
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Vibrio cholerae causes life-threatening infections in low-income countries due to the rise of antibacterial resistance. Innovative pharmacological targets have been investigated and carbonic anhydrases (CAs, EC: 4.2.1.1) encoded by V. cholerae (VchCAs) emerged as a valuable option. Recently, we developed a large library of para- and meta-benzenesulfonamides characterised by moieties with a different flexibility degree as CAs inhibitors. Stopped flow-based enzymatic assays showed strong inhibition of VchαCA for this library, while lower affinity was detected against the other isoforms. In particular, cyclic urea 9c emerged for a nanomolar inhibition of VchαCA (KI = 4.7 nM) and high selectivity with respect to human isoenzymes (SI≥ 90). Computational studies revealed the influence of moiety flexibility on inhibitory activity and isoform selectivity and allowed accurate SARs. However, although VchCAs are involved in the bacterium virulence and not in its survival, we evaluated the antibacterial activity of such compounds, resulting in no direct activity.
Graphical Abstract

Introduction
The COVID-19 pandemic represented an unanticipated global issue, responsible for over 6.8 M deaths worldwide and a big concern in Public Healthcare in relation to the impairment of human immune defences, thus unable to fight secondary microbial infections that became potentially fatalCitation1. However, besides these direct and economic burdens, the outbreak of COVID-19 was also responsible for the crowding of hospitals and the consequent slowing down of other disease treatments and surgery, such as those related to cancer patientsCitation2. Moreover, underdeveloped countries significantly suffered from the COVID-19 disruption of humanitarian aid programs, fundamental in the life-saving safe water, sanitation, and hygiene (WASH) techniques and the diagnosis, treatment, and vaccination campaigns for those diseases, such as cholera, that are a passed threat for industrialised countriesCitation3,Citation4.
Cholera, also referred to as “blue death”, is an acute diarrheal illness that became fatal if left untreated (50–70% mortality rate) and provoked seven pandemics till the 19th century and a consequent number amounting to an impressive 143 000 deaths per yearCitation5. In recent years, its incidence is most evident in those areas with low hygienic conditions and limited access to safe drinking water and food, in particular after cataclysmic events or during armed conflicts, such as sub-Saharan African countries, i.e. Niger, Nigeria, Ethiopia, and Sudan, India, and BangladeshCitation6–8. The alarming data from these areas envisaged the institutions of a worldwide strategy to control the disease named “Ending Cholera: a global roadmap to 2030” in 2017 aimed at reducing mortality by 90%. However, a significative increase in reported cholera cases was noticed during the COVID-19 pandemic compared to previous yearsCitation7.
Cholera aetiology is related to the infection by the Gram-negative curved rod-shaped Vibrio cholerae, but only two serogroups, O1 and O139, can cause illnessCitation9, characterised by a massive loss of water and electrolytes, resulting in severe dehydration and hypovolemic shock. Thus, the pharmacological therapies are usually based on intravenous rehydration and the administration of antibiotics, such as a single dose of doxycycline as first-line treatment, erythromycin, azithromycin, norfloxacin, ciprofloxacin, and trimethoprim-sulfamethoxazole combinationCitation10. However, several cases of bacterial resistance were reportedCitation11–14 and make the search for new antibiotics and their development urgently needed, although some oral vaccines (the FDA-licensed Vaxchora® and the WHO-prequalified Dukoral®, ShanChol®, and Euvichol-Plus®/Euvichol®) are already in clinical use.
In the last decades, carbonic anhydrases (CAs, E.C. 4.2.1.1) emerged for their multifaceted physio-pathological roles and, thus, their high potential as pharmacological targets in medicinal chemistry and the antibacterial fieldCitation15. CAs are ubiquitous metalloenzymes involved in the physiological balance between carbonic dioxide and bicarbonate anion in the cell, thus governing the pH homeostasis, the secretion of electrolytes, the carboxylation reactions, and other metabolic pathwaysCitation16,Citation17. Herein, the interest in inhibiting CAs with antibacterial purposes is amply justified and recently confirmed in in vivo studiesCitation18–22.
The genome of V. cholerae species encodes for three CAs, belonging to three of the eight different classes of CA isoforms: the α-, also present in mammalians, and the β- and the γ-isoenzymesCitation23,Citation24. Although the role of VchCAs has not been well established yet, accumulating evidence relates their function to the etiopathogenesis of the V. cholerae infectionCitation25. After the invasion of the host and the colonisation of the upper small intestine epithelial layer, the pathogen penetrates the mucus and attaches to the microvilli via the bacterial pili, where it releases its endotoxin (cholera toxin, CT), the main virulence factor, that triggers the increased secretion of water, sodium and potassium cations and bicarbonate anion into the lumen of the intestine, leading to severe dehydrationCitation26. The small intestine maintains an alkaline pH environment through the pancreatic release of sodium bicarbonate, an inducer of the CT expressionCitation25. However, the absence of genes encoding for bicarbonate transporters in V. cholerae lets us hypothesise the use of CAs as a bicarbonate-assimilating system in the cell.
Moreover, as assessed through the STRING web tool (https://string-db.org/ accessed on March 4th, 2022)Citation27, VchCAs participate in several functional and physical protein-protein association networks, such as proteins belonging to the sulfate permease family, those involved in the degradation of long-chain fatty acids, fumarate hydratase, uridine kinase, etc. Relevantly, VchCAs also seem to be associated with the thioredoxin system, fundamental in the physiology and pathogenesis of bacteria due to its influence on the expression of many genes, in the reduction of cytoplasmic proteins and hydrogen peroxide and, in general, cell division, energy transduction, oxidative stress response, transcriptional regulation, phage assembly and propagationCitation28.
After the isolation and characterisation of the three isoenzymes from V. choleraeCitation29,Citation30, several studies have been reported on the development of potent inhibitors, most of them selective, endowed with different chemical scaffoldsCitation31, such as benzoxaborolesCitation32, indole-based hydrazonesCitation33, sulfamides (-NHSO2NH2)Citation34, N-hydroxy-ureasCitation35, and differently substituted benzenesulfonamides designed through the tail approachCitation36–39.
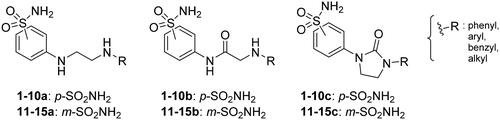
We recently developed a series of benzenesulfonamides as antitumor agents, highlighting, in some cases, a clear trend in the inhibitory profile according to the presence of specific moieties able to confer different rigidity to the chemical structure ()Citation40,Citation41.
Figure 1. Compounds investigated in this work.
However, the compounds showed no higher selectivity towards the enzymes of interest, CA IX and CA XII, with respect to the cytosolic isoforms I and II. Thus, we investigated the possible inhibition of one or more different CAs, such as those from the bacterial V. cholerae.
Results and discussion
Rationale and preparation of the library
The derivatives design was inspired by the tail approach, a MedChem strategy that has been applied for the first time in the CA inhibition field in 1999Citation42,Citation43. In the beginning, it was aimed to enhance the pharmacokinetic properties of such compoundsCitation44, then, it was largely employed to address the isoform selectivity issue. Generally, tailed CA inhibitors are composed of three elements: a zinc-binding group (ZBG, in red, ), a main scaffold with a spacer (in cyan, ), and the hydrophobic tail (in light green, ).
Figure 2. Overview of the tail approach and the design strategy used in the current work.
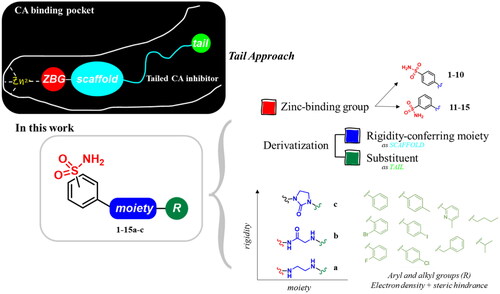
This strategy is based on the idea to allow the compound zinc-binding group to reach the enzyme metal in the binding cavity and to establish a complex and well-defined interaction network with the isoform-specific enzyme residues (most of which are hydrophobic) in the entry of the binding site through the scaffold and tail portions.
