
Abstract
β-Elemene is the major active ingredient of TCM anticancer drug elemene extracts. To further improve its antitumor activity and poor solubility, a polar HDACi pharmacophore was incorporated its scaffold. Systematic SAR studies led to the discovery of compounds 27f and 39f, which exhibited potent inhibitory activity against HDACs (HDAC1: IC50 = 22 and 9 nM; HDAC6: 8 and 14 nM, respectively). In cellular levels, 27f and 39f significantly inhibited cell proliferation of five tumour cell lines (IC50: 0.79 - 4.42 µM). Preliminary mechanistic studies indicated that 27f and 39f efficiently induced cell apoptosis. Unexpectedly, compound 39f could also stimulate cell cycle arrest in G1 phase. Further in vivo study in WSU-DLCL-2 xenografted mouse model validated the antitumor activities of 27f, without significant toxicity. The results suggest the therapeutic potential of these HDACs inhibitors in lymphoma and provide valuable insight and understanding for further structural optimisation around β-elemene scaffold.
GRAPHICAL ABSTRACT
Introduction
β-Elemene (, 1) is a sesquiterpene extracted from the rhizome of Curcuma wenyujinCitation1,Citation2. It has been used in the treatment of lung cancer, pancreatic cancer, gastric cancer, breast cancer, bladder cancer, and malignant brain gliomaCitation3–8. Studies have shown that β-elemene inhibits tumour cell growth via diverse mechanismsCitation9,Citation10, including induction of apoptosis, autophagy and cell cycle arrest, and inhibition of cell proliferation and migration, etc. However, its clinical application has been limited by poor solubility, low bioavailability, and moderate antitumor activityCitation11–14. To overcome these shortcomings, the combination therapy of β-elemene with other anticancer drugs, such as cisplatinCitation15, paclitaxelCitation16, and gefitinibCitation17 have been extensively studied. On the other hand, the structural modifications based on β-elemene scaffold such as the hydroxyl carboximates derivativesCitation18, vinylated derivativesCitation19, NO-donating derivativesCitation20 and macrocycles derivatives Citation21 have been a major trend for identifying new chemical entities with better anticancer effects and potential better druggability than β-elemene. The above mentioned cancer treatment paradigms have achieved great success and broad prospects in the treatment of solid tumours, but has poorly reported for the treatment of lymphoma.
Figure 1. The structures of β-elemene (1) and approved HDAC inhibitors (2–6).
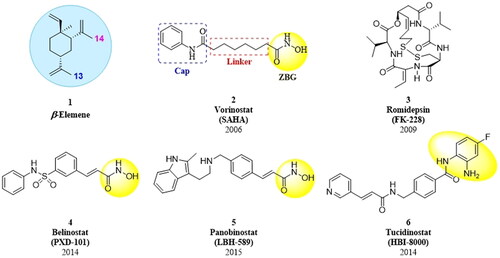
Histone deacetylases (HDACs) are a family of enzymes that catalyse the deacetylation of lysine residues of both histone and non-histone protein substratesCitation22,Citation23. The expression levels of HDACs are usually higher in tumour cells than in normal cellsCitation24. Inhibition of HDACs can lead to pleiotropic effects on cellular signalling and transcription, including cell death, differentiation, autophagy, and enhancement of immunogenicityCitation25–30. To date, five drugs are approved for the treatment of various types of haematologic malignancies: vorinostat (SAHA, 2), romidepsin (FK-228, 3), belinostat (PXD-101, 4), panobinostat (LBH-589, 5), and tucidinostat (HBI-8000, 6), as shown in . These HDAC inhibitors demonstrated excellent antitumor effect, but generally limited by a narrow antitumor spectrum, drug resistance, and undesirable side effectsCitation31–35. To address these issues, HDAC inhibitors are clinically used in combination with other antitumor agents to achieve synergistic or additive effects and reduce the potential for developing drug resistanceCitation36–38. Although drug combinations have the advantages of higher antitumor efficacy, they also suffer from enhanced adverse effects, unpredictable pharmacokinetic profiles, drug-drug interactions, and poor patient complianceCitation36. In recent years, the designed and synthesised hybrid drugs, comprising the HDAC inhibitor (HDACi) pharmacophores, are emerging as an alternative strategy to drug combinationsCitation24,Citation39. Scientific studies have shown that many hybrid agents had more potent anti-proliferation effects than the corresponding single agentCitation40–45, which encouraged and promoted us to develop the hybrid molecules of β-elemene and HDAC inhibitor.
Based on the above information and our experience in the structural modification of β-elemene, we have designed and synthesised novel HDAC inhibitors derived from the β-elemene scaffold. Herein, we reported the design, synthesis and biological evaluation of the novel HDACi derived from the β-elemene scaffold.
Results and discussion
Design
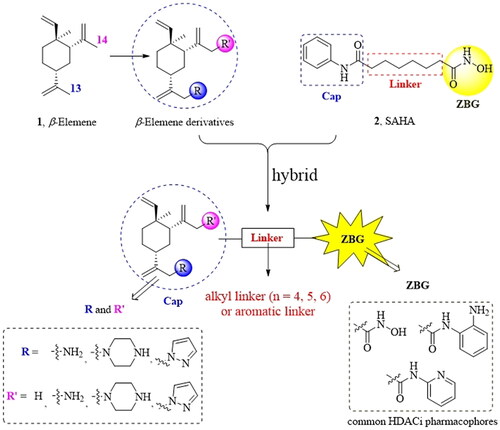
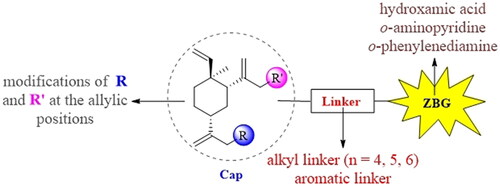
Generally, the pharmacophores of HDACi are divided into three key components: the cap group (acts as a surface binding, Cap), the linker, and the pharmacophore (zinc-binding group, ZBG) (2, ). The cap portion, which accounts for the affinity gain through hydrophobic interaction with protein, can be large or small in size and can tolerate diverse structures. We envisioned that the lipophilic antitumor β-elemene scaffold should be well tolerated as a cap portion and might potentially provide some beneficial effects to achieve better anticancer activity through “hybrid” drug concept (). The linker is typically linear and hydrophobic as well (e.g. straight carbon chain, trans- di-substituted olefin or di-substituted phenyl or heteroaryl are often used as the linker). In this study, the different carbon atoms “alkyl linker” and “aromatic linker” was used as the “Linker” to connect the β-elemene derivatives and the “ZBG” portion. The “ZBG” group (also known as pharmacophore or warhead) of many HDACi is hydroxamic acid, and those of few HDACi can also be acyl aniline, cyclic tetrapeptides, thiol, and aliphatic carboxylic acids, or their isosteric replacements. In this work, we examined three common pharmacophores as “ZBG”: hydroxamic acid, acyl aniline and acyl pyridine. As a result, six series of HDACi derived from β-elemene scaffold were designed and synthesised, and their biological activities were evaluated.
Figure 2. The designing strategy of HDACi derived from β-elemene scaffold.
Chemistry
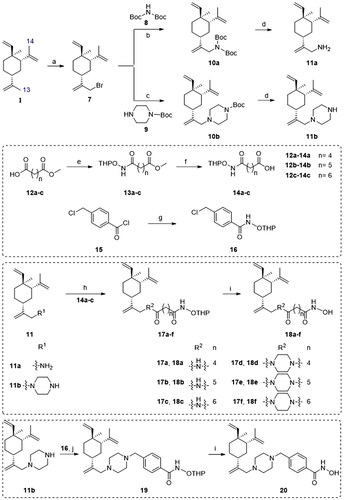
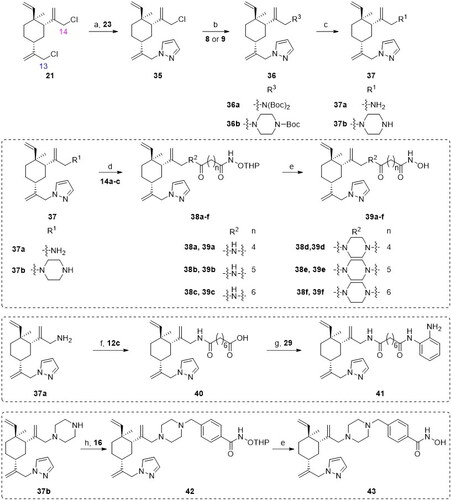
The synthesis of target compounds 18a–f and 20 is illustrated in Scheme 1. Bromination between the starting material β-elemene (1) and N-bromosuccinimide (NBS) yielded 13-Br-β-elemene (7). The reaction of 7 with different amines gave N-Boc protected intermediates 10a–b. The Boc protecting group was subsequently removed to yield amines 11a–b. Separately, the condensation between various carboxylic acids 12a–c and O-(Tetrahydro-2H-pyran-2-yl)hydroxylamine (NH2OTHP) afforded THP-protected intermediates 13a–c, which underwent ester hydrolysis to give corresponding acids 14a–c. Then, the amide coupling reaction between amines 11a–b and carboxylic acids 14a–c gave intermediates 17a–f, which underwent subsequent THP deprotection to afford the desired compounds 18a–f. On the other hand, intermediate 16 was easily obtained from commercially available acyl chloride 15 and NH2OTHP. The SN2 displacement reaction between 11b and 16 was facilitated in the presence of DIPEA under heating to afford intermediate 19, which underwent THP deprotection to give the target compound 20.
Scheme 1. Synthetic routes of the title compounds 18a–f and 20. Reaction conditions and reagents: (a) NBS, AcOH, CH2Cl2, 0 °C to rt, 9 h; (b) Cs2CO3, DMF, Bis(tert-butoxycarbonyl)amine, 60 °C, 10 h; (c) DIPEA, DMF, 1-Boc-piperazine, 60 °C, 10 h; (d) CF3CO2H, 0 °C ∼ rt, 5 h; (e) EDCI, HOBT, DIPEA, DMF, NH2OTHP, rt, 5 h; (f) NaOH (aq), CH3OH, rt, 2 h; (g) CH2Cl2, NH2OTHP, 0 °C, 3 h; (h) EDCI, HOBT, DIPEA, DMF, rt, 5 h; (i) TsOH·H2O, CH3OH, rt, 8 h; (j) DIPEA, DMF, 16, 60 °C, 6 h.
The preparation of analogues 27a–f, 31–32 and 34 is outlined in Scheme 2. First, β-elemene (1) was treated with NaClO under the mixed solvent of AcOH and CH2Cl2 to furnish 13,14-dichloro-β-elemene (21). It was discovered by us that the two allylic chloro groups had differentiated reactivity in the presence of a nucleophile. This is presumably due to the stereo effects in the chair-like conformation of cyclohexane ring, in which slightly more bulky around 14-position in 21 leads to less reactivity than 13-positionCitation46. By carefully controlling the reaction conditions, two different R3 were installed at position 13- of 21 to yield intermediate 22. Subsequently, a pyrazole group was installed at position 14- to afford intermediate 24. The removal of Boc-deprotecting group afforded key intermediates 25a–b. The transformation of 25a–b to the target molecules 27a–f could then be achieved using the similar reaction sequences described in Scheme 1. For the target molecules bearing HDACi pharmacophores other than hydroximate (i.e. 31 and 32), slightly modified reaction sequence was adopted. Finally, compound 34 was prepared using the similar procedure for compound 20 described in Scheme 1.
Scheme 2. Synthetic routes of the title compounds 27a–f, 31–32 and 34. Reaction conditions and reagents: (a) NaClO, AcOH, CH2Cl2, TBAF, 0 °C, 6 h; (b) Cs2CO3, DMF, 8 or 9, 50 °C, 8 h; (c) Cs2CO3, DMF, 23, 60 °C, 10 h; (d) HCl-Dioxane (4 M), rt, 8 h; (e) EDCI, HOBT, DIPEA, DMF, 14a–c, rt, 5 h; (f) TsOH·H2O, CH3OH, rt, 8 h. (g) (i) EDCI, HOBT, DIPEA, DMF, 12c, rt, 5 h; (ii) NaOH (aq), CH3OH, rt, 2 h; (h) EDCI, HOBT, DIPEA, DMF, rt, 5 h; (i) DIPEA, DMF, 16, 60 °C, 6 h.
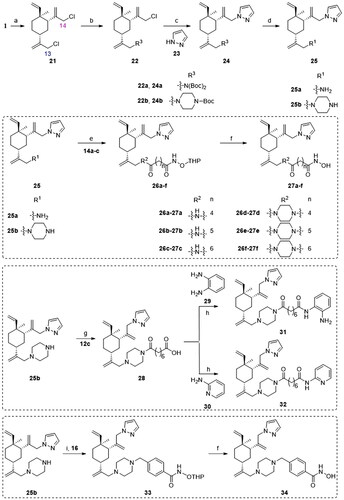
Analogues 39a–f, 41 and 43 were designed to examine the anticancer activity of different attaching positions in comparison with compounds 27a–f, 31–32, and 34. The synthetic route for preparing these compounds is depicted in Scheme 3. The reactions used are similar to those discussed in Scheme 2, except some of the sequences were reversed.
