
Abstract
Twenty-five azole compounds (P1–P25) were synthesised using regioselective base-metal catalysed and microwave-assisted approaches, fully characterised by high-resolution mass spectrometry (HRMS), nuclear magnetic resonance (NMR), and infrared spectra (IR) analyses, and evaluated for anticancer, anti-tyrosinase, and anti-oxidant activities in silico and in vitro. P25 exhibited potent anticancer activity against cells of four skin cancer (SC) lines, with selectivity for melanoma (A375, SK-Mel-28) or non-melanoma (A431, SCC-12) SC cells over non-cancerous HaCaT-keratinocytes. Clonogenic, scratch-wound, and immunoblotting assay data were consistent with anti-proliferative results, expression profiling therewith implicating intrinsic and extrinsic apoptosis activation. In a mushroom tyrosinase inhibition assay, P14 was most potent among the compounds (half-maximal inhibitory concentration where 50% of cells are dead, IC50 15.9 μM), with activity greater than arbutin and kojic acid. Also, P6 exhibited noteworthy free radical-scavenging activity. Furthermore, in silico docking and absorption, distribution, metabolism, excretion, and toxicity (ADMET) simulations predicted prominent-phenotypic actives to engage diverse cancer/hyperpigmentation-related targets with relatively high affinities. Altogether, promising early-stage hits were identified – some with multiple activities – warranting further hit-to-lead optimisation chemistry with further biological evaluations, towards identifying new skin-cancer and skin-pigmentation renormalising agents.
Graphical Abstract

Introduction
Cancer is the second-leading cause of death after cardiovascular diseases, and skin cancer (SC) is one of the most prevalent cancer types globally, with incidence and resultant mortality expected to keep growing at alarming ratesCitation1,Citation2. Non-melanoma SCs, including squamous cell carcinomas (SCCs) and basal cell carcinomas (BCCs), and melanomas affect nearly 3 million and more than 120 000 people globally each year, respectivelyCitation3. The still less-than-complete restoration of the ozone layer allows increased amounts of UVB radiation to reach the Earth’s surface, which is a significant risk factor for developing cutaneous cancersCitation4–6. Despite surgical tumour excision being among the most effective SC treatment approaches, such interventions often lead to disfigurement, requiring additional costly skin grafts to cover resultant defectsCitation7–11. Moreover, targeting proliferation and inducing apoptosis in proliferating cancer cells are effective approaches in cancer therapyCitation12–15, and significant strides have been made recently towards a better understanding of the mechanisms that trigger and sustain SCs, thus revealing new possibilities for intervention.
Tyrosinase (also known as polyphenol oxidase, PPO) is a rate-limiting binuclear copper-containing metalloenzyme that enables the biosynthesis of melanin polymers. These polymers are high-molecular-weight pigments known for protecting the skin against the deleterious effects of UV raysCitation16–19. Specifically, tyrosinase catalyses the first two steps in melanogenesis (the process of melanin synthesis). It converts l-tyrosine, viz. hydroxylation to 3,4-dihydroxyphenylalanine (l-DOPA) and effects subsequent oxidation of l-DOPA to DOPA-quinone, which spontaneously polymerises to form melanin in the melanosomes of melanocytesCitation18–27. The phosphorylated isomers of l-DOPA or l-tyrosine are vital regulatory molecules of melanogenesis within melanocytes localised in the skin epidermal basal layer and hair follicles. In the skin, every melanocyte is bound to over 30 keratinocytes (acting as melanin-recipient cells) to distribute the synthesised melanin and dictate the colour of human skin, eye (pigmented retina), and hairCitation19,Citation27–31. Diverse extrinsic and intrinsic factors such as UV exposure, α-melanocyte-stimulating hormone (α-MSH), melanocortin 1 receptor (MC1R), and pathogenic mutations modulate melanogenesis and aberrant proliferation of melanocytes. Altered melanin production, and accumulation or deficiency of melanocytes and melanin, can lead to various dermatoses and oculo-cutaneous pigmentation disorders such as albinism, age-related spots, chloasma, freckles, post-inflammatory hyperpigmentation, melasma, lentigo, malignant melanoma, and oculocutaneous albinism type 1 (OCA1)Citation24,Citation30,Citation32–39.
Pharmacological modulation of tyrosinase-dependent melanogenesis is a potential therapeutic approach for combatting melanoma as well as other pigmentation-related disordersCitation40–44. A number of naturally occurring tyrosinase inhibitors, including arbutin, azelaic acid, hydroquinone, kojic acid, and various phenolics, can reduce excessive melanin production to improve skin tone and treat dermatosesCitation30,Citation34,Citation40,Citation45–47. However, due to safety concerns related to serious adverse effects, only a few are currently in clinical/cosmeceutical useCitation48. This dearth of options provides the impetus to create and develop new anti-tyrosinase agents that are prospectively safer and more efficacious alternatives for treating pigment-related dermatological disordersCitation38,Citation49,Citation50. Compounds built on an array of natural or synthetic scaffolds, including various azoles, chalcones, flavonoids, stilbenes, and tropolones, have been reported to not only exhibit tyrosinase inhibitory activitiesCitation26,Citation38,Citation50–52, but also to provide various therapeutic augmentations, such as antioxidant activityCitation53,Citation54, and anti-inflammatory and anticancer actionsCitation55–57.
In cancers and other skin disorders such as hyperpigmentation, increased generation of reactive oxygen species (ROS) and free radicals, as well as severe perturbations of cellular redox balance, cause enhanced growth, genetic instability, and adaptive responses that promote the maintenance of malignant phenotypes by cancerous cellsCitation58. Also, hyperpigmentation increases hydrogen peroxide (H2O2) and ROS levels, resulting in elevated oxidative stress load on melanocytes. Thus, antioxidant or ROS buffering in disease conditions has long been a significant drug discovery and development focusCitation59.
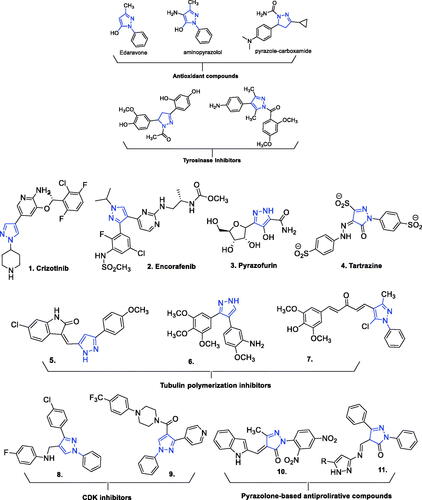
Numerous small-molecule compounds incorporating a pyrazole substructure exhibit one or more biological activities among a wide array thereof. Of medicinal relevance, these activities include analgesic, anticancer, antibacterial, antifungal, anti-inflammatory, antiviral, antidiabetic, antitubercular, antidepressant, anticonvulsant, antipyretic, anxiolytic, antimalarial, immunosuppressive, and antioxidant (reviewed in refs.Citation60–67) The large number of structurally diverse compounds elaborated from a pyrazole core includes representatives exhibiting anticancer activities of wide-ranging character and demonstrated or apparent efficacies against cancers appearing in almost every human organ or tissue, including lung, brain, colon, kidney, prostate, pancreas, and bloodCitation68,Citation69. Exemplary market-approved pyrazole-based anti-cancer drugs include crizotinib, encorafenib, pyrazofurin, and tartrazine. Various other pyrazole and pyrazolone derivatives have also exhibited antiproliferative activities mediated through their interactions with various cancer related targets, or directly ()Citation65,Citation70–75.
Figure 1. Molecular structures of previously reported antioxidant, antityrosinase, and anticancer compounds incorporating pyrazole or pyrazolone substructures.
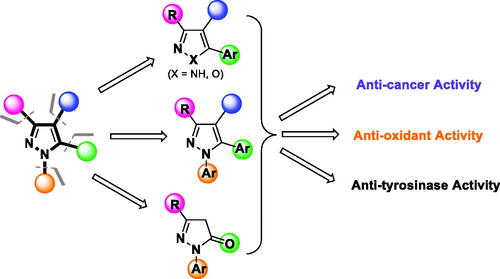
Given the broad spectrum of activities exhibited by elaborated pyrazoles, this moiety arguably merits “privileged structure” status, offering a versatile core chemotype in drug design and for compounds to be included in physical and virtual libraries for screening-based discoveryCitation76–79. The pyrazole moiety has four different sites () at which a wide array of substituents can be introduced to achieve a desired pharmacological activity, target selectivity, and other attributes needed for a biochemical/pharmacological tool or clinically/commercially useful agent through hit-to-lead and lead optimisation.
Figure 2. Structural design of biologically relevant substituted azoles and the proposed activity studies.
Well-construed in vitro screening studies hold promise for expediting discovery-to-development translation, as such studies can often quickly provide crucial information as to whether an early-stage test agent should be discarded or further pursued. This impact has now been seen in projects addressing unmet medical need for numerous human health problems, including cancers, inflammation, and pigmentation anomaliesCitation80,Citation81.
As noted above, available treatments still have serious limitations. Beyond safety and intrinsic efficacy, for agents applied dermally, low drug penetrability into the stratum corneum and diseased skin lesionsCitation14,Citation82,Citation83 can compromise the attainment of therapeutic levels at sites-of-actionCitation84. Compensating with higher applied concentrations can, for many available agents, result in skin irritation, or severe local or even systemic side effects and other safety concernsCitation85–88. As part of our effort to discover new synthetic scaffold-based entities with low toxicity, enhanced efficacy, and improved biopharmaceutic profiles against cutaneous cancers, pigmentation disorders, and various other dermal pathologies, we recently synthesised 1,3-diarylpyrazolones having potent suppressive activity against non-small-cell lung cancer cellsCitation89.
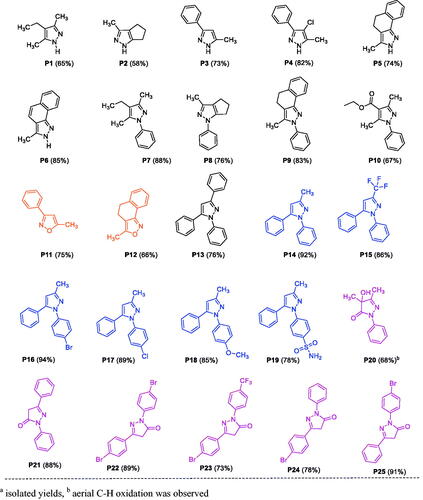
In the present work, we synthesised various pyrazoles and isoxazoles, and one set of pyrazolones, to evaluate their anti-tyrosinase, antioxidant and anticancer activities (P1–P25, ), by selecting suitable reaction partners with the primary goal of introducing a variety of substituents with diverse electronic and steric properties. A few of these compounds exhibited relatively potent anti-tyrosinase and anti-SC activities in vitro. We further explored the mode-of-action of the most-active broad-spectrum compound P25, and characterised alterations in physicochemical characteristics across the array of compounds tested (structure–property relationships). Additionally, in silico studies were carried out for targets predictions, including docking simulations to predict interactions with identified targets as well as an established non-human target for structurally similar compounds (i.e. mushroom tyrosinase), and predictive assessment of ADMET (absorption, distribution, metabolism, excretion, and toxicity) characteristics.
Figure 3. List of substituted pyrazole, isoxazole, and pyrazolone derivativesa. aIsolated yields and baerial C–H oxidation was observed.
Results and discussion
Chemistry
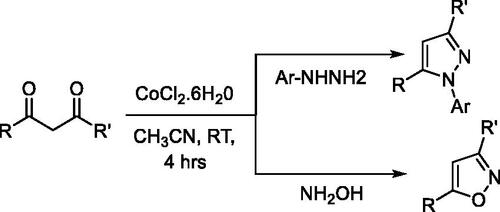
The wide range of biological activities associated with pyrazoles has made them popular synthetic targets. Thus efforts continue to develop new and more versatile synthetic approaches, resulting in expansions of such approaches as: cyclocondensation of hydrazines with carbonyl systems; dipolar cycloadditions of diazo compounds with alkynes; and the reaction of α,β-unsaturated aldehydes and ketones with hydrazinesCitation67. Some of the emergent procedures involve expensive transition-metal catalystsCitation90–92, stoichiometric amounts of harsh reagents (i.e. LiHMDS, tBuOLi, tBuOK, n-BuLi, HCl, and H2SO4)Citation93,Citation94, or specialised reagentsCitation95,Citation96. Cyclocondensation of 1,3-dicarbonyl compounds with substituted hydrazines at relatively higher temperatures offers a direct approach to access substituted pyrazoles. However, unsymmetrical 1,3-dicarbonyl synthon usage often results in the formation of regioisomeric pyrazoles, which are difficult to separate using chromatographyCitation97–100. Therefore, it is important to develop efficient, selective, and economically viable synthetic methods with potential for large-scale synthesis. Along these lines, we have developed two synthetic approaches for substituting pyrazoles using unsymmetrical or symmetrical dicarbonyl synthons. The first approach entails base metal-catalysed regioselective synthesis of N-arylpyrazoles and isoxazoles from unsymmetrical dicarbonyl derivatives. The second approach is based on microwave-assisted synthesis of pyrazoles and pyrazolones. To establish a room-temperature cobalt-catalysed approach for the regioselective synthesis of N-arylpyrazoles and isoxazoles, a set of base metal (cobalt and nickel) catalysts and solvents were screened for a reaction between phenylhydrazine and benzoylacetone. Among the tested catalysts and solvents, Co(II)Cl2·6H2O in acetonitrile produced a quantitative yield of N-phenylpyrazole (P14) with >99% regioisomeric selectivity. The same reaction conditions afforded other N-arylpyrazoles (P8, P9, P15–P19) and isoxazoles (P11–P12) with good yields (Table S6). Structural diversity was achieved by introducing a variety of substituents on the pyrazole ring through the variation of diketone and arylhydrazine synthons (Scheme 1).
Scheme 1. Co-catalysed synthesis of pyrazoles and isoxazoles.
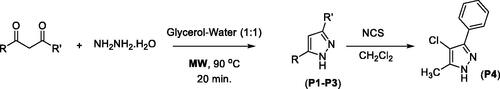
The microwave-assisted approach was found to be more efficient for pyrazoles P1–P6 as compared to the cobalt-catalysed approachCitation89. In fact, microwave irradiation facilitates the polarisation of the substrate molecules, accelerating the reactions and providing higher product yields than conventional heating procedures. In organic synthesis, microwave irradiation is considered an ecofriendly route. The microwave-assisted reaction of hydrazine with 1,3-diketones produced the pyrazole compounds P1–P3 in good yields. Moreover, pyrazole P3 was converted to the corresponding chloropyrazole (P4) via chlorination using N-chlorosuccinimideCitation101 (Scheme 2).
Scheme 2. Microwave-assisted synthesis of pyrazoles.
Tricyclic pyrazole P5 was synthesised from α-tetralone using tert-butylcarbazate as a source of hydrazine (Scheme 3); P5 was further converted to a fully aromatised indazole benznidazole (P6) using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) DDQ as an oxidising agentCitation102.
Scheme 3. Microwave-assisted synthesis of tricyclic pyrazole P5 and benznidazole P6.
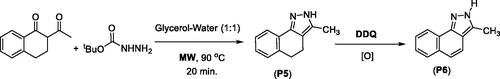
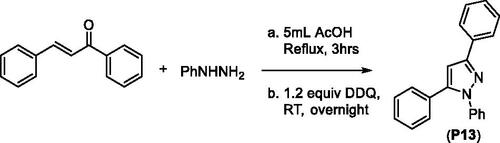
1,3,5-Triphenylpyrazole (P13) was obtained from a two-step synthesis via cyclocondensation of chalcone and phenylhydrazine under acid-catalysed reaction conditions followed by Pd-catalysed ring-oxidationCitation103 (Scheme 4).
Scheme 4. Synthesis of 1,3,5-triphenylpyrazoline from chalcone.
A set of 1,3-disubstituted pyrazolones (P20–P25) were synthesised from β-ketoesters and phenylhydrazine using our recently reported procedureCitation89. The pyrazolone P20 is a mono-aryl pyrazolone, whereas P21–P25 are diaryl pyrazolones. Moreover, P22 and P23 have para-substitutions on both benzene rings.
Tyrosinase inhibitory activities of pyrazoles, isoxazoles, and 3-pyrazolones P1–P25
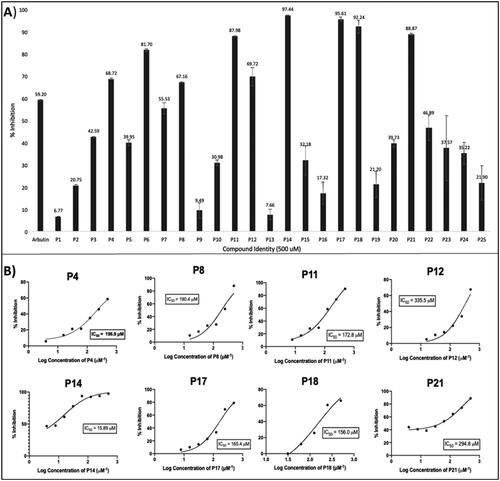
Due to the lack of an available high-resolution crystal structure of human tyrosinase, a mushroom tyrosinase with a highly similar active site protein sequenceCitation41–43,Citation104 was adopted for preliminary assessment of tyrosinase inhibition of the test compounds. The initial screening of all test compounds at 500 µM identified 10 compounds exhibiting promising tyrosinase inhibitory activity (cf. ), including P4 (68.7 ± 0.4%), P6 (81.7 ± 0.7%), P8 (67.2 ± 0.2%), P11 (88.0 ± 0.3%), P12 (69.7 ± 5.0%), P14 (97.4 ± 0.2%), P17 (95.6 ± 1.1%), P18 (92.2 ± 3.7%), and P21 (88.9 ± 0.4%). Inhibition was greater than for arbutin (59 ± 0.4%), which was used as a positive control. These compounds were further tested at various concentrations, from which half-maximal inhibitory concentration where 50% of cells are dead (IC50) values could be calculated (, Table S1). Eight of the 25 synthesised compounds (P4, P8, P11, P12, P14, P17, P18, and P21) exhibited potent-to-moderate tyrosinase inhibitory activities, with estimated IC50 values ranging from 16 µM to 340 µM, among which P14 exhibited the highest tyrosinase-inhibiting potency. Notably, the molecular weight of 11 of the evaluated compounds is relatively low, at only 200 or less (see Tables S2 and S3), including P4, P8, P11, and P12 among the compounds listed above. For all other tested compounds, the estimated IC50 value was either greater than 500 µM, or no activity was discerned (“not active” (N.A.)) (Table S1). The pyrazole derivative P14 (IC50 15.9 ± 1.2 µM), one of several relatively close analogues of celecoxib tested, was the most potent tyrosinase inhibitor (), exhibiting activity greater than the positive comparators arbutin (IC50 91 µM) and kojic acid (IC50 31 µM) (Table S1).