In the current work, the design of derivatives 1–15a–c involved the use of 4- and 3-benzenesulfonamides as zinc-binding groups (in red, ) and different derivatization elements as binary systems composed of flexibility/rigidity-conferring moiety (in blue, ), such as the amine (a), the amide (b), and the cyclic urea (c) function (in increasing order of rigidity in ), in lieu of the traditional scaffold-spacer component, and a specific aryl or alkyl substituent (R, in green, ) endowed with different electron density and steric hindrance properties as the compound tail.
Aimed at exploring the enzymatic inhibitory profile of the library of compounds 1–15a–c and discovering promising activities for further development as antibacterial agents, we evaluated their ability to inhibit the three CAs from the pathogen V. cholerae. Compounds 1–15a–c were prepared as reportedCitation40.
In vitro inhibition of VchCAs and preliminary SAR considerations
The inhibition profiles for sulfonamides 1–15a–c and the reference AAZ against α-, β-, and γ-CAs from V. cholerae were determined through the stopped-flow CO2 hydrase assayCitation45, and as a comparison, inhibitory data on the physiologically relevant hCAs I and II are reported as inhibition constants (KIs) ().
Table 1. Inhibition data of sulfonamides 1–15a–c and reference compound AAZ on hCAs I and II and VchCAs through the stopped-flow CO2 hydrase assay. 
Based on the reported inhibition data, several structure-activity-relationship (SAR) considerations can be drawn, especially regarding inhibition spectrum and selectivity. Overall, all the compounds showed a nanomolar inhibitory activity against hCAs I and IICitation40.
Furthermore, the assessment of the inhibitory activity on α-CA from V. cholerae highlights the structural differences among the binding pockets of the human and bacterial enzymes. The in vitro experiments seem to confirm the bigger size of the VchαCA binding pocket, as outlined by the good activity of the 4-iodophenyl (4a) and 2-bromophenyl (5a) derivatives. Remarkably, pyridinyl-bearing compound 7b results to be the most active among the amido library (scaffold b), while the bulky alkyl substituent of 10a confers a higher potency to the amino para-series (scaffold a). The rigidity of scaffold c contributes to reducing the KI values, resulting in notable inhibitory profiles for almost all the para-sulfonamides and monosubstituted phenyl derivatives 12–14c. In general, all tested cyclic compounds were found active in inhibiting the target enzymes, even if they failed to reach the high potency of AAZ in some cases, especially for those endowed with scaffolds a and b.
The whole set of compounds exerts a less potent inhibitory activity on the β-CA isoform of V. cholerae, as shown by the KIs in the micromolar range. Low activity was found for halophenyl derivatives characterised by a scaffold (4–6a and 14a). Also, the same trend of inhibition was observed for the acyclic derivatives, highlighting a better activity profile of the bulky 10a than the linear derivatives 9a and 15a. Regarding the aromatic moieties on scaffold b, the unsubstituted phenyl ring positively affects the activity, conferring the best inhibitory profile among the para-series (1b) and the worst among the meta-series (11b). However, the most active amide is the i-propyl compound 10b with a KI value of 4.0 µM.
Finally, the tested compounds show a very low (in the micromolar range) activity on the γ isoform. In particular, in the para-benzenesulfonamide amine series (scaffold a) the inactivity of the phenyl derivative 1a seems to be restored only in the presence of an electron donor (methyl) substituent on the ring (2a), while the introduction of one halogen in para or ortho position (3–5a) still corresponds to a lack of activity, except for the fluorophenyl compound 6a. Not relevant KI values were also detected on the other derivatives. Inactivity also for the phenyl derivative in the meta-a series (11a), but this time, restored by tolyl moiety in 12a and still improved by the chlorophenyl derivative 13a. The butyl tail improves the VchγCA inhibition in the meta-series (15a) with respect to the para- one (9a). In the b library, the only interesting compound, with a KI of 9.6 µM, bears the tolyl ring (12b) in the meta-series. The cyclic ureas (scaffold c) show notably low micromolar activities, especially for the unsubstituted phenyl derivatives (1c and 11c).
The panel of tested CAs in seems to suggest a promising development of this library of derivatives against V. cholerae enzymes. In fact, although the moderate-low activity against VchβCA and VchγCA, a relevant inhibitory profile emerged for several compounds and, in some cases, a good selectivity on the bacterial α isoform with respect to the human ones is gained. Indeed, selectivity index (SI) values on hCA I reported in show that the presence of a 4-chlorophenyl ring and the n-butyl tail are helpful to address the activity on VchαCA for all the investigated scaffolds, while the tolyl group helps to gain selectivity only in the meta-series also towards the human isoform II. Considering the aromatic groups, the hCA I/VchαCA selectivity is also reached by derivatives endowed with a phenyl ring on scaffolds b and c and the pyridinyl and benzyl functions in the para-series of scaffolds a and c. However, several compounds display a good hCA II/VchαCA selectivity, even if with a different trend. Overall, the amino compound 10a, the amido 4b, and the ureidic 9c and 12c emerged as the most selective derivatives, with higher human/bacterial SI than AAZ.
We could argue that the increase in rigidity from the most flexible amino tail (scaffold a) to the amido (b) and cyclic urea (c) functions corresponds to an enhancement of the inhibitory activity of this class of compounds, maybe due to a different binding mode. However, clear evidence of the optimal sulfonamide moiety (meta or para) position on the structural nucleus could not be found from the in vitro assay results. In general, it is not trivial to draw accurate SARs with a large amount of experimental data, and a computational structure-based study of compounds into the binding pockets of the targeted enzymes could help the understanding of their inhibition and affinity.
In silico studies
Flexibility properties calculation
The structural flexibility of studied ligands was assessed through the FAFDrugs4 webserver (FAFDrugs4, https://fafdrugs4.rpbs.univ-paris-diderot.fr/)Citation46, considering the number of rigid and flexible bonds as established by Veber rulesCitation47. Examples of the calculated values are reported in .
Table 2. Flexibility properties of compounds 1a–c and 9a–c, selected as representative of the whole library of derivatives, calculated through FAFDrugs4 webtool.
Observing data in , the same trend was noticed for both the phenyl substituted derivative 1 and the alkyl compound 9, reported as representatives of the whole library of compounds. The flexibility value decreases within the series a–b–c from the amine (a) to the urea (c) in correspondence to the increase in the number of the rigid bond. However, very different values were obtained for 1 and 9, since the aliphatic tail belonging to the latter is very flexible with respect to the aromatic ring of 1. In fact, as expected, the alkyl derivatives 9a–c hold a flexibility value higher than both 1b and 1c. In this study, we tried to better understand how this broad range of flexibility of the tails, also considering the different interactions they can establish with the enzyme residues, could affect the affinity and, thereby, the inhibitory activity of the compound.
Molecular docking and MD simulations on the VchαCA
To shed light on the binding mode of our benzenesulfonamide inhibitors, structure-based studies of all compounds in VchCAs and hCAs were conducted.
Since the 3D coordinates of VchαCA are not included in the PDB database, a homology modelling protocol has been applied. The VchαCA model was based on the experimental coordinates of Photobacterium profundus (PDB: 5HPJ), showing the highest sequence identity (64.75%). During our study, the 3D structure of this protein predicted by AlphaFold2Citation48 was made available in the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/) and was compared with the structure generated by homology modelling. The RMSD value for VchαCA was 0.838 Å underpinning the validity of our model. To optimise residue positioning around the sulfonamide group, the AAZ geometry derived from the crystallographic complex with the αCA from Helicobacter pylori (PDB: 4YGF) was placed in the binding site after the proper alignment of the three zinc-coordinating histidine residues (His104, His106, and His123). To relax the protein with the known sulfonamide, the complex of the HM-VchαCA and AAZ was minimised, and the obtained protein was used for the docking studies.
