppl):274S–2787.
- Errante F, Ledwoń P, Latajka R, Rovero P, Papini AM. 2020; Cosmeceutical peptides in the framework of sustainable wellness economy. Front Chem. 8:572923.
- Ledwoń P, Errante F, Papini AM, Rovero P, Latajka R. Peptides as active ingredients: a challenge for cosmeceutical industry. Chem. Biodiversity. 2021;18(2):cbdv.202000833.
- Lupo MP, Cole AL. Cosmeceutical peptides: cosmeceutical peptides. Dermatol Ther. 2007;20(5):343–349.
- Otvos L, Wade JD. Current challenges in peptide-based drug discovery. Front Chem. 2014;2:62.
- Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20(1):122–128.
- Ledwoń P, Papini AM, Rovero P, Latajka R. Peptides and peptidomimetics as inhibitors of enzymes involved in fibrillar collagen degradation. Materials. 2021;14(12):3217.
- Hariri R, Saeedi M, Akbarzadeh T. Naturally occurring and synthetic peptides: efficient tyrosinase inhibitors. J Pep Sci. 2021;27(7):e3329.
- Song Y, Chen S, Li L, Zeng Y, Hu X. The hypopigmentation mechanism of tyrosinase inhibitory peptides derived from food proteins: an overview. Molecules. 2022;27(9):2710.
- Hsiao N-W, Tseng T-S, Lee Y-C, Chen W-C, Lin H-H, Chen Y-R, Wang Y-T, Hsu H-J, Tsai K-C. Serendipitous discovery of short peptides from natural products as tyrosinase inhibitors. J Chem Inf Model. 2014;54(11):3099–3111.
- Mohan T, Kleinschek KS, Kargl R. Polysaccharide peptide conjugates: chemistry, properties and applications. Carbohydr Polym. 2022;280:118875.
- Xu L, Xu S, Xiang T, Liu H, Chen L, Jiang B, Yao J, Zhu H, Hu R, Chen Z. Multifunctional building elements for the construction of peptide drug conjugates. Engineered Regeneration. 2022;3(1):92–109.
- Balogh B, Ivánczi M, Nizami B, Beke-Somfai T, Mándity IM. ConjuPepDB: a database of peptide–drug conjugates. Nucleic Acids Res. 2021;49(D1):D1102–D1112.
- CONJUPEPDB. A database of drug-peptide conjugates. 2022. https://conjupepdb.ttk.hu/
- He R, Finan B, Mayer JP, DiMarchi RD. Peptide conjugates with small molecules designed to enhance efficacy and safety. Molecules. 2019;24(10):1855.
- Fu C, Yu L, Miao Y, Liu X, Yu Z, Wei M. Peptide–drug conjugates (PDCs): a novel trend of research and development on targeted therapy, hype or hope? Acta Pharmaceutica Sinica B. 2022;13(2):498–516.
- Kim H, Choi J, Cho JK, Kim SY, Lee Y-S. Solid-phase synthesis of kojic acid-tripeptides and their tyrosinase inhibitory activity, storage stability, and toxicity. Bioorg Med Chem Lett. 2004;14(11):2843–2846.
- Noh J-M, Kwak S-Y, Kim D-H, Lee Y-S. Kojic acid–tripeptide amide as a new tyrosinase inhibitor. Biopolymers. 2007;88(2):300–307.
- Upadhyay A, Chompoo J, Taira N, Fukuta M, Gima S, Tawata S. Solid-phase synthesis of mimosine tetrapeptides and their inhibitory activities on neuraminidase and tyrosinase. J Agric Food Chem. 2011;59(24):12858–12863.
- Yang J-K, Lee E, Hwang I-J, Yim D, Han J, Lee Y-S, Kim J-H. β-lactoglobulin peptide fragments conjugated with caffeic acid displaying dual activities for tyrosinase inhibition and antioxidant effect. Bioconjug Chem. 2018;29(4):1000–1005.
- Ebenezer O, Singh-Pillay A, Koorbanally NA, Singh P. Antibacterial evaluation and molecular docking studies of pyrazole–thiosemicarbazones and their pyrazole–thiazolidinone conjugates. Mol Divers. 2021;25(1):191–204.
- Hassan M, Ghaffari R, Sardari S, Farahani Y, Mohebbi S. Discovery of novel isatin-based thiosemicarbazones: synthesis, antibacterial, antifungal, and antimycobacterial screening. Res Pharm Sci. 2020;15(3):281–290.
- Sevinçli ZŞ, Duran GN, Özbil M, Karalı N. Synthesis, molecular modeling and antiviral activity of novel 5-fluoro-1H-indole-2,3-dione 3-thiosemicarbazones. Bioorg Chem. 2020;104:104202.
- Gu X, Li X, Guan M, Jiang C, Song Q, Sun N, Zou Y, Zhou Q, Chen J, Qiu J. Discovery of thiosemicarbazone-containing compounds with potent anti-proliferation activity against drug-resistant K562/A02 cells. Bioorg Med Chem Lett. 2020;30(24):127638.
- Buitrago E, Vuillamy A, Boumendjel A, Yi W, Gellon G, Hardré R, Philouze C, Serratrice G, Jamet H, Réglier M, et al. Exploring the Interaction of N/S Compounds with a Dicopper Center: Tyrosinase Inhibition and Model Studies. Inorg Chem. 2014;53(24):12848–12858.
- Xu J, Liu J, Zhu X, Yu Y, Cao S. Novel inhibitors of tyrosinase produced by the 4-substitution of TCT. Food Chem. 2017;221:1530–1538.