Figure 4. The mushroom tyrosinase inhibition screen of pyrazoles, pyrazolones, and isoxazoles P1–P25 identified several relatively active tyrosinase inhibitors (P4, P8, P11, P12, P14, P17, P18, and P21). (A) Initial single-concentration tyrosinase inhibition screening for derivatives P1–P25, with arbutin as a positive control. Ten compounds at a fixed concentration of 500 µM showed comparable or better percentage activity than arbutin. Data are expressed as mean ± SD from at least six independent experiments performed in triplicate. (B) Response vs. concentration curves for the above-listed potent pyrazole and pyrazolone compounds, with calculated 50% inhibitory activity (IC50) values. Data are expressed as mean ± SD from at least six independent experiments performed in quadruplicate.
Over the years, the mechanism-of-action of tyrosinase inhibitors has been experimentally linked to one of five main modes: competitive inhibition; reverse conversion (of ortho-dopaquinone to l-DOPA, i.e. a shift to reductase mode); non-specific enzyme inactivation (NE); specific, irreversible enzyme inactivation (SIE), in some instances more specifically entailing “suicide inhibition”; and specific reversible enzyme (SRE) inactivation (pseudo-irreversible inhibition)Citation27. Moreover, an extremely subtle relationship exists between antioxidant defence systems and tyrosinase inhibitionCitation105. However, P14 (15.9 ± 1.2 µM), the most potent tested anti-tyrosinase compound, was inactive in the DPPH antioxidant assay. It is worth mentioning that the screening of both tyrosinase inhibition and antioxidant activity in prospective studies requires further exploration.
Exploiting the availability of a high-quality X-ray structure of an inhibitor-bound tyrosinase (more specifically, a tyrosinase from Agaricus bisporus in deoxy-form, in complex with an unidentified lectin-like subunit, and with the inhibitor tropolone bound in its catalytic site; PDB ID 2Y9X), we carried out docking simulations to predict binding poses and specific molecular recognition interactions of P1–P25 within the catalytic pocket. Based on binding energies estimated with these simulations (cf. Refs.Citation106,Citation107) the eight most active of the synthesised compounds (P4, P8, P11, P12, P14, P17, P18, and P21) are predicted to bind with comparable or higher affinity than the inhibitor bound in the complex for which the PDB structure (2y9x) was obtained (tropolone: −5.90 kcal/mol) as well as one of our positive control ligands (kojic acid: −5.80 kcal/mol) ligands (). Detailed scrutiny of binding poses for the ligands predicted to bind with the highest affinity () indicates involvement of diverse interactions, including conventional H-bonds and various pi bonds at distances ranging 2.16 Å and 3.96 Å, as well as hydrophobic interactions. The comparative analysis is predicted by the simulation to exhibit relatively high binding affinities, as shown in .
Figure 5. Docking interactions and poses between compounds (P4, P8, P11, P12, P14, P17, P18, P21, P25, and kojic acid) and the target mushroom tyrosinase protein structure adopted from PDB 2Y9X. The target protein is highlighted in line format with light grey colour (A, B). The key interacting binding site amino acids are shown in black colour, while several hydrogen bonds are observed between functional groups of particular compounds and the enzyme amino acids, as summarised in , with interaction distances ranging from 2.16 Å to 3.26 Å. Left: Ligands in line and stick representation. Right: Ligands in ball and stick representation. Binding positions are proximal to the two copper ions (Cu2+) (not shown) in the active site.
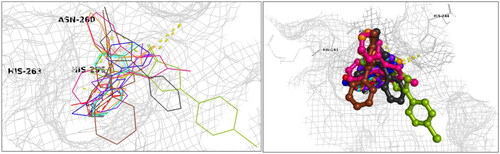
Table 1. Docking interaction energies for the active tyrosinase inhibitory compounds.
From an SAR standpoint, eight of the 25 test compounds showed moderate to potent tyrosinase inhibitory activity (≥70% inhibition). The 3-methyl-1,5-diaryl pyrazoles P14, P17, and P18 exhibited considerably greater potencies as compared to other tested compounds, which may be attributed to aryl substituents on positions 1 and 5 of the pyrazole moiety, and suggests these positions of the pyrazole ring as well as the nature of the substituent may be critical for the activity of the compound (see Table S1). Although P14, P17, and P18 showed similar % inhibition, P14 exhibited a substantially lower IC50 value (15.9 ± 1.2 µM), perhaps suggestive that the presence of para-substitutions on the N1-phenyl may not be favourable for binding. Moreover, compound P6 (a benznidazole), P11 and P12 (both isoxazoles), and P21 (a pyrazolone) displayed good to moderate anti-tyrosinase activities, although clearly, P6 and P12 are not overtly similar structurally. On the other hand (cf. ), the activities of pyrazoles P3 and P5 were significantly increased upon isosteric replacement of their pyrazole ring moiety with isoxazole (P11 and P12, respectively). Unlike other pyrazolones tested, P21 lacks substituents at the para-position of the benzene rings, suggesting that this might have accounted for the observed effect.
In vitro assessment of antiproliferative activity and preliminary structure–activity relationships
The antiproliferative effects of all the synthesised compounds (P1–P25), alongside cisplatin and celecoxib (serving as positive controls), were investigated in cultured human cutaneous melanoma (A375 and SK-MEL-28) and cutaneous SCC (A431 and SCC-12) cell lines by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. HaCaT immortalised keratinocytes were used as control noncancerous cells. As described earlierCitation108,Citation109, antiproliferative and cytotoxic activities were assessed for all the indicated five human skin-derived cell lines.
Some of the test compounds exhibited antiproliferative activities of varying degrees against the different human SC cell lines, comparing to their effect on noncancerous control cells (). Twelve compounds (P3, P9, P12, P14, P16, P17, P18, P19, P22, P23, P24, and P25) exhibited anti-SC activity against at least one among the spectrum of cancer cell lines tested, with markedly differing potencies. The most active compound, P25, displayed good-to-moderate potency across all four SC cell lines (A431: IC50 = 3.7 µM, SKMEL-28: IC50 = 7.6 µM, SCC-12: IC50 = 12.2 µM, and A375: IC50 = 14.3 µM). Importantly, P25 was also selectively active against cancer cells (A431 and SCC-12) vs. noncancerous control (HaCaT) cells, albeit with modest selectivity indexes (SIs) of only about 8.2 and 2.5, respectively. These SI values are, however, better than for cisplatin (SI of 0.8 and 0.4), a clinical anticancer agent with numerous side-effectsCitation110. Of potential interest for downstream investigation, P25 exhibited better potency against non-melanoma A431 cells than SCC-12 cells (IC50 3.7 µM vs. 12.2 µM, respectively). The next-most-active compound, P22, exhibited only moderate to marginal activities across the non-melanoma (A431: IC50 13 µM and SCC-12: IC50 19 µM) and melanoma (A375: IC50 29 µM and SK-MEL-28: IC50 31 µM) lines. Compounds P18 and P23 exhibited modest activities with some observed selectivity for cancerous over noncancerous cells and between cancerous lines.
Table 2. Cytotoxicity of pyrazole (celecoxib analogs), isoxazole, pyrazolone, and positive control compounds P1–P25 (structures, ) against cells of human cutaneous melanoma and non-melanoma skin cancer lines relative to standard control noncancerous immortalised HaCaT cells.
The anticancer activities of nonsteroidal anti-inflammatory cyclooxygenase inhibitors (“NSAIDs”), including aspirin (modestly COX1-selective) and celecoxib (a moderately COX2-selective pyrazole), prompted us to synthesise several celecoxib analogues and test their activities alongside the parent molecule in our panel of cutaneous cancerous cell linesCitation111–114. Celecoxib exhibited modest to marginal activity against the three cancerous cell lines tested (non-melanoma A431: IC50 7.4 µM, SI 6.7; melanoma SK-MEL-28: IC50 11.4 µM, SI 4.4 and A375: IC50 27 µM, SI 1.9). Amongst the celecoxib analogues (P14–P19), P15 was the only inactive compound, while the others showed marginal to moderate potencies. Compound P19 was reasonably selective with modest potency against non-melanoma A431 cells (IC50 7.6 µM, SI 7.5), values comparable to those obtained for celecoxib (vide supra) and cisplatin (IC50 7.7 µM, but with no selectivity (SI 0.8)). However, P19 exhibited poor activity against cells of the other nonmelanoma line (SCC-12), with essentially no selectivity, and also poor activity against cells of the two melanoma lines (A375 and SK-MEL-28). Marginal activity was observed for the remaining compounds, in most instances with poor selectivity against cancerous vs. control cells. However, P5 and P6 showed some selectivity against non-melanoma vs. non-cancerous control HaCaT cells.
From a screening stage structure–activity relationships (SARs) standpoint, compounds of greatest interest are pyrazoles that are celecoxib analogues, maintaining (P15) or replacing (P14 and P16–P19) the trifluoromethyl (CF3, powerfully electron-withdrawing) at position 3 of the pyrazole heterocycle with a methyl group (CH3; moderately electron-donating), and with omission (P14 and P15), substitution (P16, P17, and P18), or maintenance (P19) of the sulphonamide (SO2NH2) moiety. Only P25 exhibited activity and selectivity of some interest among the four pyrazolones. Still, no clear SAR conclusions can be drawn from these results, and non-analogous binding poses might easily be a confounding contributor. Moreover, it should be noted that the effects of various substituents on lipophilicity and, thus, cell penetrability must be considered in future studies. Given that no clear SAR emerged from this initial series of anti-proliferative screens, more insight into the relationship between the structural patterns and the activities of these compounds was sought by exploring their anti-tyrosinase (above) and antioxidant potentials, given that multi-component action might also confound interpretation of phenotypic results at this stage.
Colony formation, scratch wound healing, and apoptotic machinery investigations for phenotypic assessment of the most-active broad-spectrum anti-SC agent, pyrazolone P25
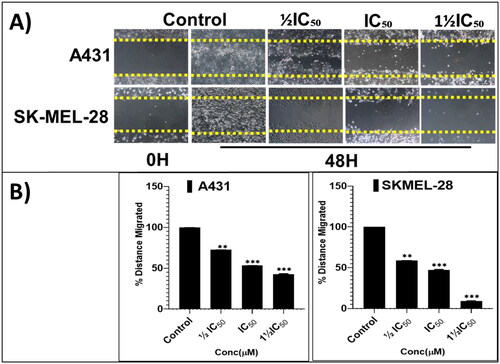
From the cell proliferation and viability experiments, the most-active and somewhat-selective broad-spectrum compound, pyrazolone P25, was selected for further characterising mode(s)-of-action phenotypically, in an attempt to identify molecular-level contributors to, and determinates of, efficacy, potency, and all-important cancer-directed selectivity. Towards these ends, anti-proliferative effects on colony formation, inhibition of scratch wound healing, and apoptosis-promoting effects were assessed. Long-term (14-days) treatment with various concentrations of P25 (0, ½IC50, IC50, and 1½IC50) revealed significant concentration-dependent reductions in the relative percentage of colonies formed by A431 and SKMEL-28 cells, as compared to untreated controls (). Further, a scratch-wound assay was carried out to scrutinise the inhibitory effects of P25 on the migration of A431 and SK-MEL-28 cells into cell-free scratch-wounded areas after 48-h treatments. P25 significantly and dose-dependently decreased the scratch wound areas in both A431 (non-melanoma) and SK-MEL-28 (melanoma) cultures as compared to untreated control cells ().
Figure 6. The potent broad-spectrum anticancer hit compound P25 significantly inhibits colony formation capacity in A431 (A) and SK-MEL-28 (B) cells after 14 days. The percentage decrease in colony formation was concentration-dependent (0, ½IC50, IC50, and 1½IC50 obtained from the antiproliferation assay) and was comparable in A431 (C, D) and SK-MEL-28 (E, F) cells. The data expressed in the bar graphs represent the mean ± SD in the P25-treated group expressed as a percentage relative to the untreated control group. Data are from three independent experiments performed in quadruplicate. Statistical significance was assessed using one-way ANOVA and Tukey’s multiple comparison tests; **p < 0.01, and ***p < 0.001 were considered significant.
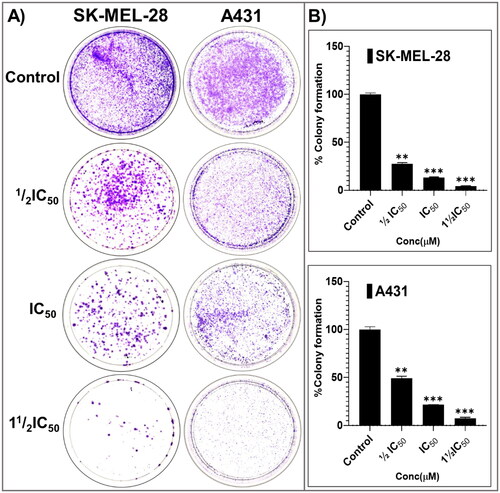
Figure 7. The screening hit compound P25 potently and dose-dependently (0, ½IC50, IC50, and 1½IC50) inhibits the migration of A431 (A; top panel) and SKMEL-28 (A; bottom panel) cells into the cell-free scratched-wounded areas of a confluent cell monolayer. This suppression of A431 and SKMEL-28 cells scratch wound closure in a concentration-dependent manner was significant. The bar graphs (B) represent the mean ± SD of covered scratch-wound area values (expressed as the migrated distance from 0-time point of scratch) after 48 h. They are expressed as a percentage of wound area at 0 h filled by migrated cells by the end of the experiment, vs. the same indicator for untreated control cells, from three independent experiments conducted in triplicate. Statistical significance was assessed using one-way ANOVA and Tukey’s multiple comparison tests; **p < 0.01, and ***p < 0.001 were considered significant.
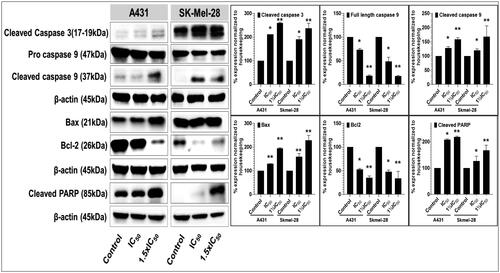
Apoptosis is an adaptive cellular mechanism that ensures replicating cells to maintain the integrity of their sourced DNA. Apoptosis is often impaired or dysfunctional in cancerous cells; consequently, the ability of a compound to restore normalcy of apoptotic triggering (and other programmed cell death pathways) constitutes an essential aspect in the screening of potential anticancer agents, and is a key prospective source of targeting selectivity (i.e. cancerous vs. non-cancerous cells and tissue)Citation115. As P25 significantly decreased cell viability and colony formation, apoptotic mechanisms of cytotoxicity were investigated by assessing the potential of this compound to modulate the intrinsic and extrinsic apoptosis pathways and function in A431 and SK-MEL-28 cell lines. The key apoptosis-related markers assessed (by western blot analysis) included the pro- and cleaved forms of caspase-3 (a member of the executioner apoptotic caspase family along with caspases 6 and 7) and caspase-9 (a member of the initiator apoptotic caspase family along with caspases 8 and 10), as well as cleaved vs. uncleaved poly(ADP-ribose) polymerase (PARP). Particular changes in these markers of pathway function are often positive indicators that a compound could serve for the successful development of an anticancer agent (more commonly, serving as a lead for further optimisation)Citation116,Citation117.
Treatment of A431 and SK-MEL-28 cells with P25 resulted in significant and concentration-dependent activation of apoptosis, as evidenced by substantially increased levels of activated caspase-3, and caspase-9, and of cleaved PARP, as compared with untreated controls (). Additionally, the involvement of the mitochondria, specifically with respect to intrinsic apoptotic pathways, was observed from the dose-dependent amplified expression of pro-apoptotic Bax with corresponding decreases in anti-apoptotic Bcl-2 protein levels in both A431 and SK-MEL-28 P25-treated cells as compared with untreated controlsCitation118. Therefore, these data strongly support that P25 induces the apoptosis of A431 and SKMEL-28 SC cells via the intrinsic mitochondrial apoptotic pathways associated with PARP activation (extrinsic pathways).