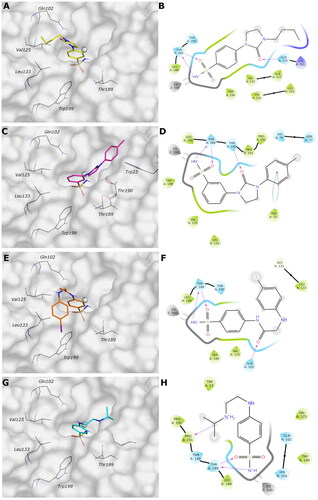
The docked poses reveal that all compounds assume a similar placement in the active site. The sulfonamide NH group, negatively charged, coordinates the zinc ion by a tetrahedral geometry and establishes an H-bond with the hydroxyl group of Thr189, while one of the sulphonyl oxygen H-bonds to the NH of the Thr189 backbone. We noticed a different binding mode depending on the position of the substituent: most para-substituted compounds point towards the VchαCA coil (residues 128–132) (), while meta-substituted compounds move in the opposite direction ().
Considering the linker between the benzenesulfonamide portion and the R-group, more constrained compounds bearing the cyclic urea (1–15c) show a further H-bond between the cyclic carbonyl and, alternatively, Gln102 for para-substituted (9c, ), or Thr190 backbone NH for meta-substituted (12c, ). Compounds containing the amide linker show the same H-bond with Gln102 for para-sulfonamides, while the H-bond with Thr190 was conserved only for 4b for meta-substituted ().
In the para-substituted series with the more flexible linker, the amine group, only compound 10a () makes an additional H-bond between the distal amine and the Pro190 carbonyl, preferring a different placement on the active site with respect to the other para-compounds. To obtain more insight into the binding process, 100 ns MD simulations on the VchαCA complexes with most selective inhibitors 9c, 12c, 4b, and 10a were carried out ().
Figure 3. (A, C, E, and G) Predicted 3D binding mode and (B, D, F, and H) corresponding ligand interaction diagram of most selective ligands (A, B) 9c yellow, (C, D) 12c magenta, (E, F) 4b orange, and (G, H) 10a cyan, within VchαCA (light grey). The compounds are represented as sticks, and the protein surface is visualised.
MD simulation analysis highlights the good stability of the studied complexes. The RMSD values calculated on the protein VchαCA are relatively stable along the simulation and lower than 2.5 Å for all complexes. The same parameter calculated for ligands shows larger values, indicating a change in all ligands’ initial binding conformation maintained along the production phase. The distal substituent to the benzene ring, exposed to the solvent, is the less stable part of the molecules during the simulation and is in charge of raising the RMSD value of the ligands as underlined by the RMSF values (Figures S1–S2 in Supporting Information). On the contrary, the benzenesulfonamide portion of all analysed ligands is firmly bound to the zinc ion and to Thr189 via a hydrogen bond. The most constrained compounds, 9c (KI = 4.7 nM) and 12c (KI = 11 nM), do not establish a π–π stacking contact with the zinc-coordinated His104 as for 4b and 10a, probably due to the steric hindrance of the 1,3-imidazolidin-2-one, but the carbonyl urea finds a water bridge interaction with Pro191 (Thr190) or Tyr25.
For ligand 9c (), the protein average RMSD is close to 1.4 Å, whereas the ligand RMSD is 2.8 Å. During the simulation, the H-bond of the carbonyl urea with Gln102 is maintained at a very low percentage and is replaced by a water-bridged interaction with persistence of 22% (). The 1,3-imidazolidin-2-one turns in the opposite direction of Gln102 to find a water bridge interaction between the carbonyl oxygen and Pro191 (53%) or Thr190 (46%), confirming its conserved involvement in HB interactions even with different residues. Few hydrophobic interactions with residues of Val125, Leu133, and Leu188 are present in line with the docking study, and the n-butyl portion is exposed to the solvent ().
Figure 4. Most representative geometry retrieved after MD simulation for the four studied ligands. (A) 9c yellow, (B) 12c magenta, (C) 4b orange, and (D) 10a cyan in the VchαCA (light grey). The compounds are represented in stick and the protein surface is visualised.
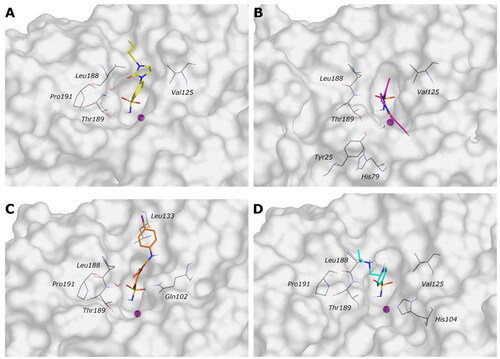
The analysis of the MD simulation of ligand 12c () reveals an average protein RMSD of 1.6 Å, whereas the average ligand RMSD is 2.6 Å. Similar to the previous inhibitor, the H-bond between the carbonyl group and Thr190 is maintained only for 20% of the simulation, while the 1,3-imidazolidin-2-one slightly turns to find a water bridge with Tyr25 (29%). The π-π stacking contact between the distal phenyl and Trp23 is similarly maintained for a very short time during the simulation, and it is replaced by a π-π stacking contact with His79 (20%, ), following the rotation of the linker.
For ligand 4b (), the average protein RMSD is 1.5 Å, whereas the ligand RMSD is 2.5 Å, with a significant fluctuation during the simulation caused mainly by the rotation of the 4-iodophenyl ring that flips between two different positions, as highlighted by the RMSD profile. As for the previous compounds, the benzenesulfonamide function coordinates the zinc ion, but there is also evidence of a π-π stacking between the benzene ring and His104 (27%). The carbonyl amide forms a water bridge interaction with Pro191 (36%) or Thr190 (24%), while the interaction with Gln102 is maintained for less than 20% of the production phase (), as for compound 9c. The distal portion of the molecule is exposed to the solvent.
For the less restricted compound 10a (), the protein average RMSD is 1.7 Å, whereas the ligand RMSD is 2.1 Å, with the benzene sulfonamide substituent moving quite enough during the simulation. Other than the expected interactions of the sulfonamide with the zinc ion and Thr189, a π–π stacking contact between the benzene ring and His104 is present, also seen in the compound with the amide linker (4b). The interaction between the distal amine and Pro191 found in the docking study is maintained only for 20% of the simulation. The few hydrophobic interactions involve the residues Val125 and Leu188, whereas the linker and the isopropyl remain exposed to the solvent ().
Docking on the hCA I and hCA II
One of the current project’s key aspects is the identification of compounds that selectively inhibit VchCAs, sparing hCAs. To find out the critical features for the selectivity over, in particular, the VchαCA, a docking protocol has been carried out on the human CA isoforms I and II (PDB ID: 6IOJ and 3K34, resolution 1.35 and 0.90 Å, respectively).
The analysis of the docking poses reveals that all compounds successfully adopt the correct geometry of the sulfonamide function that coordinates the zinc ion and a π–π stacking contact of the phenyl ring with His94 (hCA I and hCA II) for most compounds. As for VchαCA, hydrophobic interactions are not prevalent, and the linker and the tail of most compounds are exposed to the solvent.
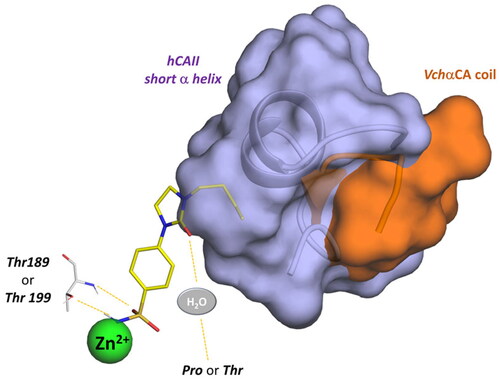
The active sites of hCA I and hCA II are superimposable to the active site of VchαCA, except for the α-helix comprising residues 124–140 for hCA I and hCA II, which is much shorter in VchαCA and forms a coil (residues 128–132)Citation49. This determines a partial restriction at the edge of the active site of hCAs, causing a different placement of the inhibitor tail ( and , Figures S3 and S4).
