
Abstract
Epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) protein tyrosine kinases co-expressed in various cancers such as ovarian, breast, colon, and prostate subtypes. Herein, new TAK-285 derivatives (9a–h) were synthesised, characterised, and biologically evaluated as dual EGFR/HER2 inhibitors. Compound 9f exhibited IC50 values of 2.3 nM over EGFR and 234 nM over HER2, which is 38-fold of staurosporine and 10-fold of TAK-285 over EGFR. Compound 9f also showed high selectivity profile when tested over a small kinase panel. Compounds 9a–h showed IC50 values in the range of 1.0–7.3 nM and 0.8–2.8 nM against PC3 and 22RV1 prostate carcinoma cell lines, respectively. Cell cycle analysis, apoptotic induction, molecular docking, dynamics, and MM-GBSA studies confirmed the plausible mechanism(s) of compound 9f as a potent EGFR/HER2 dual inhibitor with an effective antiproliferative action against prostate carcinoma.
Introduction
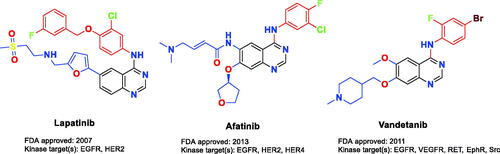
Epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) protein tyrosine kinases co-expressed in various cancers such as ovarian, breast, colon, and prostate subtypesCitation1–4. In 20–25% of human breast cancers, HER2 gene amplification and receptor overexpression are observedCitation5. EGFR/HER2 small molecule inhibitors could prevent tyrosine kinase phosphorylation, which in turn suppresses the upregulated intracellular signalling in solid tumours, consequently, the dysfunction of tumour regulation occurs. Many ATP-competitive EGFR/HER2 RTK (receptor tyrosine kinase) dual small molecule inhibitors bearing diverse chemical scaffolds are widely tested in human clinical studies for cancer therapy. The FDA-approved small molecule lapatinib () is prescribed to treat patients with HER2 overexpression metastatic breast cancer. It possesses a 4-anilinoquinazoline scaffold which is a promising chemical moiety for EGFR/HER2 dual inhibitionCitation6. In the literature, the interaction of lapatinib with the catalytic domain of EGFR/HER2 kinases has been well studied. Generally, the hinge region is hydrogen bound to the quinazoline ring, which is located at the ATP binding site. In order to establish additional hydrophobic interactions, the aniline moiety at the C4 position on the quinazoline scaffold is directed to bind with a neighbouring back pocketCitation7,Citation8. In the previous research, it has demonstrated that the size and functionality of this hydrophobic pocket have a decisive effect on the kinase inhibitor selectivity, while the substituents on C5 and C6 positions could enhance the physical properties to attain favourable pharmacokinetics of the quinazoline-based scaffold. Furthermore, several dual inhibitory candidates were designed to bind to Cys805 in HER2 and Cys773 in EGFRCitation9–13. Even though lapatinib treatment was found beneficial, many patients did not show a positive response to it or acquired resistance for diverse undiscovered causesCitation14–17. For this reason, new therapeutics using novel small molecule inhibitors are needed for EGFR/HER2 suppression.
Figure 1. FDA-approved multikinase kinase inhibitors.
Since the 1960s, 2-nitroimidazole derivatives have been used as chemotherapies and hypoxia-activated radio-sensitisation therapyCitation18. In the presence of hypoxia, the 2-nitroimidazole is reduced by nitroreductase to generate reactive radicals, and it could exhaust tumour-specific antioxidants such as glutathione (GSH), which would make tumours more susceptible to radiotherapyCitation19. Moreover, the reactive radicals build up in cells and induce lethal consequences because of their irrevocable binding to the nucleic acid and proteinCitation20.
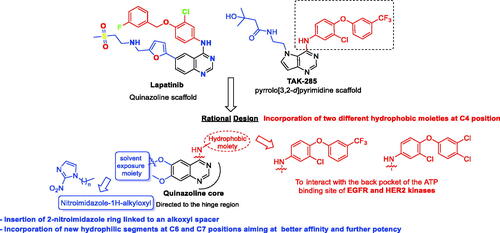
Up to date, many potential antitumor small molecule inhibitors bearing 2-nitroimidazole moiety have been discoveredCitation21–23. Recently, our group reported a series of lapatinib derivatives possessing 6-(nitroimidazole-1H-alkyloxyl) moiety with potent dual EGFR/HER2 kinase inhibitory activitiesCitation24. Consequently, we focus in this research on development of this hybrid scaffold () to selectively inhibit EGFR/HER2 tyrosine kinases as well as to assess the antiproliferative activity of this new series against prostate carcinoma cell lines. Herein, two different aniline moieties (3-chloro-4-(3-(trifluoromethyl)phenoxy)aniline and 3-chloro-4-(3,4-dichlorophenoxy)aniline) were incorporated at C4 position in a fashion similar to the potent dual inhibitor TAK-285 ()Citation25. A variety of polar/solubilising nitroimidazole moieties linked to alkoxy linkers with different lengths were added to C6 and C7 positions of the quinazoline scaffold. The inhibition profile over EGFR and HER2 kinases of all the synthesised compounds (9a–h) was assessed. The IC50 values of the highly active candidate were evaluated over both kinases. In addition, to verify the selectivity of the most active candidate, a small kinase panel was then employed. Cell-based antiproliferative evaluation was carried out over two prostate carcinoma cell lines. Moreover, comprehensive simulation analyses were accomplished to recognise the binding affinities and direction of the final small molecules.
Figure 2. Rational design of the new TAK-285 derivatives 9a–h.
Results and discussion
Chemical synthesis
Scheme 1(A) demonstrates the chemical synthesis of compounds 2a–d via reacting 2-nitroimidazole (1) with different 1,n-dibromoalkanes in DMF solvent and K2CO3 at 60 °C. In Scheme 1(B), 2-chloro-1-fluoro-4-nitrobenzene (3) reacted with 3,4-dichlorophenol in the presence of K2CO3 and acetonitrile solvent for 3 h at 85 °C to afford 1,2-dichloro-4-(2-chloro-4-nitrophenoxy)benzene (4). The nitro group of intermediate 4 was then reduced via 10% platinum on carbon and H2 gas at room temperature to afford 3-chloro-4-(3,4-dichlorophenoxy)aniline (5). The second aniline reagent (3-chloro-4-(3-(trifluoromethyl)phenoxy)aniline) was purchased. In Scheme 1(C), 4-chloro-7-methoxyquinazolin-6-yl acetate (6) reacted separately with the two aniline reagents to form intermediates 7a and 7b. Intermediates 8a and 8b were then produced by hydrolysis of the acetate group of compounds 7a and 7b utilising 28% aqueous ammonia in methanol solvent. The free phenolic group in compounds 8a and 8b was allowed to react with the imidazoles 2a–d in DMF solvent and K2CO3 at 80 °C to generate the desired TAK-285 derivatives (9a–h) ().
Scheme 1. Reagents and conditions: (i) 1,n-dibromoalkane, K2CO3, DMF, 60 °C, 4 h; (ii) 3,4-dichlorophenol, K2CO3, acetonitrile, 85 °C, 3 h; (iii) 10% Pt/C, H2 gas, methanol, rt, 18 h; (iv) aniline reagent, isopropyl alcohol, reflux, 4 h; (v) aqueous ammonia solution (28%), methanol, rt, 4 h; (vi) 1-(n-bromoalkyl)-2-nitro-1H-imidazole derivative, K2CO3, DMF, 80 °C, 4 h.
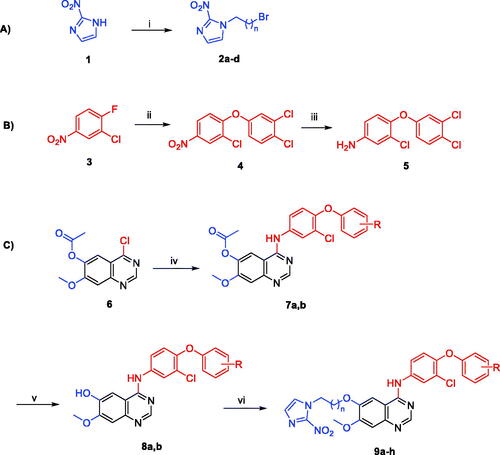
Table 1. Chemical structures and isolated yields of compounds 9a–h.
Structure elucidation of the newly synthesised TAK-285 derivatives 9a–h
To elucidate the chemical structure of compounds 9a–h, various spectroscopic approaches were used including 1H NMR, 13C NMR, and HRMS. Also, their purity was acquired via the HPLC system where they showed purity higher than 95%. Since all final compounds have a methoxy group, a singlet peak at the 3.95–3.92 ppm range was observed in the 1H NMR spectra. In addition, the specific peak of C2 position proton of quinazoline scaffold was found at 8.54–8.51 ppm (see supporting information). Furthermore, the extended alkyl linkers of all final compounds (n = 2–5) were confirmed where their protons and carbons were found in relatively low chemical shifts. In the 1H NMR spectrum, compound 9a showed the C2 of quinazoline at 8.52 ppm, and three protons of the methoxy group at 3.92 ppm as a singlet peak. Additionally, the O-alkylation reaction between the OH group and the imidazole linker (step vi) was proved by the disappearance of the OH peak. Like compound 9a, the target compounds 9b, 9c, and 9d have also shown a similar pattern, and their extending protons and carbons were found in the range of 2.04–1.34 ppm in 1H NMR spectra and 80.00–20.00 ppm in 13C NMR spectra, respectively. The 3-chloro-4-(3-(trifluoromethyl)phenoxy)aniline derivative 9e was identified by one proton of benzene ring at 8.22 ppm as a doublet peak with a J coupling constant of 4.0 Hz. In addition, there is a singlet peak at 3.95 ppm attributable to the three protons of the methoxy group in the 1H NMR spectrum, while its carbon was acquired at 56.35 ppm in the 13C NMR spectrum. These findings provided evidence proving the successful synthesis of the desired TAK-285 derivatives 9a–h.