Figure 8. Compound P25 induces apoptosis by activating the extrinsic and intrinsic apoptotic pathways in cutaneous melanoma (SK-Mel-28) and non-melanoma (A431) cancer cells in culture. The blots (left) document a concentration-dependent effect (0, ½IC50, IC50, and 1½IC50; IC50 refers to the cell viability result, cf. ) of protein expression levels of markers of apoptosis, including pro- and cleaved caspase-3, and caspase-9, and of cleaved PARP, after 48 h of treatment. The data shown are representative immunoblots from three independent experiments with similar results. β-actin was used as a loading control. At right, protein levels as normalised (mean ± SD of relative quantitative density values) are expressed as a percent increase or decrease vs. untreated cells (control) in the bar graphs. Statistical significance was assessed using one-way ANOVA and Bonferroni’s multiple comparison tests; *p < 0.05 and **p < 0.01, were considered significant.
Antioxidant activity as assessed by DPPH radical scavenging
In cancers, increased ROS generation and severe perturbation of cellular redox balance have been associated with enhanced tumour growth, genetic instability, and adaptive responses that promote malignant phenotypes in cancerous cellsCitation59. However, antioxidant or ROS buffering as a component activity of a therapeutic agent for treating certain chronic skin diseases, notably those involving inflammation or cancers, may confer greater efficacyCitation119.
The antioxidant potential of the test compounds was explored via the DPPH assay, a rapid, simple, inexpensive, and widely used method to measure the ability of compounds to act as free-radical scavengers or hydrogen donors. Initial screening of all 25 compounds at 1 mM revealed compounds P6, P21, P22, P23, P24, and P25 to be the most active, with a scavenging effect ranging from 65 to 75%, comparing favourably to quercetin as a positive control (60%; ). All of other compounds exhibited lesser scavenging activities than quercetin. The activities of all compounds were further investigated as a function of concentration, with further comparisons to the known antioxidants ascorbic acid and butylated hydroxytoluene (BHT). The six compounds listed above exhibited antioxidant IC50 values ranging from 31 to 440 µM (; Table S4), whereas the activities of the remaining 19 compounds were negligible. The tricyclic naphtho[1,2-c]pyrazole P6 (IC50 30.6 ± 2.1 µM) was the most potent, exhibiting antioxidant activity comparable to that of ascorbic acid (IC50 25.2 ± 1.4 µM), and far better than of BHT (IC50 285 ± 7 µM). From a preliminary SAR perspective, except for P6 (pyrazole), all of the other active compounds showing appreciable antioxidant activities were pyrazolones, which are able to undergo keto-enol and imine-enamine tautomerism. Among the three possible tautomers, rapid exchange can occur between OH and NH tautomers, versus a slow equilibrium involving the CH (parent pyrazolone) tautomer. Possibly the NH (enamine) and OH (enol) tautomeric forms are responsible for the antioxidant effect of pyrazolones P21–P25, via sequential electron-transfer proton-transfer (SETPT), hydrogen atom transfer (HAT) or sequential proton-loss electron-transfer mechanisms (SPLETM)Citation85. Compound P20, the only pyrazolone of those tested found to exhibit no appreciable antioxidant activity, includes a C5-alkyl rather than aryl substituent and a quaternary C4 ring carbon. This observation suggests that the presence of 2,5-diaryl substitution and/or absence of a quaternary carbon in the pyrazolone ring is essential for retaining antioxidant activity, as exemplified in previous reportsCitation119–121. The tricyclic P6 is the only pyrazole that also emerged as a free-radical scavenger, and was the most active of all compounds assessed in this assay. Its high antioxidant activity is likely due to its free N–H moiety in conjunction with its highly conjugated fused aromatic rings, which can readily support one or more antioxidant mechanisms (i.e. SETPT, HAT, and SPLETM, vide supra). The inactivity of P5 in this assay, despite its close structural analogy to P6, is concordant with these assertions.
Figure 9. The 2,2-diphenyl-1-picrylhydrazyl (DPPH) anti-oxidant (radical scavenging) activity of compounds P1–P25 identifies modest to moderately potent antioxidant activity of one tricyclic pyrazole (a naphtho[1,2-c] pyrazole) and several pyrazolones. (A) Concentration-dependent radical scavenging activities, compared to quercetin as a positive control. (B) IC50 values for appreciably active compounds. Data are expressed as mean ± SD from at least six independent experiments performed in quadruplicate.
![Figure 9. The 2,2-diphenyl-1-picrylhydrazyl (DPPH) anti-oxidant (radical scavenging) activity of compounds P1–P25 identifies modest to moderately potent antioxidant activity of one tricyclic pyrazole (a naphtho[1,2-c] pyrazole) and several pyrazolones. (A) Concentration-dependent radical scavenging activities, compared to quercetin as a positive control. (B) IC50 values for appreciably active compounds. Data are expressed as mean ± SD from at least six independent experiments performed in quadruplicate.](/cms/asset/cf94aab0-d3ff-4bc6-ade5-6b25a878c4b7/ienz_a_2205042_f0009_c.jpg)
In silico target(s) prediction, molecular docking, and biopharmaceutic properties prediction
In silico ligand-based target prediction (so-called “target fishing”) is increasingly proving its suitability in predicting pertinent protein targets underlying observed phenotypic activitiesCitation122. Accordingly, we used the SwissTargetPrediction server, which exploits molecular similarities between test compounds and large, curated libraries of compounds with known and well-document activities (≥280 000 active compounds, and ≥440 000 documented interactionsCitation123) and generates data suggesting the probability that a particular molecule will interact with a given protein as a targetCitation123–126. Of the thousands of included targets, all 25 of the synthesised compounds (P1–P25) displayed molecular interaction similarities with only 11 molecular targets, including four kinases (mitogen-activated protein kinase 14 (MAPK14/p38α), vascular endothelial growth factor receptor 1 (VEGFR1), tyrosine-protein kinase FLT3, and tyrosine protein kinase JAK1), two oxidoreductases (COX-1 and COX-2), and four other targets including alpha-2B adrenergic receptor (α2bAR), carbonic anhydrase II, a cytosolic heat shock protein isoform (Hsp90-α), and cytochrome P450s (aggregate), as well as tyrosinase (cf. and discourse above).
Table 3. Predicted binding energies of compounds to molecular targets, as compared to their crystal-structure-native and commercial ligands. The bold values are the significant binding affinities.
We next employed the molecular docking simulation protocols of the AutoDock Vina suite to estimate the binding affinities in parallel with those of well-documented high-affinity ligands. We identified prospective targets for several of these compounds through a specific reverse screening protocol. Docking simulations were carried out for all 25 compounds against the 11 protein targets identified by SwissTargetPrediction (MAPK14, VEGFR1, FLT3, JAK1, COX-1/2, α2bAR, carbonic anhydrase II, Hsp90α, cytochrome P450s, and tyrosinase). Results were scrutinised with special attention to the most-active hits from the cell proliferation and tyrosinase inhibition assays. The docking protocol was validated by re-docking the co-crystallised (native) standard ligand within the catalytic domain of the specifically selected target protein. We observed that the re-docked pose of the co-crystallised ligand retained the experimental binding conformation with an RMSD value of less than 1 Å. Key interactions essential for inhibiting the identified protein targets were considered screening parameters along with the obtained docking score for each hit against respective target proteins. Compounds were predicted to have appreciable binding affinities to only selected macromolecular targets (). Briefly, for the substituted pyrazoles P1–P3 (P2 being an aliphatic-ring-fused bicyclic) and P11 (an isoxazole), no significant interactions with any of the identified targets were predicted. For pyrazole P4, only a single but considerably selective interaction was predicted, namely with the MAPK14/p38α kinase, a rather remarkable finding given the small size of this molecule. Compounds P5–P8, P10, P14, P16, and P18, which are substituted or comparable ring-fused pyrazoles, and substituted pyrazolones P20, P21, P24, and P25, displayed appreciable interactions with multiple targets were predicted, including the four kinases (MAPK14, VEGFR1, FLT3, and JAK1), two oxidoreductases (COX-1 and COX-2), one adrenergic receptor (α2bAR), and cytochrome P450). A number of the other compounds (P9, P13, and P15–P19, which are substituted pyrazole analogues of celecoxib, as well as the substituted pyrazolones P22 and P23) were predicted to be relatively more promiscuous (less-selective). Among the predictions are good-to-strong binding interactions with VEGFR1 (P23), FLT3 (P13, P15, P17, and P23), JAK1 (P9, P19, and P23), and α2bAR (P15), as well as a stronger interaction (but with less selectivity) to the cytosolic Hsp90α for two pyrazoles (P9 and P13); only these two pyrazoles were predicted to interact significantly with Hsp90α. Importantly, while no meaningful interactions were predicted for any of the identified targets for one of the isoxazoles (P11), for the other isoxazole (P12) significant interactions with the MAPK14, FLT3, one oxidoreductase (COX-2), α2bAR and the cytochrome P450s were predicted. Here, it seems worth noting that in the cytotoxicity assay, compound P14 (A375) exhibited an IC50 comparable to that of celecoxib, whereas compound P19 (A431) exerted no discernable activity against cells of SCC-12 or SK-MEL-28 lines, in contrast to celecoxib.
Overexpression of heat shock protein 90 (Hsp90) is common in various types of cancer, including cutaneous cancersCitation127–130, as well as in chronic skin defects such as inflammation and burnsCitation131–134. Carbonic anhydrase II elevation has been reported in mycosis fungoidesCitation135, in adenomas, in glandular tumours (sweat, sebaceous, and major salivary glands), and in various tumours of the skinCitation136–139. Cytochrome P450 enzyme systems are involved in drug biotransformation, and as such, are especially noteworthy for their involvement in numerous deleterious drug–drug interactionsCitation140,Citation141. Particular P450 isoforms (isozymes; CYPs) are also known to be significant players in various inflammatory conditions, notably through altering aryl hydrocarbon receptorCitation142 and retinoid-X-receptor (RxR) gene products expressionCitation143 and in other aspects of function in the human epidermis constitutively or under drug treatmentsCitation140–144. Modulation of VEGFR1 expression has been shown to contribute to the pathogenesis of skin inflammation and cancer, including melanoma, placing VEGFR1 prominently among important dermal therapeutic targetsCitation145–149. Alterations of MAPK14 expression and associated pathways have been tied (directly and by targeted modulation) to SCs including cutaneous malignant melanoma, as well as to skin inflammationCitation150–157. Dysregulation of Janus kinase 1, a protein tyrosine kinase (JAK1), is known to be a potential contributor to cutaneous inflammation and carcinogenesis, and its targeting can lead to disease resolutionCitation158–162. Alpha-2B adrenergic receptor modulation has been shown to mitigate skin cell migration, glaucoma, and other human ailmentsCitation163–165. Deregulation of FLT3 has been implicated in disorders involving cutaneous inflammation, such as psoriasis, unwanted hair pigmentation changes, and cancers including melanoma and leukemiasCitation166–170.
Finally, using the in silico SwissADME and ProTox II drug-likeness (DL) properties and organ toxicity prediction analyses, we observed that all these hit compounds’ violations of DL rules, physicochemical properties, ADME parameters, and DL rules fall within the defined and acceptable parameters for a good drug candidate (cf. Tables S2, S3, and S5). The predictions concerning P-glycoprotein (P-gp) and P450 family member inhibition varied across individual compound structures, and any compounds moving directly forward into further preclinical assessment would require assessment in suitable physical assessments.
Discussion and conclusions
The central goal of our work here is to introduce new small molecule scaffolds into the development pipelines for SC or pigmentation disorders. Towards furthering this aim, we devised and reports herein two modified approaches for the synthesis of various pyrazoles, isoxazoles, and pyrazolones. We engaged microwave-assisted methods for synthesising unsubstituted pyrazoles (P1–P6) and pyrazolones (P20–P25), and a catalytic regioselective synthesis for making substituted pyrazoles (P7–P10, P14–P19) and isoxazoles (P11, P12) in good to high yields. These compounds were characterised and identified as a series of lead compounds with varying degrees of anti-tyrosinase, antioxidant, and anticancer activities. Melanocytes with dysregulated tyrosinase-associated function leading to excessive production of melanin have been implicated in the development of various pigmentary dermatoses, including age-related spots, chloasma, freckles, melasma, post-inflammatory hyperpigmentation, and malignant melanomaCitation24,Citation32–36,Citation39. Moreover, disease-causing mutations in the tyrosinase gene cause melanin deficiency in OCA1, an autosomal recessive disorder characterised by the lack of eye, hair, and skin pigmentationCitation30. Therefore, genetic or pharmacological modulation of melanogenesis is a potential therapeutic approach for melanoma and various skin pigmentation disordersCitation40–44. Furthermore, tyrosinase inhibition aimed at reducing melanogenesis is relevant in the agroindustry for the preservation of fruit via mitigating browning, interfering with insect moulting (pesticidal), and preventing deleterious adhesion of marine organismsCitation171. Clearly, suitably designed tyrosinase inhibitors would be of prospectively high value for an array of cosmetics, skin health, and agro-industry usesCitation49.
Cancers of the skin are in aggregate among the most prevalent of cancer types with respect to tissue-of-origin, with about 9500 diagnoses per day in the U.S. aloneCitation172–174. Available treatments are hampered by drug resistance, side-effects, low bioavailability, or high cost. Several of the compounds characterised in our studies exhibited noteworthy anti-skin-cancer activities, some superior to cisplatin’s (ranging up to eightfold greater), with better SI for cancerous over non-cancerous cells. The most-active broad-spectrum anticancer compound, P25, suppressed cell migration and colony formation in the A431 and SK-MEL-28 cell lines in vitro, likely through modulation of certain parallel pathways significantly impinging on the induction of apoptosis, as evidenced by altered apoptosis-related marker proteins, namely marked increases in cleaved caspases 3 and 9, and cleaved PARP, as well as a pronounced increase in the Bax/Bcl-2 ratio.
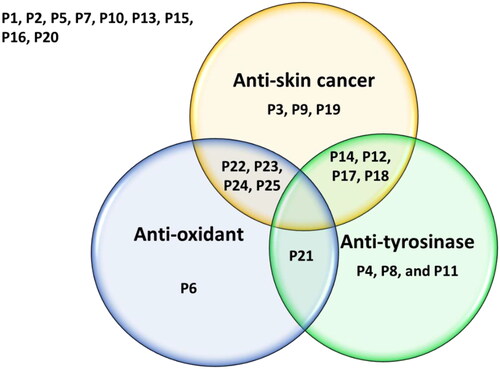
Several of the evaluated compounds also showed dual anti-tyrosinase and antioxidant, anticancer and antioxidant, or potent anti-tyrosinase with mild anticancer properties (Venn diagram, ). The anti-tyrosinase, anti-proliferative, antioxidant, anti-migratory, and pro-apoptotic effects observed among these structurally analogous compounds suggest that further elaboration and optimisation efforts could ultimately provide high-value clinical candidates, possibly with advantageous, synergistic dual-targeting attributes. To further understand the underlying molecular mechanism-of-action of these compounds, in silico simulations and analyses identified that a number of the compounds are predicted to bind to a relatively small number of targets highly relevant to cancer, inflammation, and pigmentation disorders. It is noteworthy that many of these identified targets including enzymes, kinases, proteases, and receptors have been shown to play important roles in carcinogenesis and SC progression and inflammation. Moreover, targets such as lipoxygenases are well known to catalyse the formation of corresponding hydroperoxides from polyunsaturated fatty acids such as linoleic acid and arachidonic acid. They are expressed in immune, epithelial, and tumour cells that display a variety of physiological functions, including inflammation, skin disorder, and tumorigenesisCitation175–177. In fact, SK-MEL-28, one of a series of melanoma cell lines established from patient-derived tumour samples used in this study, all believed to play an essential role in skin carcinogenesis or treatment. Therefore, further studies are warranted to assess the validity of these predictions in suitably construed physical experiments. Regardless, the data amassed in this study will be helpful in further attempts to optimise the activity of the hit molecules and the determination of their putative drug targets and potential mechanisms of action. The fact that pharmacological modulation of several of these dysregulated targets and receptors appeared to induce apoptotic death of tumorigenic epidermal cells without affecting the viability of non-transformed epidermal cellsCitation178 suggests that suitable multicomponent manipulation of various signalling pathways via multiple targets and mechanisms can be a promising strategy for the small-molecule-based management of some types of cancers, chronic inflammatory maladies, and pigmentation disorders.
Figure 10. The Venn diagram summarises the anti-proliferative (anti-skin cancer), antioxidant, and tyrosinase inhibitory effects of the 25 compounds. Dual-acting compounds were identified with noteworthy anti-proliferative and antioxidant activity (P22, P23, P24, and P25); anti-proliferative and anti-tyrosinase activity (P12, P14, P17, and P18); and antioxidant and anti-tyrosinase activity (P21). Some tested compounds exhibited only a single noteworthy activity: antioxidant (P6); anti-tyrosinase (P4, P8, and P19), and/or anti-skin-cancer (P3, P9, and P19).
Altogether, our results offer up promising prototypes for further studies towards identifying single, in-combination, or adjuvant agents for the control of melanoma and NMSCs, and tyrosinase-related pigmentary disorders.