Figure 5. (A, C, E, and G) The 2D representation of most conserved ligand–protein interactions with (B, D, F, and H) the indication of the persistence (%) along the simulation and depiction of frequency and type of ligand-protein interaction along with the MD simulation. (A, B) 9c, (C, D) 12c, (E, F) 4b, and (G, H) 10a.
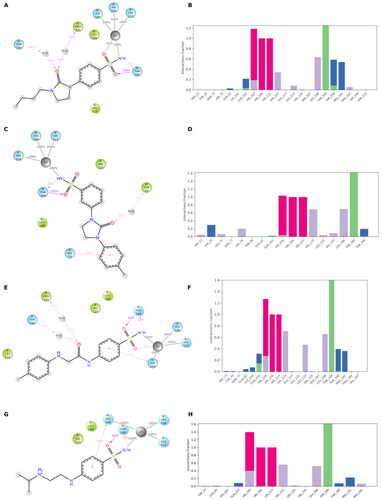
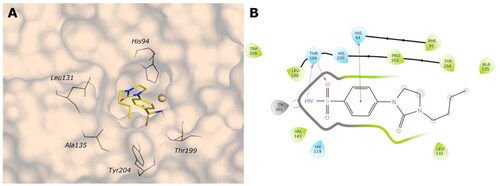
Figure 6. (A) Predicted 3D binding mode and (B) corresponding ligand interaction diagram of the most selective ligands 9c (yellow) in the hCA I (light salon). The compound is represented as the stick and the protein surface is visualised.
Docking on the VchβCA
Even if the activity of compounds results in the micromolar order on the VchβCA, we investigated the binding mode of the studied compounds in the narrower active site of this isoform. Starting from the coordinates of the “closed” form of VchβCA (PDB: 5CXK), the “open” form was generated by trimming the fundamental residue AspA44 during the Induced Fit protocol to obtain the conformational changes in the target sidechain that can resemble the “open” conformation of the VchβCA. The obtained conformation of this residue was verified by overlapping the obtained protein with the crystal structure of the “open” β-CA of Aspergillus fumigatus (PDB: 7COJ). The coordinates of the cocrystallized AAZ in complex with β-CA of Coccomyxa (PDB: 3UCJ) were used to minimise the obtained “open” form of VchβCA and optimise the residue positioning around the sulfonamide function. The minimised protein was then used to dock all ligands into the active site.
The docked poses of all compounds confirm that the sulfonamide group is coordinated and correctly oriented to the zinc ion, and the benzene ring interacts with TyrB83 by π–π stacking, reproducing the typical orientation of benzenesulfonamide inhibitors. Other fundamental contacts are with the hydrophobic pocket at chains A and B interface, involving GlyA102, GlyA103, AlaA106, ProA113, and LeuA113.
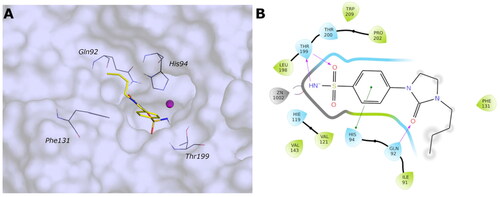
The docking results do not demonstrate different poses depending on the type of linker (amine, amide, or cyclic urea) or the para or meta position of the R-group. Even though the correct placement of the benzenesulfonamide group in the tight cleft of the VchβCA, none of the compounds can establish more favourable interactions further than hydrophobic. The predicted binding mode and the ligand interactions of 9c within the VchβCA active site are represented in .
Figure 7. (A) Predicted 3D binding mode and (B) corresponding ligand interaction diagram of the most selective ligand 9c (yellow) in the hCA II (light cyan). The compounds are represented as a stick and the protein surface is visualised.
Docking on the VchγCA
The 3D structure of VchγCA was generated by homology modelling using the 3D crystal structure of γ-CA from Escherichia coli (PDB: 3TIO, identity 64.16%) as the template. The active site, located at the interface of two chains of the trimer of the VchγCA, can exist in the “open” or “closed” conformation as already observed for the β isoform. The open form between chains D and F of 3TIO was chosen to model corresponding chains of the homologous VchγCA. The 3D structure produced by homology modelling was compared with that predicted by AlphaFold2Citation48 and is available in the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/). Also in this case, the validity of our model was supported by the low RMSD value (0.772 Å) obtained by superimposing the two VchγCA structures. As no benzenesulfonamide X-ray ligands are known to bind γ-CAs, we used the previously studied 4-(hydroxymethyl)benzenesulfonamideCitation31 as a reference to validate our model and optimise residues surrounding the sulfonamide portion. This compound was docked into VchγCA, and the best-docked pose reveals the correct coordination geometry with the zinc ion, two H-bonds between the sulfonamide group and GlnF61, the H-bond between the hydroxyl and AspD114, and hydrophobic interaction with LeuD113, MetF108, LeuF105, TyrF168, similar to those previously obtainedCitation31. The complex of the best-docked pose of 4-(hydroxymethyl)benzenesulfonamide and the HM-VchγCA was minimised to optimise residue positioning around the ligand. The obtained protein was then used to dock all compounds. The binding mode of all sulfonamide inhibitors in the active site replicates the interaction with zinc ion and GlnF61. For compounds containing the amine linker (1–15a), the residue AspD114 is involved in an H-bond with the distal or proximal amine of ligands of the series meta- and para-substituted.
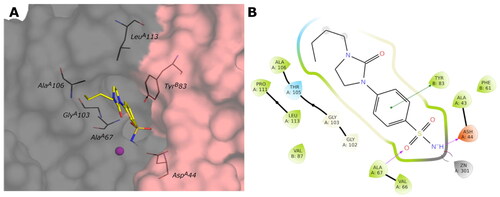
The para-substituted inhibitors with the amide linker (1–15b) deploy in the cylindrical active site interacting with the MetF108 NH by the carbonyl group of the amide linker or with the carboxyl group of AspD114 by the NH of the amide. Only compound 15b of the meta-substituted inhibitors interact with AspD114 with the amide and amine groups. The inhibitors characterised by the cyclic urea, in addition to the interactions of the sulfonamide group, engage only hydrophobic contacts with LeuF105, GlyF107, MetF108, LeuF141, MetF143, LeuD113, ProD132. shows the predicted binding mode and the ligand interactions of ligand 9c within the VchγCA active site.
Figure 8. (A) Predicted 3D binding mode and (B) corresponding ligand interaction diagram of the most selective ligand 9c (yellow) within VchβCA (grey and pink). The compound is represented as a stick and the protein surface is visualised.
In summary, the computational analysis suggests that the structural determinants contributing to ligand selectivity towards VchαCA with respect to hCAs lie in the tail portion of arylsulfonamide ligands. Arylsulfonamides are well-known binders of αCAs, interacting with the zinc ion via the negatively charged NH− group and establishing well-conserved H-bonds with a threonine residue. On the other hand, the rigidity of the linker increases the binding affinity towards all αCAs, both the human and bacterial isoenzymes. A more promising activity profile is accompanied by an H-bond acceptor in the rigid linker that directs the tail portion towards the VchαCA coil (residues 128–134). The corresponding region in the human CAs is occupied by a short α-helix comprising residues 121–140 and constituting the most diverse region between human and bacterial αCAs ().
Figure 9. (A) Predicted 3D binding mode and (B) corresponding ligand interaction diagram of the most selective ligand 9c (yellow) within VchβCA (grey and pink). The compound is represented as a stick and the protein surface is visualised.