Scheme 3. Synthetic routes of the title compounds 39a–f, 41 and 43. Reaction conditions and reagents: (a) Cs2CO3, DMF, 23, 45 °C, 10 h; (b) Cs2CO3, DMF, 8 or 9, 80 °C, 8 h; (c) HCl-Dioxane (4 M), rt, 8 h; (d) EDCI, HOBT, DIPEA, DMF, 14a–c, rt, 5 h; (e) TsOH·H2O, CH3OH, rt, 8 h; (f) (i) EDCI, HOBT, DIPEA, DMF, 12c, rt, 5 h; (ii) NaOH (aq), CH3OH, rt, 2 h; (g) EDCI, HOBT, DIPEA, DMF, 29, rt, 5 h; (h) DIPEA, DMF, 16, 60 °C, 6 h.
Biological evaluation
In vitro HDAC inhibition assay
With several different series of target molecules in hand, assays to evaluate enzymatic inhibitions against HDAC were established. Among all HDAC subtypes, HDAC1 and HDAC6 represent Class I and II, respectively. Small molecules inhibiting both HDAC1 and HDAC6 are typically considered as pan-HDAC inhibitors. On the other hand, when the inhibitors have higher activity against HDAC6 than HDAC1, they are considered as HDAC6-selective inhibitors. Therefore, HDAC1 and HDAC6 were selected for the evaluation of the target molecules (). Most of the compounds showed moderate to excellent inhibitory activities against HDAC1 and HDAC6. For the compounds bearing alkyl linkers and hydroxamic acid as “ZBG” (i.e. 18a–f, 27a–f and 39a–21f), the length of alkyl linker had a great influence on the HDAC1 and HDAC6 inhibitory activities. For instance, four methylene linker analogs (18a, 18d, 27a, 27d, 39a and 39d) had poor HDAC1 and HDAC6 inhibitory activities. As the linker length increased, the activities of target molecules improved gradually (18c ≈ 18b > 18a, 18f > 18e > 18d, 27c > 27b > 27a, 27f > 27e > 27d, 39c > 39b > 39a and 39f > 39e > 39d). Interestedly, compounds with aromatic linkers (i.e. 20, 34 and 43) are potent HDAC6-selective inhibitors with 20–64-fold selectivity over HDAC1 (351 nM ≤ IC50 ≤ 1120 nM for HDAC1; 11 nM ≤ IC50 ≤ 55 nM for HDAC6). It’s known that HDAC6-selective inhibitors could have potential therapeutic benefits for lower toxicity and side effectsCitation23, which is worth further investigation in the future.
Table 1. The structures and corresponding IC50 values of the target compounds against HDAC1 and HDAC6. 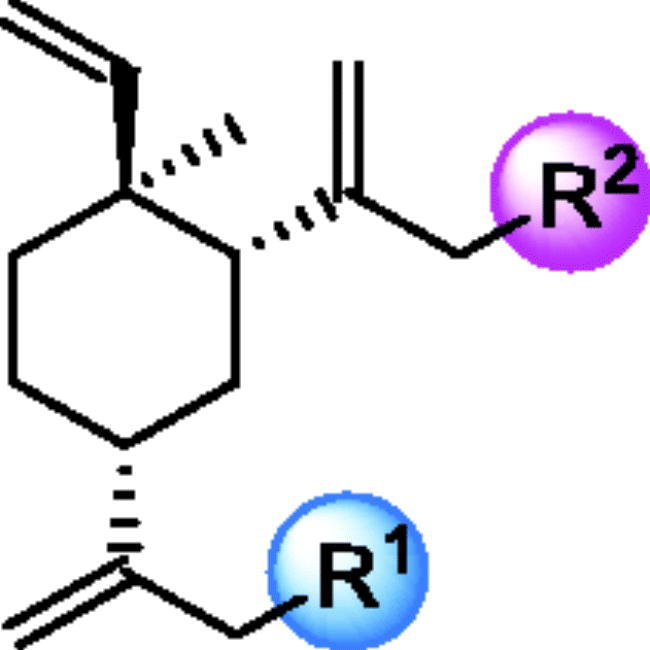
Subsequently, the influence of the different “Cap” groups on the inhibitory activities of HDAC was evaluated. When compounds had the identical “Linker” and “ZBG” group, the following phenomena were observed: (1) Compounds using 11a as the “Cap” group exhibited better inhibitory activities against HDAC1 and HDAC6 than those compounds using 11b as the “Cap” group (18a–c vs 18d–f). (2) Compounds using 25a as “Cap” group exhibited comparable inhibitory activities against HDAC1 and HDAC6 compared to those compounds using 25b as the “Cap” group. The above two series have one common point the pharmacophore and the linker were attached to the 13-position of β-elemene. On the other hand, when the pharmacophore and the linker attached to the 14-position of β-elemene, the structure-activity relationships (SARs) were varied from case to case. For instance, compound 39a using 37a as the “Cap” group showed 5-fold more potent against HDAC1 and HDAC6 than 39d. Both 39a and 39d had identical aliphatic linkers, i.e. four methylenes length. For other aliphatic linkers such as five or six methylenes length linkers, the trend was reversed, i.e. the activities for 39b–39c were slightly weaker than that for 39e–39f (i.e. 39b–39c, IC50 ≤ 136 nM for HDAC1/6; 39e–39f, IC50 ≤ 70 nM for HDAC1/6). Overall, the “Cap” group variation had minimal impact on the inhibitory activities of HDAC.
We further investigated the effects of the “ZBG” group on the inhibitory activities of HDAC. The result suggested that target compounds with hydroxamic acid as the “ZBG” group had better HDAC inhibitory potency than those with o-phenylenediamine or o-aminopyridine as the “ZBG” group. In fact, compounds 31, 32, and 41 all have micromole range IC50 values indicating weak inhibitory activity against HDAC1 and HDAC6.
After the completion of the evaluation for HDAC enzymatic activity of all compounds, SARs were developed and summarised in . To sum up, (1) The “Cap” group, bearing different R and R’ group, had no dramatic influence on HDAC inhibitory activities; (2) Proper linker length was significant for retaining HDAC inhibitory activities and the target compounds with linker length five or six methylene groups appeared to be optimal for obtaining potent inhibitory activities against HDAC; (3) The compounds bearing aromatic structures as a part of the linker showed up to 64-fold HDAC6-selectivity over HDAC1; (4) “Hydroxamic acid” appears to be the best “ZBG” group than others such as “o-aminopyridine” and the “o-phenylenediamine”.
Figure 3. SARs summary of histone deacetylase inhibitors derived from β-elemene scaffold.
In vitro anti-proliferative assay
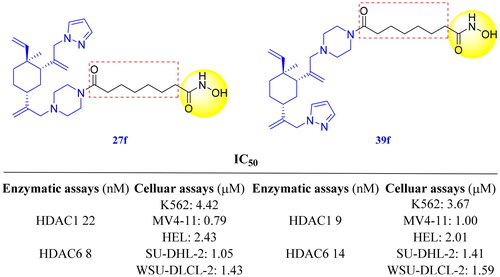
Compounds with good activities against HDAC1 (IC50 ≤ 53) and HDAC6 (IC50 ≤ 15 nM) were selected to perform anti-proliferative activities experiment against three different leukaemia cells (K562, MV4-11 and HEL cells) using the standard CCK8 test method (). The results suggested that compounds 18b, 27c, 27f and 39f were potent in inhibiting the proliferation of three types of leukaemia cells. Among all compounds tested against MV4-11 cell proliferation, 27f showed the best cytotoxicity with an IC50 value of 0.79 µM, which was over 250-fold more potent than β-elemene. The IC50 values for compounds 18b, 27c and 39f were all less than 1.65 µM. To further identify whether these four molecules had broad-spectrum antitumor activities, we determined their inhibitory activities against two other lymphoma cells: SU-DHL-2 and WSU-DLCL-2 (). Strikingly, the anti-proliferative activities of compounds 18b, 27c, 27f and 39f against SU-DHL-2 cell were < 1.55 µM, which was significantly superior to compounds 1 (IC50 > 200 µM) and 2 (IC50 = 1.84 µM). For WSU-DLCL-2 cells, these four compounds displayed over 88-fold more potent than their parent β-elemene. The above results suggested that (1) The anti-proliferative activities of the combination drug were stronger than that of β-elemene and SAHA alone, which was consistent with our expectation; (2) Compared with their parent β-elemene, our tested compounds showed more potent anti-proliferative activities against five cell lines; (3) The anti-proliferation activities of 27f were comparable with those of 39f and slightly stronger those of 18b and 27c in all tested tumour cells. Considering the anti-enzyme activities and tumour cell proliferation inhibitory activities, we finally chose compounds 27f and 39f as the representative compounds for follow-up research.
Table 2. In vitro anti-proliferative evaluation of target compounds against selected cell lines.
The solubility and solubility levels predictions of the selected compounds
Accelrys Discovery Studio (DS) software was used to assess the solubility level of target compounds (). The results suggested that compounds 18b, 27c, 27f and 39f were expected to have better aqueous solubility compared to the β-elemene group. Compounds 27f and 39f are slightly better than 18b and 27c.
Table 3. The solubility and solubility levels predictions of the selected compounds.
In vitro induced cell apoptosis
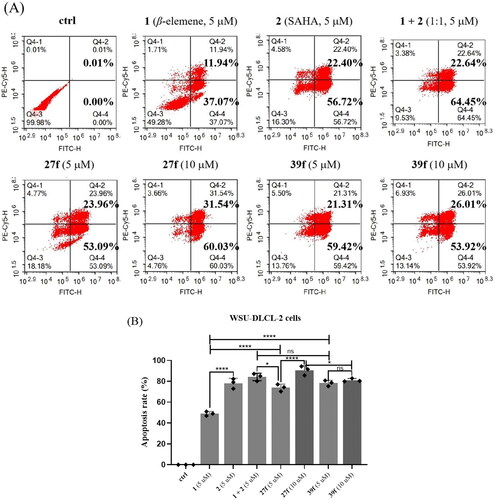
Based on inhibitory activities against HDAC1 and HDAC6, anti-proliferative activities against tumour cells, and calculated aqueous solubility level, compounds 27f and 39f were selected for further studies on their pro-apoptotic activity using flow cytometry (). Compounds 27f and 39f exhibited same magnitude of activity in the five cancer cell lines tested above. Since WSU-DLCL-2 is one of aggressive forms of diffuse large B-cell lympoma (DLBCL) with fewer effective treatment option, it was selected for flow cytometry. The results indicated that the ability of compounds 27f and 39f to induce WSU-DLCL-2 cells apoptosis at the concentration of 5 µΜ was comparable to SAHA, but was significantly stronger than β-elemene. Interestingly, SAHA possessed a certain level of synergistic pro-apoptotic effects when combined with β-elemene at the concentration of 5 µΜ (the percentage of cell apoptosis: 49.01%, 79.12%, and 87.09%, β-elemene, SAHA, and β-elemene + SAHA (1:1) respectively). Intriguingly, compounds 27f and 39f could significantly induced WSU-DLCL-2 cell apoptosis. Compound 27f, in particular, showed a much higher apoptosis rate at 10 uM than at 5 uM (91.57% vs 77.05%) in a dose-dependent manner. The above results indicated that compounds 27f and 39f could effectively induce WSU-DLCL-2 cell apoptosis, which are more potent than β-elemene.
Figure 4. Effects of compounds 27f and 39f on cell apoptosis. (A) WSU-DLCL-2 cells were treated with 5 μM and 10 μM of compounds 27f and 39f for 72 h. Cells treated with DMSO were used for comparison and cells treated with 1, 2 and 1 + 2 (1:1) were used for positive control. Data were represented as mean standard deviation from three independent experiments; (B) The histogram of compounds 1, 2, 1 + 2 (1:1), 27f and 39f on WSU-DLCL-2 cells apoptosis. Data are expressed as the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA test and Turkey’s comparison test, ns: not statistically.
In vitro arrested cell cycle
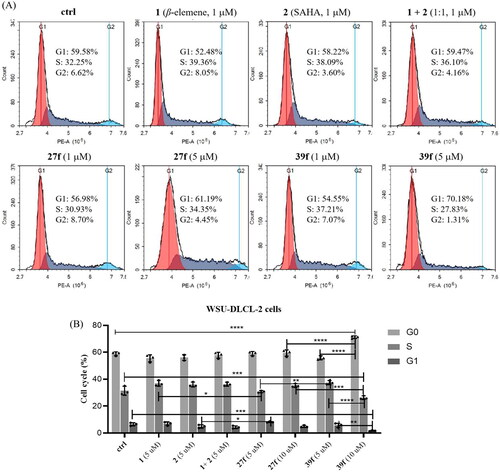
In addition to studying the effects of compounds 27f and 39f on cell apoptosis, we also explored their effects on cell cycle progression by flow cytometry. As shown in , DMSO, β-elemene, SAHA and β-elemene + SAHA were used as the blank and positive control, respectively. WSU-DLCL-2 cells were treated with compounds 27f and 39f at 1 µM and 5 µM for 72 h, respectively. The results revealed that β-elemene, SAHA, β-elemene + SAHA, 27f and 39f groups had hardly or slight effects on cell cycle at 1 µM. When the concentration of compound 27f was increased to 5 µM, the cell proportion of G1, G2 and S phase did not significantly change, compared with the control group and 27f group at 1 µM. However, the cell proportion of G1 phase increased dramatically (G1: 54.55% vs 70.18%) and the cell proportion of S phase and G2 phase decreased accordingly (S: 37.21% vs 27.83%; G2: 7.07% vs 1.31%), when the concentration of compound 39f was increased from 1 µM to 5 µM. The above results indicated that compound 39f could arrest the WSU-DLCL-2 cell cycle at the G1 phase in a concentration-dependent manner.