Materials and experimental methods
Chemistry
Synthesis and characterisation of pyrazole derivatives
General information: All the chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and TCI (Portland, OR). All the reagents were commercial grade, and solvents were purified according to established procedures. Organic extracts were dried over anhydrous sodium sulphate. After completion of the reactions, solvents were removed in a rotary evaporator under reduced pressure using a Buchi Rotavapor equipped with a vacuum controller and vacuum pump. Silica gel (60–120 mesh size) was used for column chromatography. Reactions were monitored by thin-layer chromatography (TLC) on silica gel precoated aluminium cards with fluorescent indicator visualisable at 254 nm (0.25 mm). Developed plates were visualised by a Spectroline ENF 260C/FE UV apparatus. Nuclear magnetic resonance (NMR) spectra were recorded on a JEOL 400 MHz FT spectrometer in the indicated solvent. Chemical shifts are expressed in δ units (ppm) from tetramethylsilane (TMS) as the internal standard for 1H NMR (400 MHz) and for 13C NMR (100 MHz). Infrared spectra (IR) were run on a SpectrumOne FT-ATR spectrophotometer (Perkin Elmer, Waltham, MA). Band position and absorption ranges are given in cm−1. Gas chromatography–mass spectrometry (GC–MS) analysis was carried out on an Agilent GC-MS (7890A-5975C VL MSD) system.
Microwave-assisted reactions were performed with a Discover S-Class (CEM) single-mode microwave reactor, where the instrument settings were controlled with PC-running Synergy 1.4 software. All experiments were carried out in microwave reaction vials using a stirring bar (length 20 mm, diameter 6 mm). Stirring, temperature, irradiation power, PowerMAX (in situ cooling during the microwave irradiation), ramp, and hold times were set as indicated. The temperature of the reaction was monitored by a built-in infrared sensor. After completion of the reaction, the mixture was cooled to 25 °C via air-jet cooling. Most products were purified using column chromatography, and some were isolated in pure form through recrystallisation. All the synthesised compounds were characterised by NMR, GC–MS, and HRMS analysis.
General synthesis procedures
Microwave assisted synthesis of pyrazoles P1, P2, P3, and P5
A 10 mL microwave reaction vial, equipped with a stir bar, charged with hydrazine hydrate (1 mmol), 1,3-diketone (1 mmol), 1:1 water and glycerol (2 mL) and added potassium carbonate was mixed by stirring the solution at room temperature. The vial was then sealed with a Teflon-lined silicone cap and placed in the microwave synthesiser cavity. Microwave irradiation of 200 W was used, and the temperature ramped from 25 °C to 80 °C. Once the temperature reached 80 °C, the reaction continued for 10–20 min at the same temperature while stirring. Upon completion, the reaction mixture was cooled to room temperature, extracted with ethyl acetate (EtOAc), and evaporated to collect the solid product mixture, which was either recrystallised or purified by column chromatography to obtain a pure product.
Synthesis of 4-chloro-5-methyl-3-phenyl-1H-pyrazole (P4)Citation101
To a solution of 5-methyl-3-phenyl-1H-pyrazole (P3) (1 mmol) in acetonitrile (5 mL), N-chlorosuccinimide (2 mmol) was added, and the solution stirred continuously at 40 °C for 3 h. After completion, the acetonitrile solvent was removed, and the mixture was washed with saturated sodium thiosulphate (2 × 20 mL) and water (20 mL), and the product was extracted with EtOAc solvent.
Synthesis of pyrazole P6Citation102
We used a slightly modified method developed by Hilt et al. To the solution of P5 (1 mmol) in 5 mL of acetonitrile, DDQ (1.2 mmol) was added, and the mixture was stirred continuously at room temperature for 5 h. After filtration through a short pad of silica, the crude product was purified by column chromatography to yield pure pyrazole P6.
Cobalt(II)-catalysed synthesis of pyrazoles, isoxazoles (P7–P12 and P14–P19)
To a solution of cobalt(II) chloride hexahydrate and arylhydrazine/hydroxylamine hydrochloride (1 mmol) in acetonitrile, a 1,3-diketone (1 mmol) was added, and the mixture was stirred at room temperature. The reaction was monitored using TLC and GC–MS analysis. After completion, the reaction mixture was filtered through celite and washed with EtOAc. Products were purified by recrystallisation or column chromatography using EtOAc and hexane mixture as eluent.
Microwave-assisted synthesis of 1,3-diarylpyrazolones (P20–P25)Citation89
A 10 mL microwave reaction vial equipped with a stir bar was charged with the substituted β-ketoester (2 mmol), the appropriate arylhydrazine (2 mmol), glycerol (1 mL), and water (1 mL). The vial was sealed with a Teflon-lined silicone cap and placed in the microwave reactor. Microwave irradiation of 300 W was used, and the temperature ramped from room temperature to 100 °C. The reaction continued for 20 min at the same temperature while stirring. After completion, the reaction mixture was cooled to room temperature, extracted with EtOAc, dried with sodium sulphate, and the solvent was removed under vacuum to extract the crude product. When the crude product was a solid, it was recrystallised using acetonitrile solvent to obtain the pure product in crystalline form. In some cases, the products were purified by column chromatography using hexane–EtOAc as eluent.
Synthesis of 1,3,5-triphenyl pyrazole (P13)
We initially synthesised a chalcone from benzaldehyde and acetophenone under microwave reaction conditions that we had developed previouslyCitation179. The isolated chalcone was further converted to 1,3,5-triphenyl pyrazole via cyclocondensation with phenyl hydrazine, followed by an oxidation reaction using Pd/C as the oxidising agent. We have slightly modified the procedure reported by Hayashi and coworkers for the oxidation stepCitation103. To the solution of chalcone (1 mmol) in 5 mL of acetic acid, phenyl hydrazine (1.2 mmol) was added and refluxed for 3 h. The pyrazoline product was extracted using EtOAc used directly for the next step. To the solution of pyrazoline in dichloromethane, 272 mg (1.2 mmol) of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was added and stirred overnight at room temperature. The final product was then extracted with EtOAc (40 mL × 3). After usual work-up, the obtained residue was column chromatographed on silica-gel to afford 1,3,5-triphenylpyrazole (P13).
Structures assignment based on 1H and 13C NMR, IR, GC–MS, and HRMS analysis
All the structures of synthesised azoles were confirmed by 1H NMR, 13C NMR, IR, GC–MS, and HRMS (ESI-MS) analysis.
4-Ethyl-3,5-dimethyl-1H-pyrazole (P1)Citation180: 1H NMR (400 MHz, CDCl3): 9.96 (brs, 1H), 2.34 (q, 2H, J = 7.3 Hz), 2.20 (s, 6H), and 1.04 (t, 3H, J = 7.3 Hz). 13C NMR (100 MHz; CDCl3): 141.5, 117.2, 16.3, 15.2, 10.8, and 10.7. IR (KBr) cm−1: 3202, 3154, 3092, 2962, 2927, 1665, 1592, 1451, 1419, 1301, 1159, 1007, and 796. High-resolution mass spectrometry (HRMS) (ESI) calcd. for C7H12N2 (M + H+), 125.1073; found, 125.1073.
3-Methyl-1,4,5,6-tetrahydrocyclopenta[c]pyrazole (P2)Citation181: 1H NMR (400 MHz, CDCl3): 10.14 (brs, 1H), 2.67 (t, 2H, J = 6.9 Hz), 2.52 (t, 2H, J = 6.9 Hz), 2.38–2.43 (m, 2H), and 2.20 (s, 3H). 13C NMR (100 MHz; CDCl3): 159.9, 133.9, 122.9, 30.6, 24.6, 22.3, and 10.7. HRMS (ESI) calcd. for C7H10N2 (M + H+), 123.0917; found, 123.0919.
5-Methyl-3-phenyl-1H-pyrazole (P3)Citation182: 1H NMR (400 MHz, CDCl3): 10.90 (brs, 1H), 7.74 (d, 2H, J = 7.2 Hz), 7.40 (t, 2H, J = 7.2 Hz), 7.33 (t, 2H, J = 7.2 Hz), 6.37 (s, 1H), and 2.33 (s, 3H). 13C NMR (100 MHz; CDCl3): 149.9, 143.2, 132.6, 128.7, 127.8, 125.8, 102.1, and 11.6. HRMS (ESI) calcd. for C10H10N2 (M + H+), 159.0917; found, 159.0915.
4-Chloro-5-methyl-3-phenyl-1H-pyrazole (P4): m.p.: 119–120 °C. 1H NMR (400 MHz, CDCl3): 10.93 (brs, 1H), 7.73 (d, 2H, J = 8.2 Hz), 7.33–7.38 (m, 3H), and 2.10 (s, 3H). 13C NMR (100 MHz; CDCl3): 144.1, 141.7, 130.4, 128.7, 128.6, 127.3, 106.8, and 10.0. HRMS (ESI) calcd. for C10H9ClN2 (M + H+), 193.0527; found, 193.0524.
3-Methyl-4,5-dihydro-2H-benzo[g]indazole (P5): m.p.: 137–138 °C. 1H NMR (400 MHz, CDCl3): 10.5 (brs, 1H), 7.72–7.73 (m, 1H), 7.16–7.23 (m, 3H), 2.92 (t, 2H, J = 7.3 Hz), 2.63 (t, 2H, J = 7.3 Hz), and 2.22 (s, 3H). 13C NMR (100 MHz; CDCl3): 146.2, 137.7, 136.7, 129.1, 128.4, 127.6, 126.8, 122.3, 113.8, 29.8, 18.6, and 10.1. HRMS (ESI) calcd. for C12H12N2 (M + H+), 185.1073; found, 185.1074.
3-Methyl-2H-benzo[g]indazole (P6)Citation183: 1H NMR (400 MHz, CDCl3): 8.17–8.20 (m, 1H), 7.87–7.90 (m, 1H), 7.50–7.53 (m, 2H), 7.39 (d, 1H, J = 8.7 Hz), 7.38 (d, 1H, J = 8.7 Hz), and 2.50 (s, 3H). 13C NMR (100 MHz; CDCl3): 143.2, 138.8, 132.6, 128.7, 126.5, 126.2, 121.8, 121.6, 120.5, 118.42, 118.39, and 11.9. HRMS (ESI) calcd. for C12H10N2 (M + H+), 183.0917; found, 183.0923.
4-Ethyl-3,5-dimethyl-1-phenyl-1H-pyrazole (P7): m.p.: 69–70 °C. 1H NMR (400 MHz, CDCl3): 7.38–7.44 (m, 4H), 7.30–7.34 (m, 1H), 2.40 (q, 2H, J = 7.8 Hz), 2.28 (s, 3H), 2.22 (s, 3H), and 1.11 (t, 3H, J = 7.8 Hz). 13C NMR (100 MHz; CDCl3): 162.8, 147.2, 139.4, 136.2, 129.2, 127.5, 124.8, 119.9, 16.7, 15.1, 11.6, and 10.7. HRMS (ESI) calcd. for C13H16N2 (M + H+), 201.1386; found, 201.1391.
3-Methyl-1-phenyl-1,4,5,6-tetrahydrocyclopenta[c]pyrazole (P8)Citation184: m.p.: 51–52 °C. 1H NMR (400 MHz, CDCl3): 7.56 (d, 2H, J = 7.3 Hz), 7.35 (t, 2H, J = 7.3 Hz), 7.15 (t, 1H, J = 7.3 Hz), 2.92 (m, 2H), 2.55 (t, 4H, J = 4.1 Hz), and 2.25 (s, 3H). 13C NMR (100 MHz; CDCl3): 149.0, 143.9, 140.4, 129.3, 128.4, 125.1, 118.6, 31.0, 26.9, 22.4, and 12.8. HRMS (ESI) calcd. for C13H14N2 (M + H+), 199.1230; found, 199.1228.
3-Methyl-2-phenyl-4,5-dihydro-2H-benzo[g]indazole (P9)Citation185: 1H NMR (400 MHz, CDCl3): 7.34–7.39 (m, 2H), 7.33 (t, 2H, J = 7.2 Hz), 7.27 (d, 1H, J = 7.6 Hz), 7.16 (d, 1H, J = 7.2 Hz), 7.01 (t, 1H, J = 7.6 Hz), 6.87 (t, 1H, J = 7.6 Hz), 6.73 (d, 1H, J = 7.6 Hz), 2.87 (t, 2H, J = 7.6 Hz), 2.55 (t, 2H, J = 7.6 Hz), and 2.21 (s, 3H). 13C NMR (100 MHz; CDCl3): 146.0, 140.9, 138.1, 137.2, 129.2, 128.6, 127.8, 127.3, 127.2, 126.3, 125.6, 123.1, 118.6, 30.7, 19.3, and 11.8. HRMS (ESI) calcd. for C18H16N2 (M + H+), 261.1386; found, 261.1383.
Ethyl 3,5-dimethyl-1-phenyl-1H-pyrazole-4-carboxylate (P10)Citation186: 1H NMR (400 MHz, CDCl3): 7.36–7.39 (m, 2H), 7.28–7.32 (m, 3H), 4.22 (q, 2H, J = 7.2 Hz), 2.42 (s, 3H), 2.41 (s, 3H), and 1.28 (t, 3H, J = 7.2 Hz). 13C NMR (100 MHz; CDCl3): 164.5, 151.4, 144.4, 138.8, 129.2, 128.4, 125.6, 110.8, 59.7, 14.4, 14.3, and 12.6. HRMS (ESI) calcd. for C14H16N2O2 (M + H+), 245.1285; found, 245.1285.
5-Methyl-3-phenylisoxazole (P11)Citation187: 1H NMR (400 MHz, CDCl3): 7.69–7.72 (m, 2H), 7.36–7.40 (m, 3H), 6.31 (s, 1H), and 2.30 (s, 3H). 13C NMR (100 MHz; CDCl3): 169.7, 160.4, 130.1, 129.0, 127.6, 125.8, 100.3, and 11.6. HRMS (ESI) calcd. for C10H9NO (M + H+), 160.0757; found, 160.0756.
3-Methyl-4,5-dihydronaphtho[1,2-c]isoxazole (P12): m.p.: 56–57 °C. 1H NMR (400 MHz, CDCl3): 7.53–7.55 (m, 1H), 7.17–7.20 (m, 3H), 2.92 (t, 2H, J = 7.8 Hz), 2.56 (t, 2H, J = 7.8 Hz), and 2.20 (s, 3H). 13C NMR (100 MHz; CDCl3): 164.9, 157.9, 136.4, 129.4, 128.4, 127.0, 125.4, 121.7, 112.4, 28.9, 17.8, and 10.0. HRMS (ESI) calcd. for C12H11NO (M + H+), 186.0913; found, 186.0920.
1,3,5-Triphenyl-1H-pyrazole (P13)Citation188: m.p.: 136–137 °C. 1H NMR (400 MHz, CDCl3): 7.85 (d, 2H, J = 7.6 Hz), 7.35 (t, 2H, J = 7.6 Hz), 7.17–7.31 (m, 11H), and 6.75 (s, 1H). 13C NMR (100 MHz; CDCl3): 152.0, 144.5, 140.1, 133.0, 130.6, 128.9, 128.8, 128.7, 128.5, 128.4, 128.1, 127.5, 125.9, 125.4, and 105.3. GC–MS calcd. for C21H16N2, 296.3; found, 296.1.
3-Methyl-1,5-diphenyl-1H-pyrazole (P14)Citation188: 1H NMR (400 MHz, CDCl3): 7.24–7.31 (m, 8H), 7.19–7.21 (m, 2H), 6.31 (s, 1H), and 2.39 (s, 3H). 13C NMR (100 MHz; CDCl3): 149.4, 144.0, 139.7, 130.4, 129.0, 128.7, 128.5, 128.4, 127.5, 125.3, 107.8, and 13.5. HRMS (ESI) calcd. for C16H14N2 (M + H+), 235.1230; found, 235.1229.
1,5-Diphenyl-3-(trifluoromethyl)-1H-pyrazole (P15)Citation189: m.p.: 90–91 °C. 1H NMR (400 MHz, CDCl3): 7.35–7.38 (m, 8H), 7.24–7.28 (m, 2H), and 6.78 (s, 1H). 13C NMR (100 MHz; CDCl3): 144.7, 143.4 (q, C-F coupling, J = 31 Hz), 139.2, 129.2, 129.1, 129.0, 128.8, 128.7, 128.5, 125.5, and 105.6. HRMS (ESI) calcd. for C16H11F3N2 (M + H+), 289.0947; found, 289.0940.
1-(4-Bromophenyl)-3-methyl-5-phenyl-1H-pyrazole (P16)Citation190: m.p.: 85–86 °C. 1H NMR (400 MHz, CDCl3): 7.44–7.42 (d, 2H, J = 6.8 Hz), 7.29–7.33 (m, 3H), 7.15–7.18 (m, 4H), 6.34 (s, 1H), and 2.44 (s, 3H). 13C NMR (100 MHz; CDCl3): 149.1, 145.5, 136.1, 132.7, 129.9, 129.2, 128.9, 128.4, 127.3, 123.1, 108.4, and 12.8. HRMS (ESI) calcd. for C16H13BrN2 (M + H+), 313.0335; found, 313.0327.
1-(4-Chlorophenyl)-3-methyl-5-phenyl-1H-pyrazole (P17)Citation190: m.p.: 102–103 °C. 1H NMR (400 MHz, CDCl3): 7.27–7.29 (m, 3H), 7.24–7.26 (m, 2H), 7.17–7.19 (m, 4H), 6.28 (s, 1H), and 2.36 (s, 3H). 13C NMR (100 MHz; CDCl3): 149.9, 144.2, 138.4, 133.1, 130.4, 129.3, 128.9, 128.8, 128.7, 126.4, 108.4, and 13.6. HRMS (ESI) calcd. for C16H13ClN2 (M + H+), 269.0840; found, 269.0838.
1-(4-Methoxyphenyl)-3-methyl-5-phenyl-1H-pyrazole (P18)Citation190: m.p.: 107–108 °C. 1H NMR (400 MHz, CDCl3): 7.24–7.26 (m, 3H), 7.16–7.18 (m, 4H), 6.80 (d, 2H, J = 7.4 Hz), 6.29 (s, 1H), 3.76 (s, 3H), and 2.38 (s, 3H). 13C NMR (100 MHz; CDCl3): 159.1, 148.9, 144.3, 132.7, 130.3, 128.8, 128.7, 128.5, 126.9, 114.4, 107.4, 55.7, and 13.5. HRMS (ESI) calcd. for C17H16N2O (M + H+), 265.1335; found, 265.1330.