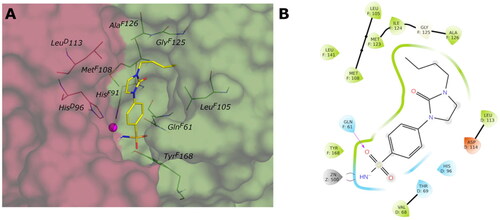
Figure 10. Schematic representation of the ligand contacts producing the selectivity towards VchαCA with respect to hCAs. The compound (9c) is represented as sticks and the VchαCA coil and the short alpha helix hCAII (similar to hCAI) are visualised as cartoon and surface.
Physicochemical and pharmacokinetic properties calculation
Parameters affecting the drug-likeness and bioavailability of the studied sulfonamides were predicted by using QikProp calculationsCitation50 (Table S1 in Supporting Information).
Considering these compounds should act locally in the intestinal tract, we focussed on properties favouring their low absorption. It is known from the literature that compounds working locally in the gutCitation51 are usually polar, with a high molecular weight (MW) and polar surface area (PSA). In this case, focussing on the most selective inhibitors, it is possible to highlight their compliance with Lipinski’s Rule of five, their limited permeability of the gut–blood barrier, and the blood-brain barrier (BBB). Some concerns are due to the possible binding to HERG. shows the essential properties for the most selective compounds.
Table 3. Physicochemical and pharmacokinetic properties of the studied ligands.
Direct-acting antibacterial activity
Most of the compounds (1a, 2c, 3a–c, 4–5c, 6b–c, 7c, 8–9a–c, 10c, 11a–c, 12c, 13a–c, 14a, 15a) and AAZ were tested on three different clinical isolates of V. cholerae, namely SI-Vc22, SI-Vc71, and SI-Vc912, to assess their direct antibacterial activity. Using an agar diffusion method (ampicillin, chloramphenicol, and ciprofloxacin were used as reference antibiotics), only twelve (amines 8–9a, 11a, and 13a, amides 3b, 9b, and 11b, ureas 4–7c and 9c) out of 28 tested compounds showed a moderate growth inhibition against at least one of the clinical isolates. Five compounds (amines 9a, 11a, and ureas 4c, 6c, 9c) were found to be moderately active on all the isolates (inhibition zone diameter in the range of 3–5 mm; data not shown).
However, the determination of the minimum inhibitory concentrations (MICs) (broth microdilution method) confirmed the very low direct-acting activity of these compounds, as MIC values were ≥64 µg/mL. These data are not entirely unexpected considering that the inhibition of VchCAs would not result to be to be detrimental for the bacterium in vitro, but would be important in vivo, especially for the onset of virulence, whose study would require far more complicated biological investigations. However, and since we can now confirm the lack of a direct antibacterial activity for all the derivatives, it would be extremely interesting to assess whether these compounds could inhibit the production of the active exotoxin (e.g. with gene reporting, biochemical or cellular assays) to verify this hypothesisCitation52.
Conclusions
Herein, we reported the design strategy of CA inhibitors bearing conformationally restricted alkyl/aryl amines and amides into imidazolidinones. These para- or meta-benzenesulfonamides are ad hoc characterised by differential rotational features, leading to highly potent (nanomolar) and selective compounds acting preferentially against VchαCA with respect to β- and γ-isoforms.
Stopped flow-based enzymatic assays showed that all the compounds, with properly inter-series differences, are able to strongly inhibit VchαCA, with the following progressive inhibitory potency: meta-benzenesulfonamides < para-benzenesulfonamides and amine < amide < cyclic urea.
Although the above-mentioned trend was not completely respected and it resulted to be quite different for some substituents, we could notice that the more the linker is rigid (urea) the more the inhibition is strong. Thus, by performing these preliminary SAR considerations, we could assume that the activity profile of the derivatives library is strictly linked to the different rigidity of the compounds tail. After a preliminary calculation of the flexibility properties of representative compounds, we find out how they affect the affinity of the whole set of compounds for the different enzyme catalytic sites and, thereby, their inhibitory activities. In particular, we investigated their binding poses in both the human and bacterial enzymes through an in-depth structure-based computational study. Moreover, MD simulations of the most selective inhibitors of VchαCA revealed the high stability of the benzenesulfonamide core in the interaction with the zinc ion, assuming the usual coordination geometry. The structural determinants able to guarantee a proper interaction with the active site were found to be (i) the presence of the urea carbonyl group in the linker (9c), able to establish a water-mediated hydrogen bond with Pro191 or Thr190, and (ii) the constraint of the linker in cyclic urea, producing the highest selectivity over the hCA I and II.
In silico, ADME was also investigated to assess the physicochemical properties of the benzenesulfonamide derivatives and the compounds were found to be in accordance with Lipinski’s rule, even if they seem to suffer from a low oral bioavailability. Keeping into consideration the localisation of our target, this property can be considered fruitful to avoid systemic distribution and off-target interaction.
Although not entirely unexpected, antibacterial susceptibility assays on three different epidemiologically unrelated V. cholerae strains confirmed the lack of direct-acting antibacterial activity of these compounds. Further characterisation of these compounds would be required to evaluate their potential inhibition of the production of virulence determinants, of primary importance in the host environment, although this study is beyond the scope of the present work.
In summary, a combined analysis of inhibitory enzymatic activity and in silico simulations of a large library of benzenesulfonamide derivatives revealed and rationalised specific SARs regarding the rigidity/flexibility properties of the compounds tail. Thus, thanks to our protocol, we screened 45 benzenesulfonamides decorated with three different moieties (amine, amide, and cyclic urea) and a number of decorated tails against the three CA expressed by V. cholerae pathogen though the versatile and high-throughput stopped-flow technique. The obtained KI values highlighted interesting activity profile for several derivatives and notable isoform selectivity was found for at least one compound for series, such as the 4-benzenesulfonamides with amino (10a), amido (4b), and the cyclic urea (9c) moieties and only one 3-benzenesulfonamide derivative (12c).
Further efforts will be focussed on the assessment of the antibacterial behaviour of these compounds in an infection model to evaluate their activity towards the virulence and pathogenicity of the bacterium.
Material and methods
Preparation of the derivative libraries
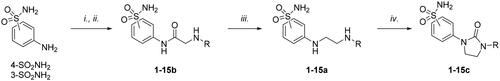
The compounds were prepared as reported according to the general synthetic approach reported in Scheme 1Citation40.
Scheme 1. Synthesis of compounds 1-15a–c. Reagents and conditions: (i) chloroacetyl chloride, dry acetone, N2, 0 °C, 0.5 h; (ii) appropriately substituted aniline, KI, sealed tube, dry THF, N2, 110 °C, 24 h; or 2-amino-6-methylpyridine, dry TEA, abs EtOH, N2, ref., 24 h; or benzylamine, dry TEA, dry ACN, N2, 24 h; or amine, KI, dry THF, N2, 24 h; (iii) 1 M BH3·THF, dry THF, N2, r.t., 24 h; or LiAlH4, dry THF, N2, 0–70 °C, 24 h; (iv). triphosgene, dry TEA, dry THF, N2, r.t., 2 h.
In vitro carbonic anhydrase inhibition assay
The CA-catalyzed CO2 hydration activity was performed on an Applied Photophysics stopped-flow instrument using Phenol Red, at a concentration of 0.2 mM, as a pH indicator working at the maximum absorbance of 557 nmCitation53,Citation54 with 20 mM HEPES (pH 7.40 for α- and 8.40 for β-CAs and γ-CAs) as the buffer, 20 mM Na2SO4 to maintain constant ionic strength, and following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10 − 100 s and The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. Enzyme concentrations ranged between 5 and 12 nM. For each inhibitor, at least six traces of the initial 5 − 10% of the reaction have been used to determine the initial velocity. The uncatalyzed reaction rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water, and dilutions up to 0.01 nM were prepared. Solutions containing inhibitor and enzyme were preincubated for 15 min at room temperature prior to performing the assay to allow the formation of the E − I complex. The inhibition constants were obtained by nonlinear least-squares methods using PRISM 3Citation29 and the Cheng-Prusoff equation as reported earlier and represent the mean from at least three different determinations. All CA isoforms are recombinant and obtained in-house, as reported earlierCitation29.