Biological evaluation
Kinase assay of TAK-285 derivatives 9a–h over EGFR and HER2
All the newly synthesised TAK-285 derivatives (9a–h) were assessed over EGFR and HER2 kinases at Reaction Biology Co. (Malvern, PA) via “HotSpotSM” assay at 10 µM concentration in the presence of 10 µM of ATP. The results were obtained as the % remaining kinase activity of test samples in comparison to the DMSO vehicle. The rates of % kinase inhibition of compounds 9a–h over both kinases were computed and described in . In brief, the results revealed promising inhibitory activities of all TAK-285 derivatives 9a–h over EGFR kinase. Incorporation of 3-chloro-4-(3,4-dichlorophenoxy)anilino group in compounds 9a–d demonstrated an inhibitory range of 88.90–94.25%, which is lower than the inhibitory range expressed by derivatives with the hydrophobic moiety of TAK-285 (3-chloro-4-(3-(trifluoromethyl)phenoxy)aniline, 9e–h) that demonstrated an outstanding range of % inhibition over EGFR (96.75–99.33%). In the case of HER2, it was observed that most tested compounds showed lower inhibition rates. Nevertheless, a comparable SAR model was revealed over HER2 kinase. While derivatives 9a–d exhibited a modest inhibitory range of 69.83–80.26%, TAK-285 derivatives 9e–h revealed a higher inhibitory activity range (81.74–97.95%). Among all, compound 9f exhibited the best dual inhibition values over EGFR and HER2 with 99.33 and 97.95% inhibition over both kinases, respectively. Accordingly, compound 9f was selected for further investigations.
Table 2. % Inhibition results of EGFR and HER2 kinases by compounds 9a–h at 10 µM.
Dose-dependent assessment of TAK-285 derivative 9f over EGFR and HER2
The primary results over the molecular level of both tyrosine receptors (EGFR and HER2) encouraged us to do further assessment of compound 9f. A dose-dependent evaluation was carried out to assess its IC50 values over both kinases (). The IC50 values of compound 9f were found to be 2.3 nM over EGFR and 234 nM over HER2. Compared to staurosporine and TAK-285, compound 9f showed 38- and 10-fold of potency over EGFR, respectively.
Table 3. IC50 values (µM) of compound 9f, staurosporine, and TAK-285 over EGFR and HER2 tyrosine kinases.
Kinase selectivity assessment of TAK-285 derivative 9f
To assess the selectivity and the kinase inhibition profile of the most active dual inhibitor of this new series (9f), an in vitro screening assay was performed over a small panel of cancer-related kinases including fibroblast growth factor receptor 1 (FGFR1), vascular endothelial growth factor receptor 2 (VEGFR2), cyclin-dependent kinase 2 (CDK2), c-mesenchymal-epithelial transition factor (c-MET), and p38α mitogen-activated protein kinase (MAPK14) in a single-dose concentration of 10 µM. As shown in , modest inhibitory activities were detected over the tested enzymes. These findings clearly showed a good selectivity of compound 9f with its nanomolar potency against both kinases (EGFR and HER2) compared to the other tested kinase targets.
Table 4. % Inhibition of TAK-285 derivative 9f over a small kinase panel at 10 µM.
In vitro cytotoxic activity against PC3 and 22RV1 cells
Some inhibitors of the ErbB (EGFR/HER2/ErbB3/ErbB4) family, notably the dual EGFR/HER2 inhibitor lapatinib, failed in phase II clinical trials despite overexpression of this family in castration-resistant prostate cancer (CRPC)Citation26,Citation27. Accordingly, we aimed to investigate the cytotoxic potential of this new series against prostate carcinoma cell lines. The in vitro cytotoxicity of the newly synthesised TAK-285 derivatives (9a–h) was measured using MTT assay against two human prostate carcinoma cell lines (PC3 and 22RV1). indicates that compounds 9a–h showed IC50 values in the range of 1.0–7.3 nM and 0.8–2.8 nM against PC3 and 22RV1, respectively. Compound 9f was the most potent derivative over PC3 cell line with an IC50 value of 1.0 nM, while compound 9e showed the best IC50 value over 22 RV1 cell line (0.8 nM). The potent nanomolar cell-based activity of this series of compounds over the tested prostate carcinoma cell line could be attributed to other possible targets in addition to EGFR and HER2 receptors. The presence of the 2-nitroimidazole moiety in the chemical structure of compounds 9a–h could develop possible covalent bonds between the nitroimidazole moiety and some cellular proteinsCitation28.
Table 5. The cytotoxic effect and IC50 value assessment of TAK-285 derivatives 9a–h against PC3 and 22RV1 prostate cancer cell lines by MTT assay.
Three independent experiments were performed in triplicate. Standard deviation (±SD) of the mean was also obtained for all experiments along with the IC50 values.
Cell cycle analysis
Cell cycle progression is responsible for normal cell growth and proliferation. DNA damage can result in apoptosis, which causes cell death, or DNA repair. At specific checkpoints that serve as control mechanisms to guarantee correct cell division, the state of the cells is evaluated. Checkpoints in the cell cycle include the G1 (restriction), S (metaphase), and G2/MCitation29. Anticancer medications’ function is to halt cell division at these checkpoints. Treatment with potent cytotoxic (as an anticancer) agents can determine at which phase apoptosis occurs in the cell cycle. As a result, the most potent derivative, 9f, was chosen for testing its outcomes on the cell cycle profile and apoptosis. PC3 and 22RV1 cells were treated with compound 9f at its IC50. The comparison data in and indicate that compound 9f (test 2) arrested the cell cycle of 22RV1 and PC3 cells at the G2/M phase by 62.74% and 49.43%, respectively (). Also, the cell population in G1 and S phases decreases after treatment (test 2) compared to a negative control (test 1). The comparison data showed the control sample has arrested the cell cycle at G0/G1 phases while 9f treated sample has arrested it at G2/M phases as indicated by higher number of counts (%) in these phases of both cell cycle studies.
Figure 3. The effect of compound 9f (test 2) on the phases of the cell cycle compared with control (test 1); 22RV1 cells (A) and PC3 (B).
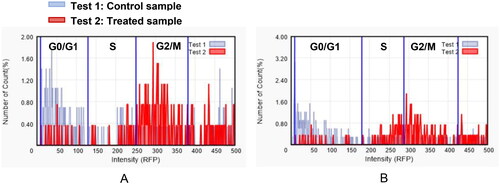
Table 6. The effect of compound 9f on the different phases of cell cycle of PC3 and 22RV1 cell lines.
Apoptosis analysis
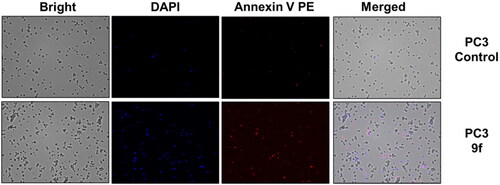
The control PC3 and 22RV1 cells and compound-treated cells were stained with PE-Annexin V and DAPI for a more appropriate cell death examination, and the cellular fluorescence analysis was then performed using ADAMII LS (). Apoptosis is a type of programmed cell death that can be detected using Annexin V and the DAPI reagent. On plasma membranes, annexin V binds to phosphatidyl amine and DAPI can bind to DNA in cells. The dot plot and image data detect early and late apoptotic cells using these two fluorophores. The dot plot results revealed that a large portion of compound 9f-treated 22RV1 cells underwent early apoptosis (13.20%), and simultaneously, some cells had prominently progressed to the late apoptosis phase (15.38%) (). We found that most of the cancer cells in the control groups (about 92.12% and 96.03%, respectively) were alive (). Moreover, compound 9f-treated PC3 cells went through early apoptosis (6.27%), and late apoptosis phase (16.94%) ( and ). Likewise, the fluorescence images for 22RV1 () and PC3 () cells complement the dot plot data, as higher fluorescence of DAPI and RF images clearly distinguished compound 9f-treated cells from control. Therefore, compound 9f exerted noticeable damage to 22RV1 and PC3 cells when treated with compound doses around its IC50 value. The results of the cell cycle and apoptosis studies suggest that compound 9f has remarkable anticancer properties for chemotherapy.
Figure 4. Dot plots showing apoptosis analysis of 22RV1 cells (A, B) and PC3 (C, D) induced by compound 9f along its control.
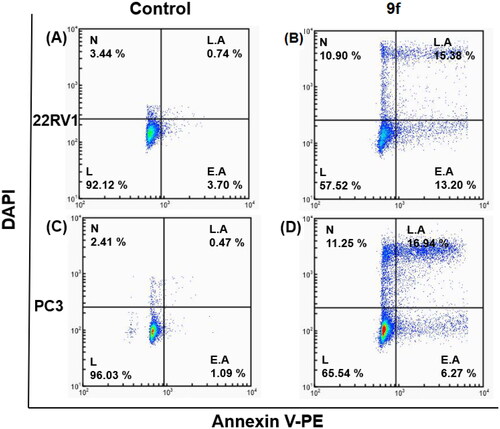
Figure 5. Cellular fluorescence images of 22RV1 cell line treated with compound 9f for 24 h. Bright-field images, fluorescence images (DAPI: 4′,6-diamidino-2-phenylindole, Annexin V PE), and merged images were assigned to the 22RV1 prostate cancer cells with control (without any compound treatment), compound 9f-treated, showing apoptotic cells.