4-(3-Methyl-5-phenyl-1H-pyrazol-1-yl)benzenesulfonamide (P19)Citation191: m.p.: 201–203 °C. 1H NMR (400 MHz, CDCl3): 7.84 (d, 2H, J = 7.6 Hz), 7.41 (d, 2H, J = 7.6 Hz), 7.36–7.38 (m, 3H), 7.23–7.25 (m, 2H), 6.36 (s, 1H), 4.98 (brs, 2H), and 2.41 (s, 3H). 13C NMR (100 MHz; CDCl3): 150.8, 144.1, 143.4, 139.7, 130.2, 128.8, 128.7, 127.3, 124.6, 109.4, and 13.6. HRMS (ESI) calcd. for C16H15N3O2S (M + H+), 314.0958; found, 314.0952.
Compounds P20–P25 were prepared according to our previous reportCitation89.
Biological evaluation assays
Antibodies, chemicals, and reagents
Bovine serum albumin (BSA), dimethyl sulphoxide (DMSO), 4′,6-diamidino-2-phenylindole (DAPI-#DUO82040), MTT dye (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 98% TLC) in situ mounting media, 4-hydroxyphenyl β-d-glucopyranoside (arbutin), mushroom tyrosinase (EC 1.14.18.1), and l-tyrosine were purchased from Sigma-Aldrich Chemical Company (St. Louis, MO) and MP Biomedicals (Irvine, CA). The antibodies for immunoblotting and immunocytochemistry, including Bcl-2 (2876S), Bax (2772S), caspase-3 (9662S), caspases 9 (#9502), PARP antibody #9542, mTOR, β-actin (13E5), and horseradish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies, were all obtained from Cell Signaling Technologies (Beverly, MA). Antibody to peIF4E (Ser209) (76256) was from Abcam (Cambridge, UK). A 2% (w/v) aqueous solution of gentian violet and crystal violet were from Ricca Chemical Company (Arlington, TX). Radioimmunoprecipitation assay (RIPA) buffer and Pierce Bicinchoninic Acid (BCA™) protein assay kit were purchased from Thermo Fisher Scientific (Rockford, IL). Mini-protean precast Tris-Glycine Gels (TGX) were from Bio-Rad (Bio-Rad Laboratories Inc., Hercules, CA). The SuperSignal™ West Pico PLUS Chemiluminescent substrate detection system was from Thermo Fisher Scientific (Waltham, MA). Dulbecco’s modified Eagle’s medium (DMEM) and Roswell Park Memorial Institute Medium (RPMI 1640) were from Corning (Corning, Manassas, VA). Cascade Biologics® PMA-free Human Melanocyte Growth Supplement (HMGS-2, # S0165), and Medium 254 (# M-254-500) were purchased from Gibco™ Invitrogen Cell culture (Thermo Fisher Scientific, Rockford, IL). Epi-Life® Growth Medium with 60 µM calcium and Cascade Biologist human keratinocyte growth supplement (HKGS) 100X (S-001-5) were from Life Technologies (Grand Island, NY). The quick coating solution (cAP-01) was purchased from Angio-Proteomie (Boston, MA). The Dulbecco’s phosphate-buffered saline (DPBS), phosphate-buffered saline (PBS) 1X, defined trypsin inhibitor (DTI) 1X (R007-100), trypsin EDTA 0.25%, 1X(R25200-072), trypsin neutraliser (TN) 1X (R-002-100), penicillin–streptomycin (Pen Strep, 15140-122) (PEST) 100X were purchased from Gibco, Thermo Fisher Scientific (Rockford, IL), and HKGS 100X (S-001-5) were procured from VWR Corporation (Missouri, TX). Quercetin and 5-hydroxy-2-(hydroxymethyl)-4H-pyran-4-one (kojic acid) were purchased from Cayman Chemical (Ann Arbor, MI). USDA-approved Origin Fetal Bovine Serum (FBS) was from VWR Seradigm Life Science (Missouri, TX). The organic solvents, including ethanol (6183-10) and methanol (BDH 1135-4LP), were acquired from Macron Chemicals (Radnor, PA) and VWR (Missouri, TX), respectively.
Cell lines, cell cultures, cytotoxicity, and viability assessment
Human-derived GFP-expressing melanoma A375 and epidermoid carcinoma A431 cell lines were purchased from Angio-Proteomie (Boston, MA). Human melanoma carcinoma SK-MEL-28 and human immortalised keratinocytes (HaCaT) cell lines were acquired from American Type Culture Collection (ATCC; Manassas, VA). Human cutaneous SCC cell line SCC-12Citation192 was generously provided by Dr. James G. Rheinwald to Dr. Tatiana Efimova. Except SK-MEL-28, which was cultured in an RPMI-1640 medium supplemented with 5% FBS, all other human immortalised cell lines were grown and maintained in DMEM containing varying percentages of FBS; 5% FBS for A431, SCC-12, and HaCaT lines, and 10% FBS for A375. All culture media were routinely supplemented with 1% PEST 100X (100 U/mL penicillin, 100 μg/mL streptomycin), and cells were cultured in incubators maintained at 37 °C under an atmosphere of 95% humidity, 20% O2, and 5% CO2. The growth media of incubated cells were changed every 2–3 days until they attained 70–80% confluence, after which they were sub-cultured for experiments and re-passaging. DMSO was used as the vehicle to prepare a 10 mM stock solution of the test compounds. Control cells were treated with the vehicle (DMSO) at concentrations of 0.01–0.2%, which did not affect cell viability.
Stock solutions (10 mM) were prepared by dissolving each compound to be screened in DMSO. From these stock solutions, escalating concentrations were prepared using the corresponding cell growth media as a diluent, and the cells (65–75% confluent) were treated with (0–40 µM) or without the drug in sext- to octuplicate and incubated for 48 h. All treatment protocols and controls were prepared as previously describedCitation109.
The potency and cytotoxicity of the test compounds against two cutaneous melanoma (A375 and SK-MEL-28) and two non-melanoma (A431 and SCC-12) SC cell lines in addition to a control immortalised normal human epidermal keratinocytes (HaCaT) cell line were assessed by MTT assay as previously describedCitation109. The results from increasing concentrations of each test and control (cisplatin and celecoxib) agent were analysed using the zero drug control group as the relative comparator and are expressed as a percentile according to the following equation: ((absorbance in treatment group/absorbance in control) × 100%), and data were computed as earlier describedCitation109.
Colony formation assay
SCC-12 and SK-MEL-28 cells were seeded in T25 flasks in triplicate and allowed to adapt for 24 h before treatment with different concentrations of P25 (0, ½IC50, IC50, and 1½IC50) for 48 h. Cells were then trypsinised, and 1000 viable cells from each treated group were seeded on 10 cm2 plates in 10 mL of the medium, and incubated for an additional 14 days before fixation and crystal violet staining, imaging, and colonies assessment as previously describedCitation109,Citation116. The plates were scanned, and the area of clones was quantified with the free software Image J (Bethesda, MD) (https://imagej.nih.gov/ij/).
In vitro wound healing assay
The scratch wound healing assay was performed to evaluate compound P25’s ability to limit cell migration and scratch wound closure following our previously described protocolCitation109. Briefly, cells were seeded in wells of a 24-well plate and incubated to 100% confluent monolayer, following which a sterile 200 µL Gilson pipette tip was used to scratch a straight line across the centre. Cells were washed gently to remove debris and detached cells and were treated (or not) with increasing concentrations (0, ½IC50, IC50, and 1½IC50) of P25. The wounded and re-epithelialisation areas were imaged at 0 h, 24 h, and 48 h using an inverted phase-contrast Olympus IX51 microscope (Olympus America Inc., Center Valley, PA) attached to an Infinity 2 digital camera. Images were processed with an Infinity analysis software version 6.5.6 (Teledyne Lumenera, Ottawa, Canada) and analysed with Adobe Photoshop. The ImageJ software (NIH, Bethesda, MD) was used to measure the migratory area of each treatment group, with results expressed as remaining percentage of unfilled wound surface at 0 hCitation108.
Immunoblotting
Immunoblotting was performed according to standard procedures of the Bio-Rad electrophoresis system (Bio-Rad, Hercules, CA) as described earlierCitation109. Briefly, protein extracts were obtained by lysing cells in RIPA buffer containing protease inhibitors, and were resolved by 4–12% polyacrylamide ready mini-PROTEAN TGX gel electrophoresis with sodium dodecyl sulphate (SDS) running buffer. Gels were electrotransferred to 0.45 μm nitrocellulose membranes (Bio-Rad, Hercules, CA) in Tris-glycine (TG) buffer using a Trans-Blot Turbo Transfer Pack according to manufacturer’s instructions. Membranes were incubated for 1 h in 5–7% non-fat powdered milk dissolved in TBST (Tris-buffered saline containing 0.05% Tween 20) or 5% BSA for phosphorylated proteins, to saturate nonspecific binding sites, followed by an overnight incubation (4 °C) with primary antibodies. Membranes were washed 5× with TBST and treated with suitable HRP-conjugated secondary antibodies (Amersham Life Science Inc., Arlington Height, IL) for another 2 h at room temperature, followed by additional wash steps and chemiluminescence-based detection with the Amersham ECL Prime detection reagent kit and the Bio-Rad Quantity One ChemiDoc™ detection and imaging analysis software systems as described earlierCitation108,Citation109.
Antioxidant activity by the DPPH free radical scavenging assay
The antioxidant potential of the synthesised derivatives was determined based on their ability to scavenge the 2,2′-diphenyl-1-picrylhydrazyl (DPPH) radical as previously reportedCitation193, and the assays were conducted in a 96-well format. An initial screen was done of all 25 derivatives at a concentration of 1 mM. Compounds exhibiting greater than 50% scavenging at 1 mM were then serial-diluted using 100 µL aliquots (ranging from 0.125 mM to 7.8 µM). Methanol was used as the diluent, and 100 µL of DPPH (200 µM; Sigma-Aldrich, St. Louis, MO) was added to each well. Absorbance was determined after 30 min at 517 nm, using a microplate reader (SpectraMax M2, Molecular Devices Corp., Sunnyvale, CA, operated by SoftmaxPro v.4.6 software). Experimentally, it has been determined that absorbance constantly decreases and stabilises after preparing the sample of test compounds. The scavenging capacity (SC) was calculated as SC% = ((ΔAcontrol – ΔAsample)/ΔAcontrol) × 100%, where ΔAcontrol was the absorbance of the reagent blank, and ΔAsample was the absorbance of the test samples. The control contained all reagents except the compounds, and all tests were performed in triplicate. Quercetin served as a positive control. The DPPH methanolic solution (1 mL) mixed with DMSO (5 µL) served as a negative control. Antioxidant activity was evaluated by calculating the IC50 values denoting the concentration (in µM) of the sample required to scavenge 50% of formed DPPH free radicals, based on absorbance measurements (Table S4).
In vitro mushroom tyrosinase inhibition assay
The anti-tyrosinase potential of all the synthesised derivatives was determined by an enzymatic tyrosinase inhibition assay based on their ability to inhibit the substrate, l-tyrosine, from binding to the tyrosinase enzymeCitation194. Briefly, mushroom tyrosinase, l-tyrosine, and positive control, arbutin and the more potent kojic acid (a fungal secondary metabolite used as a skin whitening agent) were used in the tyrosinase inhibition assays, which were performed in a 96-well microplate format using a SpectraMax M2 microplate reader, operated by SoftmaxPro v.4.6 software (Molecular Devices, Sunnyvale, CA). Test samples were dissolved in DMSO at a concentration of 10 mM and then diluted to different concentrations with phosphate buffer (0.1 M, pH 6.8). Each well contained 40 µL of the sample with 80 µL of phosphate buffer solution (0.1 M, pH 6.8), 40 µL of tyrosinase enzyme (100 units/mL), and 40 µL l-tyrosine (2.5 mM). The mixture was incubated for 30 min at room temperature, and absorbance was measured at 490 nm. Each sample was accompanied by a sample blank containing all components except tyrosinase. Arbutin and kojic acid were used as the positive controls. The results were compared with a negative control consisting of 0.1 M PBS in place of the sample. The percentage of tyrosinase inhibition was calculated as follows: ((ΔAcontrol – ΔAsample)/ΔAcontrol) × 100. IC50 values denoted the sample concentration required to inhibit 50% tyrosinase activity (Table S1).
In silico computational screening studies
In silico target(s) fishing, physicochemical and drug-likeness properties predictions, and bioavailability (absorbability) predictions
The SwissTargetPrediction database (http://www.swisstargetprediction.ch/) web server, which predicts the targets of bioactive molecules based on a combination of two-dimensional (2D) and 3D similarity measures with known ligandsCitation123,Citation195, was employed to screen and estimate the putative macromolecular targets of all 25 derivatives synthesised. Briefly, each compound’s canonical Simplified Molecular Input-Line Entry System (SMILES) structure formats were individually converted using the ChemDraw tool before importing into the SwissTargetPrediction network database. After the SMILES structures were uploaded to the database, with the species set as “Homo sapiens,” a report of probable matched targets data was obtained. The 3D structures of the target proteins used for molecular docking were selected from the available Protein Data Bank (PDB). During target prediction, compounds possessing the highest probability to regulate identified targets >88.8% probability, and the specific targets were selected based on the predicted score value, and all PDB codes are provided in the master excel sheet as supplementary data.
In silico molecular docking simulation methodology
Molecular docking simulations were used in this study to predict the binding interactions between ligands and their protein targets and the ligand conformations and orientations (poses) in the functionally relevant sites of target proteinsCitation196. Using this technique, a molecule is docked into the ligand-binding domain of the target protein associated with the potential activity to identify a specific ligand that can fit within a specific protein ligand-binding domainCitation197. The credibility of the connection between the identified targets and the molecules screened was evaluated by docking each compound with the predicted target protein. The molecular docking data were evaluated by selecting the native and clinical ligands of the studied targets and docking them as the positive longitudinal control. First, the SMILES structures of native (commercially available natural or clinically functional) ligands used in the study were retrieved from the PubChem network database (https://pubchem.ncbi.nlm.nih.gov/)Citation198, compounds to be screened, and the SMILES structures for those and for the synthesised compounds to be screened were converted into 3D conformations predicted using OpenBabel software (v2.2.1) (https://pyrx.sourceforge.io/). A cross-docking protocol with numbers associated with each selected proteinCitation199,Citation200 was used. The 3D crystallographic structure of the protein targets, including the mushroom tyrosinase structure (PDB ID: 2Y9X; resolution: 1.33 Å), was retrieved from the RCSB PDB web (https://www.rcsb.org/)Citation201, and retrieved proteins (receptors) were in complex with other hetero-atoms and water molecules. PyMOL was used to process the downloaded PDB structures further by removing water molecules and heteroatoms (e.g. chloride or sodium ions), and separating the structure of the protein and structure-native ligand (inhibitor, for enzymes) from the complex. The separated native ligand was considered as a positive test control for the docking simulations. Target and ligand structures were then converted to PDBQT files using AutoDock Tools (ADT) from the open-source software suite MGL tools 1.5.6Citation202 (Molecular Graphics Laboratory package from The Scripps Research Institute, http://autodock.scripps.edu/). During processing (to prepare a file suitable for docking), polar hydrogen was added to the separated protein structure before converting it to the PDBQT file. There was no need to add polar hydrogen to any ligands, but the torsion angle of each ligand was defined. While OpenBabel software (v2.2.1) (https://pyrx.sourceforge.io/) was used for converting the SMILES format of all the ligands to generate their 3D conformations in PDB format, the program RaccoonCitation203. Once the torsion angle was found, it was converted and saved in PDBQT file format and was later used for docking as the ligand.
The grid box was set centred in Cartesian coordinate (11.25–19.88x-, 10.88–23.62y-, and 11.0018.36z) of the docking site, defined according to the pre-complexed ligand (native ligand) of the PDB structures for each target after removing water molecules and hetero atoms for each of the native-ligand-bound:target complex, with a default grid point spacing of 0.375 Å. After preparing all the ligands and targets, the binding affinities were predicted using the graphic user interface (GUI) AutoDock Vina software (v.1.2.0.)Citation204 in Linux OS. After the initial docking simulation, 10 other conformations of the docked ligands were obtained, and each ligand with the minimum binding energy and RMSD value was chosen. The ligand:target complexes (ligand–protein interactions) were then visualised using PyMOLCitation205 and processed using Discovery Studio (version 3.5, Dassault Systèmes BIOVIA, San Diego, CA). As an example, tyrosinase docking was similar to all other docking simulations. In brief, prior to docking of the test ligands, tropolone, the co-crystallised ligand in the PDB structure, was removed and redocked to the active site. The obtained binding mode was compared with the co-crystallised configuration by determining the root-mean-square deviation (RMSD) as a means of assessing the robustness and validity of the methodology. It was confirmed that the binding pose for tropolone was comparable to that of the native configuration of the PDB structure for the ligand:target complex (RMSD: 1.33 Å).
Predicted binding energies were taken as an index for evaluating potential for a compound to bind specifically to one of the identified targets with appreciable and pharmacologically significant affinity. Values ≤ −8 were considered to be predictive of high-affinity binding, irrespective of the calculated value for the native and/or clinical ligands. The low binding energy (< −8.00 kcal/mol) indicates good binding strength, sufficient to move forward with the predicted target for each derivative.