In silico studies
Calculation of flexibility properties of the derivatives
Flexibility and numbers of rotable/rigid bonds were calculated through FAFDrugs4 (Free ADME-Tox Filtering Tool)Citation46.
3D protein structure retrieval
Molecular modelling studies were performed on Schrödinger Life-Sciences Suite 2021–4Citation50. All ligands were drawn as 2D structures from Maestro and prepared by using LigPrep to generate the 3D geometry and find all possible tautomers and protonation states at pH 7.0 ± 0.4 with EpikCitation55,Citation56. The three-dimensional X-ray structures of hCA I and hCA II were retrieved from the Protein Data Bank (PDB ID: 6IOJ and 3K34, resolution 1.35 and 0.90 Å, respectively)Citation57,Citation58. The Protein Preparation workflow was used to correct, optimise and minimise the crystal structures. The crystal structure of VchβCA was retrieved from the Protein Data Bank (PDB ID: 5CXK, resolution 1.90 Å)Citation59. The “open” catalytic site of VchβCA was realised starting from the crystallographic “closed” form of the crystal structure. The Induced-fit protocolCitation60–62 was used by employing GlideCitation63–65 and PrimeCitation66,Citation67 software with the OLPS4 force field, and the fundamental residue AspA44 was trimmed to obtain the conformational changes in the target sidechain that can resemble the “open” conformation of the VchβCA. The resulting conformation of the AspA44 side chain was checked by overlapping it to the “open” VchβCA of the fungal pathogen Aspergillus fumigatus (PDB: 7COJ)Citation68.
Homology modelling
The 3D structures of VchαCA and VchγCA were obtained by homology modelling. The primary sequences of VchαCA (Uniprot ID: A0A0H6VI20, 249 aa) and VChγCA (Uniprot ID: A0A0H6TVJ0, 184 aa) were retrieved from the UniProt KnowledgeBase (UniProtKB) databaseCitation69. The protein sequence was used as a query sequence for homology modelling using Prime. This tool used BLASTCitation70 to identify suitable templates from Protein Data Bank (PDB) using a single template protocol. The homology models of VchαCA and VchγCA were obtained on the structure of αCA from Photobacterium profundus (PDB: 5HPJ, identity 64.75%) and the structure of γ-CA from Escherichia coli γCA (PDB: 3TIO, identity 64.16%) as the template, respectivelyCitation71,Citation72. For VchγCA, the chains D-F were used as the template as they present the catalytic site in the “open” form. The crystallographic AAZ ligand coordinates taken from the complex with Helicobacter pylori αCA (PDB: 4YGF) and the 4-(hydroxymethyl)benzenesulfonamideCitation31 were added as ligands to the homology models of α and γ isoforms, respectively. The protein-ligand complexes were fully minimised by using MacroModel applying the OPLS4 force field, 5000 steps of PRCG minimisation algorithm with a convergence criterion of 0.05 KJ/mol Å, to optimise the residue positioning around the sulfonamide function.
Docking calculations
Molecular docking analyses were performed using the Glide software. The Glide Grids were generated by positioning the enclosing boxes on the centre of mass of the respective sulfonamide ligands. The SP docking protocol was used by setting 5000 poses per ligand for the initial phase and 400 poses per ligand for energy minimisation with the OPLS4 forcefield. Rotatable groups were defined for each protein: Thr189 for VchαCA, AspA44 for VchβCA, ThrD69 for VchγCA, and Thr199 for hCAs. Additionally, a core constraint on the sulfonamide positioning was applied in the docking protocol into the VchαCA and VchβCA.
Molecular Dynamics
Molecular Dynamics simulation was carried out using DesmondCitation50,Citation73. The complexes of VchαCA with the docked poses of compounds 9c, 12c, 4b, and 10a were embedded in an orthorhombic box of TIP4P water molecules resulting in systems of 28 787, 28 824, 28 748, 28 840 atoms, respectively. In order to balance the system charge, sodium, and chlorine ions were added. The systems were relaxed by applying the default relaxation protocol before the production phase. The simulation duration was set to 100 ns registering frames every 100 ps. The OPLS4 force field, a normal pressure-temperature (NPT) ensemble with a Nose–Hoover thermostat set to 300 K and a Martyna–Tobias–Klein barostat set to 1.01325 bar pressure were used. Electrostatic interactions were examined by applying the smooth particle mesh Ewald method. Figures were generated using Maestro and PyMoLCitation74.
QikProp calculations
Physico-chemical and pharmacokinetic parameters were calculated using QikPropCitation50 and applying the default parameters.
Antibacterial susceptibility testing
Epidemiologically unrelated clinical isolates of V. cholerae (SI-Vc22, SI-Vc71, and SI-Vc-912) were obtained from the collection of the Department of Medical Biotechnologies (University of Siena, Italy). Compounds were resuspended in DMSO at a final concentration of 50 mg/mL. For insoluble compounds, DMSO was further added to lower the concentration until complete solubility of the compound (final concentrations, 25, 12.5, or 6.25 mg/mL). The direct antibacterial activity was evaluated using an agar diffusion-based methodCitation75. Briefly, Mueller-Hinton agar plates were inoculated with a bacterial suspension containing ≈1.5 × 108 CFU/mL of the tested strain. 2 μL of each compound solution were spotted on the surface of the inoculated medium and incubated at 37 °C for 24 h. Controls included the vehicle (100% DMSO) or 2-µL spots of an antibiotic (ampicillin, chloramphenicol and ciprofloxacin) solution in sterile milliQ water (5 mg/mL, i.e. each spot contained 10 µg of the antibiotic). The results were recorded as the diameter of the growth inhibition zone.
MIC values of the compounds were determined using the broth microdilution method as recommended by Clinical Laboratory Standards InstituteCitation76. Bacterial inoculum was 5 × 104 CFU/well. MICs were recorded after 18 h of incubation at 35 °C.
Authors contributions
The manuscript was written through the contributions of all authors. All authors approved the final version of the manuscript.
Supplemental Material
Download PDF (1 MB)Acknowledgements
This research was partially supported by the Italian Ministry for University and Research (MIUR) grant number FISR2019_04819 BacCAD to S.C., C.C., and C.T.S. This work (J.D.D.) was supported also in part by the Italian MUR (Ministero dell’Università e Ricerca) in the frame of the PNRR PE-13 (“Piano Nazionale di Ripresa e Resilienza, Partenariato Esteso 13, Malattie infettive emergenti”) INF-ACT project (One Health Basic and Translational Research Actions addressing Unmet Needs on Emerging Infectious Diseases). Thanks are due to Dr. Stefania Cresti (Department of Medical Biotechnologies, University of Siena & Azienda Ospedaliero- Universitaria Senese, Siena, Italy) and to Tiziana Di Maggio and Prof. Lucia Pallecchi (Department of Medical Biotechnologies, University of Siena, Italy) and Prof. Gian Maria Rossolini (Department of Experimental and Clinical Medicine, University of Florence, Florence, Italy; Clinical Microbiology and Virology Unit, Careggi University Hospital, Florence, Italy) for providing the bacterial strains used in this work.
Disclosure statement
C.T. Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript.
Additional information
Funding
References
- Silva DL, Lima CM, Magalhães VCR, Baltazar LM, Peres NTA, Caligiorne RB, Moura AS, Fereguetti T, Martins JC, Rabelo LF, et al. Fungal and bacterial coinfections increase mortality of severely ill COVID-19 patients. J Hosp Infect. 2021;113:145–154.