Figure 5. Effects of compounds 27f and 39f on cell cycle. (A) WSU-DLCL-2 cells were treated with 1 μM and 5 μM of compounds 27f and 39f for 72 h. Cells treated with DMSO were used for comparison and cells treated with 1, 2 and 1 + 2 (1:1) were used for positive control. Data were represented as mean standard deviation from three independent experiments; (B) The histogram of compounds 1, 2, 1 + 2 (1:1), 27f and 39f on WSU-DLCL-2 cells cycle. Data are expressed as the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA test and Turkey’s comparison test.
Molecular docking study
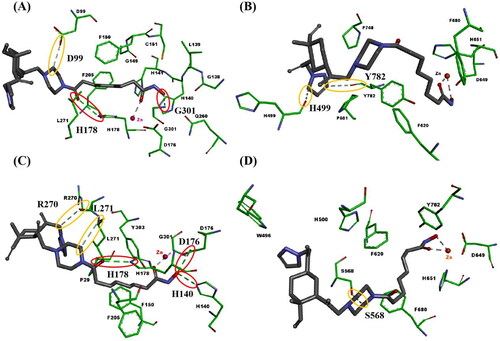
To better understand the binding modes of compounds 29f and 39f with HDAC1, and HDAC6, we performed molecular docking studies using the solved crystal structures of hHDAC1Citation47 (PDB ID 5ICN) and hHDAC6Citation48 (PDB ID 5EDU), as shown in . Compounds 27f and 39f bound with HDAC, mainly through formed metal-acceptor bonding interactions with zinc ions and hydrogen bonds interactions with amino acid residues on the surface of HDAC. In studying the molecular docking of compounds 27f and 37f with the active pocket of HDAC1, it was found that both 27f and 37f could form metal-acceptor bonding interactions with Zn2+. The difference is that 27f formed three hydrogen bonds with D99, H178 and G301, respectively (). In comparison, 39f formed five hydrogen bonds with R270, L271, H178, D176 and H140, respectively (). This may be the reason that compound 27f had a slightly weaker inhibitory activity against HDAC1 than 39f. A similar phenomenon was observed in the molecular docking analysis of 27f and 39f in the active pocket of HDAC6. The hydroxamic acid moiety of 27f and 39f were stretched into the catalytic pocket, forming a coordination contact with Zn2+. The difference is that 27f formed interaction with H499 and Y782 respectively (), while 39f formed interactions with S568 (). This may be the reason that compound 27f displayed more potent inhibitory activity against HDAC6 than 39f. The docking results proposed the favourable binding mode of our synthesised HDAC inhibitors 27f and 39f and explained their good inhibitory activities against HDAC1 and HDAC6.
Figure 6. Binding modes of compounds 27f or 39f with hHDAC1 (PDB ID 5ICN) and hHDAC6 (PDB ID 5EDU). (A) Binding model of compound 27f with hHDAC1; (B) Binding model of compound 27f with hHDAC6; (C) Binding model of compound 39f with hHDAC1; (D) Binding model of compound 39f with hHDAC6. Only surrounding amino acid residues are shown for clarity. The zinc ion is representative as red sphere, and hydrogen bonds are indicated with dashed lines. The conventional hydrogen bond is representative as red ellipse. The carbon hydrogen bond is representative as yellow ellipse. The carbons of compounds 27f and 39f are coloured in black. The oxygen atoms are coloured in red, and nitrogen atoms in blue.
Oral pharmacokinetics study
Due to its excellent HDAC inhibitory activities, potent anti-proliferative and pro-apoptosis capacities and good solubility level, compound 27f was progressed into an in vivo pharmacokinetic (PK) study. Compound 27f was administered intravenously (i.v.) at 5 mg/kg or orally (p.o.) at 10 mg/kg to female ICR mice. As shown in , 27f was rapidly cleared with the clearance (CL) of 65.1 ml/(h·kg) and the half-life (T1/2) of 0.361 h, after intravenous administration. The oral bioavailability of 27f was low (3.15%), presumably due to the rodent-specific PK profiles of HDACs pharmacophore (similar to SAHA). These data suggested that oral administration was not a suitable dosing route for compound 27f.
Table 4. In vivo pharmacokinetic properties of 27fTable Footnotea.
In vivo antitumor activity against WSU-DLCL2 xenografts
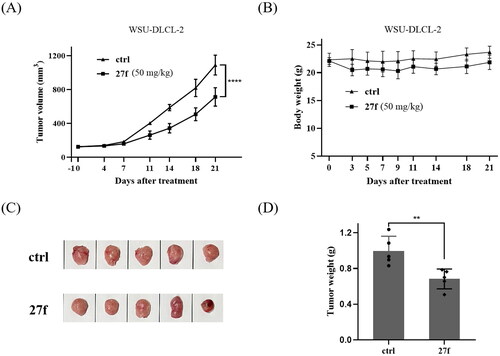
In a proof of concept study, 27f was intraperitoneally administrated to WSU-DLCL-2 xenografted mice model to preliminarily evaluate its in vivo antitumor potency. First, WSU-DLCL-2 cells (4 × 106) were implanted in the right flanks subcutaneously in female nude mice. When the implanted tumour reached a volume of 100 ∼ 150 mm3, the animals were randomly divided into groups of 2. In one of the groups, animals were treated with compound 27f intraperitoneally at 50 mg/kg once a day (QD) for 21 consecutive days. In another group, animals were treated with vehicles. As shown in , 27f showed a moderate in vivo antitumor potency with TGI of 31.5%. The tumour growth curve and the final tumour tissue size are presented in , respectively, which explicitly demonstrates the superior in vivo potency of compound 27f group.
Figure 7. Tumour growth inhibition of compound 27f in WSU-DLCL-2 xenografted mice model. (A) The efficacy of compound 27f in the WSU-DLCL-2 xenograft model. (B) Average body weights for 27f and vehicle-treated mice groups. (C) Photo of dissected WSU-DLCL-2 tumour tissues. (D) Tumour weight of dissected WSU-DLCL-2 tumour tissues. Single asterisks indicate p < 0.05, double asterisks indicate p < 0.01, and triple asterisks indicate p < 0.001 versus the control group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by two-way ANOVA test.
Conclusion and discussion
To improve antitumor activities of β-elemene and expand its scope of clinical application, we designed and synthesised a novel series of novel HDACi based on β-elemene structure in this study. Most of the prepared compounds exhibited potent inhibitory activities against HDACs and significant inhibitory effects on the proliferation of K562 and MV4-11. In particular, the representative compounds 27f and 39f demonstrated excellent in vitro anti-enzyme (IC50 values of 22 nM and 9 nM for HDAC1 and 8 nM and 14 nM for HDAC6, respectively) and broad spectrum in vitro anti-proliferative activities (IC50 values ranging from 0.79 to 4.42 µM against K562, MV4-11, HEL, SU-DHL-2 and WSU-DLCL-2 cell lines). Preliminary mechanistic studies revealed that compounds 27f and 39f could efficiently induce cell apoptosis. Unexpectedly, compound 39f could also stimulate cell cycle arrest in G1 phase. Further in vivo study in WSU-DLCL-2 xenograft mouse model validated the antitumor activities of 27f, without significant toxicity.
In summary, we have reported a series of novel compounds with potent HDAC inhibitory activity by installing HDACi pharmacophore onto β-elemene scaffold. Despite good progress has been obtained, more work is needed to better understand the SAR and to guide the future structural optimisation. For example, it could be interested to evaluate the compounds against other cancer cell lines including both solid tumour cells and haematologic cells. Further optimisation of lead 27f could also help to identify compounds with improved oral bioavailability. In addition, in-depth mechanism studies should be done with some of the most potent derivatives to elucidate the pathway and target proteins involved in antitumor effects. Nevertheless, our results reported herein should provide useful information for the future studies in this field.
Experimental section
Chemistry
General information
All of the chemical materials were purchased from commercial suppliers. The melting points of the compounds were determined using Büchi B-540 capillary melting point instrument. NMR spectra were recorded on a Bruker instrument at 400 MHz for 1H NMR and 100 MHz for 13C NMR, using CDCl3, CD3OD or DMSO-d6 as the deuterated solvent. Chemical shifts (δ) were reported in parts per million (ppm) relative to residual solvent as an internal reference. Low-resolution mass spectra were registered with Agilent 1260 InfinityII/6125. High-resolution mass spectra (HRMS) were measured on a Bruker MICR OTOF-Q II instrument.
Synthesis of the crucial intermediate 7
To a solution of β-elemene (1, 6.01 g, 29.46 mmol) in AcOH (20 ml) was added N-bromosuccinimide (NBS, 6.29 g, 35.35 mmol) at 0 °C. After addition, the mixture was stirred at room temperature for 9 h. The reaction was monitored by TLC. Upon completion, the mixture was then neutralised with saturated aqueous NaHCO3 solution and extracted with petroleum ether (PE, 3 × 100 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via flash column chromatography (PE as eluent) to give 13-Br-β-elemene 7 (2.51 g, yield 30.1%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 5.89–5.76 (m, 1H), 5.21 (s, 1H), 5.04 (t, J = 1.1 Hz, 1H), 4.97–4.81 (m, 3H), 4.59 (dt, J = 1.9, 0.9 Hz, 1H), 4.04 (d, J = 0.7 Hz, 2H), 2.33–2.17 (m, 1H), 2.06 (dd, J = 12.6, 3.5 Hz, 1H), 1.74–1.70 (m, 3H), 1.69–1.39 (m, 6H), 1.01 (s, 3H).
Synthesis of intermediate 10a
A solution of intermediate 7 (1.72 mmol), bis(tert-butoxycarbonyl)amine (8, 4.47 mmol) and Cs2CO3 (2.58 mmol) in N,N-dimethylformamide (DMF, 5 ml) was stirred at 60 °C for 10 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with ethyl acetate (EA, 3 × 30 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via column chromatography (PE/EA 4:1, v/v) to give 10a (575.93 mg, yield 79.8%) as a light yellow liquid.
Synthesis of intermediate 10b
A solution of intermediate 7 (0.40 mmol), 1-Boc-piperazine (0.48 mmol) and DIPEA (0.48 mmol) in DMF (5 ml) was stirred at 60 °C for 10 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with EA (3 × 50 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via column chromatography (PE/EA 3:1, v/v) to give 10b (122.40 mg, yield 78.8%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 5.76 (dd, J = 17.9, 10.4 Hz, 1H), 4.94–4.77 (m, 4H), 4.77–4.72 (m, 1H), 4.51 (s, 1H), 3.35 (s, 4H), 2.86 (q, J = 11.6, 9.8 Hz, 2H), 2.26 (s, 4H), 2.08–1.89 (m, 2H), 1.64 (s, 3H), 1.62–1.40 (m, 6H), 1.39 (s, 9H), 0.94 (s, 3H).
General procedure for the synthesis of intermediate 11
To a solution of intermediate 10 (1.37 mmol) in dichloromethane (DCM, 3 ml) in an ice-cooled bath was slowly added CF3CO2H (1.5 ml) solution. The mixture was gradually warmed to room temperature and stirred for 5 h. The reaction was monitored by TLC. Upon completion, the reaction solution was slowly added to the saturated NaHCO3 solution (until no bubbles were formed) at 0 °C. Then the mixture was extracted with EA (3 × 50 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was directly used in the next reaction without purification.
General procedure for the synthesis of intermediates 13a–c
A solution of H2N − OTHP (1.63 mmol), DIPEA (4.08 mmol), EDCI (3.54 mmol), HOBt (1.77 mmol) and the corresponding acids (12, 1.36 mmol) in DMF (5 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with EA (3 × 50 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via column chromatography (DCM/MeOH 9:1, v/v) to give the products as a colourless liquid.
Methyl 6-oxo-6-(((tetrahydro-2H-pyran-2-yl)oxy)amino)hexanoate (13a)
Colourless liquid, yield 81.0%. 1H NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 4.95 (s, 1H), 4.04–3.87 (m, 1H), 3.67 (s, 3H), 3.66–3.61 (m, 1H), 2.35 (td, J = 6.7, 6.2, 3.4 Hz, 2H), 2.18 (d, J = 30.1 Hz, 2H), 1.93–1.47 (m, 10H).
Methyl 7-oxo-7-(((tetrahydro-2H-pyran-2-yl)oxy)amino)heptanoate (13b)
Colourless liquid, yield 92.0%. 1H NMR (500 MHz, CDCl3) δ 8.48 (s, 1H), 4.94 (s, 1H), 3.95 (s, 1H), 3.67 (s, 3H), 3.66–3.61 (m, 1H), 2.32 (t, J = 7.5 Hz, 2H), 2.13 (s, 2H), 1.90–1.78 (m, 3H), 1.73–1.53 (m, 7H), 1.42–1.32 (m, 2H).
Methyl 8-oxo-8-(((tetrahydro-2H-pyran-2-yl)oxy)amino)octanoate (13c)
Colourless liquid, yield 86.4%. 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 4.95 (s, 1H), 3.93 (d, J = 10.6 Hz, 1H), 3.67 (s, 3H), 3.66–3.60 (m, 1H), 2.31 (t, J = 7.4 Hz, 2H), 2.11 (m, 2H), 1.90–1.73 (m, 3H), 1.67–1.54 (m, 7H), 1.41–1.29 (m, 4H).
General procedure for the synthesis of intermediates 14a–c
A solution of intermediates 13 (1.03 mmol) and cooled NaOH aqueous solution (1 N, 3 ml) in CH3OH (3 ml) was stirred at room temperature for 2 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then the residue was neutralised by HCl solution (1 M) to pH 6 ∼ 7 and was extracted with EA (3 × 20 ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was directly used in the next reaction without purification.