In silico predictions for drug-likeness, ADME/pharmacokinetics, and toxicity
The SwissADME server was used for the computational predictions of absorption, distribution, metabolism, and excretion (ADME) and DL properties of each of the newly synthesised compounds (P1–P25) as described earlierCitation206,Citation207. Gastrointestinal absorption (GA), DL, and other parameters such as oral bioavailability were considered to define the bioactive compoundsCitation123,Citation206. The SwissADME online server predictor program (Swiss Institute of Bioinformatics, Écublens, Switzerland) reports various druglikeness indices, including molecular weight, number of H-bond donors and acceptors, total polar surface area (TPSA), number of rotatable bonds, and aggregate Lipinski Rule-of-Fives assessment, as well as further predictions of partition coefficient (as milogP) and even general pharmacokinetic characteristicsCitation123,Citation206,Citation208. For toxicity prediction, the web-tool Pro Tox II (https://tox-new.charite.de/protox_II) was employed for as described earlierCitation209,Citation210. Herein, each ligand was assessed in silico with respect to predicted hepatotoxicity, carcinogenicity, immunotoxicity, cytotoxicity, or mutagenicity, particular organ toxicities, and other endpoints of toxicological interest and prospective concern; this tool even generates LD50 predictions. Data generated with this tool are included with the supplementary data provided in the master Excel spreadsheet.
Statistical analysis
All statistical analyses were performed using GraphPad Prism Software version 9.1 (GraphPad Prism Inc., San Diego, CA) as previously describedCitation109, and quantitative and grouped data were expressed as means ± standard deviation (SD) or ± standard error of the mean (SEM) from at least three independent experiments. The significance was analysed by one-way ANOVA, Student’s t-test, or ANOVA with Turkey’s or Bonferroni’s multiple comparison post hoc tests for the difference between two or more groups. Statistical significance is denoted as: *p values <0.05, **p values < 0.01, ***p values <0.001, and ****p values <0.0001.
Author contributions
The manuscript was written through the contributions of all authors: J.C.C. and S.M. conceived the research idea, designed and administered the study; R.A.H., N.B.M., H.M., and M.S. contributed to the study design; T.R., S.T.B., K.T., S.B., A.D.A., E.N., and D.A. performed in silico molecular docking, ADME, targets prediction, biological evaluations, and acquired data; U.O., D.B., D.E., A.S., and S.M. performed the chemical synthesis and acquired data; S.T.B., T.R., K.T., S.B., M.S., S.M., H.M., and J.C.C. analysed and interpreted the data; S.T.B., T.R., R.A.H., M.S., and J.C.C. drafted the original manuscript and figure presentation; S.T.B., T.R., K.T., N.P.M., T.E., M.S., H.M., N.B.M., A.N.F., R.A.H., S.M., and J.C.C. co-wrote, edited, and critically revised the manuscript and provided important intellectual input; T.E., B.M.N., and M.S. provided research supplies; T.E., M.S., H.M., S.M., and J.C.C. supervised the study and provided guidance and critical insights in study planning; S.M. and J.C.C. acquired funding; all authors revised, edited, and approved the final version of the manuscript submitted.
Supplemental Material
Download PDF (5.3 MB)Acknowledgements
NMR Spectroscopy data were acquired using Bruker 400 MHz instrument located at the NMR facility in the College of Engineering and Science at Louisiana Tech University and JEOL 400 MHz instrument located at the NMR facility in the College of Pharmacy at University of Louisiana Monroe. The authors would like to acknowledge the assistance of Dr. Elisabeth Fatila (Department of Chemistry, Louisiana Tech University) for NMR data acquisition and Danny D. Dang and William Pierre Henry Leddet for their assistance with cell culture work, drug screening and testing. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data presented in this study are available at reasonable request from the corresponding authors.
Additional information
Funding
References
- Leiter U, Keim U, Garbe C. Epidemiology of skin cancer: update 2019. Adv Exp Med Biol. 2020;1268:123–139.
- Wright CY, Millar DA, Wright CY, Du PDJ, Du PDJ, Norval M. The epidemiology of skin cancer and public health strategies for its prevention in Southern Africa. Int J Environ Res Public Health. 2020;17(3):1017.
- Karimkhani C, Green AC, Nijsten T, Weinstock MA, Dellavalle RP, Naghavi M, Fitzmaurice C. The global burden of melanoma: results from the Global Burden of Disease Study 2015. Br J Dermatol. 2017;177(1):134–140.
- Yousefi H, Mohammadzadeh S, Irompour A, Shenasifam N, Roshandel E, Jalili A. Overall study on molecular pathways of skin cancer derived from ultraviolet radiation as an environmental threat. Environ Conserv J. 2015;16(SE):547–555.
- Rivas M, Rojas E, Araya MC, Calaf GM. Ultraviolet light exposure, skin cancer risk and vitamin D production. Oncol Lett. 2015;10(4):2259–2264.
- Rigby CM, Roy S, Deep G, Guillermo-Lagae R, Jain AK, Dhar D, Orlicky DJ, Agarwal C, Agarwal R. Role of p53 in silibinin-mediated inhibition of ultraviolet B radiation-induced DNA damage, inflammation and skin carcinogenesis. Carcinogenesis. 2017;38(1):40–50.
- Pavel TI, Chircov C, Rădulescu M, Grumezescu AM. Regenerative wound dressings for skin cancer. Cancers. 2020;12(10):2954.
- Mulvaney PM, Schmults CD. Molecular prediction of metastasis in cutaneous squamous cell carcinoma. Curr Opin Oncol. 2020;32(2):129–136.
- Robertson BF, Wokes JET, Siddiqui H. Management of incompletely excised skin tumors: our experience. Dermatol Surg. 2018;44(3):365–369.
- Adefusika JA, Pimentel JD, Chavan RN, Brewer JD. Primary mucinous carcinoma of the skin: the Mayo Clinic experience over the past 2 decades. Dermatol Surg. 2015;41(2):201–208.
- Lu Y-g, Wang Y-y, Yang Y-d, Zhang X-c, Gao Y, Yang Y, Zhang J-b, Li G-l. Efficacy of topical ALA-PDT combined with excision in the treatment of skin malignant tumor. Photodiagn Photodyn Ther. 2014;11(2):122–126.
- Zhi D, Yang T, Zhang T, Yang M, Zhang S, Donnelly RF. Microneedles for gene and drug delivery in skin cancer therapy. J Control Release. 2021;335:158–177.
- Hwang S-Y, Chae J-I, Kwak A-W, Lee M-H, Shim J-H. Alternative options for skin cancer therapy via regulation of AKT and related signaling pathways. Int J Mol Sci. 2020;21(18):6869.
- Jain R, Sarode I, Singhvi G, Dubey SK. Nanocarrier based topical drug delivery – a promising strategy for treatment of skin cancer. Curr Pharm Des. 2020;26(36):4615–4623.
- Ascierto PA, Garbe C. Updates and new perspectives in nonmelanoma skin cancer therapy: highlights from 'Immunotherapy Bridge’. Immunotherapy. 2020;12(3):167–174.
- Solomon EI, Sundaram UM, Machonkin TE. Multicopper oxidases and oxygenases. Chem Rev. 1996;96(7):2563–2606.
- Saeedi M, Khezri K, Seyed Zakaryaei A, Mohammadamini H. A comprehensive review of the therapeutic potential of α-arbutin. Phytother Res. 2021;35(8):4136–4154.
- Li H, DaSilva NA, Liu W, Xu J, Dombi GW, Dain JA, Li D, Chamcheu JC, Seeram NP, Ma H. Thymocid(®), a standardized black cumin (Nigella sativa) seed extract, modulates collagen cross-linking, collagenase and elastase activities, and melanogenesis in murine B16F10 melanoma cells. Nutrients. 2020;12(7):2146.
- Pillaiyar T, Namasivayam V, Manickam M, Jung SH. Inhibitors of melanogenesis: an updated review. J Med Chem. 2018;61(17):7395–7418.
- Li Y, Huang J, Lu J, Ding Y, Jiang L, Hu S, Chen J, Zeng Q. The role and mechanism of Asian medicinal plants in treating skin pigmentary disorders. J Ethnopharmacol. 2019;245:112173.
- Liu-Smith F, Meyskens FL. Molecular mechanisms of flavonoids in melanin synthesis and the potential for the prevention and treatment of melanoma. Mol Nutr Food Res. 2016;60(6):1264–1274.
- Nirmal NP, Benjakul S, Ahmad M, Arfat YA, Panichayupakaranant P. Undesirable enzymatic browning in crustaceans: causative effects and its inhibition by phenolic compounds. Crit Rev Food Sci Nutr. 2015;55(14):1992–2003.
- Sánchez-Ferrer A, Rodríguez-López JN, García-Cánovas F, García-Carmona F. Tyrosinase: a comprehensive review of its mechanism. Biochim Biophys Acta. 1995;1247(1):1–11.
- Min K, Park GW, Yoo YJ, Lee JS. A perspective on the biotechnological applications of the versatile tyrosinase. Bioresour Technol. 2019;289:121730.
- Olivares C, Solano F. New insights into the active site structure and catalytic mechanism of tyrosinase and its related proteins. Pigment Cell Melanoma Res. 2009;22(6):750–760.
- Ioniţă E, Aprodu I, Stănciuc N, Râpeanu G, Bahrim G. Advances in structure–function relationships of tyrosinase from Agaricus bisporus – investigation on heat-induced conformational changes. Food Chem. 2014;156:129–136.
- Chang TS. An updated review of tyrosinase inhibitors. Int J Mol Sci. 2009;10(6):2440–2475.
- Pillaiyar T, Manickam M, Jung SH. Downregulation of melanogenesis: drug discovery and therapeutic options. Drug Discov Today. 2017;22(2):282–298.
- Brenner M, Hearing VJ. The protective role of melanin against UV damage in human skin. Photochem Photobiol. 2008;84(3):539–549.
- Ray K, Chaki M, Sengupta M. Tyrosinase and ocular diseases: some novel thoughts on the molecular basis of oculocutaneous albinism type 1. Prog Retin Eye Res. 2007;26(4):323–358.
- Hearing VJ, Tsukamoto K. Enzymatic control of pigmentation in mammals. FASEB J. 1991;5(14):2902–2909.
- Costin GE, Hearing VJ. Human skin pigmentation: melanocytes modulate skin color in response to stress. FASEB J. 2007;21(4):976–994.
- Videira IF, Moura DF, Magina S. Mechanisms regulating melanogenesis. An Bras Dermatol. 2013;88(1):76–83.
- Ghanem G, Fabrice J. Tyrosinase related protein 1 (TYRP1/gp75) in human cutaneous melanoma. Mol Oncol. 2011;5(2):150–155.
- Gradilone A, Cigna E, Aglianò AM, Frati L. Tyrosinase expression as a molecular marker for investigating the presence of circulating tumor cells in melanoma patients. Curr Cancer Drug Targets. 2010;10(5):529–538.
- Jawaid S, Khan TH, Osborn HM, Williams NA. Tyrosinase activated melanoma prodrugs. Anticancer Agents Med Chem. 2009;9(7):717–727.
- Lee SY, Baek N, Nam TG. Natural, semisynthetic and synthetic tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2016;31(1):1–13.
- Pillaiyar T, Manickam M, Namasivayam V. Skin whitening agents: medicinal chemistry perspective of tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2017;32(1):403–425.
- Ando H, Ichihashi M, Hearing VJ. Role of the ubiquitin proteasome system in regulating skin pigmentation. Int J Mol Sci. 2009;10(10):4428–4434.
- Zolghadri S, Bahrami A, Hassan Khan MT, Munoz-Munoz J, Garcia-Molina F, Garcia-Canovas F, Saboury AA. A comprehensive review on tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2019;34(1):279–309.
- Zhao Y, Hu JJ, Bai XL, Liu HP, Qi XW, Liao X. Fast screening of tyrosinase inhibitors from traditional Chinese medicinal plants by ligand fishing in combination with in situ fluorescent assay. Anal Bioanal Chem. 2022;414(6):2265–2273.
- Chidambaram S, Ali D, Alarifi S, Gurusamy R, Radhakrishnan S, Akbar I. Tyrosinase-mediated synthesis of larvicidal active 1,5-diphenyl pent-4-en-1-one derivatives against Culex quinquefasciatus and investigation of their ichthyotoxicity. Sci Rep. 2021;11(1):20730.
- Blaschek L, Pesquet E. Phenoloxidases in plants—how structural diversity enables functional specificity. Front Plant Sci. 2021;12:754601.
- Pacelli C, Cassaro A, Maturilli A, Timperio AM, Gevi F, Cavalazzi B, Stefan M, Ghica D, Onofri S. Multidisciplinary characterization of melanin pigments from the black fungus Cryomyces antarcticus. Appl Microbiol Biotechnol. 2020;104(14):6385–6395.
- Dolinska MB, Wingfield PT, Sergeev YV. Purification of recombinant human tyrosinase from insect larvae infected with the baculovirus vector. Curr Protoc Protein Sci. 2017;89:6.15.1–6.15.12.
- Cordes P, Sun W, Wolber R, Kolbe L, Klebe G, Röhm KH. Expression in non-melanogenic systems and purification of soluble variants of human tyrosinase. Biol Chem. 2013;394(5):685–693.
- Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M. Crystallographic evidence that the dinuclear copper center of tyrosinase is flexible during catalysis. J Biol Chem. 2006;281(13):8981–8990.
- Kim YJ, Uyama H. Tyrosinase inhibitors from natural and synthetic sources: structure, inhibition mechanism and perspective for the future. Cell Mol Life Sci. 2005;62(15):1707–1723.
- Parvez S, Kang M, Chung HS, Bae H. Naturally occurring tyrosinase inhibitors: mechanism and applications in skin health, cosmetics and agriculture industries. Phytother Res. 2007;21(9):805–816.
- Goenka S, Johnson F, Simon SR. Novel chemically modified curcumin (CMC) derivatives inhibit tyrosinase activity and melanin synthesis in B16F10 mouse melanoma cells. Biomolecules. 2021;11(5):674.
- Sendovski M, Kanteev M, Ben-Yosef VS, Adir N, Fishman A. First structures of an active bacterial tyrosinase reveal copper plasticity. J Mol Biol. 2011;405(1):227–237.
- Bijelic A, Pretzler M, Molitor C, Zekiri F, Rompel A. The structure of a plant tyrosinase from walnut leaves reveals the importance of "substrate-guiding residues" for enzymatic specificity. Angew Chem Int Ed Engl. 2015;54(49):14677–14680.
- Qu Y, Zhan Q, Du S, Ding Y, Fang B, Du W, Wu Q, Yu H, Li L, Huang W. Catalysis-based specific detection and inhibition of tyrosinase and their application. J Pharm Anal. 2020;10(5):414–425.
- McLarin MA, Leung IKH. Substrate specificity of polyphenol oxidase. Crit Rev Biochem Mol Biol. 2020;55(3):274–308.
- Fenoll LG, Peñalver MJ, Rodríguez-López JN, Varón R, García-Cánovas F, Tudela J. Tyrosinase kinetics: discrimination between two models to explain the oxidation mechanism of monophenol and diphenol substrates. Int J Biochem Cell Biol. 2004;36(2):235–246.
- Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999;1(1):55–68.
- Bocci G, Carosati E, Vayer P, Arrault A, Lozano S, Cruciani G. ADME-Space: a new tool for medicinal chemists to explore ADME properties. Sci Rep. 2017;7(1):6359.
- Eid AM, Hawash M, Amer J, Jarrar A, Qadri S, Alnimer I, Sharaf A, Zalmoot R, Hammoudie O, Hameedi S, et al. Synthesis and biological evaluation of novel isoxazole-amide analogues as anticancer and antioxidant agents. Biomed Res Int. 2021;2021:6633297.
- Gabrič A, Hodnik Ž, Pajk S. Oxidation of drugs during drug product development: problems and solutions. Pharmaceutics. 2022;14(2):325.
- Ramadan M, Aly AA, El-Haleem LEA, Alshammari MB, Bräse S. Substituted pyrazoles and their heteroannulated analogs-recent syntheses and biological activities. Molecules. 2021;26(16):4995.
- Farghaly TA, Dawood KM. Inhibitory activities of pyrazolo-oxazine heterocyclic derivatives. Mini Rev Med Chem. 2021;22(9):1256–1267.
- Mantzanidou M, Pontiki E, Hadjipavlou-Litina D. Pyrazoles and pyrazolines as anti-inflammatory agents. Molecules. 2021;26(11):3439.
- Kerru N, Gummidi L, Maddila S, Gangu KK, Jonnalagadda SB. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules. 2020;25(8):1909.
- Dhameliya TM, Chudasma SJ, Patel TM, Dave BP. A review on synthetic account of 1,2,4-oxadiazoles as anti-infective agents. Mol Divers. 2022;26(5):2967–2980.
- Costa RF, Turones LC, Cavalcante KVN, Rosa Júnior IA, Xavier CH, Rosseto LP, Napolitano HB, Castro PFdS, Neto MLF, Galvão GM, et al. Heterocyclic compounds: pharmacology of pyrazole analogs from rational structural considerations. Front Pharmacol. 2021;12:666725.
- Sharma R, Chawla PA, Chawla V, Verma R, Nawal N, Gupta V. A therapeutic journey of 5-pyrazolones as a versatile scaffold: a review. Mini Rev Med Chem. 2021;21(13):1770–1795.
- Karrouchi K, Radi S, Ramli Y, Taoufik J, Mabkhot Y, Al-Aizari F, Ansar M. Synthesis and pharmacological activities of pyrazole derivatives: a review. Molecules. 2018;23(1):134.