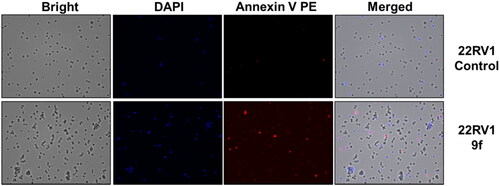
Figure 6. Cellular fluorescence images of PC3 cell line treated with compound 9f for 24 h. Bright-field images, fluorescence images (DAPI: 4′,6-diamidino-2-phenylindole, Annexin V PE), and merged images were assigned to the PC3 prostate cancer cells with control (without any compound treatment), compound 9f-treated, showing apoptotic cells.
Annexin V conjugated to fluorochromes (PE) retains its high affinity for phosphatidylserine (PS), making it a sensitive probe for flow cytometric analysis of apoptotic cells. PE Annexin V staining occurs prior to the loss of membrane integrity that occurs in the final stages of cell death caused by either apoptotic or necrotic processes. As a result, PE Annexin V staining is typically used in conjunction with a vital dye such as propidium iodide (PI) or DAPI to allow the investigator to identify early apoptotic cells (DAPI negative, PE Annexin V positive). Viable cells with intact membranes exclude DAPI, whereas dead and damaged cells’ membranes are permeable to DAPI. When apoptosis is measured over time, cells can often be tracked from PE Annexin V and DAPI negative (viable, or no measurable apoptosis), to PE Annexin V positive and DAPI negative (early apoptosis), and finally to PE Annexin V and DAPI positive (end stage apoptosis and death). The progression of cells through these three stages suggests apoptosis.
Molecular docking
To validate the dual inhibitory activities of the newly designed TAK-285 derivatives (9a–h) towards EGFR (PDB ID: 1M17Citation30) and HER2 (PDB ID: 3RCDCitation25) receptors, molecular docking studies were carried out using the MOE 2019.0102Citation31,Citation32. Besides, the co-crystallised inhibitors (4-anilinoquinazoline (AQ4) and pyrrolo[3,2-d]pyrimidine (03P)) of EGFR and HER2 were inserted as reference standards. Since all docked compounds (9a–h) showed promising results, derivatives 9f and 9g, which were biologically superior, were selected for deep investigations compared to the co-crystallised inhibitor in each case.
The binding pocket of EGFR receptor (PDB ID: 1M17Citation30) showed that Met769, Gln767, Cys773, Cys751, Lys692, Thr766, and Thr830 are very crucial to produce the antagonistic activity. The docked AQ4 inhibitor bound Cys773 through a pi–H bond (4.68 Å) with a binding score of −7.47 kcal/mol (RMSD = 1.01). However, compound 9f showed the formation of one H-bond with Lys692 (2.95 Å) and three pi–H bonds with Cys773, Val702, and Arg817 (4.55, 4.33, and 4.65 Å, respectively). Its binding score was found to be −8.79 kcal/mol (RMSD = 1.13), besides compound 9g binding score was −8.46 kcal/mol (RMSD = 1.45), indicating superior binding affinities for both derivatives compared to that of the docked AQ4 inhibitor. Also, compound 9g formed one H-bond with Met769 (3.16 Å) and two pi–H interactions with Cys773 and Leu694 (4.33 and 4.61 Å, respectively), as depicted in .
Table 7. 3D binding interactions and positioning of the docked co-crystallised AQ4 inhibitor, 9f, and 9g candidates within the EGFR (PDB ID: 1M17) binding pocket.
On the other hand, the binding site of the HER2 receptor (PDB ID: 3RCDCitation25) clarified that Met801, Leu726, and Lys753 represent the most important amino acids to produce the antagonistic activity. Herein, the docked 03P inhibitor formed two H-bonds with Met801 and Lys753 (3.08 and 3.04 Å, respectively) with a binding score of −11.52 kcal/mol (RMSD = 1.76). Furthermore, compound 9f (S = −10.64 kcal/mol and RMSD = 1.74) achieved two H-bonds with Met801 and Cys805 (3.43 and 3.70 Å, respectively). Besides, compound 9g (S = −10.26 kcal/mol and RMSD = 1.94) got stabilised through the formation of four H-bonds with Met801 (1), Lys753 (2), and Phe731 (1) at 3.52, 3.01, 3.20, and 3.39 Å, respectively, as represented in . Based on the above fact, we can conclude the very promising inhibitory activities of the newly designed candidates (especially 9f and 9g members) towards the binding pockets of both EGFR and HER2 receptors.
Table 8. 3D binding interactions and positioning of the docked co-crystallised 03P inhibitor, 9f, and 9g candidates within the HER2 (PDB ID: 3RCD) binding pocket.
Molecular dynamics (MD) simulations
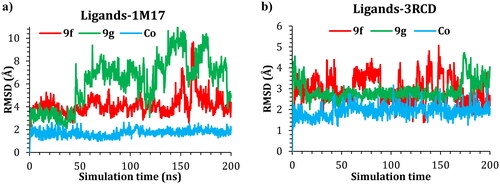
Molecular dynamic simulations were applied to simulate the behaviour of the hit compounds in a cell-like environment. Compounds 9f and 9g were selected, and their performance was studied inside the active site of both the EGFR (1M17) and HER2 (3RCD) tyrosine kinase domains. The protein conformational change was monitored via the difference in the position of the Cα atoms of the protein backbone and was reported in Å. The positional change of the Cα for both protein complexes was plotted as a function of simulation time in .
Figure 7. The RMSD of 9f, 9g, and co-crystal ligand in complex with (a) 1M17 and (b) 3RCD.
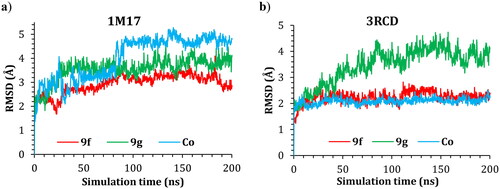
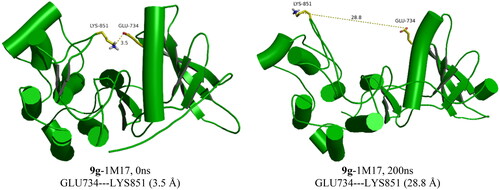
The RMSD of the 1M17 complexes is plotted in , and as seen from the figure, the 9f-1M17 complex showed an RMSD of about 3.00 Å, which is considered acceptable for such protein. The 9g-1M17 and the Co-1M17 complexes showed a fluctuation at around 4.00 and 5.00 Å, respectively; such instability is due to the presence of the hinge as it is playing a critical rule whenever the ligand tries to orient itself inside the active site, the salt bridge between the residue Glu734 and Lys851 will break, the protein subunits will move apart, and the hinge will open which results in higher fluctuation. The distance between the Glu734 and Lys851 for the 9g-1M17 complex is depicted in , and for 9f, the co-crystal is illustrated in Figure SI 1 (SI).
Figure 8. The GLU734–LYS851 distances in the 9g-1M17 complex during the simulations time.
In the case of the 3RCD complexes, the 9f-3RCD and the Co-3RCD were stable and fluctuated at around 2.00 Å. On the other hand, the 9g-3RCD complex fluctuates at around 4.00 Å, which comes from the unfolded loop of the Arg756, Glu757, Asn758, Thr759, and Ser760, coloured red in . On the other hand, the ligand’s behaviour was also monitored via the RMSD with respect to their initial position, and the RMSD plotted as a function of simulation time for ligand–1M17 complexes in and ligand–3RCD complexes in .
Figure 9. Plots of RMSD for ligand atoms (Å) with respect to the initial structure vs. simulation time (ns) for all the complexes.
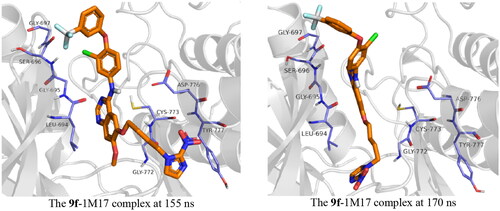
With regards to 1M17, compound 9g was unstable inside the active site. This instability is a result of the moving of the imidazole ring out of the active site, and this leads to relocating the ligand within the active site. The butyl bridge rotatability plays a critical role in this instability. Compound 9f, on the other hand, showed much more stability; the compound was stable during the simulation time, at around 155–170 ns, a fluctuation of 8.00 Å was observed; this fluctuation is again due to the presence of the alkyl bridge, the imidazole group moved out of the protein active site during this period, before it goes back inside the active site as described in . The co-crystal ligand showed stability inside the active site with an RMDS of almost 2.00 Å.
Figure 10. Snapshot of 9f-1M17 at 155 and 170 ns of simulation time showing the moving of the imidazole group out of the active site at 155 ns.
In the case of 3RCD, compound 9f showed a fluctuation between 2.00 and 4.00 Å; the compound imidazole group was shifting its position inside the active site, which led to this RMSD. Compound 9g also showed similar behaviour, and the pentyl bridge gives the imidazole flexibility to move out/in the active site, which results in an RMSD of 3.00–4.00 Å. The co-crystal ligand holds tight inside the active site with an RMSD of ∼2.00 Å during the simulation.
Further, a deep analysis of the interactions of the compounds 9f-1M17 and 9g-3RCD with the active site residues was carried out. The interactions of these ligands with protein residue were plotted using the simulation interaction diagram panel of Maestro software.