Synthesis of intermediate 16
To a solution of H2N − OTHP (17.46 mmol) and Et3N (31.75 mmol) in DCM (15 ml) was added dropwise DCM solution (15 ml) containing compound 15 (15.87 mmol) at 0 °C. After addition, the mixture was stirred at room temperature for 2 ∼ 3 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with DCM (3 × 50 ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via column chromatography (PE/EA 3:2, v/v) to give 16 (1.51 g, yield 35.4%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 11.66 (s, 1H), 7.77 (d, J = 8.2 Hz, 2H), 7.53 (d, J = 8.2 Hz, 2H), 5.00 (d, J = 3.1 Hz, 1H), 4.81 (s, 2H), 4.05 (t, J = 9.7 Hz, 1H), 3.61–3.44 (m, 1H), 1.64 (dtd, J = 86.9, 11.2, 9.3, 4.9 Hz, 6H).
General procedure for the synthesis of intermediates 17a–f
A solution of 11 (0.70 mmol), DIPEA (2.1 mmol), EDCI (1.82 mmol), HOBt (0.91 mmol) and the corresponding acids 14 (0.84 mmol) in DMF (5 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with EA (3 × 50 ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via column chromatography (DCM/MeOH 99:1 ∼ 9:1, v/v) to give the products as a light yellow liquid.
General procedure for the synthesis of the target compounds 18a–f
To a solution of 17 (0.45 mmol) in MeOH (3 ml) was added TsOH·H2O (0.90 mmol) and stirred at room temperature for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the residue was diluted in H2O (10 ml) and was extracted with DCM (3 × 15 ml). The organic layer was separated, washed with saturated NaHCO3 solution and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via C18 column chromatography (MeCN/H2O 1:4 ∼ 4:1, v/v) to afford the products.
N1-hydroxy-N6-(2-((1R,3S,4S)-4-methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl)adipamide (18a)
White solid, yield 63.1%, m.p. 95.6–96.7 °C. 1H NMR (500 MHz, CDCl3) δ 6.78 (t, J = 5.9 Hz, 1H), 5.80 (dd, J = 17.7, 10.5 Hz, 1H), 4.97–4.84 (m, 4H), 4.81 (s, 1H), 4.57 (s, 1H), 3.82 (d, J = 5.6 Hz, 2H), 2.22 (d, J = 40.7 Hz, 4H), 2.04–1.89 (m, 2H), 1.69 (s, 3H), 1.68–1.52 (m, 7H), 1.52–1.39 (m, 3H), 0.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.76, 171.43, 150.40, 150.00, 147.41, 112.29, 110.07, 108.18, 52.65, 42.99, 42.69, 39.87, 39.77, 35.87, 33.08, 32.33, 27.21, 24.97, 24.92, 24.87, 16.61. HRMS (ESI) m/z: calcd for C21H34N2O3 [M + H]+, 363.2642; found, 363.2641.
N1-hydroxy-N7-(2-((1R,3S,4S)-4-methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl)heptanediamide (18b)
Colourless viscous colloid, yield 58.9%, m.p. 55.7–58.1 °C. 1H NMR (500 MHz, CDCl3) δ 6.35 (s, 1H), 5.80 (dd, J = 17.7, 10.5 Hz, 1H), 4.95–4.87 (m, 3H), 4.86 (s, 1H), 4.82 (t, J = 1.7 Hz, 1H), 4.57 (d, J = 1.9 Hz, 1H), 3.85 (d, J = 5.6 Hz, 2H), 2.29–2.11 (m, 4H), 2.04–1.90 (m, 2H), 1.70 (s, 3H), 1.68–1.53 (m, 5H), 1.52–1.40 (m, 3H), 1.40–1.24 (m, 4H), 0.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.67, 171.44, 150.52, 149.99, 147.40, 112.27, 110.06, 108.32, 52.67, 43.00, 42.72, 39.85, 39.77, 36.16, 33.11, 32.36, 28.32, 27.20, 25.08, 24.89, 24.84, 16.61. HRMS (ESI) m/z: calcd for C22H36N2O3 [M + H]+, 377.2799; found, 377.2798.
N1-hydroxy-N8-(2-((1R,3S,4S)-4-methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl)octanediamide (18c)
Colourless viscous colloid, yield 72.3%, m.p. 58.6–59.3 °C. 1H NMR (500 MHz, CDCl3) δ 6.04 (s, 1H), 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 4.96–4.76 (m, 5H), 4.58 (s, 1H), 3.86 (s, 2H), 2.27–2.09 (m, 4H), 2.02–1.91 (m, 2H), 1.70 (s, 3H), 1.64–1.30 (m, 14H), 1.00 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.77, 171.50, 150.55, 150.00, 147.37, 112.28, 110.05, 108.30, 52.68, 42.98, 42.70, 39.86, 39.76, 36.37, 33.10, 29.69, 28.61, 28.40, 27.20, 25.53, 25.17, 24.84. HRMS (ESI) m/z: calcd for C23H38N2O3 [M + H]+, 391.2955; found, 391.2955.
N-hydroxy-6–(4-(2-((1R,3S,4S)-4-methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl)piperazin-1-yl)-6-oxohexanamide (18d)
Colourless liquid, yield 70.3%. 1H NMR (500 MHz, CDCl3) δ 8.29 (s, 1H), 5.80 (dd, J = 17.8, 10.5 Hz, 1H), 5.12 (d, J = 10.0 Hz, 2H), 4.96–4.86 (m, 2H), 4.82 (s, 1H), 4.57 (s, 1H), 3.68 (d, J = 46.1 Hz, 4H), 3.28 (d, J = 15.4 Hz, 2H), 2.76 (d, J = 40.0 Hz, 4H), 2.36 (t, J = 6.6 Hz, 2H), 2.20 (t, J = 5.9 Hz, 2H), 2.04 (ddd, J = 37.4, 12.2, 6.2 Hz, 2H), 1.70 (s, 3H), 1.69–1.35 (m, 10H), 0.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.96, 166.12, 149.99, 147.48, 147.05, 115.06, 112.21, 110.07, 62.15, 52.58 (d, J = 18.0 Hz), 52.05, 44.13, 41.63, 40.26, 39.76 (d, J = 2.0 Hz), 33.08, 32.38, 29.69, 27.14, 24.82 (d, J = 11.0 Hz), 24.19, 16.55. HRMS (ESI) m/z: calcd for C25H41N3O3 [M + H]+, 432.3221; found, 432.3222.
N-hydroxy-7–(4-(2-((1R,3S,4S)-4-methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl)piperazin-1-yl)-7-oxoheptanamide (18e)
White solid, yield 60.7%, m.p. 126.7–127.5 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.65 (s, 1H), 5.82 (dd, J = 17.9, 10.5 Hz, 1H), 4.98–4.83 (m, 4H), 4.78 (p, J = 1.5 Hz, 1H), 4.58 (d, J = 2.2 Hz, 1H), 3.41 (q, J = 5.8, 5.3 Hz, 4H), 2.90 (q, J = 13.4 Hz, 2H), 2.33–2.22 (m, 6H), 2.11–1.99 (m, 2H), 1.93 (t, J = 7.4 Hz, 2H), 1.67 (s, 3H), 1.65–1.55 (m, 2H), 1.53–1.32 (m, 8H), 1.29–1.20 (m, 2H), 0.97 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 170.93, 169.50, 150.83, 150.52, 147.63, 112.69, 111.53, 110.44, 63.18, 53.56, 53.07, 52.38, 45.46, 42.06, 41.52, 33.17, 32.63, 28.84, 27.21, 25.45, 25.01 (d, J = 5.3 Hz), 16.75. HRMS (ESI) m/z: calcd for C26H43N3O3 [M + H]+, 446.3377; found, 446.3378.
N-hydroxy-8–(4-(2-((1R,3S,4S)-4-methyl-3-(prop-1-en-2-yl)-4-vinylcyclohexyl)allyl)piperazin-1-yl)-8-oxooctanamide (18f)
White solid, yield 73.2%, m.p. 144.3–144.9 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.65 (s, 1H), 5.82 (dd, J = 17.9, 10.5 Hz, 1H), 4.96–4.82 (m, 4H), 4.78 (p, J = 1.5 Hz, 1H), 4.58 (d, J = 2.3 Hz, 1H), 3.41 (q, J = 5.8 Hz, 4H), 2.90 (q, J = 13.4 Hz, 2H), 2.34–2.19 (m, 6H), 2.11–1.98 (m, 2H), 1.93 (t, J = 7.4 Hz, 2H), 1.67 (s, 3H), 1.65–1.54 (m, 2H), 1.52–1.31 (m, 8H), 1.30–1.18 (m, J = 4.8, 4.0 Hz, 4H), 0.97 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 170.98, 169.56, 150.82, 150.53, 147.63, 112.69, 111.54, 110.45, 63.18, 53.58, 53.08, 52.38, 45.48, 42.06, 41.52, 33.17, 32.70, 28.93 (d, J = 6.7 Hz), 27.21, 25.50, 25.18, 25.03, 16.75. HRMS (ESI) m/z: calcd for C27H45N3O3 [M + H]+, 460.3534; found, 460.3538.
Synthesis of the intermediate 19
A solution of 11b (0.52 mmol), DIPEA (0.78 mmol) and the benzyl chloride 16 (0.62 mmol) in DMF (4 ml) was stirred at 60 °C for 6 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (40 ml) and was extracted with EA (3 × 30 ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (DCM/MeOH 49:1, v/v) to give 19 (203.48 mg, yield 75.0%) as a colourless liquid. 1H NMR (400 MHz, CDCl3) δ 8.75 (s, 1H), 7.63 (d, J = 7.9 Hz, 2H), 7.33 (d, J = 7.9 Hz, 2H), 5.75 (dd, J = 17.6, 10.7 Hz, 1H), 5.01 (s, 1H), 4.89–4.79 (m, 4H), 4.75 (t, J = 1.8 Hz, 1H), 4.54–4.48 (m, 1H), 3.99–3.89 (m, 1H), 3.64–3.55 (m, 1H), 3.47 (s, 2H), 2.91–2.79 (m, 2H), 2.37 (s, 8H), 1.98 (ddd, J = 20.6, 11.6, 4.3 Hz, 2H), 1.90–1.75 (m, 3H), 1.71–1.32 (m, 12H), 0.93 (s, 3H).
Synthesis of the target compound 20
To a solution of 19 (0.38 mmol) in MeOH (3 ml) was added TsOH·H2O (0.76 mmol) and stirred at room temperature for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the residue was diluted in H2O (15 ml) and was extracted with DCM (3 × 15 ml). The organic layer was separated, washed with saturated NaHCO3 solution and brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified via C18 column chromatography (MeCN/H2O 3:2, v/v) to afford 20 (141.35 mg, yield 85.1%) as a white solid, m.p. 99.4–100.2 °C. 1H NMR (500 MHz, DMSO-d6) δ 7.70 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 8.3 Hz, 2H), 5.82 (dd, J = 17.9, 10.5 Hz, 1H), 4.91–4.84 (m, 4H), 4.77 (t, J = 1.8 Hz, 1H), 4.57 (d, J = 2.2 Hz, 1H), 3.32 (d, J = 10.9 Hz, 2H), 2.88 (q, J = 13.3 Hz, 2H), 2.36 (s, 8H), 2.08–1.95 (m, 2H), 1.66 (s, 3H), 1.64–1.51 (m, 2H), 1.50–1.39 (m, 3H), 1.36–1.31 (m, 1H), 0.96 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 151.13, 150.53, 147.65, 142.07, 131.86, 129.10, 127.23, 112.65, 111.33, 110.42, 63.41, 62.10, 53.24 (d, J = 7.1 Hz), 52.40, 42.20, 33.18, 27.17, 25.02, 16.75. HRMS (ESI) m/z: calcd for C27H39N3O2 [M + H]+, 438.3115; found, 438.3117.
Synthesis of the crucial intermediate 21
To a solution of β-elemene (1, 29.36 mmol) and tetrabutylammonium fluoride (TBAF, 1.0 mol/l THF) in DCM and AcOH was stirred at 0 °C. Then, NaClO solution (available chlorine ≥ 5.2%) was slowly added dropwise with a micro-injection pump within 5 h at 0 °C. The reaction was monitored by TLC. Upon completion, the reaction solution was slowly added to the saturated NaHCO3 solution (until no bubbles were formed) at 0 °C. Then the mixture was extracted three times with PE (3 × 150 ml). The organic layer was separated, washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified via column chromatography (PE) to afford 21 (2.50 g, yield 31.17%) as a colourless liquid. 1H NMR (400 MHz, CDCl3) δ 5.79 (dd, J = 17.2, 11.1 Hz, 1H), 5.28 (s, 1H), 5.18 (s, 1H), 5.04 (s, 1H), 4.99–4.89 (m, 3H), 4.13–4.08 (m, 4H), 3.98 (d, J = 11.7 Hz, 1H), 3.78–3.66 (m, 2H), 2.36–2.21 (m, 2H), 1.78–1.40 (m, 10H), 0.99 (s, 3H).