- Bennani FE, Doudach L, Cherrah Y, Ramli Y, Karrouchi K, Ansar M, Faouzi MEA. Overview of recent developments of pyrazole derivatives as an anticancer agent in different cell line. Bioorg Chem. 2020;97:103470.
- Mor S, Khatri M, Punia R, Sindhu S. Recent progress in anticancer agents incorporating pyrazole scaffold. Mini Rev Med Chem. 2022;22(1):115–163.
- Hawash MM, Kahraman DC, Eren F, Cetin Atalay R, Baytas SN. Synthesis and biological evaluation of novel pyrazolic chalcone derivatives as novel hepatocellular carcinoma therapeutics. Eur J Med Chem. 2017;129:12–26.
- Jing L, Wang L, Zhao Y, Tan R, Xing X, Liu T, Huang W, Luo Y, Li Z. Synthesis, crystal structure and evaluation of cancer inhibitory activity of 4-[indol-3-yl-methylene]-1H-pyrazol-5(4H)-one derivatives. J Chem Res. 2012;36(12):691–696.
- Huang S, Lin R, Yu Y, Lu Y, Connolly PJ, Chiu G, Li S, Emanuel SL, Middleton SA. Synthesis of 3-(1H-benzimidazol-2-yl)-5-isoquinolin-4-ylpyrazolo[1,2-b]pyridine, a potent cyclin dependent kinase 1 (CDK1) inhibitor. Bioorg Med Chem Lett. 2007;17(5):1243–1245.
- Baytas SN, Inceler N, Yılmaz A. Synthesis, cytotoxicity, and molecular properties prediction of novel 1,3-diarylpyrazole derivatives. Med Chem Res. 2013;22(10):4893–4908.
- Doan NQH, Nguyen NTK, Duong VB, Nguyen HTT, Vong LB, Duong DN, Nguyen N-TT, Nguyen TLT, Do TTH, Truong TN. Synthesis, biological evaluation, and molecular modeling studies of 1-aryl-1H-pyrazole-fused curcumin analogues as anticancer agents. ACS Omega. 2022;7(38):33963–33984.
- Hawash M. Recent advances of tubulin inhibitors targeting the colchicine binding site for cancer therapy. Biomolecules. 2022;12(12):1843.
- Taher AT, Mostafa Sarg MT, El-Sayed Ali NR, Hilmy Elnagdi N. Design, synthesis, modeling studies and biological screening of novel pyrazole derivatives as potential analgesic and anti-inflammatory agents. Bioorg Chem. 2019;89:103023.
- Ran F, Liu Y, Zhang D, Liu M, Zhao G. Discovery of novel pyrazole derivatives as potential anticancer agents in MCL. Bioorg Med Chem Lett. 2019;29(9):1060–1064.
- Faisal M, Saeed A, Hussain S, Dar PM, Larik FA. Recent developments in synthetic chemistry and biological activities of pyrazole derivatives. J Chem Sci. 2019;131(8):70.
- He B, Lu C, Zheng G, He X, Wang M, Chen G, Zhang G, Lu A. Combination therapeutics in complex diseases. J Cell Mol Med. 2016;20(12):2231–2240.
- van Vlijmen H, Ortholand JY, Li VM, de Vlieger JSB. The European lead factory: an updated HTS compound library for innovative drug discovery. Drug Discov Today. 2021;26(10):2406–2413.
- Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432(7019):855–861.
- Tran PHL, Duan W, Lee B-J, Tran TTD. Nanogels for skin cancer therapy via transdermal delivery: current designs. Curr Drug Metab. 2019;20(7):575–582.
- Ma X, Qu Q, Zhao Y. Targeted delivery of 5-aminolevulinic acid by multifunctional hollow mesoporous silica nanoparticles for photodynamic skin cancer therapy. ACS Appl Mater Interfaces. 2015;7(20):10671–10676.
- Feoktistova M, Panayotova-Dimitrova D. Overcoming cell death resistance in skin cancer therapy: novel translational perspectives. Exp Dermatol. 2017;26(10):854–857.
- Dong L, Li Y, Li Z, Xu N, Liu P, Du H, Zhang Y, Huang Y, Zhu J, Ren G, et al. Au nanocage-strengthened dissolving microneedles for chemo-photothermal combined therapy of superficial skin tumors. ACS Appl Mater Interfaces. 2018;10(11):9247–9256.
- Liu J, Sun L, Li L, Zhang R, Xu ZP. Synergistic cancer photochemotherapy via layered double hydroxide-based trimodal nanomedicine at very low therapeutic doses. ACS Appl Mater Interfaces. 2021;13(6):7115–7126.
- Silk T, Wu J. Cutaneous toxicities of targeted therapies in the treatment of hepatocellular carcinoma. Hepatoma Res. 2020;6(Spec. Iss.):1–16.
- Hawash M, Jaradat N, Eid AM, Abubaker A, Mufleh O, Al-Hroub Q, Sobuh S. Synthesis of novel isoxazole-carboxamide derivatives as promising agents for melanoma and targeted nano-emulgel conjugate for improved cellular permeability. BMC Chem. 2022;16(1):47.
- Dahal A, Lo M, Singh S, Vo H, ElHage D, Jois SD, Murru S. 1,3-Diarylpyrazolones as potential anticancer agents for non-small cell lung cancer: synthesis and antiproliferative activity evaluation. Chem Biol Drug Des. 2022;99(4):620–633.
- Mohamed Ahmed MS, Kobayashi K, Mori A. One-pot construction of pyrazoles and isoxazoles with palladium-catalyzed four-component coupling. Org Lett. 2005;7(20):4487–4489.
- Dissanayake AA, Odom AL. Single-step synthesis of pyrazoles using titanium catalysis. Chem Commun. 2012;48(3):440–442.
- Ding Z, Tan Q, Gao M, Xu B. Copper-catalyzed aerobic cascade cycloamination and acyloxylation: a direct approach to 4-acyloxy-1H-pyrazoles. Org Biomol Chem. 2015;13(16):4642–4646.
- Harigae R, Moriyama K, Togo H. Preparation of 3,5-disubstituted pyrazoles and isoxazoles from terminal alkynes, aldehydes, hydrazines, and hydroxylamine. J Org Chem. 2014;79(5):2049–2058.
- Heller ST, Natarajan SR. 1,3-Diketones from acid chlorides and ketones: a rapid and general one-pot synthesis of pyrazoles. Org Lett. 2006;8(13):2675–2678.
- Kirkham JD, Edeson SJ, Stokes S, Harrity JP. Synthesis of ynone trifluoroborates toward functionalized pyrazoles. Org Lett. 2012;14(20):5354–5357.
- Zhang G, Ni H, Chen W, Shao J, Liu H, Chen B, Yu Y. One-pot three-component approach to the synthesis of polyfunctional pyrazoles. Org Lett. 2013;15(23):5967–5969.
- Deng X, Mani NS. Reaction of N-monosubstituted hydrazones with nitroolefins: a novel regioselective pyrazole synthesis. Org Lett. 2006;8(16):3505–3508.
- Peruncheralathan S, Yadav AK, Ila H, Junjappa H. Highly regioselective synthesis of 1-aryl-3 (or 5)-alkyl/aryl-5 (or 3)-(N-cycloamino)pyrazoles. J Org Chem. 2005;70(23):9644–9647.
- Persson T, Nielsen J. Synthesis of N-methoxy-N-methyl-beta-enaminoketoesters: new synthetic precursors for the regioselective synthesis of heterocyclic compounds. Org Lett. 2006;8(15):3219–3222.
- Peruncheralathan S, Khan TA, Ila H, Junjappa H. Regioselective synthesis of 1-aryl-3,4-substituted/annulated-5-(methylthio)pyrazoles and 1-aryl-3-(methylthio)-4,5-substituted/annulated pyrazoles. J Org Chem. 2005;70(24):10030–10035.
- Bernhammer JC, Huynh HV. Correlation of spectroscopically determined ligand donor strength and nucleophilicity of substituted pyrazoles. Dalton Trans. 2012;41(28):8600–8608.
- Pünner F, Schieven J, Hilt G. Synthesis of fluorenone and anthraquinone derivatives from aryl- and aroyl-substituted propiolates. Org Lett. 2013;15(18):4888–4891.
- Nakamichi N, Kawashita Y, Hayashi M. Oxidative aromatization of 1,3,5-trisubstituted pyrazolines and Hantzsch 1,4-dihydropyridines by Pd/C in acetic acid. Org Lett. 2002;4(22):3955–3957.
- Rzepecki LM, Waite JH. A chromogenic assay for catecholoxidases based on the addition of l-proline to quinones. Anal Biochem. 1989;179(2):375–381.
- Wang Y, Hao M-M, Sun Y, Wang L-F, Wang H, Zhang Y-J, Li H-Y, Zhuang P-W, Yang Z. Synergistic promotion on tyrosinase inhibition by antioxidants. Molecules. 2018;23(1):106.
- Ashraf Z, Rafiq M, Nadeem H, Hassan M, Afzal S, Waseem M, Afzal K, Latip J. Carvacrol derivatives as mushroom tyrosinase inhibitors; synthesis, kinetics mechanism and molecular docking studies. PLOS One. 2017;12(5):e0178069.
- Pintus F, Matos MJ, Vilar S, Hripcsak G, Varela C, Uriarte E, Santana L, Borges F, Medda R, Di Petrillo A, et al. New insights into highly potent tyrosinase inhibitors based on 3-heteroarylcoumarins: anti-melanogenesis and antioxidant activities, and computational molecular modeling studies. Bioorg Med Chem. 2017;25(5):1687–1695.
- Chamcheu JC, Esnault S, Adhami VM, Noll AL, Banang-Mbeumi S, Roy T, Singh SS, Huang S, Kousoulas KG, Mukhtar H, et al. Fisetin, a 3,7,3′,4′-tetrahydroxyflavone Inhibits the PI3K/Akt/mTOR and MAPK pathways and ameliorates psoriasis pathology in 2D and 3D organotypic human inflammatory skin models. Cells. 2019;8(9):1089.
- Roy T, Boateng ST, Banang-Mbeumi S, Singh PK, Basnet P, Chamcheu R-CN, Ladu F, Chauvin I, Spiegelman VS, Hill RA, et al. Synthesis, inverse docking-assisted identification and in vitro biological characterization of flavonol-based analogs of fisetin as c-Kit, CDK2 and mTOR inhibitors against melanoma and non-melanoma skin cancers. Bioorg Chem. 2021;107:104595.
- Liu H, Li X, Ji M, Wang N, Xu Y, Kong Y, Gou J, Yin T, He H, Zhang Y, et al. Two-step fabricating micelle-like nanoparticles of cisplatin with the 'real’ long circulation and high bioavailability for cancer therapy. Colloids Surf B Biointerfaces. 2022;210:112225.
- de la Cour CD, von Buchwald C, Dehlendorff C, Garset-Zamani M, Grønhøj C, Carlander A-LF, Friis S, Kjaer SK. Low-dose aspirin use and mortality risk in patients with head and neck cancer: a nationwide cohort study of 10 770 patients. Int J Cancer. 2022;150(6):969–975.
- Liebman TN, Stein JA, Polsky D. Cyclo-oxygenase-2 inhibitors for chemoprevention of nonmelanoma skin cancer: is there a role for these agents? J Am Acad Dermatol. 2013;68(1):173–176.
- Elmets CA, Viner JL, Pentland AP, Cantrell W, Lin H-Y, Bailey H, Kang S, Linden KG, Heffernan M, Duvic M, et al. Chemoprevention of nonmelanoma skin cancer with celecoxib: a randomized, double-blind, placebo-controlled trial. J Natl Cancer Inst. 2010;102(24):1835–1844.
- Sultania M, Imaduddin M, Deo SSV, Kar M, Muduly DK, Kumar S, Sharma A, Mishra A, Majumdar SKD, Adhya AK, et al. Role of metronomic therapy for advanced oral cancers and predictors of response: multi-institutional feasibility study. Head Neck. 2022;44(1):104–112.
- Li Y, Zhou M, Hu Q, Bai X-C, Huang W, Scheres SHW, Shi Y. Mechanistic insights into caspase-9 activation by the structure of the apoptosome holoenzyme. Proc Natl Acad Sci U S A. 2017;114(7):1542–1547.
- Brzozowska B, Gałecki M, Tartas A, Ginter J, Kaźmierczak U, Lundholm L. Freeware tool for analysing numbers and sizes of cell colonies. Radiat Environ Biophys. 2019;58(1):109–117.
- Vickers CJ, Gonzalez-Paez GE, Wolan DW. Selective detection and inhibition of active caspase-3 in cells with optimized peptides. J Am Chem Soc. 2013;135(34):12869–12876.
- Liu D, Tian Z, Yan Z, Wu L, Ma Y, Wang Q, Liu W, Zhou H, Yang C. Design, synthesis and evaluation of 1,2-benzisothiazol-3-one derivatives as potent caspase-3 inhibitors. Bioorg Med Chem. 2013;21(11):2960–2967.
- Hawash M, Jaradat N, Abualhasan M, Thaher M, Sawalhi R, Younes N, Shanaa A, Nuseirat M, Mousa A. In vitro and in vivo assessment of the antioxidant potential of isoxazole derivatives. Sci Rep. 2022;12(1):18223.
- Kumara K, Prabhudeva MG, Vagish CB, Vivek HK, Lokanatha Rai KM, Lokanath NK, Ajay Kumar K. Design, synthesis, characterization, and antioxidant activity studies of novel thienyl-pyrazoles. Heliyon. 2021;7(7):e07592.
- Ali SA, Awad SM, Said AM, Mahgoub S, Taha H, Ahmed NM. Design, synthesis, molecular modelling and biological evaluation of novel 3-(2-naphthyl)-1-phenyl-1H-pyrazole derivatives as potent antioxidants and 15-lipoxygenase inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):847–863.
- Cereto-Massagué A, Ojeda MJ, Valls C, Mulero M, Pujadas G, Garcia-Vallve S. Tools for in silico target fishing. Methods. 2015;71:98–103.
- Daina A, Michielin O, Zoete V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019;47(W1):W357–W364.
- Armstrong MS, Finn PW, Morris GM, Richards WG. Improving the accuracy of ultrafast ligand-based screening: incorporating lipophilicity into ElectroShape as an extra dimension. J Comput Aided Mol Des. 2011;25(8):785–790.
- Armstrong MS, Morris GM, Finn PW, Sharma R, Moretti L, Cooper RI, Richards WG. ElectroShape: fast molecular similarity calculations incorporating shape, chirality and electrostatics. J Comput Aided Mol Des. 2010;24(9):789–801.
- Johnson M, Lajiness M, Maggiore G. Molecular similarity: a basis for designing drug screening programs. Prog Clin Biol Res. 1989;291:167–171.
- Qin F, Wang Y, Jiang X, Wang Y, Zhang N, Wen X, Wang L, Jiang Q, He G. Design, synthesis and molecular mechanisms of novel dual inhibitors of heat shock protein 90/phosphoinositide 3-kinase alpha (Hsp90/PI3Kα) against cutaneous melanoma. J Enzyme Inhib Med Chem. 2019;34(1):909–926.
- Egger ME, Huang JS, Yin W, McMasters KM, McNally LR. Inhibition of autophagy with chloroquine is effective in melanoma. J Surg Res. 2013;184(1):274–281.
- Mielczarek-Lewandowska A, Hartman ML, Czyz M. Inhibitors of HSP90 in melanoma. Apoptosis. 2020;25(1–2):12–28.
- Zhao Q, Zhu HP, Xie X, Mao Q, Liu YQ, He XH, Peng C, Jiang Q-L, Huang W. Novel HSP90-PI3K dual inhibitor suppresses melanoma cell proliferation by interfering with HSP90-EGFR interaction and downstream signaling pathways. Int J Mol Sci. 2020;21(5):1845.
- Xiang X, Tu C, Li Q, Wang W, Huang X, Zhao Z, Xiong H, Mei Z. Oxymatrine ameliorates imiquimod-induced psoriasis pruritus and inflammation through inhibiting heat shock protein 90 and heat shock protein 60 expression in keratinocytes. Toxicol Appl Pharmacol. 2020;405:115209.
- Dickel H, Gambichler T, Kamphowe J, Altmeyer P, Skrygan M. Standardized tape stripping prior to patch testing induces upregulation of Hsp90, Hsp70, IL-33, TNF-α and IL-8/CXCL8 mRNA: new insights into the involvement of 'alarmins’. Contact Dermatitis. 2010;63(4):215–222.
- Zhang Y, Bai X, Wang Y, Li N, Li X, Han F, Su L, Hu D. Role for heat shock protein 90α in the proliferation and migration of HaCaT cells and in the deep second-degree burn wound healing in mice. PLOS One. 2014;9(8):e103723.
- Tukaj S, Kleszczyński K, Vafia K, Groth S, Meyersburg D, Trzonkowski P, Ludwig RJ, Zillikens D, Schmidt E, Fischer TW, et al. Aberrant expression and secretion of heat shock protein 90 in patients with bullous pemphigoid. PLOS One. 2013;8(7):e70496.
- Cengiz FP, Beyaztas S, Gokce B, Arslan O, Guler OO. Catalase, carbonic anhydrase and xanthine oxidase activities in patients with mycosis fungoides. J Enzyme Inhib Med Chem. 2015;30(2):212–215.
- Noda Y, Oosumi H, Morishima T, Tsujimura T, Mori M. Immunohistochemical study of carbonic anhydrase in mixed tumours and adenomas of sweat and sebaceous glands. J Cutan Pathol. 1987;14(5):285–290.