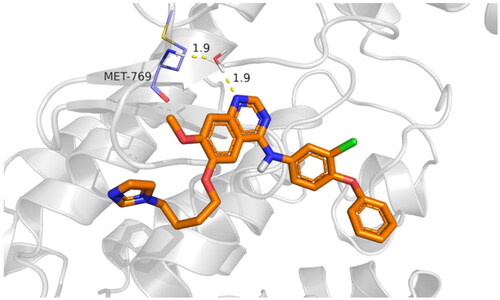
The active site cavity of the 1M17 is quite big, and hence, the stability of ligands inside the active side is dependent on water bridge H-bonding, especially to residue Met769, as observed in the case of the reference compound, Figure SI 3. Compound 9f interactions histogram ( and ) shows that multiple water bridge H-bonds were formed, including residue Met769 (70%), Asp776 (40%), and Pro770 (30%). In addition, residues Leu694 (60%) and Phe699 (32%) were able to form lipophilic interactions with 9f during the simulation time. The residues that were able to develop interactions more than 30% of the simulation time are reported in Figure SI 4.
Figure 11. The histogram of 9f-1M17 contact throughout the trajectory.
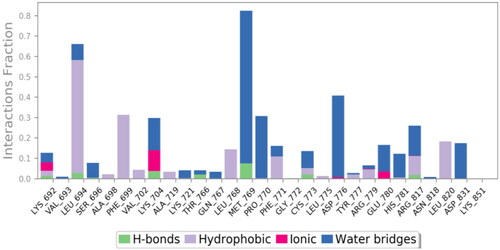
Figure 12. Compound 9f water bridge H-bond interactions with Met769 residue in 1M17.
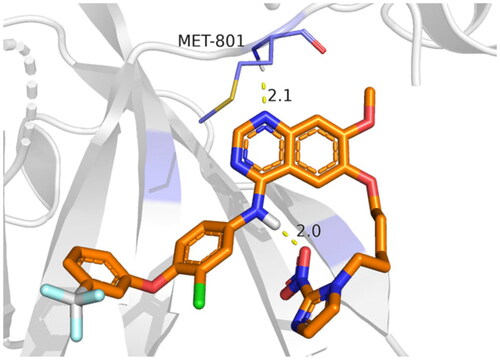
In the case of 3RCD, compound 9g showed much more stability inside the active site of the 3RCD, as seen in the interaction histogram (), the Met801 residue playing an essential role via forming an H-bond interaction almost 100% of the simulation time (). Compound 9g was able to develop lipophilic interactions with Leu726 (50%), Phe731 (95%), Ala751 (75%), Phe1004 (45%), and Leu852 (40%). In addition, Thr862 formed a water bridge hydrogen bond almost 65% of the simulation time, along with Lys753 (35%). The residues that were able to form interactions more than 30% of the simulation time are reported in Figure SI 5.
Figure 13. The histogram of 9g-3RCD contact throughout the trajectory.
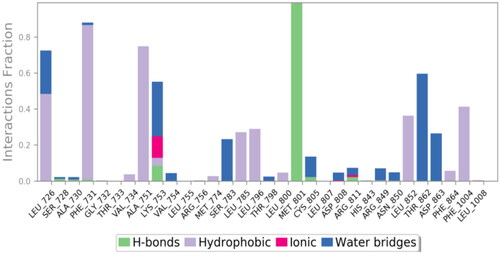
Figure 14. Compound 9g H-bond interactions with 3RCD residues.
MM-GBSA study
The average MM-GBSA binding energy over the last 50 ns was generated using the thermal_mmgbsa.py python script provided by Schrödinger, which also generates Coulomb energy, covalent binding energy, van der Waals energy, lipophilic energy, generalised Born electrostatic solvation energy, and hydrogen-bonding energy. All the obtained data are shown in . MM-GBSA free binding energy reveals that compounds 9f and 9g showed lower binding energy compared to the co-crystal ligand in the case of 1M17, while in the case of 3RCD, compound 9g showed superiority over 9f, which in turn, showed higher binding energy compared to the co-crystal ligand.
Table 9. Prime MM-GBSA energies for ligands binding at the active sites of EGFR (1M17) and HER2 (3RCD) receptors.
Experimental
Chemistry
The general protocols utilised for the chemical synthesis, structure elucidation, and purity of the synthesised compounds were provided in the Supplementary File. Compounds 2a–d were synthesised in our recently published reportCitation24. Compounds 4 and 5 were synthesised following the earlier reportCitation33, and their 1H NMR spectra were added in the Supplementary File.
Synthesis of compounds 7a and 7b
A round-bottom flask was charged with 4-chloro-7-methoxyquinazolin-6-yl acetate (6, 1.0 equiv.) and the appropriate aniline reagent (1.2 equiv.) in isopropyl alcohol (i-PrOH). The mixture was refluxed for 4 h and cooled to rt after completion of the reaction. The reaction mixture was filtered using i-PrOH to give the target intermediate, which was used in the next step without further purification.
4-((3-Chloro-4-(3,4-dichlorophenoxy)phenyl)amino)-7-methoxyquinazolin-6-yl acetate (7a). Ivory solid. Yield: 100% (1.99 g, 3.96 mmol). 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H, NH), 8.92 (s, 1H, quinazoline-C2-H), 8.61 (s, 1H, quinazoline-C8-H), 8.13 (d, J = 2.4 Hz, 1H, Ar-H), 7.76 (dd, J = 8.8, 2.4 Hz, 1H, Ar-H), 7.64 (d, J = 8.0 Hz, 1H, Ar-H), 7.43 (s, 1H, quinazoline-C5-H), 7.35 (d, J = 8.0 Hz, 1H, Ar-H), 7.31 (d, J = 4.0 Hz, 1H, Ar-H), 6.97 (dd, J = 8.8, 2.8 Hz, 1H, Ar-H), 3.99 (s, 3H, OCH3), and 2.38 (s, 3H, OAc-H).
4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)amino)-7-methoxyquinazolin-6-yl acetate (7b). Ivory solid. Yield: 100% (1.45 g, 2.89 mmol). 1H NMR (400 MHz, DMSO-d6) δ 10.99 (s, 1H, NH), 8.91 (s, 1H, quinazoline-C2-H), 8.57 (s, 1H, quinazoline-C8-H), 8.15 (d, J = 2.4 Hz, 1H, Ar-H), 7.78 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 7.63 (t, J = 8.0 Hz, 1H, Ar-H), 7.51 (d, J = 8.0 Hz, 1H, Ar-H), 7.41 (s, 1H, quinazoline-C5-H), 7.36 (d, J = 8.0 Hz, 1H, Ar-H), 7.29–7.24 (m, 2H, Ar-H), 3.99 (s, 3H, OCH3), and 2.38 (s, 3H, OAc-H).
Synthesis of compounds 8a and 8b
The appropriate acetate intermediate (7, 3–4 mmol) and an excess amount of aqueous ammonia solution (28% in H2O) in methanol (CH3OH) were put into the round-bottom flask and stirred for 4 h at rt. The excess solvent of the resulting mixture was partially concentrated, filtered with chilled methanol, and dried to give the required intermediates without further purification.
4-((3-Chloro-4-(3,4-dichlorophenoxy)phenyl)amino)-7-methoxyquinazolin-6-ol (8a). Ivory solid. Yield: 94.3% (1.72 g, 3.72 mmol). 1H NMR (400 MHz, DMSO-d6) δ 9.70 (s, 1H, OH), 9.52 (s, 1H, NH), 8.48 (s, 1H, quinazoline-C2-H), 8.29 (d, J = 4.0 Hz, 1H, Ar-H), 7.91 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 7.79 (s, 1H, quinazoline-C8-H), 7.60 (d, J = 8.0 Hz, 1H, Ar-H), 7.29 (d, J = 8.0 Hz, 1H, Ar-H), 7.22–7.21 (m, 2H, quinazoline-C5-H and Ar-H), 6.91 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), and 3.96 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 157.25 (quinazoline-C4), 156.21 (quinazoline-C7), 154.41 (Ar-C), 152.35 (quinazoline-C2), 147.25 (quinazoline-C6), 146.73 (quinazoline-C8a), 145.12 (Ar-C), 138.83 (Ar-C), 132.46 (Ar-C), 132.00 (Ar-C), 125.27 (Ar-C), 125.23 (Ar-C), 123.10 (Ar-C), 122.95 (Ar-C), 121.96 (Ar-C), 118.70 (Ar-C), 117.05 (Ar-C), 110.10 (quinazoline-C4a), 107.65 (quinazoline-C8), 105.74 (quinazoline-C5), and 56.40 (OCH3). HRMS (ESI) m/z calculated for C21H15Cl3N3O3 [M + H]+: 462.0179, found: 462.0170.
4-((3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)amino)-7-methoxyquinazolin-6-ol (8b). Ivory solid. Yield: 64% (0.85 g, 1.85 mmol). 1H NMR (400 MHz, DMSO-d6) δ 9.71 (s, 1H, OH), 9.53 (s, 1H, NH), 8.49 (s, 1H, quinazoline-C2-H), 8.30 (d, J = 2.4 Hz, 1H, Ar-H), 7.92 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 7.79 (s, 1H, quinazoline-C8-H), 7.60 (t, J = 8.0 Hz, 1H, Ar-H), 7.45 (d, J = 8.0 Hz, 1H, Ar-H), 7.30 (d, J = 8.0 Hz, 1H, Ar-H), 7.21–7.18 (m, 3H, quinazoline-C5 and Ar-H), 3.96 (s, 3H, OCH3). 13C NMR (100 MHz, DMSO-d6) δ 158.23 (quinazoline-C4), 156.23 (quinazoline-C7), 154.41 (Ar-C), 152.36 (quinazoline-C2), 147.25 (quinazoline-C6), 146.73 (quinazoline-C8a), 145.11 (Ar-C), 138.79 (Ar-C), 131.85 (Ar-C), 131.28 (Ar-C), 125.43 (Ar-C), 123.14 (Ar-C), 123.08 (Ar-C), 122.81 (Ar-C), 122.04 (Ar-C), 120.49 (Ar-C), 119.76 (Ar-C), 113.11 (Ar-C), 110.10 (quinazoline-C4a), 107.65 (quinazoline-C8), 105.75 (quinazoline-C5), and 56.39 (OCH3). HRMS (ESI) m/z calculated for C22H16ClF3N3O3 [M + H]+: 462.0832, found: 462.0821.