Synthesis of intermediate 22a
To a solution of 13,14-Cl-β-elemene (21, 3.66 mmol) in DMF (10 ml) was added Cs2CO3 (4.39 mmol) and bis(tert-butoxycarbonyl)amine (4.39 mmol) successively at room temperature. Then mixture was stirred at 50 °C for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (100 ml) and was extracted with EA (3 × 100 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (PE/EA 1:4, v/v) to give 22a (1244.71 mg, yield 74.9%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 5.83–5.73 (m, 1H), 5.26 (d, J = 1.0 Hz, 1H), 4.98–4.86 (m, 4H), 4.77 (s, 1H), 4.20 (d, J = 1.7 Hz, 2H), 3.96 (dd, J = 11.6, 0.8 Hz, 1H), 2.98–2.87 (m, 1H), 2.31–2.20 (m, 1H), 2.00 (dd, J = 11.1, 4.0 Hz, 1H), 1.74–1.61 (m, 3H), 1.59–1.50 (m, 2H), 1.48 (s, 18H), 1.46–1.41 (m, 1H), 0.98 (s, 3H).
Synthesis of intermediate 22b
To a solution of 13,14-Cl-β-elemene (21, 1.77 mmol) in DMF (10 ml) was added Cs2CO3 (2.12 mmol) and 1-Boc-piperazine (2.12 mmol) successively at room temperature. Then mixture was stirred at 50 °C for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (100 ml) and was extracted with EA (3 × 100 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (PE/EA 1:3, v/v) to give 22b (558.51 mg,yield 81.2%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 5.79 (dd, J = 17.4, 10.8 Hz, 1H), 5.27 (s, 1H), 4.97–4.86 (m, 5H), 4.10 (dd, J = 11.6, 1.1 Hz, 1H), 3.97 (d, J = 11.7 Hz, 1H), 3.41 (t, J = 4.9 Hz, 4H), 2.98–2.86 (m, 2H), 2.32 (q, J = 4.4, 3.8 Hz, 4H), 2.30–2.25 (m, 1H), 2.14 (ddd, J = 11.6, 8.9, 6.1 Hz, 1H), 1.69–1.57 (m, 3H), 1.57–1.47 (m, 3H), 1.46 (s, 9H), 0.98 (s, 3H). LCMS m/z [M + H]+: 423.5.
Synthesis of intermediate 24a
To a solution of 22a (2.70 mmol) in DMF (10 ml) was added Cs2CO3 (4.05 mmol) and 1H-pyrazole (23, 3.24 mmol) successively at room temperature. Then mixture was stirred at 60 °C for 10 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (100 ml) and was extracted with EA (3 × 100 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (PE/EA 3:1, v/v) to give 24a (993.97 mg, yield 75.8%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 1.8 Hz, 1H), 7.31 (d, J = 2.3 Hz, 1H), 6.25 (t, J = 2.1 Hz, 1H), 5.83 (dd, J = 17.4, 10.8 Hz, 1H), 5.05–4.96 (m, 2H), 4.85 (d, J = 14.9 Hz, 2H), 4.78 (s, 1H), 4.73 (dd, J = 8.8, 7.0 Hz, 2H), 4.65 (d, J = 15.6 Hz, 1H), 4.13 (d, J = 1.9 Hz, 2H), 1.94–1.80 (m, 2H), 1.72–1.49 (m, 6H), 1.47 (s, 18H), 1.02 (s, 3H).
Synthesis of intermediate 24b
To a solution of 22b (2.22 mmol) in DMF (10 ml) was added Cs2CO3 (3.33 mmol) and 1H-pyrazole (23, 2.66 mmol) successively at room temperature. Then mixture was stirred at 60 °C for 10 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (100 ml) and was extracted with EA (3 × 100 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (PE/EA 3:1, v/v) to give 24b (789.31 mg, yield 78.2%) as a light yellow liquid. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 1.9 Hz, 1H), 7.33 (d, J = 2.2 Hz, 1H), 6.25 (t, J = 2.1 Hz, 1H), 5.84 (dd, J = 17.4, 10.8 Hz, 1H), 5.04–4.96 (m, 2H), 4.95–4.88 (m, 2H), 4.87 (s, 1H), 4.78 (s, 1H), 4.71 (d, J = 1.4 Hz, 1H), 4.66 (d, J = 15.7 Hz, 1H), 3.39 (s, 4H), 2.88 (q, J = 13.5 Hz, 2H), 2.36–2.25 (m, 4H), 2.00 (m, 1H), 1.93 (dd, J = 12.7, 3.4 Hz, 1H), 1.60–1.48 (m, 3H), 1.47 − 1.36 (m, 13H), 1.03 (s, 3H). LCMS m/z [M + H]+: 455.5.
General procedure for the synthesis of intermediates 25a–b
To a solution of 24 (1.74 mmol) in MeOH (1.5 ml) in an ice-cooled bath was added a solution of hydrochloride in dioxane (4 M, 6 ml). The mixture was gradually warmed to room temperature and stirred for 8 h. The reaction was monitored by TLC. Upon completion, the solvent was removed under reduced pressure to give the intermediates 25 (1.74 mmol, yield 100%) as a white solid. The residue was used for the next reaction without further purification.
General procedure for the synthesis of intermediates 26a–f
A solution of 25 (0.52 mmol), DIPEA (1.56 mmol), EDCI (1.35 mmol), HOBt (0.68 mmol) and the corresponding acids (14, 0.62 mmol) in DMF (5 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with EA (3 × 50 ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via column chromatography (DCM/MeOH 99:1 ∼ 9:1, v/v) to give the products as a light yellow liquid.
General procedure for the synthesis of the target compounds 27a–f
To a solution of 26 (0.41 mmol) in MeOH (3 ml) was added TsOH·H2O (0.82 mmol) and stirred at room temperature for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the residue was diluted in H2O (10 ml) and was extracted with DCM (3 × 10 ml). The organic layer was separated, washed with saturated NaHCO3 solution and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via C18 column chromatography (MeCN/H2O 1:4 ∼ 4:1, v/v) to afford the products.
N1-(2-((1R,3R,4S)-3–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-4-methyl-4-vinylcyclohexyl)allyl)-N6-hydroxyadipamide (27a)
White solid, yield 50.6%, m.p. 65.6–66.8 °C. 1H NMR (500 MHz, CDCl3) δ 7.48 (d, J = 1.8 Hz, 1H), 7.33 (d, J = 2.3 Hz, 1H), 6.25 (t, J = 2.1 Hz, 1H), 5.80 (dd, J = 17.4, 10.8 Hz, 1H), 5.04–4.95 (m, 2H), 4.84 (t, J = 5.9 Hz, 3H), 4.67 (d, J = 5.0 Hz, 3H), 3.79 (d, J = 5.3 Hz, 2H), 2.19 (d, J = 40.0 Hz, 4H), 1.96–1.80 (m, 2H), 1.73–1.36 (m, 10H), 1.00 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.54, 150.18, 149.34, 147.43, 139.13, 129.85, 113.93, 111.41, 108.73, 105.73, 58.73, 48.08, 42.99, 42.30, 39.81, 39.65, 35.87, 33.33, 26.88, 25.02 (d, J = 7.6 Hz), 15.81. HRMS (ESI) m/z: calcd for C24H36N4O3 [M + H]+, 429.2860; found, 429.2858.
N1-(2-((1R,3R,4S)-3–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-4-methyl-4-vinylcyclohexyl)allyl)-N7-hydroxyheptanediamide (27b)
Colourless viscous colloid, yield 42.8%, m.p. 47.3–49.0 °C. 1H NMR (500 MHz, CDCl3) δ 7.48 (s, 1H), 7.33 (d, J = 2.3 Hz, 1H), 6.25 (t, J = 2.0 Hz, 1H), 5.80 (dd, J = 17.4, 10.8 Hz, 1H), 5.10–4.94 (m, 2H), 4.85 (t, J = 4.1 Hz, 3H), 4.78–4.61 (m, 3H), 3.80 (s, 2H), 2.32–2.06 (m, 4H), 1.97–1.81 (m, 2H), 1.76–1.18 (m, 12H), 1.01 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.52, 150.27, 149.31, 147.39, 139.14, 129.76, 114.13, 111.43, 108.78, 105.74, 58.81, 48.07, 42.94, 42.38, 39.82, 39.65, 36.12, 33.32, 29.70, 26.90, 25.07 (d, J = 23.7 Hz), 15.82. HRMS (ESI) m/z: calcd for C25H38N4O3 [M + H]+, 443.3017; found, 443.3021.
N1-(2-((1R,3R,4S)-3–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-4-methyl-4-vinylcyclohexyl)allyl)-N8-hydroxyoctanediamide (27c)
Light yellow solid, yield 53.5%, m.p. 54.7–55.9 °C. 1H NMR (500 MHz, CDCl3) δ 7.48 (d, J = 2.0 Hz, 1H), 7.33 (d, J = 1.8 Hz, 1H), 6.25 (t, J = 2.1 Hz, 1H), 5.80 (dd, J = 17.4, 10.8 Hz, 1H), 5.04–4.95 (m, 2H), 4.85 (d, J = 5.9 Hz, 3H), 4.67 (dd, J = 28.0, 15.6 Hz, 3H), 3.81 (s, 2H), 2.28–2.03 (m, 4H), 1.95–1.81 (m, 2H), 1.72–1.16 (m, 14H), 1.01 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.64, 150.26, 149.30, 147.36, 139.10, 129.71, 114.20, 111.44, 108.63, 105.74, 58.82, 48.04, 42.91, 42.35, 39.81, 39.65, 36.29, 33.31, 29.69, 28.55 (d, J = 17.1 Hz), 26.89, 25.52, 25.27, 15.81. HRMS (ESI) m/z: calcd for C26H40N4O3 [M + H]+, 457.3173; found, 457.3172.
6–(4-(2-((1R,3R,4S)-3–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-4-methyl-4-vinylcyclohexyl)allyl)piperazin-1-yl)-N-hydroxy-6-oxohexanamide (27d)
Light yellow viscous colloid, yield 58.9%, m.p. 71.7–72.2 °C. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 1.8 Hz, 1H), 7.34 (d, J = 2.2 Hz, 1H), 6.25 (t, J = 2.1 Hz, 1H), 5.83 (dd, J = 17.4, 10.8 Hz, 1H), 5.10–4.83 (m, 5H), 4.81–4.62 (m, 3H), 3.57 (s, 2H), 3.43 (s, 2H), 2.89 (q, J = 13.4 Hz, 2H), 2.47–2.13 (m, 8H), 2.07–1.89 (m, 2H), 1.80–1.40 (m, 10H), 1.03 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.66, 149.99, 149.46, 147.60, 139.12, 129.49, 114.20, 111.37 (d, J = 22.0 Hz), 105.60, 63.26, 59.01, 53.17, 52.81, 48.09, 45.64, 41.83 (d, J = 7.3 Hz), 39.90, 39.70, 33.66, 32.61, 29.69, 26.92, 24.89, 24.34, 15.82. HRMS (ESI) m/z: calcd for C28H43N5O3 [M + H]+, 498.3439; found, 498.3436.
7–(4-(2-((1R,3R,4S)-3–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-4-methyl-4-vinylcyclohexyl)allyl)piperazin-1-yl)-N-hydroxy-7-oxoheptanamide (27e)
Colourless viscous colloid, yield 51.5%, m.p. 55.3–56.7 °C. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 1.9 Hz, 1H), 7.34 (d, J = 2.2 Hz, 1H), 6.25 (t, J = 2.1 Hz, 1H), 5.83 (dd, J = 17.4, 10.8 Hz, 1H), 5.07–4.84 (m, 5H), 4.80–4.63 (m, 3H), 3.65–3.36 (m, 4H), 2.90 (q, J = 13.4 Hz, 2H), 2.48–2.09 (m, 8H), 2.04–1.90 (m, 2H), 1.75–1.30 (m, 12H), 1.03 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.78, 171.14, 149.97, 149.46, 147.59, 139.12, 129.51, 114.20, 111.49, 111.28, 105.60, 63.24, 59.02, 53.18, 52.81, 48.09, 45.67, 41.85, 39.90, 39.69, 33.67, 32.77, 32.33, 28.44, 26.90, 25.02, 24.47, 15.82. HRMS (ESI) m/z: calcd for C29H45N5O3 [M + H]+, 512.3595; found, 512.3597.
7–(4-(2-((1R,3R,4S)-3–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-4-methyl-4-vinylcyclohexyl)allyl)piperazin-1-yl)-N-hydroxy-7-oxoheptanamide (27f)
Colourless viscous colloid, yield 68.7%, m.p. 59.6–60.0 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.69 (s, 1H), 7.60 (d, J = 2.2 Hz, 1H), 7.42 (d, J = 1.8 Hz, 1H), 6.24 (t, J = 2.1 Hz, 1H), 5.87 (dd, J = 17.5, 10.8 Hz, 1H), 5.02–4.88 (m, 4H), 4.81 (s, 1H), 4.64 (s, 3H), 3.40 (d, J = 15.5 Hz, 4H), 2.86 (s, 2H), 2.24 (dt, J = 18.1, 6.1 Hz, 6H), 1.94 (dt, J = 14.7, 5.4 Hz, 4H), 1.68–1.32 (m, 10H), 1.32–1.17 (m, J = 4.7, 3.5 Hz, 4H), 0.99 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 172.00, 171.16, 149.38, 148.88, 147.58, 139.09, 129.68, 113.97, 112.75, 111.32, 105.61, 62.90, 58.88, 53.10, 52.58, 48.12, 45.22, 41.69, 41.16, 39.85, 39.63, 33.60, 32.90, 32.60, 31.91, 30.30, 29.66 (d, J = 4.5 Hz), 28.50 (d, J = 20.2 Hz), 28.42, 26.83, 25.01 (d, J = 23.7 Hz), 15.82. HRMS (ESI) m/z: calcd for C30H47N5O3 [M + H]+, 526.3752; found, 526.3755.