- Bardet A, Fraslin AM, Marghadi J, Borget I, Faron M, Honoré C, Delaloge S, Albiges L, Planchard D, Ducreux M, et al. Impact of COVID-19 on healthcare organisation and cancer outcomes. Eur J Cancer. 2021;153:123–132.
- Uwishema O, Okereke M, Onyeaka H, Hasan MM, Donatus D, Martin Z, Oluwatomisin LA, Mhanna M, Olumide AO, Sun J, et al. Threats and outbreaks of cholera in Africa amidst COVID-19 pandemic: a double burden on Africa’s health systems. Trop Med Health. 2021;49(1):93.
- D'Mello-Guyett L, Gallandat K, Van den Bergh R, Taylor D, Bulit G, Legros D, Maes P, Checchi F, Cumming O. Prevention and control of cholera with household and community water, sanitation and hygiene (WASH) interventions: A scoping review of current international guidelines. PLOS One. 2020;15(1):e0226549.
- WHO. [accessed 2023 Jan 31]. https://www.who.int/news-room/fact-sheets/detail/cholera.
- ECDC. [accessed 2023 Jan 31]. https://www.ecdc.europa.eu/en/all-topics-z/cholera/surveillance-and-disease-data/cholera-monthly.
- Owoicho O, Abechi P, Olwal CO. Cholera in the era of COVID-19 pandemic: a worrying trend in Africa? Int J Public Health. 2021;66:1604030.
- Oladapo RK, Oyetola AB, Obidiro OP, Olajide A, Osuagwu-Nwogu E, Olaitan OM, Ngokere C, Monisola I, Ibraheem B, Afolayan A. Rising cholera cases: harnessing the momentum of COVID-19 to strengthen Nigeria’s health systems. Int J Health Plann Manage. 2021;36(6):2030–2034.
- Faruque SM, Ahmed KM, Siddique AK, Zaman K, Alim AR, Albert MJ. Molecular analysis of toxigenic Vibrio cholerae O139 Bengal strains isolated in Bangladesh between 1993 and 1996: evidence for emergence of a new clone of the Bengal vibrios. J Clin Microbiol. 1997;35(9):2299–2306.
- CDC. [accessed 2023 Jan 31]. https://www.cdc.gov/cholera/treatment/antibiotic-treatment.html. (accessed on 31/01/2023).
- Das B, Verma J, Kumar P, Ghosh A, Ramamurthy T. Antibiotic resistance in Vibrio cholerae: understanding the ecology of resistance genes and mechanisms. Vaccine. 2020;38(Suppl 1):A83–A92.
- Ahmadi MH. Global status of tetracycline resistance among clinical isolates of Vibrio cholerae: a systematic review and meta-analysis. Antimicrob Resist Infect Control. 2021;10(1):115.
- Gupta P, Modgil V, Kant V, Kaur H, Narayan C, Mahindroo J, Verma R, Mohan B, Taneja N. Phenotypic and genotypic characterization of antimicrobial resistance in clinical isolates of Vibrio cholerae over a decade (2002–2016). Indian J Med Microbiol. 2022;40(1):24–29.
- Mashe T, Domman D, Tarupiwa A, Manangazira P, Phiri I, Masunda K, Chonzi P, Njamkepo E, Ramudzulu M, Mtapuri-Zinyowera S, et al. Highly resistant cholera outbreak strain in Zimbabwe. N Engl J Med. 2020;383(7):687–689.
- Campestre C, De Luca V, Carradori S, Grande R, Carginale V, Scaloni A, Supuran CT, Capasso C. Carbonic anhydrases: new perspectives on protein functional role and inhibition in Helicobacter pylori. Front Microbiol. 2021;12:629163.
- Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032.
- De Luca V, Carginale V, Supuran CT, Capasso C. gram-negative bacterium Escherichia coli as a model for testing the effect of carbonic anhydrase inhibition on bacterial growth. J Enzyme Inhib Med Chem. 2022;37(1):2092–2098.
- Hewitt CS, Abutaleb NS, Elhassanny AEM, Nocentini A, Cao X, Amos DP, Youse MS, Holly KJ, Marapaka AK, An AW, et al. Structure-activity relationship studies of acetazolamide-based carbonic anhydrase inhibitors with activity against Neisseria gonorrhoeae. ACS Infect Dis. 2021;7(7):1969–1984.
- Abutaleb NS, Elkashif A, Flaherty DP, Seleem MN. In Vivo antibacterial activity of acetazolamide. Antimicrob Agents Chemother. 2021;65(4):e01715-e01720.
- Kaur J, Cao X, Abutaleb NS, Elkashif A, Graboski AL, Krabill AD, AbdelKhalek AH, An W, Bhardwaj A, Seleem MN, et al. Optimization of acetazolamide-based scaffold as potent inhibitors of vancomycin-resistant Enterococcus. J Med Chem. 2020;63(17):9540–9562.
- Abutaleb NS, Elhassanny AEM, Flaherty DP, Seleem MN. In vitro and in vivo activities of the carbonic anhydrase inhibitor, dorzolamide, against vancomycin-resistant enterococci. Peer J. 2021;9:e11059.
- Abutaleb NS, Elhassanny AEM, Seleem MN. In vivo efficacy of acetazolamide in a mouse model of Neisseria gonorrhoeae infection. Microb Pathog. 2022;164:105454.
- Mancuso F, De Luca L, Bucolo F, Vrabel M, Angeli A, Capasso C, Supuran CT, Gitto R. 4-sulfamoylphenylalkylamides as inhibitors of carbonic anhydrases expressed in Vibrio cholerae. ChemMedChem. 2021;16(24):3787–3794.
- Akgul O, Angeli A, Selleri S, Capasso C, Supuran CT, Carta F. Taurultams incorporating arylsulfonamide: first in vitro inhibition studies of α-, β- and γ-class Carbonic Anhydrases from Vibrio cholerae and Burkholderia pseudomallei. Eur J Med Chem. 2021;219:113444.
- Abuaita BH, Withey JH. Bicarbonate Induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect Immun. 2009;77(9):4111–4120.
- Butler SM, Camilli A. Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat Rev Microbiol. 2005;3(8):611–620.
- Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–D612.
- Zeller T, Klug G. Thioredoxins in bacteria: functions in oxidative stress response and regulation of thioredoxin genes. Naturwissenschaften. 2006;93(6):259–266.
- Del Prete S, De Luca V, Scozzafava A, Carginale V, Supuran CT, Capasso C. Biochemical properties of a new α-carbonic anhydrase from the human pathogenic bacterium, Vibrio cholerae. J Enzyme Inhib Med Chem. 2014;29(1):23–27.
- Del Prete S, Vullo D, De Luca V, Carginale V, Osman SM, AlOthman Z, Supuran CT, Capasso C. Comparison of the sulfonamide inhibition profiles of the α-, β- and γ-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem Lett. 2016;26(8):1941–1946.
- Bonardi A, Nocentini A, Osman SM, Alasmary FA, Almutairi TM, Abdullah DS, Gratteri P, Supuran CT. Inhibition of α-, β- and γ-carbonic anhydrases from the pathogenic bacterium Vibrio cholerae with aromatic sulphonamides and clinically licenced drugs – a joint docking/molecular dynamics study. J Enzyme Inhib Med Chem. 2021;36(1):469–479.
- Bonardi A, Nocentini A, Cadoni R, Del Prete S, Dumy P, Capasso C, Gratteri P, Supuran CT, Winum JY. Benzoxaboroles: new potent inhibitors of the carbonic anhydrases of the pathogenic bacterium Vibrio cholerae. ACS Med Chem Lett. 2020;11(11):2277–2284.
- Demir-Yazıcı K, Güzel-Akdemir Ö, Angeli A, Supuran CT, Akdemir A. Novel indole-based hydrazones as potent inhibitors of the α-class carbonic anhydrase from pathogenic bacterium Vibrio cholerae. IJMS. 2020;21(9):3131.