Synthesis of the final TAK-285 derivatives 9a–h
To the intermediate (8a, 1.0 equiv.) in DMF, potassium carbonate (2.0 equiv.) and the appropriate bromoalkyl imidazole (2a–d, 1.2 equiv.) were added at rt. The reaction mixture was stirred for 4 h at 80 °C and partitioned using EtOAc and water. The collected organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The crude was purified by flash column chromatography (CH2Cl2:CH3OH = 20:1) to afford the desired final TAK-285 derivatives.
N-(3-Chloro-4-(3,4-dichlorophenoxy)phenyl)-7-methoxy-6-(3-(2-nitro-1H-imidazol-1-yl)propoxy)quinazolin-4-amine (9a). Yellow solid (101.9 mg, 0.17 mmol). m.p.: 237–238 °C. HPLC purity: 96.95% (retention time, RT = 14.885 min). 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H, NH), 8.52 (s, 1H, quinazoline-C2-H), 8.18 (d, J = 2.8 Hz, 1H, Ar-H), 7.85 (dd, J = 8.8, 2.4 Hz, 1H, Ar-H), 7.81 (s, 1H, quinazoline-C8-H), 7.67 (d, J = 0.8 Hz, 1H, imidazole-C5-H), 7.62 (d, J = 12.0 Hz, 1H, Ar-H), 7.32 (d, J = 8.0 Hz, 1H, Ar-H), 7.23 (d, J = 2.8 Hz, 1H, imidazole-C4-H), 7.21 (s, 1H, quinazoline-C5-H), 7.17 (d, J = 0.8 Hz, 1H, Ar-H), 6.93 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 4.62 (t, J = 6.0 Hz, 2H, propyl-CH2), 4.19 (t, J = 6.0 Hz, 2H, propyl-CH2), 3.92 (s, 3H, OCH3), 2.42–2.35 (m, 2H, propyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 157.14 (quinazoline-C4), 156.38 (quinazoline-C7), 155.02 (Ar-C), 153.18 (imidazole-C2), 148.40 (quinazoline-C2), 147.66 (quinazoline-C6), 145.60 (quinazoline-C8a), 138.35 (Ar-C), 132.48 (Ar-C), 132.04 (Ar-C), 128.34 (Ar-C), 128.24 (imidazole-C5), 125.36 (imidazole-C4), 125.27 (Ar-C), 123.69 (Ar-C), 122.87 (Ar-C), 122.47 (Ar-C), 118.87 (Ar-C), 117.21 (Ar-C), 109.23 (quinazoline-C4a), 107.81 (quinazolie-C8), 103.35 (quinazoline-C5), 66.54 (propyl-CH2), 56.36 (OCH3), 47.43 (propyl-CH2), and 29.61 (propyl-CH2). HRMS (ESI) m/z calculated for C27H22Cl3N6O5 [M + H]+: 615.0717, found: 615.0723.
N-(3-Chloro-4-(3,4-dichlorophenoxy)phenyl)-7-methoxy-6-(4-(2-nitro-1H-imidazol-1-yl)butoxy)quinazolin-4-amine (9b). Yellow solid (74 mg, 0.12 mmol). m.p.: 101–102 °C. HPLC purity: 96.17% (RT = 15.333 min). 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H, NH), 8.52 (s, 1H, quinazoline-C2-H), 8.19 (d, J = 4.0 Hz, 1H, Ar-H), 7.86 (dd, J = 9.2, 2.4 Hz, 1H, Ar-H), 7.81 (s, 1H, quinazoline-C8-H), 7.76 (d, J = 4.0 Hz, 1H, imidazole-C5-H), 7.62 (d, J = 12.0 Hz, 1H, Ar-H), 7.32 (d, J = 8.0 Hz, 1H, imidazole-C4-H), 7.24 (d, J = 4.0 Hz, 1H, Ar-H), 7.21 (s, 1H, quinazoline-C5-H), 7.19 (d, J = 0.8 Hz, 1H, Ar-H), 6.93 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 4.50 (t, J = 8.0 Hz, 2H, butyl-CH2), 4.18 (t, J = 6.0 Hz, 2H, butyl-CH2), 3.92 (s, 3H, OCH3), 2.04–1.97 (m, 2H, butyl-CH2), and 1.86–1.79 (m, 2H, butyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 157.14 (quinazoline-C4), 156.35 (quinazoline-C7), 155.00 (Ar-C), 153.07 (imidazole-C2), 148.66 (quinazoline-C2), 147.49 (quinazoline-C6), 145.59 (quinazoline-C8a), 138.36 (Ar-C), 132.48 (Ar-C), 132.04 (Ar-C), 128.35 (Ar-C), 128.31 (imidazole-C5), 125.36 (imidazole-C4), 125.26 (Ar-C), 123.71 (Ar-C), 122.85 (Ar-C), 122.50 (Ar-C), 118.86 (Ar-C), 117.21 (Ar-C), 109.30 (quinazoline-C4a), 107.77 (quinazoline-C8), 103.15 (quinazoline-C5), 68.96 (butyl-CH2), 56.36 (OCH3), 49.70 (butyl-CH2), 27.15 (butyl-CH2), and 25.88 (butyl-CH2). HRMS (ESI) m/z calculated for C28H24Cl3N6O5 [M + H]+: 629.0874, found: 629.0881.
N-(3-Chloro-4-(3,4-dichlorophenoxy)phenyl)-7-methoxy-6-((5-(2-nitro-1H-imidazol-1-yl)pentyl)oxy)quinazolin-4-amine (9c). Yellow solid (98.3 mg, 0.15 mmol). m.p.: 94–95 °C. HPLC purity: 97.95% (RT = 15.902 min). 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H, NH), 8.51 (s, 1H, quinazoline-C2-H), 8.19 (d, J = 4.0 Hz, 1H, Ar-H), 7.89 (dd, J = 8.8, 2.8 Hz, 1H, Ar-H), 7.80 (s, 1H, quinazoline-C8-H), 7.71 (d, J = 0.4 Hz, 1H, imidazole-C5-H), 7.62 (d, J = 12.0 Hz, 1H, Ar-H), 7.32 (d, J = 8.0 Hz, 1H, imidazole-C4-H), 7.24 (d, J = 4.0 Hz, 1H, Ar-H), 7.20 (s, 1H, quinazoline-C5-H), 7.18 (d, J = 0.8 Hz, 1H, Ar-H), 6.93 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 4.43 (t, J = 6.0 Hz, 2H, pentyl-CH2), 4.13 (t, J = 8.0 Hz, 2H, pentyl-CH2), 3.92 (s, 3H, OCH3), 1.92–1.83 (m, 4H, pentyl-2CH2), and 1.51–1.44 (m, 2H, pentyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 157.15 (quinazoline-C4), 156.33 (quinazoline-C7), 154.98 (Ar-C), 153.02 (imidazole-C2), 148.79 (quinazoline-C2), 147.45 (quinazoline-C6), 145.57 (quinazoline-C8a), 138.38 (Ar-C), 132.48 (Ar-C), 132.04 (Ar-C), 128.35 (Ar-C), 128.30 (imidazole-C5), 125.35 (imidazole-C4), 125.26 (Ar-C), 123.72 (Ar-C), 122.85 (Ar-C), 122.51 (Ar-C), 118.85 (Ar-C), 117.20 (Ar-C), 109.31 (quinazoline-C4a), 107.74 (quinazoline-C8), 102.89 (quinazoline-C5), 69.04 (pentyl-CH2), 56.33 (OCH3), 49.75 (pentyl-CH2), 30.00 (pentyl-CH2), 28.56 (pentyl-CH2), and 23.00 (pentyl-CH2). HRMS (ESI) m/z calculated for C29H26Cl3N6O5 [M + H]+: 643.1030, found: 643.1031.
N-(3-Chloro-4-(3,4-dichlorophenoxy)phenyl)-7-methoxy-6-((6-(2-nitro-1H-imidazol-1-yl)hexyl)oxy)quinazolin-4-amine (9d). Yellow solid (81 mg, 0.12 mmol). m.p.: 90–91 °C. HPLC purity: 95.31% (RT = 16.266 min). 1H NMR (400 MHz, DMSO-d6) δ 9.55 (s, 1H, NH), 8.51 (s, 1H, quinazoline-C2-H), 8.19 (d, J = 4.0 Hz, 1H, Ar-H), 7.87 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 7.79 (s, 1H, quinazoline-C8-H), 7.69 (s, 1H, Ar-H), 7.61 (d, J = 8.0 Hz, 1H, imidazole-C5-H), 7.32 (d, J = 12.0 Hz, 1H, imidazole-C4-H), 7.23 (d, J = 4.0 Hz, 1H, Ar-H), 7.19 (s, 1H, quinazoline-C5-H), 7.16 (s, 1H, Ar-H), 6.92 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 4.38 (t, J = 8.0 Hz, 2H, hexyl-CH2), 4.12 (t, J = 8.0 Hz, 2H, hexyl-CH2), 3.92 (s, 3H, OCH3), 1.86–1.77 (m, 4H, hexyl-2CH2), 1.54–1.46 (m, 2H, hexyl-CH2), and 1.41–1.34 (m, 2H, hexyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 157.15 (quinazoline-C4), 156.33 (quinazoline-C7), 154.99 (Ar-C), 153.00 (imidazole-C2), 148.85 (quinazoline-C2), 147.44 (quinazoline-C6), 145.57 (quinazoline-C8a), 138.39 (Ar-C), 132.48 (Ar-C), 132.04 (Ar-C), 128.28 (Ar-C), 128.25 (imidazole-C5), 125.35 (imidazole-C4), 125.25 (Ar-C), 123.72 (Ar-C), 122.85 (Ar-C), 122.52 (Ar-C), 118.85 (Ar-C), 117.21 (Ar-C), 109.32 (quinazoline-C4a), 107.74 (quinazoline-C8), 102.86 (quinazoline-C5), 69.12 (hexyl-CH2), 56.33 (OCH3), 49.80 (hexyl-CH2), 30.17 (hexyl-CH2), 28.92 (hexyl-CH2), 26.05 (hexyl-CH2), and 25.56 (hexyl-CH2). HRMS (ESI) m/z calculated for C30H28Cl3N6O5 [M + H]+: 657.1187, found: 657.1184.