Synthesis of intermediate 28
(i) A solution of 25b (1.00 mmol), DIPEA (3.00 mmol), EDCI (2.60 mmol), HOBt (1.30 mmol) and the acid 12c (1.20 mmol) in DMF (6 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (60 ml) and was extracted with EA (3 × 50 ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the residue was purified via column chromatography (DCM/MeOH 19:1, v/v) to give the ester (384.13 mg, yield 73.2%) as a light yellow liquid. 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 2.0 Hz, 1H), 7.27 (d, J = 2.2 Hz, 1H), 6.18 (t, J = 2.1 Hz, 1H), 5.77 (dd, J = 17.5, 10.8 Hz, 1H), 5.02–4.78 (m, 5H), 4.66 (dd, J = 32.9, 17.4 Hz, 3H), 3.59 (s, 3H), 3.51 (s, 2H), 3.35 (s, 2H), 2.91–2.73 (m, 2H), 2.33–2.16 (m, 8H), 2.00–1.78 (m, 2H), 1.66–1.33 (m, 14H), 0.96 (s, 3H).
(ii) A solution of the ester (0.73 mmol) and cooled NaOH aqueous solution (1 N, 3 ml) in MeOH (3 ml) was stirred at room temperature for 2 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the residue was neutralised by HCl solution (1 M) to pH 6 ∼ 7 and was extracted with EA (3 × 20ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure, and the resulted crude product (28, yield 90.6%) as a light yellow liquid, was directly used in the next reaction without purification.
Synthesis of the target compound 31
A solution of 28 (0.33 mmol), DIPEA (0.99 mmol), EDCI (0.86 mmol), HOBt (0.43 mmol) and o-phenylenediamine 29 (1.65 mmol) in DMF (6 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (60 ml) and was extracted with EA (3 × 50 ml). The organic layer was separated, washed with water and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via column chromatography (DCM/MeOH 19:1, v/v) to give the target compound 31 (90.61 mg, yield 45.7%) as a colourless viscous colloid, m.p. 59.7–60.5 °C. 1H NMR (500 MHz, CDCl3) δ 7.87 (s, 1H), 7.43 (d, J = 1.9 Hz, 1H), 7.26 (d, J = 2.3 Hz, 1H), 7.10 (dd, J = 7.8, 1.5 Hz, 1H), 6.95 (td, J = 7.6, 1.5 Hz, 1H), 6.73–6.64 (m, 2H), 6.18 (t, J = 2.1 Hz, 1H), 5.76 (dd, J = 17.5, 10.8 Hz, 1H), 5.00–4.77 (m, 5H), 4.73–4.54 (m, 3H), 3.50 (t, J = 5.0 Hz, 2H), 3.34 (t, J = 5.0 Hz, 2H), 2.89–2.73 (m, 2H), 2.37–2.13 (m, 8H), 2.00–1.80 (m, 2H), 1.68–1.27 (m, 14H), 0.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.06, 170.57, 148.99, 148.45, 146.60, 139.84, 138.12, 128.40, 125.94, 124.26, 123.46, 118.25, 117.01, 113.23, 110.45, 110.26, 104.56, 62.23, 58.05, 52.22, 51.85, 47.06, 44.65, 40.87, 40.61, 38.88, 38.68, 35.63, 32.64, 32.04, 28.67, 27.77 (d, J = 13.2 Hz), 25.88, 24.55, 23.96, 14.79. HRMS (ESI) m/z: calcd for C35H50N6O2 [M + H]+, 587.4068; found, 587.4066.
Synthesis of the target compound 32
A solution of 28 (0.31 mmol), DIPEA (0.93 mmol), EDCI (0.81 mmol), HOBt (0.40 mmol) and o-aminopyridine 30 (3.10 mmol) in DMF (5 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with EA (3 × 50 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via column chromatography (DCM/MeOH 97:3, v/v) to give the target compound 32 (91.14 mg, yield 50.1%) as a brown viscous colloid, m.p. 62.3–63.7 °C. 1H NMR (500 MHz, CDCl3) δ 8.62 (s, 1H), 8.27–8.16 (m, 2H), 7.67 (ddd, J = 8.8, 7.4, 1.9 Hz, 1H), 7.47 (d, J = 1.9 Hz, 1H), 7.31 (d, J = 2.1 Hz, 1H), 7.00 (ddd, J = 7.4, 4.9, 1.0 Hz, 1H), 6.22 (t, J = 2.1 Hz, 1H), 5.81 (dd, J = 17.4, 10.8 Hz, 1H), 5.04–4.93 (m, 2H), 4.92–4.82 (m, 3H), 4.75 (s, 1H), 4.73–4.61 (m, 2H), 3.56 (t, J = 5.0 Hz, 2H), 3.39 (t, J = 5.0 Hz, 2H), 2.86 (q, J = 13.4 Hz, 2H), 2.40–2.24 (m, 8H), 2.03–1.88 (m, 2H), 1.73–1.55 (m, 6H), 1.49–1.33 (m, 8H), 1.00 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.92, 171.47, 151.62, 150.07, 149.46, 147.59 (d, J = 4.8 Hz), 139.12, 138.43, 129.36, 119.59, 114.20 (d, J = 13.5 Hz), 111.31 (d, J = 15.2 Hz), 105.56, 63.28, 59.07, 53.26, 52.89, 48.05, 45.66, 41.87, 41.60, 39.89, 39.69, 37.51, 33.66, 33.14, 29.68, 29.10, 28.90, 26.89, 25.12 (d, J = 7.7 Hz), 15.80. HRMS (ESI) m/z: calcd for C36H52N6O2 [M + H]+, 601.4225; found, 601.4227.
Synthesis of intermediate 33
A solution of intermediate 25b (0.34 mmol), DIPEA (0.51 mmol) and the benzyl chloride 16 (0.41 mmol) in DMF (2.5 ml) was stirred at 60 °C for 6 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (25 ml) and was extracted with EA (3 × 25 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (DCM/MeOH 19:1, v/v) to give 33 (131.80 mg, yield 65.9%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 9.00 (s, 1H), 7.71 (d, J = 7.9 Hz, 2H), 7.51 (d, J = 1.8 Hz, 1H), 7.39 (d, J = 7.9 Hz, 2H), 7.33 (d, J = 2.3 Hz, 1H), 6.25 (t, J = 2.1 Hz, 1H), 5.83 (dd, J = 17.4, 10.8 Hz, 1H), 5.08 (d, J = 3.7 Hz, 1H), 5.04–4.95 (m, 2H), 4.93–4.89 (m, 1H), 4.86 (d, J = 2.5 Hz, 2H), 4.77–4.60 (m, 3H), 4.06–3.96 (m, 1H), 3.69–3.61 (m, 1H), 3.55 (s, 2H), 2.94–2.82 (m, 2H), 2.42 (s, 8H), 2.10–1.83 (m, 2H), 1.81–1.37 (m, 12H), 1.02 (s, 3H).
Synthesis of the target compound 34
To a solution of 33 (0.22 mmol) in MeOH (2 ml) was added TsOH·H2O (0.44 mmol) and stirred at room temperature for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the residue was diluted in H2O (10 ml) and was extracted with DCM (3 × 10 ml). The organic layer was separated, washed with saturated NaHCO3 solution and brine, dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via C18 column chromatography (MeCN/H2O 3:2, v/v) to afford the target compound 34 (74.58 mg, yield 67.3%) as a white solid, m.p. 111.8–112.6 °C. 1H NMR (500 MHz, CDCl3) δ 7.66 (d, J = 7.7 Hz, 2H), 7.48 (d, J = 1.6 Hz, 1H), 7.34–7.28 (m, 3H), 6.24 (t, J = 2.1 Hz, 1H), 5.79 (dd, J = 17.4, 10.8 Hz, 1H), 5.02–4.81 (m, 5H), 4.76–4.60 (m, 3H), 3.52 (s, 2H), 2.88 (q, J = 13.4 Hz, 2H), 2.46 (s, 8H), 2.04–1.87 (m, 2H), 1.63–1.36 (m, 6H), 1.00 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 165.94, 150.16, 149.53, 147.65, 141.44, 139.12, 130.56, 129.55 (d, J = 5.5 Hz), 127.02, 113.98, 111.29 (d, J = 21.2 Hz), 105.62, 63.25, 62.44, 58.88, 52.85 (d, J = 16.7 Hz), 48.18, 41.95, 39.79 (d, J = 23.2 Hz), 33.64, 26.90, 15.81. HRMS (ESI) m/z: calcd for C30H41N5O2 [M + H]+, 504.3333; found, 504.3333.
Synthesis of intermediate 35
To a solution of 13,14-Br-β-elemene (21, 4.93 mmol) in DMF (10 ml) was added Cs2CO3 (5.92 mmol) and 1H-pyrazole (23, 5.92 mmol) successively at room temperature. Then the mixture was stirred at 45 °C for 10 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (100 ml) and was extracted with EA (3 × 100 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (PE/EA 4:1, v/v) to give 35 (1037.18 mg, yield 69.3%) as a light yellow liquid. 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 1.6 Hz, 1H), 7.40 (d, J = 2.2 Hz, 1H), 6.28 (t, J = 2.1 Hz, 1H), 5.75 (dd, J = 17.3, 10.9 Hz, 1H), 5.26 (s, 1H), 5.02 (d, J = 1.2 Hz, 1H), 4.96–4.85 (m, 3H), 4.78 (s, 3H), 4.06 (dd, J = 11.7, 1.1 Hz, 1H), 3.95 (dd, J = 11.6, 0.8 Hz, 1H), 2.22 (dd, J = 12.5, 3.7 Hz, 1H), 1.95 (tt, J = 11.6, 3.7 Hz, 1H), 1.68–1.38 (m, 6H), 0.96 (s, 3H).
Synthesis of intermediate 36a
To a solution of intermediate 35 (2.89 mmol) in DMF (10 ml) was added Cs2CO3 (3.47 mmol) and bis(tert-butoxycarbonyl)amine (3.47 mmol) successively at room temperature. Then the mixture was stirred at 80 °C for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (100 ml) and was extracted with EA (3 × 100 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (PE/EA 7:3, v/v) to give 36a (1056.24 mg, yield 75.5%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 7.51 (d, J = 1.8 Hz, 1H), 7.39 (d, J = 2.2 Hz, 1H), 6.27 (t, J = 2.1 Hz, 1H), 5.85–5.76 (m, 1H), 5.01 (s, 1H), 4.94 (q, J = 1.3 Hz, 1H), 4.92–4.89 (m, 1H), 4.86 (d, J = 1.8 Hz, 1H), 4.74 (d, J = 7.0 Hz, 4H), 4.22 (dt, J = 17.0, 2.0 Hz, 1H), 3.89 (d, J = 17.0 Hz, 1H), 1.94–1.83 (m, 2H), 1.72–1.53 (m, 3H), 1.48 (s, 3H), 1.47 (s, 18H), 0.99 (s, 3H).
Synthesis of intermediate 36b
To a solution of intermediate 35 (1.98 mmol) in DMF (7 ml) was added Cs2CO3 (2.38 mmol) and 1-Boc-piperazine (2.38 mmol) successively at room temperature. Then the mixture was stirred at 80 °C for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (70 ml) and was extracted with EA (3 × 70 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (PE/EA 3:1, v/v) to give 36b (781.72 mg, yield 86.8%) as a light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 1.8 Hz, 1H), 7.42 (d, J = 2.3 Hz, 1H), 6.31 (t, J = 2.1 Hz, 1H), 5.80 (dd, J = 17.5, 10.7 Hz, 1H), 5.06 (d, J = 8.0 Hz, 2H), 4.94–4.86 (m, 2H), 4.80 (d, J = 3.8 Hz, 4H), 3.43 (s, 4H), 3.06 (d, J = 13.8 Hz, 1H), 2.64 (d, J = 13.7 Hz, 1H), 2.42–2.31 (m, 2H), 2.22 (s, 2H), 1.91 (d, J = 11.1 Hz, 1H), 1.80 (s, 1H), 1.69–1.56 (m, 2H), 1.55–1.50 (m, 1H),1.50–1.425 (m, 12H), 1.00 (s, 3H). LCMS m/z [M + H]+: 454.9.
General procedure for the synthesis of intermediates 37a–b
To a solution of intermediate 36 (1.55 mmol) in MeOH (1.5 ml) in an ice-cooled bath was added a solution of hydrochloride in dioxane (4 M, 6 ml). The mixture was gradually warmed to room temperature and stirred for 8 h. The reaction was monitored by TLC. Upon completion, the solvent was removed under reduced pressure to give intermediates 37 (1.55 mmol, yield 100%) as a white solid. The residue was used for the next reaction without further purification.
General procedure for the synthesis of intermediates 38a–f
A solution of 37 (0.41 mmol), DIPEA (1.23 mmol), EDCI (1.07 mmol), HOBt (0.53 mmol) and the corresponding acids 14 (0.49 mmol) in DMF (5 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (50 ml) and was extracted with EA (3 × 50 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via column chromatography (DCM/MeOH 99:1 ∼ 9:1, v/v) to give the products as a light yellow liquid.
General procedure for the synthesis of the target compounds 39a–f
To a solution of intermediates 38 (0.37 mmol) in MeOH (3 ml) was added TsOH·H2O (0.74 mmol) in room temperature for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the mixture was diluted in H2O (10 ml) and was extracted with DCM (3 × 10 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via C18 column chromatography (MeCN/H2O 1:4 ∼ 4:1, v/v) to afford the products.