- Song S, You A, Chen Z, Zhu G, Wen H, Song H, Yi W. Study on the design, synthesis and structure-activity relationships of new thiosemicarbazone compounds as tyrosinase inhibitors. Eur J Med Chem. 2017;139:815–825.
- Xie J, Dong H, Yu Y, Cao S. Inhibitory effect of synthetic aromatic heterocycle thiosemicarbazone derivatives on mushroom tyrosinase: Insights from fluorescence, 1 H NMR titration and molecular docking studies. Food Chem. 2016;190:709–716.
- Dong H, Liu J, Liu X, Yu Y, Cao S. Molecular docking and QSAR analyses of aromatic heterocycle thiosemicarbazone analogues for finding novel tyrosinase inhibitors. Bioorg Chem. 2017;75:106–117.
- Dong H, Liu J, Liu X, Yu Y, Cao S. Combining molecular docking and QSAR studies for modeling the anti-tyrosinase activity of aromatic heterocycle thiosemicarbazone analogues. J Mol Struct. 2018;1151:353–365.
- Liu J, Li M, Yu Y, Cao S. Novel inhibitors of tyrosinase produced by the 4-substitution of TCT (П). Int J Biol Macromol. 2017;103:1096–1106.
- Zhu T-H, Cao S-W, Yu Y-Y. Synthesis, characterization and biological evaluation of paeonol thiosemicarbazone analogues as mushroom tyrosinase inhibitors. Int J Biol Macromol. 2013;62:589–595.
- Hałdys K, Goldeman W, Anger-Góra N, Rossowska J, Latajka R. Monosubstituted Acetophenone Thiosemicarbazones as Potent Inhibitors of Tyrosinase: Synthesis, Inhibitory Studies, and Molecular Docking. Pharmaceuticals. 2021;14(1):74.
- Hałdys K, Goldeman W, Jewgiński M, Wolińska E, Anger-Góra N, Rossowska J, Latajka R. Halogenated aromatic thiosemicarbazones as potent inhibitors of tyrosinase and melanogenesis. Bioorg Chem. 2020;94:103419.
- Hałdys K, Goldeman W, Jewgiński M, Wolińska E, Anger N, Rossowska J, Latajka R. Inhibitory properties of aromatic thiosemicarbazones on mushroom tyrosinase: Synthesis, kinetic studies, molecular docking and effectiveness in melanogenesis inhibition. Bioorg Chem. 2018;81:577–586.
- Merrifield RB. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963;85(14):2149–2154.
- Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972;37(22):3404–3409.
- Bellei B, Pitisci A, Izzo E, Picardo M. Inhibition of melanogenesis by the pyridinyl imidazole class of compounds: possible involvement of the Wnt/β-catenin signaling pathway, Slominski AT, editor. PLoS One. 2012;7(3):e33021.
- Gaussian 16, Revision C.01. 2016.
- Ismaya WT, Rozeboom HJ, Weijn A, Mes JJ, Fusetti F, Wichers HJ, Dijkstra BW. Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry. 2011;50(24):5477–5486.
- Anandakrishnan R, Aguilar B, Onufriev AV. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012;40(Web Server issue):W537–W541.
- Onufriev A, Anandakrishnan R, Aguilar B. H++, VirginiaTech [Internet]. 2023. http://newbiophysics.cs.vt.edu/H++/
- Venkatachalam TK, Pierens GK, Reutens DC. Synthesis, NMR structural characterization and molecular modeling of substituted thiosemicarbazones and semicarbazones using DFT calculations to prove the syn/anti isomer formation: Synthesis and molecular modeling of thiosemicarbazones and semicarbazones. Magn Reson Chem. 2014;52(3):98–105.
- Fatondji HR, Kpoviessi S, Gbaguidi F, Bero J, Hannaert V, Quetin-Leclercq J, Poupaert J, Moudachirou M, Accrombessi GC. Structure–activity relationship study of thiosemicarbazones on an African trypanosome: Trypanosoma brucei brucei. Med Chem Res. 2013;22(5):2151–2162.
- Thanigaimalai P, Lee K-C, Sharma VK, Roh E, Kim Y, Jung S-H. Ketonethiosemicarbazones: structure-activity relationships for their melanogenesis inhibition. Bioorg Med Chem Lett. 2011;21(12):3527–3530.
- Mann T, Gerwat W, Batzer J, Eggers K, Scherner C, Wenck H, Stäb F, Hearing VJ, Röhm K-H, Kolbe L. Inhibition of human tyrosinase requires molecular motifs distinctively different from mushroom tyrosinase. J Invest Dermatol. 2018;138(7):1601–1608.
- Oyama T, Yoshimori A, Ogawa H, Shirai Y, Abe H, Kamiya T, Tanuma S. The structural differences between mushroom and human tyrosinase cleared by investigating the inhibitory activities of stilbenes. J Mol Struct. 2023;1272:134180.
- GraphPad Prism version 8.0.0 for Windows, GraphPad Software. www.graphpad.com
- Jackman MP, Hajnal A, Lerch K. Albino mutants of Streptomyces glaucescens tyrosinase. Biochem J. 1991;274(3):707–713.
- Chang T-S. Natural melanogenesis inhibitors acting through the down-regulation of tyrosinase activity. Materials. 2012;5(9):1661–1685.
- Lehraiki A, Abbe P, Cerezo M, Rouaud F, Regazzetti C, Chignon-Sicard B, Passeron T, Bertolotto C, Ballotti R, Rocchi S. Inhibition of melanogenesis by the antidiabetic metformin. J Invest Dermatol. 2014;134(10):2589–2597.
- Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. J Mol Biol. 1997;267(3):727–748.