N-(3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)-7-methoxy-6-(3-(2-nitro-1H-imidazol-1-yl)propoxy)quinazolin-4-amine (9e). Yellow solid (97.2 mg, 0.16 mmol). m.p.: 232–233 °C. HPLC purity: 98.08% (RT = 14.336 min). 1H NMR (400 MHz, DMSO-d6) δ 9.58 (s, 1H, NH), 8.54 (s, 1H, quinazoline-C2-H), 8.22 (d, J = 4.0 Hz, 1H, Ar-H), 7.88 (dd, J = 9.0, 2.6 Hz, 1H, Ar-H), 7.84 (s, 1H, quinazoline-C8-H), 7.69 (s, 1H, Ar-H), 7.63 (t, J = 8.0 Hz, 1H, Ar-H), 7.49 (d, J = 8.0 Hz, 1H, imidazole-C5-H), 7.36 (d, J = 6.0 Hz, 1H, imidazole-C4-H), 7.25–7.23 (m, 3H, quinazoline-C5-H and Ar-H), 7.19 (d, J = 0.8 Hz, 1H, Ar-H), 4.64 (t, J = 8.0 Hz, 2H, propyl-CH2), 4.22 (t, J = 6.0 Hz, 2H, propyl-CH2), 3.95 (s, 3H, OCH3), and 2.43–2.38 (m, 2H, propyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 158.14 (quinazoline-C4), 156.39 (quinazoline-C7), 155.02 (Ar-C), 153.18 (imidazole-C2), 148.39 (quinazoline-C2), 147.65 (quinazoline-C6), 145.58 (quinazoline-C8a), 145.20 (Ar-C), 138.31 (Ar-C), 131.89 (Ar-C), 131.30 (Ar-C), 130.98 (imidazole-C5), 128.34 (imidazole-C4), 128.23 (Ar-C), 125.45 (CF3), 123.75 (Ar-C), 123.02 (Ar-C), 122.55 (Ar-C), 120.65 (Ar-C), 119.87 (Ar-C), 113.22 (quinazoline-C4a), 109.23 (Ar-C), 107.80 (quinazoline-C8), 103.34 (quinazoline-C5), 66.54 (propyl-CH2), 56.35 (OCH3), 47.43 (propyl-CH2), and 29.61 (propyl-CH2). HRMS (ESI) m/z calculated for C28H23ClF3N6O5 [M + H]+: 615.1371, found: 615.1371.
N-(3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)-7-methoxy-6-(4-(2-nitro-1H-imidazol-1-yl)butoxy)quinazolin-4-amine (9f). Yellow solid (53.1 mg, 0.08 mmol). m.p.: 103–104 °C. HPLC purity: 95.07% (RT = 14.794 min). 1H NMR (400 MHz, DMSO-d6) δ 9.57 (s, 1H, NH), 8.52 (s, 1H, quinazoline-C2-H), 8.20 (d, J = 4.0 Hz, 1H, Ar-H), 7.87 (dd, J = 9.2, 2.8 Hz, 1H, Ar-H), 7.82 (s, 1H, quinazoline-C8-H), 7.75 (s, 1H, Ar-H), 7.61 (t, J = 8.0 Hz, 1H, Ar-H), 7.47 (d, J = 4.0 Hz, 1H, imidazole-C5-H), 7.34 (d, J = 12.0 Hz, 1H, imidazole-C4-H), 7.23–7.19 (m, 4H, quinazoline-C5-H and Ar-H), 4.50 (t, J = 8.0 Hz, 2H, butyl-CH2), 4.18 (t, J = 8.0 Hz, 2H, butyl-CH2), 3.92 (s, 3H, OCH3), 2.03–1.97 (m, 2H, butyl-CH2), and 1.85–1.79 (m, 2H, butyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 157.60 (quinazoline-C4), 155.83 (quinazoline-C7), 154.47 (Ar-C), 152.55 (imidazole-C2), 148.12 (quinazoline-C2), 146.98 (quinazoline-C6), 145.04 (quinazoline-C8a), 144.52 (Ar-C), 137.78 (Ar-C), 131.35 (Ar-C), 130.75 (Ar-C), 130.43 (imidazole-C5), 127.80 (imidazole-C4), 127.76 (Ar-C), 124.89 (CF3), 123.24 (Ar-C), 122.47 (Ar-C), 122.06 (Ar-C), 120.11 (Ar-C), 119.33 (Ar-C), 112.62 (quinazoline-C4a), 108.75 (Ar-C), 107.26 (quinazoline-C8), 102.62 (quinazoline-C5), 68.41 (butyl-CH2), 55.82 (OCH3), 49.15 (butyl-CH2), 26.60 (butyl-CH2), and 25.33 (butyl-CH2). HRMS (ESI) m/z calculated for C29H25ClF3N6O5 [M + H]+: 629.1527, found: 629.1528.
N-(3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)-7-methoxy-6-((5-(2-nitro-1H-imidazol-1-yl)pentyl)oxy)quinazolin-4-amine (9g). Yellow solid (103.4 mg, 0.16 mmol). m.p.: 90–91 °C. HPLC purity: 95.59% (RT = 15.254 min). 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H, NH), 8.51 (s, 1H, quinazoline-C2-H), 8.20 (d, J = 4.0 Hz, 1H, Ar-H), 7.87 (dd, J = 8.8, 2.4 Hz, 1H, Ar-H), 7.80 (s, 1H, quinazoline-C8-H), 7.71 (d, J = 0.4 Hz, 1H, Ar-H), 7.61 (t, J = 8.0 Hz, 1H, Ar-H), 7.47 (d, J = 8.0 Hz, 1H, imidazole-C5-H), 7.34 (d, J = 12.0 Hz, 1H, imidazole-C4-H), 7.22–7.18 (m, 4H, quinazoline-C5-H and Ar-H), 4.43 (t, J = 8.0 Hz, 2H, pentyl-CH2), 4.13 (t, J = 8.0 Hz, 2H, pentyl-CH2), 3.92 (s, 3H, OCH3), 1.91–1.83 (m, 4H, pentyl-2CH2), and 1.52–1.44 (m, 2H, pentyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 158.15 (quinazoline-C4), 156.34 (quinazoline-C7), 154.98 (Ar-C), 153.03 (imidazole-C2), 148.79 (quinazoline-C2), 147.45 (quinazoline-C6), 145.56 (quinazoline-C8a), 145.00 (Ar-C), 138.35 (Ar-C), 131.89 (Ar-C), 131.30 (Ar-C), 130.98 (imidazole-C5), 128.34 (imidazole-C4), 128.29 (Ar-C), 125.43 (CF3), 123.78 (Ar-C), 123.00 (Ar-C), 122.60 (Ar-C), 120.65 (Ar-C), 119.86 (Ar-C), 113.20 (quinazoline-C4a), 109.31 (Ar-C), 107.75 (quinazoline-C8), 102.90 (quinazoline-C5), 69.04 (pentyl-CH2), 56.32 (OCH3), 49.75 (pentyl-CH2), 30.00 (pentyl-CH2), 28.56 (pentyl-CH2), and 23.00 (pentyl-CH2). HRMS (ESI) m/z calculated for C30H27ClF3N6O5 [M + H]+: 643.1684, found: 643.1693.
N-(3-Chloro-4-(3-(trifluoromethyl)phenoxy)phenyl)-7-methoxy-6-((6-(2-nitro-1H-imidazol-1-yl)hexyl)oxy)quinazolin-4-amine (9h). Yellow solid (134.3 mg, 0.20 mmol). m.p.: 84–85 °C. HPLC purity: 96.10% (RT = 15.657 min). 1H NMR (400 MHz, DMSO-d6) δ 9.56 (s, 1H, NH), 8.51 (s, 1H, quinazoline-C2-H), 8.20 (d, J = 2.4 Hz, 1H, Ar-H), 7.88 (dd, J = 8.0, 4.0 Hz, 1H, Ar-H), 7.81 (s, 1H, quinazoline-C8-H), 7.69 (s, 1H, Ar-H), 7.61 (t, J = 8.0 Hz, 1H, Ar-H), 7.47 (d, J = 8.0 Hz, 1H, imidazole-C5-H), 7.33 (d, J = 8.0 Hz, 1H, imidazole-C4-H), 7.22–7.20 (m, 3H, quinazoline-C5-H and Ar-H), 7.16 (s, 1H, Ar-H), 4.38 (t, J = 6.0 Hz, 2H, hexyl-CH2), 4.13 (t, J = 6.0 Hz, 2H, hexyl-CH2), 3.93 (s, 3H, OCH3), 1.87–1.78 (m, 4H, hexyl-2CH2), 1.54–1.47 (m, 2H, hexyl-CH2), and 1.42–1.34 (m, 2H, hexyl-CH2). 13C NMR (100 MHz, DMSO-d6) δ 158.15 (quinazoline-C4), 156.33 (quinazoline-C7), 154.97 (Ar-C), 153.00 (imidazole-C2), 148.84 (quinazoline-C2), 147.44 (quinazoline-C6), 145.54 (quinazoline-C8a), 144.98 (Ar-C), 138.36 (Ar-C), 131.87 (Ar-C), 131.30 (Ar-C), 130.98 (imidazole-C5), 128.28 (imidazole-C4), 128.23 (Ar-C), 125.43 (CF3), 123.77 (Ar-C), 122.97 (Ar-C), 122.59 (Ar-C), 120.63 (Ar-C), 119.88 (Ar-C), 113.16 (quinazoline-C4a), 109.32 (Ar-C), 107.73 (quinazoline-C8), 102.84 (quinazoline-C5), 69.11 (hexyl-CH2), 56.30 (OCH3), 49.80 (hexyl-CH2), 30.17 (hexyl-CH2), 28.92 (hexyl-CH2), 26.06 (hexyl-CH2), and 25.56 (hexyl-CH2). HRMS (ESI) m/z calculated for C31H29ClF3N6O5 [M + H]+: 657.1840, found: 657.1841.