N1-(2-((1R,2S,5R)-5–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-2-methyl-2-vinylcyclohexyl)allyl)-N6-hydroxyadipamide (39a)
White solid, yield 70.4%, m.p. 61.7–63.3 °C. 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 7.40 (d, J = 2.3 Hz, 1H), 6.28 (t, J = 2.1 Hz, 1H), 5.76 (dd, J = 17.4, 10.8 Hz, 1H), 5.08–4.62 (m, 8H), 3.83 (dd, J = 15.6, 6.4 Hz, 1H), 3.61 (dd, J = 15.8, 4.6 Hz, 1H), 2.19 (d, J = 28.1 Hz, 4H), 1.93 (d, J = 10.9 Hz, 1H), 1.83 (s, 1H), 1.72–1.38 (m, 10H), 0.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.40, 149.46, 149.14, 147.90, 139.12, 129.66, 112.34, 111.63, 110.98, 106.00, 56.17, 48.97, 46.09, 41.57, 39.68, 39.41, 35.90, 33.45, 32.32, 26.76, 24.93 (d, J = 14.8 Hz), 16.04. HRMS (ESI) m/z: calcd for C24H36N4O3 [M + H]+, 429.2860; found, 429.2863.
N1-(2-((1R,2S,5R)-5–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-2-methyl-2-vinylcyclohexyl)allyl)-N7-hydroxyheptanediamide (39b)
Colourless viscous colloid, yield 72.5%, m.p. 63.8–64.9 °C. 1H NMR (500 MHz, CDCl3) δ 7.49 (s, 1H), 7.40 (d, J = 2.2 Hz, 1H), 6.27 (s, 1H), 5.76 (dd, J = 17.4, 10.8 Hz, 1H), 4.97 (d, J = 11.2 Hz, 2H), 4.89 (dd, J = 14.2, 10.5 Hz, 2H), 4.73 (q, J = 9.9, 8.5 Hz, 4H), 3.80 (d, J = 17.2 Hz, 1H), 3.63 (d, J = 13.7 Hz, 1H), 2.16 (d, J = 36.6 Hz, 4H), 1.98–1.79 (m, 2H), 1.72–1.37 (m, 8H), 1.36–1.20 (m, 4H), 0.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.46, 149.47, 149.18, 147.95, 139.16, 129.61, 111.61, 110.95, 105.94, 56.11, 49.13, 45.89, 41.70, 39.67, 39.40, 36.11, 33.33, 29.70, 26.78, 16.12. HRMS (ESI) m/z: calcd for C25H38N4O3 [M + H]+, 443.3017; found, 443.3014.
N1-(2-((1R,2S,5R)-5–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-2-methyl-2-vinylcyclohexyl)allyl)-N8-hydroxyoctanediamide (39c)
White solid, yield 59.7%, m.p. 55.4–57.9 °C. 1H NMR (500 MHz, CDCl3) δ 10.16 (s, 1H), 7.43 (d, J = 1.9 Hz, 1H), 7.33 (d, J = 2.2 Hz, 1H), 6.21 (t, J = 2.1 Hz, 1H), 6.08 (s, 1H), 5.70 (dd, J = 17.4, 10.8 Hz, 1H), 4.97–4.79 (m, 4H), 4.75–4.62 (m, 4H), 3.78 (dd, J = 15.8, 6.5 Hz, 1H), 3.56 (dd, J = 15.8, 4.7 Hz, 1H), 2.09 (dt, J = 26.4, 7.4 Hz, 4H), 1.86 (dd, J = 12.5, 3.3 Hz, 1H), 1.82–1.72 (m, 1H), 1.59–1.34 (m, 10H), 1.24 (dd, J = 7.9, 4.0 Hz, 4H), 0.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 173.42, 171.15, 149.42, 149.15, 148.02, 139.12, 129.57, 112.18, 111.68, 110.98, 105.97, 56.14, 49.09, 45.97, 41.63, 39.68, 39.46, 36.29, 33.41, 32.62, 28.42, 28.30, 26.74, 25.41, 25.12, 21.06, 16.03, 14.20. HRMS (ESI) m/z: calcd for C26H40N4O3 [M + H]+, 457.3173; found, 457.3177.
6–(4-(2-((1R,2S,5R)-5–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-2-methyl-2-vinylcyclohexyl)allyl)piperazin-1-yl)-N-hydroxy-6-oxohexanamide (39d)
Light yellow solid, yield 72.5%, m.p. 100.2–102.3 °C. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 1.9 Hz, 1H), 7.40 (d, J = 2.2 Hz, 1H), 6.27 (t, J = 2.1 Hz, 1H), 5.75 (dd, J = 17.5, 10.8 Hz, 1H), 5.02 (d, J = 11.3 Hz, 2H), 4.91–4.83 (m, 2H), 4.76 (d, J = 1.8 Hz, 4H), 3.58 (s, 2H), 3.50–3.34 (m, 2H), 3.02 (d, J = 13.8 Hz, 1H), 2.60 (d, J = 13.8 Hz, 1H), 2.43–2.12 (m, 9H), 1.90 (dt, J = 11.4, 6.9 Hz, 1H), 1.71–1.21 (m, 10H), 0.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.75, 149.75, 149.70, 147.71, 139.16, 129.35, 114.09, 111.60, 110.42, 105.79, 66.01, 56.31, 53.09, 52.89, 47.77, 45.64, 41.90, 39.77 (d, J = 7.9 Hz), 36.50, 33.57, 32.68, 26.97, 25.28, 24.63, 15.86. HRMS (ESI) m/z: calcd for C28H43N5O3 [M + H]+, 498.3439; found, 498.3440.
7–(4-(2-((1R,2S,5R)-5–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-2-methyl-2-vinylcyclohexyl)allyl)piperazin-1-yl)-N-hydroxy-7-oxoheptanamide (39e)
Light yellow solid, yield 70.8%, m.p. 60.2–61.3 °C. 1H NMR (500 MHz, CDCl3) δ 7.43 (d, J = 1.9 Hz, 1H), 7.33 (d, J = 2.3 Hz, 1H), 6.21 (t, J = 2.1 Hz, 1H), 5.69 (dd, J = 17.5, 10.8 Hz, 1H), 4.96 (d, J = 10.5 Hz, 2H), 4.89–4.63 (m, 6H), 3.61–3.28 (m, 4H), 2.98 (d, J = 13.6 Hz, 1H), 2.55 (d, J = 13.7 Hz, 1H), 2.41–2.04 (m, 9H), 1.83 (ddt, J = 11.6, 8.2, 3.3 Hz, 1H), 1.66–1.23 (m, 12H), 0.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 170.80, 170.08, 148.66 (d, J = 4.8 Hz), 146.59, 138.13, 128.37, 113.38, 110.70, 109.44, 104.83, 65.08, 55.36, 52.09, 51.79, 46.76, 44.67, 40.75 (d, J = 9.0 Hz), 38.74 (d, J = 2.3 Hz), 32.62, 31.76, 31.36, 27.41, 25.93, 23.98, 23.45, 14.81. HRMS (ESI) m/z: calcd for C29H45N5O3 [M + H]+, 512.3595; found, 512.3596.
8–(4-(2-((1R,2S,5R)-5–(3-(1H-pyrazol-1-yl)prop-1-en-2-yl)-2-methyl-2-vinylcyclohexyl)allyl)piperazin-1-yl)-N-hydroxy-8-oxooctanamide (39f)
Colourless viscous colloid, yield 60.3%, m.p. 58.7–59.5 °C. 1H NMR (500 MHz, CDCl3) δ 7.50 (d, J = 1.8 Hz, 1H), 7.40 (d, J = 2.2 Hz, 1H), 6.28 (t, J = 2.1 Hz, 1H), 5.76 (dd, J = 17.5, 10.7 Hz, 1H), 5.03 (d, J = 10.4 Hz, 2H), 4.95–4.83 (m, 2H), 4.83–4.68 (m, 4H), 3.58 (s, 2H), 3.45 (tdd, J = 15.2, 7.5, 4.0 Hz, 2H), 3.04 (d, J = 13.6 Hz, 1H), 2.61 (d, J = 13.7 Hz, 1H), 2.49–2.10 (m, 9H), 1.96–1.84 (m, 1H), 1.74–1.18 (m, 14H), 0.97 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 171.89, 149.72, 149.68, 147.66, 139.15, 129.37, 114.31, 111.71, 110.44, 105.84, 66.12, 56.38, 53.19, 52.84, 47.78, 45.75, 41.82, 41.70, 39.76 (d, J = 2.4 Hz), 33.64, 32.95, 32.44, 32.42, 28.47 (d, J = 18.9 Hz), 26.96, 25.01 (d, J = 17.6 Hz), 15.84. HRMS (ESI) m/z: calcd for C30H47N5O3 [M + H]+, 526.3752; found, 526.3752.
Synthesis of the compound 40
(i) A solution of 37a (0.35 mmol), DIPEA (1.05 mmol), EDCI (0.91 mmol), HOBt (0.46 mmol) and the acid (12c, 0.42 mmol) in DMF (3 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the residue was diluted in H2O (30 ml) and was extracted with EA (3 × 30 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via column chromatography (DCM/MeOH 49:1, v/v) to give the corresponding ester (153.00 mg, yield 95.7%) as a light yellow liquid.
(ii) A solution of the ester (0.28 mmol) and cooled NaOH aqueous solution (1 N, 3 ml) in MeOH (3 ml) was stirred at room temperature for 2 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the residue was neutralised by HCl solution (1 M) to pH 6 ∼ 7 and was extracted with EA (3 × 20 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure to afford the crude product 40 (yield 92.3%) as a light yellow liquid. It was directly used in the next reaction without purification.
Synthesis of the target compound 41
A solution of 40 (0.12 mmol), DIPEA (0.36 mmol), EDCI (0.31 mmol), HOBt (0.16 mmol) and o-phenylenediamine 29 (0.60 mmol) in DMF (3 ml) was stirred at room temperature for 5 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (30 ml) and was extracted with EA (3 × 30 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via C18 column chromatography (MeCN/H2O 2:3 ∼ 1:1, v/v) to afford 41 (56.11 mg, yield 88.0%) as a light yellow solid, m.p. 98.2–100.3 °C. 1H NMR (500 MHz, CDCl3) δ 7.83 (s, 1H), 7.51 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 2.2 Hz, 1H), 7.16 (dd, J = 7.7, 1.4 Hz, 1H), 7.03 (td, J = 7.7, 1.5 Hz, 1H), 6.76 (t, J = 7.6 Hz, 2H), 6.28 (t, J = 2.1 Hz, 1H), 5.75 (dd, J = 17.5, 10.8 Hz, 1H), 4.99 (s, 1H), 4.97–4.86 (m, 3H), 4.79–4.69 (m, 4H), 3.84 (dd, J = 15.8, 6.8 Hz, 1H), 3.62 (dd, J = 15.8, 5.0 Hz, 1H), 2.37 (t, J = 7.5 Hz, 2H), 2.19 (t, J = 7.4 Hz, 2H), 1.94–1.80 (m, 2H), 1.77–1.49 (m, 5H), 1.48–1.24 (m, 9H), 0.96 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 172.90, 172.03, 149.41, 149.15, 148.09, 140.83, 139.19, 129.42, 126.98, 125.24, 124.47, 119.31, 118.10, 112.11, 111.70, 110.98, 105.88, 56.18, 49.12, 45.89, 41.73, 39.69, 39.48, 36.65, 36.39, 33.38, 28.62, 28.23, 26.80, 25.51, 25.29, 15.98. HRMS (ESI) m/z: calcd for C32H45N5O2 [M + H]+, 532.3646; found, 532.3647.
Synthesis of intermediate 42
A solution of 37b (0.37 mmol), DIPEA (0.44 mmol) and the benzyl chloride 16 (0.44 mmol) in DMF (3 ml) was stirred at 60 °C for 6 h. The reaction was monitored by TLC. Upon completion, the mixture was diluted in H2O (30 ml) and was extracted with EA (3 × 30 ml). The combined organic layers were washed with water and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via flash column chromatography (DCM/MeOH 91:9, v/v) to give 42 (196.39 mg, yield 90.3%) as a colourless liquid. 1H NMR (500 MHz, CDCl3) δ 7.76–7.67 (m, 2H), 7.49 (d, J = 1.9 Hz, 1H), 7.42–7.34 (m, 3H), 6.25 (t, J = 2.1 Hz, 1H), 5.74 (dd, J = 17.5, 10.8 Hz, 1H), 5.10–5.03 (m, 2H), 4.99 (s, 1H), 4.88–4.71 (m, 6H), 4.00 (td, J = 10.4, 9.1, 2.9 Hz, 1H), 3.63 (dd, J = 10.4, 4.9 Hz, 1H), 3.60 (s, 2H), 3.09 (d, J = 13.5 Hz, 1H), 2.68 (d, J = 13.6 Hz, 1H), 2.62–2.22 (m, 8H), 2.15 (dd, J = 12.6, 3.6 Hz, 1H), 1.96–1.76 (tdd, J = 17.0, 12.5, 9.5 Hz, 4H), 1.75–1.35 (m, 9H), 0.94 (s, 3H).