- Bua S, Berrino E, Del Prete S, Murthy VS, Vijayakumar V, Tamboli Y, Capasso C, Cerbai E, Mugelli A, Carta F, et al. Synthesis of novel benzenesulfamide derivatives with inhibitory activity against human cytosolic carbonic anhydrase I and II and Vibrio cholerae α- and β-class enzymes. J Enzyme Inhib Med Chem. 2018;33(1):1125–1136.
- Berrino E, Bozdag M, Del Prete S, Alasmary FAS, Alqahtani LS, AlOthman Z, Capasso C, Supuran CT. Inhibition of α-, β-, γ-, and δ-carbonic anhydrases from bacteria and diatoms with N'-aryl-N-hydroxy-ureas. J Enzyme Inhib Med Chem. 2018;33(1):1194–1198.
- Ali M, Angeli A, Bozdag M, Carta F, Capasso C, Farooq U, Supuran CT. Benzylaminoethylureido-tailed benzenesulfonamides show potent inhibitory activity against bacterial carbonic anhydrases. ChemMedChem. 2020;15(24):2444–2447.
- Ceruso M, Del Prete S, Alothman Z, Capasso C, Supuran CT. Sulfonamides with potent inhibitory action and selectivity against the α-carbonic anhydrase from Vibrio cholerae. ACS Med Chem Lett. 2014;5(7):826–830.
- De Vita D, Angeli A, Pandolfi F, Bortolami M, Costi R, Di Santo R, Suffredini E, Ceruso M, Del Prete S, Capasso C, et al. Inhibition of the α-carbonic anhydrase from Vibrio cholerae with amides and sulfonamides incorporating imidazole moieties. J Enzyme Inhib Med Chem. 2017;32(1):798–804.
- Mancuso F, De Luca L, Angeli A, Berrino E, Del Prete S, Capasso C, Supuran CT, Gitto R. In silico-guided identification of new potent inhibitors of carbonic anhydrases expressed in Vibrio cholerae. ACS Med Chem Lett. 2020;11(11):2294–2299.
- Liguori F, Carradori S, Ronca R, Rezzola S, Filiberti S, Carta F, Turati M, Supuran CT. Benzenesulfonamides with different rigidity-conferring linkers as carbonic anhydrase inhibitors: an insight into the antiproliferative effect on glioblastoma, pancreatic, and breast cancer cells. J Enzyme Inhib Med Chem. 2022;37(1):1857–1869.
- Combs J, Bozdag M, Cravey LD, Kota A, McKenna R, Angeli A, Carta F, Supuran CT. New insights into conformationally restricted carbonic anhydrase inhibitors. Molecules. 2023;28(2):890.
- Scozzafava A, Menabuoni L, Mincione F, Briganti F, Mincione G, Supuran CT. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: Is the tail more important than the ring? J Med Chem. 1999;42(14):2641–2650.
- Bozdag M, Ferraroni M, Nuti E, Vullo D, Rossello A, Carta F, Scozzafava A, Supuran CT. Combining the tail and the ring approaches for obtaining potent and isoform-selective carbonic anhydrase inhibitors: solution and X-ray crystallographic studies. Bioorg Med Chem. 2014;22(1):334–340.
- Wilkinson BL, Bornaghi LF, Houston TA, Innocenti A, Supuran CT, Poulsen S-A. A novel class of carbonic anhydrase inhibitors: glycoconjugate benzene sulfonamides prepared by “click-tailing. J Med Chem. 2006;49(22):6539–6548.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- Lagorce D, Bouslama L, Becot J, Miteva MA, Villoutreix BO. FAF-drugs4: free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics. 2017;33(22):3658–3660.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623.
- Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, Bridgland A, Cowie A, Meyer C, Laydon A, et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021;596(7873):590–596.
- Del Prete S, Isik S, Vullo D, De Luca V, Carginale V, Scozzafava A, Supuran CT, Capasso C. DNA cloning, characterization, and inhibition studies of an α-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. J Med Chem. 2012;55(23):10742–10748.
- Schrödinger Release 2021-4. Maestro, Glide, Protein Preparation Wizard, Epik, SiteMap, QikProp, MacroModel, Desmond, Prime. New York (NY): Schrödinger, LLC; 2021.
- Filipski KJ, Varma MV, El-Kattan AF, Ambler CM, Ruggeri RB, Goosen TC, Cameron KO. Intestinal targeting of drugs: rational design approaches and challenges. Curr Top Med Chem. 2013;13(7):776–802.
- Anthouard R, DiRita VJ. Small-molecule inhibitors of toxT expression in Vibrio cholerae. mBio. 2013;4(4):e00403–e00413.
- Vullo D, Del Prete S, De Luca V, Carginale V, Ferraroni M, Dedeoglu N, Osman SM, AlOthman Z, Capasso C, Supuran CT. Anion inhibition studies of the β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem Lett. 2016;26(5):1406–1410.
- Del Prete S, Vullo D, De Luca V, Carginale V, Ferraroni M, Osman SM, AlOthman Z, Supuran CT, Capasso C. Sulfonamide inhibition studies of the β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Bioorg Med Chem. 2016;24(5):1115–1120.
- Greenwood JR, Calkins D, Sullivan AP, Shelley JC. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comput Aided Mol Des. 2010;24(6-7):591–604.
- Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK (a) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 2007;21(12):681–691.
- Bozdag M, Ferraroni M, Ward C, Carta F, Bua S, Angeli A, Langdon SP, Kunkler IH, Al-Tamimi AS, Supuran CT. Carbonic anhydrase inhibitors based on sorafenib scaffold: Design, synthesis, crystallographic investigation and effects on primary breast cancer cells. Eur J Med Chem. 2019;182:111600.
- Behnke CA, Le Trong I, Godden JW, Merritt EA, Teller DC, Bajorath J, Stenkamp RE. Atomic resolution studies of carbonic anhydrase II. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 5):616–627.
- Ferraroni M, Del Prete S, Vullo D, Capasso C, Supuran CT. Crystal structure and kinetic studies of a tetrameric type II β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr D Biol Crystallogr. 2015;71(Pt 12):2449–2456.
- Farid R, Day T, Friesner RA, Pearlstein RA. New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg Med Chem. 2006;14(9):3160–3173.
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49(2):534–553.
- Sherman W, Beard HS, Farid R. Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des. 2006;67(1):83–84.
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196.
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759.
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749.
- Jacobson MP, Pincus DL, Rapp CS, Day TJ, Honig B, Shaw DE, Friesner RA. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55(2):351–367.
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320(3):597–608.
- Kim S, Yeon J, Sung J, Jin MS. Crystal structure of β-carbonic anhydrase CafA from the fungal pathogen Aspergillus fumigatus. Mol Cells. 2020;43(9):831–840.
- UniProt Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 2023;51(D1):D523–D531.
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421.
- Somalinga V, Buhrman G, Arun A, Rose RB, Grunden AM. A high-resolution crystal structure of a psychrohalophilic α-carbonic anhydrase from Photobacterium profundum reveals a unique dimer interface. PLOS One. 2016;11(12):e0168022.
- Park HM, Park JH, Choi JW, Lee J, Kim BY, Jung CH, Kim JS. Structures of the γ-class carbonic anhydrase homologue YrdA suggest a possible allosteric switch. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 8):920–926.
- Bowers KJ. Scalable algorithms for molecular dynamics simulations on commodity clusters. SC '06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing; Tampa, FL, USA; 2006, p. 43.
- PyMOL. The PyMOL Molecular Graphics System, Version 2.5 Schrödinger, LLC, 2021.
- Mugnaini C, Sannio F, Brizzi A, Del Prete R, Simone T, Ferraro T, De Luca F, Corelli F, Docquier JD. Screen of unfocused libraries identified compounds with direct or synergistic antibacterial activity. ACS Med Chem Lett. 2020;11(5):899–905.
- Clinical and Laboratory Standards Institute (CLSI). 2015. M07-A10: Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard. 10th ed. Wayne (PA): Clinical and Laboratory Standards Institute.