In vitro kinase assays
Enzyme inhibitory assays were carried out as described in our previous reports in the literatureCitation33–35. Briefly, the Kinase HotSpotSM service from Reaction Biology Co. (Malvern, PA) was used to screen the tested compounds. The HotSpot assay platform includes specific kinase/substrate pairs and required cofactors. The base reaction buffer consisted of 20 mM Hepes (pH 7.5), 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, and 1% DMSO. The tested compounds were dissolved in 100% DMSO to a specific concentration, and serial dilution was conducted. The reaction mixture containing the tested compound and 33P-ATP was incubated at room temperature for 2 h, and radioactivity was detected using the filter-binding method.
Cell culture
Prostate cancer cell lines (PC3 and 22RV1) of American Type Culture Collection (ATCC) were obtained from Korean Cell Center (KCL). PC3 and 22RV1 cell lines were cultured in Roswell Park Memorial Institute Medium (RPMI) 1640 and Dulbecco’s modified Eagle’s medium (DMEM) (GenDepot, Barker, TX) supplemented with 1% penicillin–streptomycin and 10% foetal bovine serum (FBS). The cells were maintained at 37 °C in a 5% CO2 with a 95% humid atmosphere.
MTT assay
To further analyse the intracellular cytotoxicity effect of the as-synthesised compounds, MTT assay was performed. Moreover, the experiments were divided into subcategories. In each group, cancer cells were first seeded in 96-well plates (4 × 104 cells per well) and then incubated for 24 h at 37 °C in a 5% CO2-containing atmosphere. Subsequently, the medium from the culture plate was removed, and 100 µL of RPMI media containing different concentrations of compounds (0.01, 0.005, 0.0025, 0.00125, and 0.000625 µM) were then added into the cell lines in each well and incubated for 24 h in a 5% CO2 humidified incubator at 37 °C. Afterwards, MTT solution (150 μL, 1 mg/mL) was added to each well, replacing the media-containing compounds. After 4 h of incubation, the MTT reagent was removed, and 200 μL of DMSO was then added to dissolve the formazan crystals, and colour intensity was measured at a wavelength of 540 nm using a BioTek Synergy H1 instrument (BioTek, Winooski, VT). IC50 was calculated according to the equation of Boltzmann sigmoidal concentration–response curve using Graph Pad Prism 5 (La Jolla, CA).
Analysis of the cell cycle distribution
To determine the effect of the tested compound on the cell cycle distribution of PC3 and 22RV1 cell lines. Cell cycles can be analysed with checking the cell cycle change compared to the control group. Control cells without any treatment were used as a reference point for determining the cell cycle arrest phase for the test sample. Cell cycle analysis was performed using the Cell Cycle Analysis Kit (ADAMII LS, NanoEntek, Seoul, South Korea). Briefly, cancer cells were first seeded in six-well plates (0.8 × 106 cells per well) and then incubated for 24 h at 37 °C in a 5% CO2-containing atmosphere. Subsequently, the medium from the culture plate was removed, and 3 mL of RPMI and DMEM media containing concentrations of compound 9f at its IC50 value were then added into the cells in each well and incubated for 24 h in a 5% CO2 humidified incubator at 37 °C. Afterward, the media was removed following the washing once with PBS and the 25 µL cell sample mixtures were stained with 25 µL PI stain. 25 µL of sample mixture was loaded into ADAMII LS assay slide, then the slide was incubated at room temperature for 1 min in the dark and run on the ADAMII LS fluorescence cell analyser. Cell cycle distribution was calculated using ADAMII LS software (ADAMII LS, NanoEntek, Seoul, South Korea).
Apoptosis analysis
The apoptotic effect of a potent compound on PC3 and 22RV1 cell lines was assessed using apoptosis analysis with Annexin V-PE, DAPI solution (ADAMII LS, NanoEntek, Seoul, South Korea). Early and late apoptotic effects were analysed compared to the control. Cells were seeded in a six-well plate with cells density 0.8 × 105 cells/well, in a volume of 3 mL RPMI and DMEM medium and incubated for 24 h at 37 °C in a 5% CO2-containing atmosphere. Subsequently, the medium from the culture plate was replaced by 3 mL of RPMI and DMEM media containing compound 9f at its IC50 value and incubated for 24 h at 37 °C and in the presence of 5% CO2. The apoptosis assay was performed using the ADAMII LS apoptosis analysis kit. Briefly, the apoptosis-induced cell sample was prepared using a cell scraper (alfa aesar) after washing with phosphate-buffered saline (PBS). The cell was resuspended in 100 µL 1X Annexin V binding buffer, and 5 µL Annexin V-PE reagent was added afterward and incubated for 15 min at room temperature. The sample was subsequently centrifuged, and the pallet was resuspended again with 500 µL 1X Annexin V binding buffer and added 1.25 µL DAPI reagent. The slide loaded with the prepared sample was incubated at room temperature for 1 min in the dark and run on the ADAMII LS fluorescence cell analyser. The comparative data including dot plot graphs and images were calculated and analysed using ADAMII LS software (NanoEntek, Seoul, South Korea).
Statistical analysis
The statistical analysis of the data was done by standard deviations and all values represent the mean ± SD of three independent experiments.
Molecular docking
Two molecular docking processes were performed for the newly designed and synthesised derivatives (9a–h) towards the two target receptors (EGFR and HER2) using the MOE 2019.0102Citation31,Citation32. Besides, the co-crystallised ligands were incorporated as reference controls. The designed candidates (9a–h) were sketched using the ChemDraw program, transferred into the MOE window, and prepared to be inserted into the same database besides the co-crystallised inhibitor in each case, as previously discussedCitation36,Citation37. The X-ray structures of the target receptors (EGFR and HER2) with PDB IDs: 1M17Citation30 and 3RCDCitation25, respectively, were downloaded, protonated, and prepared for the docking process as described earlierCitation38,Citation39. Then, two docking processes (general subtypes) were performed within the EGFR and HER2 receptors in the presence of the corresponding prepared database in each case. The items of the program specifications were adjusted to the general docking subtype process, as mentioned beforeCitation40,Citation41. Finally, we redocked the native co-crystallised inhibitors of both EGFR and HER2 receptors within their binding pockets to validate the applied forcefield. Herein, the validity was confirmed by observing the same binding modes of the superimposed docked co-crystallised inhibitor in each case over its native oneCitation42. Moreover, promising root mean square deviation (RMSD) values were obtained for EGFR and HER2 receptors (1.32 and 1.76 Å, respectively).
Molecular dynamics simulations and MM-GBSA study
The MD simulations and MM-GBSA study were performed following same procedures found in our earlier reportCitation24.
Conclusions
EGFR and HER2 have been co-expressed and recognised in numerous solid tumours. Successfully, new TAK-285 derivatives (9a–h) were synthesised and assessed as EGFR/HER2 dual inhibitors. Applying “HotSpotSM” assay at 10 µM concentration in the presence of 10 µM of ATP, all compounds 9a–h exhibited encouraging percentage inhibition ranges against both kinases. The most active candidate (9f) showed nanomolar IC50 values of 2.3 nM over EGFR and 234 nM over HER2. A kinase selectivity panel of derivative 9f indicated a potential selective profile. Potent nanomolar IC50 values of compounds were obtained against PC3 and 22RV1 cell lines. The cell cycle of 22RV1 and PC3 cells was inhibited by compound 9f by 62.74 and 49.43% in the G2/M phase, respectively. Also, cells treated with compound 9f underwent early apoptosis. Molecular simulation studies revealed insights about the binding mode of the designed compounds. Overall, we report compound 9f as a new potent dual EGFR/HER2 inhibitor with significant cytotoxic activity against 22RV1 and PC3 prostate carcinoma cell lines and could be a potent anticancer drug for future medication.
Supplemental Material
Download PDF (4.4 MB)Acknowledgements
A.E. expresses his gratitude to the Korea Institute of Science and Technology (KIST) for partially supporting this work through the "2022 KIST School Partnership Project," and he would like to thank the Technology Innovation Commercial Office (TICO) at Mansoura University for their highly effective contribution to the project’s success. The authors gratefully thank the resources provided by the Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R127), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dowsett M, Cooke T, Ellis I, Gullick WJ, Gusterson B, Mallon E, Walker R. Assessment of HER2 status in breast cancer: why, when and how? Eur J Cancer. 2000;36(2):170–176.
- Kim H, Muller WJ. The role of the epidermal growth factor receptor family in mammary tumorigenesis and metastasis. Exp Cell Res. 1999;253(1):78–87.