- Noda Y, Takai Y, Iwai Y, Meenaghan MA, Mori M. Immunohistochemical study of carbonic anhydrase in mixed tumours from major salivary glands and skin. Virchows Arch A Pathol Anat Histopathol. 1986;408(5):449–459.
- Kamsteeg M, Jansen PAM, van Vlijmen-Willems IMJJ, van Erp PEJ, Rodijk-Olthuis D, van der Valk PG, Feuth T, Zeeuwen PLJM, Schalkwijk J. Molecular diagnostics of psoriasis, atopic dermatitis, allergic contact dermatitis and irritant contact dermatitis. Br J Dermatol. 2010;162(3):568–578.
- Suri BK, Verma NK, Schmidtchen A. Toll-like receptor 3 agonist, polyinosinic–polycytidylic acid, upregulates carbonic anhydrase II in human keratinocytes. Acta Derm Venereol. 2018;98(8):762–765.
- Carfagna MA, Young KM, Susick RL. Sex differences in rat hepatic cytolethality of the protein kinase C inhibitor safingol: role of biotransformation. Toxicol Appl Pharmacol. 1996;137(2):173–181.
- Wang J, Wang YM, Ahn HY. Biological products for the treatment of psoriasis: therapeutic targets, pharmacodynamics and disease—drug-drug interaction implications. AAPS J. 2014;16(5):938–947.
- Kyoreva M, Li Y, Hoosenally M, Hardman-Smart J, Morrison K, Tosi I, Tolaini M, Barinaga G, Stockinger B, Mrowietz U, et al. CYP1A1 enzymatic activity influences skin inflammation via regulation of the AHR pathway. J Invest Dermatol. 2021;141(6):1553–1563.e3.
- Pavez Loriè E, Cools M, Borgers M, Wouters L, Shroot B, Hagforsen E, Törmä H, Vahlquist A. Topical treatment with CYP26 inhibitor talarozole (R115866) dose dependently alters the expression of retinoid-regulated genes in normal human epidermis. Br J Dermatol. 2009;160(1):26–36.
- Hawerkamp HC, Kislat A, Gerber PA, Pollet M, Rolfes KM, Soshilov AA, Denison MS, Momin AA, Arold ST, Datsi A, et al. Vemurafenib acts as an aryl hydrocarbon receptor antagonist: implications for inflammatory cutaneous adverse events. Allergy. 2019;74(12):2437–2448.
- Heo M-J, Lee C, Choi SY, Choi YM, An I-S, Bae S, An S, Jung JH. Nintedanib ameliorates animal model of dermatitis. Sci Rep. 2020;10(1):4493.
- Grun D, Adhikary G, Eckert RL. VEGF-A acts via neuropilin-1 to enhance epidermal cancer stem cell survival and formation of aggressive and highly vascularized tumors. Oncogene. 2016;35(33):4379–4387.
- de Almeida CM, de Jesus SF, Poswar FdO, Gomes ESB, Fraga CAdC, Farias LC, Santos SHS, Feltenberger JD, de Paula AMB, Guimarães ALS, et al. Increasing demonstration of angiogenic markers in skin neoplastic lesions. Pathol Res Pract. 2016;212(2):101–105.
- Decoster L, Broek IV, Neyns B, Majois F, Baurain JF, Rottey S, Rorive A, Anckaert E, De Mey J, De Brakeleer S, et al. Biomarker analysis in a phase II study of sunitinib in patients with advanced melanoma. Anticancer Res. 2015;35(12):6893–6899.
- Mahalingam D, Malik L, Beeram M, Rodon J, Sankhala K, Mita A, Benjamin D, Ketchum N, Michalek J, Tolcher A, et al. Phase II study evaluating the efficacy, safety, and pharmacodynamic correlative study of dual antiangiogenic inhibition using bevacizumab in combination with sorafenib in patients with advanced malignant melanoma. Cancer Chemother Pharmacol. 2014;74(1):77–84.
- Wen S-Y, Cheng S-Y, Ng S-C, Aneja R, Chen C-J, Huang C-Y, Kuo W-W. Roles of p38α and p38β mitogen-activated protein kinase isoforms in human malignant melanoma A375 cells. Int J Mol Med. 2019;44(6):2123–2132.
- Choo MK, Kraft S, Missero C, Park JM. The protein kinase p38α destabilizes p63 to limit epidermal stem cell frequency and tumorigenic potential. Sci Signal. 2018;11(551):eaau0727.
- Liu K, Yu D, Cho Y-Y, Bode AM, Ma W, Yao K, Li S, Li J, Bowden GT, Dong Z, et al. Sunlight UV-induced skin cancer relies upon activation of the p38α signaling pathway. Cancer Res. 2013;73(7):2181–2188.
- Zheng T, Zhao W, Li H, Xiao S, Hu R, Han M, Liu H, Liu Y, Otsu K, Liu X, et al. p38α signaling in Langerhans cells promotes the development of IL-17-producing T cells and psoriasiform skin inflammation. Sci Signal. 2018;11(521):eaao1685.
- Liu Q, Zhang S, Chen G, Zhou H. E3 ubiquitin ligase Nedd4 inhibits AP-1 activity and TNF-α production through targeting p38α for polyubiquitination and subsequent degradation. Sci Rep. 2017;7(1):4521.
- Ondet T, Muscatelli-Groux B, Coulouarn C, Robert S, Gicquel T, Bodin A, Lagente V, Grimaud J-A. The release of pro-inflammatory cytokines is mediated via mitogen-activated protein kinases rather than by the inflammasome signalling pathway in keratinocytes. Clin Exp Pharmacol Physiol. 2017;44(7):827–838.
- Lilleholt LL, Johansen C, Arthur JS, Funding A, Bibby BM, Kragballe K, Iversen L. Role of p38 mitogen-activated protein kinase isoforms in murine skin inflammation induced by 12-O-tetradecanoylphorbol 13-acetate. Acta Derm Venereol. 2011;91(3):271–278.
- Theivanthiran B, Kathania M, Zeng M, Anguiano E, Basrur V, Vandergriff T, Pascual V, Wei W-Z, Massoumi R, Venuprasad K, et al. The E3 ubiquitin ligase Itch inhibits p38α signaling and skin inflammation through the ubiquitylation of Tab1. Sci Signal. 2015;8(365):ra22.
- Xu Y, Chen S, Zhang L, Chen G, Chen J. The anti-inflammatory and anti-pruritus mechanisms of Huanglian Jiedu decoction in the treatment of atopic dermatitis. Front Pharmacol. 2021;12:735295.
- Martins C, Migayron L, Drullion C, Jacquemin C, Lucchese F, Rambert J, Merhi R, Michon P, Taieb A, Rezvani H-R, et al. Vitiligo skin T cells are prone to produce type 1 and type 2 cytokines to induce melanocyte dysfunction and epidermal inflammatory response through Jak signaling. J Invest Dermatol. 2021;142(4):1194–1205.e7.
- Nakashima C, Yanagihara S, Otsuka A. Innovation in the treatment of atopic dermatitis: emerging topical and oral Janus kinase inhibitors. Allergol Int. 2022;71(1):40–46.
- Deeks ED, Duggan S. Abrocitinib: first approval. Drugs. 2021;81(18):2149–2157.
- Cartron AM, Nguyen TH, Roh YS, Kwatra MM, Kwatra SG. Janus kinase inhibitors for atopic dermatitis: a promising treatment modality. Clin Exp Dermatol. 2021;46(5):820–824.
- Yang HY, Steenhuis P, Glucksman AM, Gurenko Z, La TD, Isseroff RR. Alpha and beta adrenergic receptors modulate keratinocyte migration. PLOS One. 2021;16(7):e0253139.
- Nocentini A, Supuran CT. Adrenergic agonists and antagonists as antiglaucoma agents: a literature and patent review (2013–2019). Expert Opin Ther Pat. 2019;29(10):805–815.
- Feng X, Guan W, Zhao Y, Wang C, Song M, Yao Y, Yang T, Fan H. Dexmedetomidine ameliorates lipopolysaccharide-induced acute kidney injury in rats by inhibiting inflammation and oxidative stress via the GSK-3β/Nrf2 signaling pathway. J Cell Physiol. 2019;234(10):18994–19009.
- Yan H-X, Li W-W, Zhang Y, Wei X-W, Fu L-X, Shen G-B, Yin T, Li X-Y, Shi H-S, Wan Y, et al. Accumulation of FLT3(+) CD11c (+) dendritic cells in psoriatic lesions and the anti-psoriatic effect of a selective FLT3 inhibitor. Immunol Res. 2014;60(1):112–126.
- Moss KG, Toner GC, Cherrington JM, Mendel DB, Laird AD. Hair depigmentation is a biological readout for pharmacological inhibition of KIT in mice and humans. J Pharmacol Exp Ther. 2003;307(2):476–480.
- Krasnov GS, Ghukasyan LG, Abramov IS, Nasedkina TV. Determination of the subclonal tumor structure in childhood acute myeloid leukemia and acral melanoma by next-generation sequencing. Mol Biol. 2021;55(5):727–741.
- Xu Q, He S, Yu L. Clinical benefits and safety of FMS-like tyrosine kinase 3 inhibitors in various treatment stages of acute myeloid leukemia: a systematic review, meta-analysis, and network meta-analysis. Front Oncol. 2021;11:686013.
- Karagounis TK, Rotemberg V, Geskin LJ, Jurcic JG, Niedt G. NPM1 and FLT3-TKD mutations are enriched in patients with leukemia cutis. Dermatol Online J. 2020;26(7).
- Brotzman N, Xu Y, Graybill A, Cocolas A, Ressler A, Seeram NP, Ma H, Henry GE. Synthesis and tyrosinase inhibitory activities of 4-oxobutanoate derivatives of carvacrol and thymol. Bioorg Med Chem Lett. 2019;29(1):56–58.
- Guy GP, Jr., Machlin SR, Ekwueme DU, Yabroff KR. Prevalence and costs of skin cancer treatment in the U.S., 2002–2006 and 2007–2011. Am J Prev Med. 2015;48(2):183–187.
- Guy GP, Thomas CC, Thompson T, Watson M, Massetti GM, Richardson LC, Centers for Disease Control and Prevention (CDC). Vital signs: melanoma incidence and mortality trends and projections – United States, 1982–2030. MMWR Morb Mortal Wkly Rep. 2015;64(21):591–596.
- Stern RS. Prevalence of a history of skin cancer in 2007: results of an incidence-based model. Arch Dermatol. 2010;146(3):279–282.
- Spanbroek R, Stark HJ, Janssen-Timmen U, Kraft S, Hildner M, Andl T, Bosch FX, Fusenig NE, Bieber T, Rådmark O, et al. 5-lipoxygenase expression in Langerhans cells of normal human epidermis. Proc Natl Acad Sci U S A. 1998;95(2):663–668.
- Katikaneni A, Jelcic M, Gerlach GF, Ma Y, Overholtzer M, Niethammer P. Lipid peroxidation regulates long-range wound detection through 5-lipoxygenase in zebrafish. Nat Cell Biol. 2020;22(9):1049–1055.
- Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015;6:297–310.
- Casanova ML, Blázquez C, Martínez-Palacio J, Villanueva C, Fernández-Aceñero MJ, Huffman JW, Jorcano JL, Guzmán M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J Clin Invest. 2003;111(1):43–50.
- Roy T, Boateng ST, Banang-Mbeumi S, Singh PK, Basnet P, Chamcheu R-CN, Ladu F, Chauvin I, Spiegelman VS, Hill RA, et al. Identification of new fisetin analogs as kinase inhibitors: data on synthesis and anti-skin cancer activities evaluation. Data Brief. 2021;35:106858.
- Fuhrmann F, Seichter W, Mazik M. Selective recognition of ammonium over potassium ion with acyclic receptor molecules bearing 3,4,5-trialkylpyrazolyl groups. Org Mater. 2022;4(03):61–72.
- Polo-Cuadrado E, Acosta-Quiroga K, Rojas-Peña C, Rodriguez-Nuñez YA, Duarte Y, Brito I, Cisterna J, Gutiérrez M. Molecular modeling and structural analysis of some tetrahydroindazole and cyclopentanepyrazole derivatives as COX-2 inhibitors. Arab J Chem. 2022;15(2):103540.
- Bartholomew GL, Carpaneto F, Sarpong R. Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion. J Am Chem Soc. 2022;144(48):22309–22315.
- Rheingold AL, Yap G, Trofimenko S. Do poly(indazolyl)borates have abnormal regiochemistry? Inorg Chem. 1995;34(4):759–760.
- Fuse S, Suzuki K, Kuchimaru T, Kadonosono T, Ueda H, Sato S, Kizaka-Kondoh S, Nakamura H. Design, synthesis, and evaluation of indeno[2,1-c]pyrazolones for use as inhibitors against hypoxia-inducible factor (HIF)-1 transcriptional activity. Bioorg Med Chem. 2020;28(1):115207.
- Abdelsalam EA, Zaghary WA, Amin KM, Abou Taleb NA, Mekawey AAI, Eldehna WM, Abdel-Aziz HA, Hammad SF. Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg Chem. 2019;89:102985.
- Neumann JJ, Suri M, Glorius F. Efficient synthesis of pyrazoles: oxidative C–C/N–N bond-formation cascade. Angew Chem Int Ed Engl. 2010;49(42):7790–7794.
- Moriya O, Urata Y, Endo T. Dehydrochlorination of hydroximic acid chlorides by the use of organotin compounds: an application for synthesis of isoxazolines and isoxazoles. J Chem Soc Chem Commun. 1991;1(1):17–18.
- Tiwari MK, Iqubal A, Das P. Intramolecular oxidative C–N bond formation under metal-free conditions: one-pot global functionalization of pyrazole ring. Tetrahedron. 2022;126:133059.
- Muzalevskiy VM, Rulev AY, Romanov AR, Kondrashov EV, Ushakov IA, Chertkov VA, Nenajdenko VG. Selective, metal-free approach to 3- or 5-CF3-pyrazoles: solvent switchable reaction of CF3-Ynones with hydrazines. J Org Chem. 2017;82(14):7200–7214.
- Kolla ST, Somanaboina R, Bhimapaka CR. TBHP/Cu(OAc)2 mediated oxidation of pyrazolines: a convenient method for the preparation of pyrazoles. Synth Commun. 2021;51(9):1425–1432.
- Zhu Y-F, Wei B-L, Wei J-J, Wang W-Q, Song W-B, Xuan L-J. Synthesis of pyrazolones and pyrazoles via Pd-catalyzed aerobic oxidative dehydrogenation. Tetrahedron Lett. 2019;60(17):1202–1205.
- Rheinwald JG, Beckett MA. Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res. 1981;41(5):1657–1663.
- Ma H, Johnson S, Liu W, DaSilva N, Meschwitz S, Dain J, Seeram N. Evaluation of polyphenol anthocyanin-enriched extracts of blackberry, black raspberry, blueberry, cranberry, red raspberry, and strawberry for free radical scavenging, reactive carbonyl species trapping, anti-glycation, anti-β-amyloid aggregation, and microglial neuroprotective effects. Int J Mol Sci. 2018;19(2):461.
- Ma H, Xu J, DaSilva NA, Wang L, Wei Z, Guo L, Johnson SL, Lu W, Xu J, Gu Q, et al. Cosmetic applications of glucitol-core containing gallotannins from a proprietary phenolic-enriched red maple (Acer rubrum) leaves extract: inhibition of melanogenesis via down-regulation of tyrosinase and melanogenic gene expression in B16F10 melanoma cells. Arch Dermatol Res. 2017;309(4):265–274.
- Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. 2014;42(Web Server issue):W32–W38.
- Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157.
- Kumar Singh P, Silakari O. In silico guided development of imine-based inhibitors for resistance-deriving kinases. J Biomol Struct Dyn. 2019;37(10):2593–2599.
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, et al. PubChem substance and compound databases. Nucleic Acids Res. 2016;44(D1):D1202–D1213.
- O'Boyle NM, Morley C, Hutchison GR. Pybel: a Python wrapper for the OpenBabel cheminformatics toolkit. Chem Cent J. 2008;2:5.
- O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33.
- Burley SK, Berman HM, Christie C, Duarte JM, Feng Z, Westbrook J, Young J, Zardecki C. RCSB Protein Data Bank: sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018;27(1):316–330.
- Harris R, Olson AJ, Goodsell DS. Automated prediction of ligand-binding sites in proteins. Proteins Struct Funct Bioinf. 2008;70(4):1506–1517.
- Forli S, Huey R, Pique ME, Sanner MF, Goodsell DS, Olson AJ. Computational protein–ligand docking and virtual drug screening with the AutoDock suite. Nat Protoc. 2016;11(5):905–919.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
- Rigsby RE, Parker AB. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem Mol Biol Educ. 2016;44(5):433–437.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
- Boateng ST, Roy T, Agbo ME, Banang-Mbeumi S, Chamcheu R-CN, Bramwell M, Pham LK, Jackson KE, Hill R, Nagalo BM, et al. Identification of potential inhibitors of cutaneous melanoma and non-melanoma skin cancer cells through in-vitro and in-silico screening of a small library of phenolic compounds. SSRN Electron J. 2022.
- Liang Y, Liang B, Wu XR, Chen W, Zhao LZ. Network pharmacology-based systematic analysis of molecular mechanisms of Dingji Fumai decoction for ventricular arrhythmia. Evid Based Complement Alternat Med. 2021;2021:5535480.
- Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):W257–W263.
- Banerjee P, Dehnbostel FO, Preissner R. Prediction is a balancing act: importance of sampling methods to balance sensitivity and specificity of predictive models based on imbalanced chemical data sets. Front Chem. 2018;6:362.