Synthesis of the target compound 43
To a solution of 42 (0.26 mmol) in MeOH (2 ml) was added TsOH·H2O (0.52 mmol) and stirred at room temperature for 8 h. The reaction was monitored by TLC. Upon completion, the mixture was concentrated in vacuo. Then, the residue was diluted in H2O (10 ml) and was extracted with DCM (3 × 10 ml). The organic layer was separated, washed with saturated NaHCO3 solution and brine, and dried over Na2SO4. The drying agent was filtered off. The filtrate was concentrated under reduced pressure and the residue was purified via C18 column chromatography (MeCN/H2O 1:1, v/v) to afford the target compound 43 (95.73 mg, yield 73.1%) as a white solid, m.p. 103.2–105.3 °C. 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 7.7 Hz, 2H), 7.48 (d, J = 1.9 Hz, 1H), 7.38 (d, J = 2.3 Hz, 1H), 7.32 (d, J = 7.7 Hz, 2H), 6.25 (t, J = 2.1 Hz, 1H), 5.74 (dd, J = 17.5, 10.8 Hz, 1H), 5.00 (d, J = 11.1 Hz, 2H), 4.90–4.69 (m, 6H), 3.53 (s, 2H), 3.02 (d, J = 13.6 Hz, 1H), 2.61 (d, J = 13.6 Hz, 1H), 2.45 (s, 8H), 2.14 (dd, J = 11.7, 4.4 Hz, 1H), 1.89 (dq, J = 11.8, 4.4, 4.0 Hz, 1H), 1.64–1.35 (m, 6H), 0.94 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 149.72 (d, J = 4.0 Hz), 147.70, 139.17, 129.50 (d, J = 11.8 Hz), 126.95, 114.32, 111.50, 110.35, 105.83, 66.20, 62.49, 56.34, 52.87 (d, J = 14.0 Hz), 47.94, 41.81, 39.69, 33.64, 26.90, 15.85. HRMS (ESI) m/z: calcd for C30H41N5O2 [M + H]+, 504.3333; found, 504.3337.
Biological evaluation
HDAC inhibition assay
All HDAC enzymes were purchased from BPS Bioscience. The various concentrations of the tested compounds and HDAC isoforms were mixed into a 384-well plate and incubated at 37 °C for 10 min. Then the diluted HDAC substrate was added into each well and incubated at 37 °C for 30 min. The reaction was stopped by adding developer, and the mixture was incubated at 37 °C for another 20 min. The fluorescence intensity was measured at the excitation and emission wavelengths of 355 nm and 460 nm with a microplate reader. The IC50 values were calculated according to the dose dependent curves. All the tests were repeated in three independent experiments.
In vitro anti-proliferative assay
The anti-proliferative activities of the target compounds were determined using CCK8 assay kit. 80 µL of cell suspensions (5.0 × 104 cell/ml) (K562, MV4-11, HEL, SU-DHL-2, WSU-DLCL-2 and U87MG) were added to a 96-well cell culture plate and incubated for 24 h at 37 °C under an atmosphere of 5% CO2. The tested compounds were dissolved in the culture medium with 0.5% DMSO at the different concentrations. The cells were treated with the drug solution for another 72 h. Then 10 µL of cell counting kit-8 (CCK8) solution was added to each well and the plate was incubated for an additional 1 h. The IC50 values were calculated according to the dose dependent curves. All the tests were repeated in three independent experiments.
Apoptosis assay
WSU-DLCL-2 cells were seeded into 6-well plates and incubated at 37 °C for 24 h, and then treated with the tested compounds at the different concentrations or without the tested compound for another 72 h. The cells were then harvested by trypsinization and washed twice with cold PBS. After the centrifugation and removal of the supernatants, cells were re-suspended in 500 µL of a 1 × binding buffer, which was then added to 5 µL of annexin V-FITC and 10 µL of PI, and incubated at room temperature for 15 min in the dark. The stained cells were analysed by a flow cytometer.
Cell cycle assay
WSU-DLCL-2 cells were seeded into 6-well plates and incubated at 37 °C for 24 h, and then treated with the tested compounds at the different concentrations or without the tested compound for another 72 h. After treatment, cells were collected and fixed with 70% pre-cold ethanol in PBS and stored at −20 °C overnight. Then washed the cells with PBS twice, and incubated with 100 µg/ml RNase A at 37 °C for 1 h, stained with propidium iodide for 30 min avoid light at room temperature. The stained cells were analysed by a flow cytometer.
Molecular docking
The molecular docking was performed using energetics (CDOCER) software (Discovery Studio® 2020). The two-dimensional structure of HDAC1 (PDB code: 5ICN) and HDAC6 (PDB code: 5EDU) was performed from the Protein Data Bank. The binding water and ligands were deleted and the polar hydrogen was added. The molecular structures of compounds 27f and 39f were optimised using Tripos force field. The molecular docking of 27f and 39f with HDAC1 and HDAC6 was carried out with Discovery Studio® 2020.
Pharmacokinetics study
The experimental procedures and the animal use and care protocols were approved by the Committee on Ethical Use of Animals of Hangzhou Normal University. Compound 27f was subjected to PK studies in ICR (SPF) female mice. Compound 27f was dissolved in the solution of 0.5% methylcellulose (containing 0.4% Tween 80) and then administrated via the oral route at 10 mg/kg (twelve rats). Compound 27f was dissolved in the solution of 5% dimethyl sulfoxide (DMSO), 5% Solutol, and 90% saline and then administrated via the intravenous route at 5 mg/kg (twelve rats). At time points 0 (prior to dosing), 5, 15, 30, 60, 120, 240, 480, and 1440 min after dosing, the blood sample was collected from each animal, and the plasma was separated from the blood by centrifugation and stored in a freezer at −80 °C. All samples for the tested compound were analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Supplemental Material
Download PDF (5.7 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Zhai B, Zhang N, Han X, et al. Molecular targets of β-elemene, a herbal extract used in traditional Chinese medicine, and its potential role in cancer therapy: a review. Biomed Pharmacother. 2019;114:108812.
- Xie T. Elemene antitumor drugs: molecular compatibility theory and its applications in new drug development and clinical practice. 1st ed. Amsterdam: Elsevier; 2022.
- Zheng F, Tang Q, Zheng X, et al. Inactivation of Stat3 and crosstalk of miRNA155-5p and FOXO3a contribute to the induction of IGFBP1 expression by beta-elemene in human lung cancer. Exp Mol Med. 2018;50:121.
- Long J, Liu Z, Hui L. Anti-tumor effect and mechanistic study of elemene on pancreatic carcinoma. BMC Complem Altern M. 2019;19:133.
- Xu L, Guo T, Qu X, et al. β-Elemene increases the sensitivity of gastric cancer cells to TRAIL by promoting the formation of DISC in lipid rafts. Cell Biol Int. 2018;42:1377.
- Xie L, Zhang J, Yan H, et al. β-Elemene induced apoptosis and senescence of triple-negative breast cancer cells through IGF1/IGF1R pathway. Tissue Cell. 2022;79:101914.
- Cai B, Ma L, Nong S, et al. β-Elemene induced anticancer effect in bladder cancer through upregulation of PTEN and suppression of AKT phosphorylation. Oncol Lett. 2018;16:6019.
- Li C-L, Chang L, Guo L, et al. β-Elemene induces caspasedependent apoptosis in human glioma cells in vitro through the upregulation of Bax and Fas/FasL and downregulation of Bcl-2. Asian Pac J Cancer Prev. 2014;15:10407.
- Wang G, Li X, Huang F, et al. Antitumor effect of beta-elemene in non-small-cell lung cancer cells is mediated via induction of cell cycle arrest and apoptotic cell death. Cell Mol Life Sci. 2005;62:881.
- Liu J, Zhang Y, Qu J, et al. β-Elemene-induced autophagy protects human gastric cancer cells from undergoing apoptosis. BMC Cancer. 2011;11:183.
- Chen J, Wang T, Xu S, et al. Discovery of novel antitumor nitric oxide-donating β-elemene hybrids through inhibiting the PI3K/Akt pathway. Eur J Med Chem. 2017;135:414.
- Sun Y, Liu G, Zhang Y, et al. Synthesis and in vitro anti-proliferative activity of β-elemene monosubstituted derivatives in HeLa cells mediated through arrest of cell cycle at the G1 phase. Bioorg Med Chem. 2009;17:1118.
- Wang B, Peng X, Sun R, et al. Systematic review of β-Elemene injection as adjunctive treatment for lung cancer. Chin J Integr Med. 2012;18:813.
- Hu Q, Bai L, Zhu Z, et al. β-Elemene-loaded polymeric micelles intensify anti-carcinoma efficacy and alleviate side effects. Chinese Chem Lett. 2020;31:915.
- Jiang L, Li A, Tan Y, et al. Clinical observation of elemene injection combined with cisplatin in the treatment of malignant pleural and peritoneal effusion. Med Sci J Central South China. 2020;48:428.
- Wang Z, Li Y, Zhou F, et al. β-Elemene enhances anticancer bone neoplasms efficacy of paclitaxel through regulation of GPR124 in bone neoplasms cells. Oncol Lett. 2018;16(2):2143.
- Cheng H, Ge X, Zhuo S, et al. β-Elemene synergizes with gefitinib to inhibit stem-like phenotypes and progression of lung cancer via down-regulating EZH2. Front Pharmacol. 2018;9:1413.
- Gao Y, Mao N-D, Che H, et al. Novel hydroxyl carboximates derived from β-elemene: design, synthesis and anti-tumour activities evaluation. J Enzym Inhib Med Chem. 2022;37:2403.
- Ye Y, Qi X, Xu B, et al. Nickel-catalyzed cross-electrophile allylation of vinyl bromides and the modification of anti-tumour natural medicine β-elemene. Chem Sci. 2022;13:6959.
- Bai R, Zhu J, Bai Z, et al. Second generation β-elemene nitric oxide derivatives with reasonable linkers: potential hybrids against malignant brain glioma. J Enzym Inhib Med Chem. 2022;37:379.
- Qi X, Jiang S, Hui Z, et al. Design, synthesis and antitumor efficacy evaluation of a series of novel β-elemene-based macrocycles. Bioorgan Med Chem. 2022;74:117049.
- Kumar V, Kundu S, Singh A, et al. Understanding the role of histone deacetylase and their inhibitors in neurodegenerative disorders: current targets and future perspective. Curr Neuropharmacol. 2022;20:158.
- He X, Li Z, Zhuo X-T, et al. Novel selective histone deacetylase 6 (HDAC6) inhibitors: a patent review (2016-2019). Recent Pat anti-Cancer Drug Dis. 2020;15:32.
- Gao Y, Zhang H, Lirussi F, et al. Dual inhibitors of histone deacetylases and other cancer-related targets: a pharmacological perspective. Biochem Pharmacol. 2020;182:114224.
- Newbold A, Falkenberg KJ, Prince HM, Johnstone RW. How do tumor cells respond to HDAC inhibition? FEBS J. 2016;283(22):4032–4046.
- Marks PA, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194.
- Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287.
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38.
- Barneda-Zahonero B, Parra M. Histone deacetylases and cancer. Mol Oncol. 2012;6:579.
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541.
- Li Y, Seto E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6:a026831.
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769.
- Eckschlager T, Plch J, Stiborova M, et al. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:1414.
- Gryder BE, Sodji QH, Oyelere AK. Targeted cancer therapy: giving histone deacetylase inhibitors all they need to succeed. Future Med Chem. 2012;4:505.
- Shah MH, Binkley P, Chan K, et al. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12:3997.
- Suraweera A, O’byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92.
- Diyabalanage HV, Granda ML, Hooker JM. Combination therapy: histone deacetylase inhibitors and platinum-based chemotherapeutics for cancer. Cancer Lett. 2013;329(1):1–8.
- Thurn KT, Thomas S, Moore A, et al. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol. 2011;7:263.
- Luan Y, Li J, Bernatchez JA, et al. Kinase and histone deacetylase hybrid inhibitors for cancer therapy. J Med Chem. 2019;62:3171.
- Li Y, Luo X, Guo Q, et al. Discovery of N1-(4-((7-Cyclopentyl-6-(dimethylcarbamoyl) -7H-pyrrolo[2,3-d]pyrimidin-2-yl)amino)phenyl)-N8-hydroxyoctanediamide as a novel inhibitor targeting cyclin-dependent kinase 4/9 (CDK4/9) and histone deacetlyase1 (HDAC1) against malignant cancer. J Med Chem. 2018;61:3166.
- Huang Y, Dong G, Li H, et al. Discovery of janus kinase 2 (JAK2) and histone deacetylase (HDAC) dual inhibitors as a novel strategy for the combinational treatment of leukemia and invasive fungal infections. J Med Chem. 2018;61:6056.
- Zhang K, Lai F, Lin S, et al. Design, synthesis, and biological evaluation of 4-methyl quinazoline derivatives as anticancer agents simultaneously targeting phosphoinositide 3-kinases and histone deacetylases. J Med Chem. 2019;62:6992.
- Rangasamy L, Ortín I, Zapico JM, et al. New dual CK2/HDAC1 inhibitors with nanomolar inhibitory activity against both enzymes. ACS Med Chem Lett. 2020;11:713.
- Liu J, Qian C, Zhu Y, et al. Design, synthesis and evaluate of novel dual FGFR1 and HDAC inhibitors bearing an indazole scaffold. Bioorgan Med Chem. 2018;26:747.
- Zang J, Liang X, Huang Y, et al. Discovery of novel pazopanib-based HDAC and VEGFR dual inhibitors targeting cancer epigenetics and angiogenesis simultaneously. J Med Chem. 2018;61:5304.
- He X, Zhuo X-T, Gao Y, et al. β-Elemene derivatives produced from SeO2-mediated oxidation reaction. Royal Soc Open Sci. 2020;7:200038.
- Watson PJ, Millard CJ, Riley AM, et al. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat Commun. 2016;7:11262.
- Miyake Y, Keusch JJ, Wang L, et al. Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat Chem Biol. 2016;12:748.