- Koeppen HKW, Wright BD, Burt AD, Quirke P, McNicol AM, Dybdal NO, Sliwkowski MX, Hillan KJ. Overexpression of HER2/neu in solid tumours: an immunohistochemical survey. Histopathology. 2001;38(2):96–104.
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137.
- Tiwari SR, Mishra P, Abraham J. A novel HER2-targeted tyrosine kinase inhibitor. Clin Breast Cancer. 2016;16(5):344–348.
- Johnston SR, Leary A. Lapatinib: a novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs Today. 2006;42(7):441–453.
- Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, Smith DJ, Landolfi S, Ramon y Cajal S, Arribas J, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28(6):803–814.
- Wood ER, Truesdale AT, McDonald OB, Yuan D, Hassell A, Dickerson SH, Ellis B, Pennisi C, Horne E, Lackey K, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64(18):6652–6659.
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500.
- Engelman JA, Zejnullahu K, Gale C-M, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, et al. PF00299804, an irreversible Pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67(24):11924–11932.
- Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792.
- Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong K-K, Tenen DG, Halmos B. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65(16):7096–7101.
- Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075.
- Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, Halsey W, Sathe GM, Martin A-M, Gilmer TM. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69(17):6871–6878.
- Wang Q, Quan H, Zhao J, Xie C, Wang L, Lou L. RON confers lapatinib resistance in HER2-positive breast cancer cells. Cancer Lett. 2013;340(1):43–50.
- Rexer BN, Ham AJL, Rinehart C, Hill S, de Matos Granja-Ingram N, Gonzalez-Angulo AM, Mills GB, Dave B, Chang JC, Liebler DC, et al. Phosphoproteomic mass spectrometry profiling links Src family kinases to escape from HER2 tyrosine kinase inhibition. Oncogene. 2011;30(40):4163–4174.
- Eichhorn PJA, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–9230.
- Lancini GC, Arioli V, Lazzari E, Bellani P. Synthesis and relationship between structure and activity of 2-nitroimidazole derivatives. J Med Chem. 1969;12(5):775–780.
- Noss MB, Panicucci R, McClelland RA, Rauth AM. 1-Methyl-2-nitrosoimidazole: cytotoxic and glutathione depleting capabilities. Int J Radiat Oncol Biol Phys. 1989;16(4):1015–1019.
- Hoigebazar L, Jeong JM, Lee JY, Shetty D, Yang BY, Lee YS, Lee DS, Chung JK, Lee MC. Syntheses of 2-nitroimidazole derivatives conjugated with 1,4,7-triazacyclononane-N,N′-diacetic acid labeled with F-18 using an aluminum complex method for hypoxia imaging. J Med Chem. 2012;55(7):3155–3162.
- Kumar P, Shustov G, Liang H, Khlebnikov V, Zheng W, Yang XH, Cheeseman C, Wiebe LI. Design, synthesis, and preliminary biological evaluation of 6-O-glucose-azomycin adducts for diagnosis and therapy of hypoxic tumors. J Med Chem. 2012;55(13):6033–6046.
- van Loon J, Janssen MH, Ollers M, Aerts HJ, Dubois L, Hochstenbag M, Dingemans AM, Lalisang R, Brans B, Windhorst B, et al. PET imaging of hypoxia using [18F]HX4: a phase I trial. Eur J Nucl Med Mol Imaging. 2010;37(9):1663–1668.
- Cheng W, Zhu S, Ma X, Qiu N, Peng P, Sheng R, Hu Y. Design, synthesis and biological evaluation of 6-(nitroimidazole-1H-alkyloxyl)-4-anilinoquinazolines as efficient EGFR inhibitors exerting cytotoxic effects both under normoxia and hypoxia. Eur J Med Chem. 2015;89:826–834.
- Elkamhawy A, Son S, Lee HY, El-Maghrabey MH, Hamd MAE, Alshammari SO, Abdelhameed AA, Alshammari QA, Abdeen A, Ibrahim SF, et al. Design, synthesis, biological evaluation, and molecular dynamics studies of novel lapatinib derivatives. Pharmaceuticals. 2022;16(1):43.
- Ishikawa T, Seto M, Banno H, Kawakita Y, Oorui M, Taniguchi T, Ohta Y, Tamura T, Nakayama A, Miki H, et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J Med Chem. 2011;54(23):8030–8050.
- Whang YE, Armstrong AJ, Rathmell WK, Godley PA, Kim WY, Pruthi RS, Wallen EM, Crane JM, Moore DT, Grigson G, et al. A phase II study of lapatinib, a dual EGFR and HER-2 tyrosine kinase inhibitor, in patients with castration-resistant prostate cancer. Urol Oncol. 2013;31(1):82–86.
- Jathal MK, Steele TM, Siddiqui S, Mooso BA, D'Abronzo LS, Drake CM, Whang YE, Ghosh PM. Dacomitinib, but not lapatinib, suppressed progression in castration-resistant prostate cancer models by preventing HER2 increase. Br J Cancer. 2019;121(3):237–248.
- Rashed FB, Diaz-Dussan D, Mashayekhi F, Macdonald D, Nation PN, Yang XH, Sokhi S, Stoica AC, El-Saidi H, Ricardo C, et al. Cellular mechanism of action of 2-nitroimidazoles as hypoxia-selective therapeutic agents. Redox Biol. 2022;52:102300.
- Soliman AM, Alqahtani AS, Ghorab MM. Novel sulfonamide benzoquinazolinones as dual EGFR/HER2 inhibitors, apoptosis inducers and radiosensitizers. J Enzyme Inhib Med Chem. 2019;34(1):1030–1040.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–46272.
- Chemical Computing Group Inc. Molecular operating environment (MOE). Montreal: Chemical Computing Group Inc.; 2016. p. 1010.
- Madbouly E, Lashine E-S, Al-Karmalawy AA, Sebaiy M, Pratsinis H, Kletsas D, Metwally K. Design and synthesis of novel quinazolinone-chalcone hybrids as potential apoptotic candidates targeting caspase-3 and PARP-1: in vitro, molecular docking, and SAR studies. New J Chem. 2022;46(46):22013–22029.
- Elkamhawy A, Farag AK, Viswanath AN, Bedair TM, Leem DG, Lee KT, Pae AN, Roh EJ. Targeting EGFR/HER2 tyrosine kinases with a new potent series of 6-substituted 4-anilinoquinazoline hybrids: design, synthesis, kinase assay, cell-based assay, and molecular docking. Bioorg Med Chem Lett. 2015;25(22):5147–5154.
- Lee K, Nada H, Byun HJ, Lee CH, Elkamhawy A. Hit identification of a novel quinazoline sulfonamide as a promising EphB3 inhibitor: design, virtual combinatorial library, synthesis, biological evaluation, and docking simulation studies. Pharmaceuticals. 2021;14(12):1247.
- Elsherbeny MH, Ammar UM, Abdellattif MH, Abourehab MAS, Abdeen A, Ibrahim SF, Abdelrahaman D, Mady W, Roh EJ, Elkamhawy A. 2-(3-Bromophenyl)-8-fluoroquinazoline-4-carboxylic acid as a novel and selective aurora a kinase inhibitory lead with apoptosis properties: design, synthesis, in vitro and in silico biological evaluation. Life. 2022;12(6):876.
- Salem MA, El-Shiekh RA, Aborehab NM, Al-Karmalawy AA, Ezzat SM, Alseekh S, Fernie AR. Metabolomics driven analysis of Nigella sativa seeds identifies the impact of roasting on the chemical composition and immunomodulatory activity. Food Chem. 2023;398:133906.
- Kutkat O, Moatasim Y, Al-Karmalawy AA, Abulkhair HS, Gomaa MR, El-Taweel AN, Abo Shama NM, GabAllah M, Mahmoud DB, Kayali G, et al. Robust antiviral activity of commonly prescribed antidepressants against emerging coronaviruses: in vitro and in silico drug repurposing studies. Sci Rep. 2022;12(1):12920.
- Mahmoud A, Mostafa A, Al-Karmalawy AA, Zidan A, Abulkhair HS, Mahmoud SH, Shehata M, Elhefnawi MM, Ali MA. Telaprevir is a potential drug for repurposing against SARS-CoV-2: computational and in vitro studies. Heliyon. 2021;7(9):e07962.
- Hammoud MM, Khattab M, Abdel-Motaal M, Van der Eycken J, Alnajjar R, Abulkhair H, Al‐Karmalawy AA. Synthesis, structural characterization, DFT calculations, molecular docking, and molecular dynamics simulations of a novel ferrocene derivative to unravel its potential antitumor activity. J Biomol Struct Dyn. 2022;1–18.
- Ezz Eldin RR, Saleh MA, Alotaibi MH, Alsuair RK, Alzahrani YA, Alshehri FA, Mohamed AF, Hafez SM, Althoqapy AA, Khirala SK, et al. Ligand-based design and synthesis of N′-Benzylidene-3,4-dimethoxybenzohydrazide derivatives as potential antimicrobial agents; evaluation by in vitro, in vivo, and in silico approaches with SAR studies. J Enzyme Inhib Med Chem. 2022;37(1):1098–1119.
- El-Masry RM, Al-Karmalawy AA, Alnajjar R, Mahmoud SH, Mostafa A, Kadry HH, Abou-Seri SM, Taher AT. Newly synthesized series of oxoindole–oxadiazole conjugates as potential anti-SARS-CoV-2 agents: in silico and in vitro studies. New J Chem. 2022;46(11):5078–5090.
- Mansour KA, Elbermawi A, Al-Karmalawy AA, Lahloub M-F, El-Neketi M. Cytotoxic effects of extracts obtained from plants of the Oleaceae family: bio-guided isolation and molecular docking of new secoiridoids from Jasminum humile. Pharm Biol. 2022;60(1):1374–1383.