
Abstract
Heat shock protein 90 (Hsp90) is considered an attractive therapeutic target for cancer treatment due to its high expression in many cancers. In this study, four potent Hsp90 inhibitors (HPs 1–4) were identified using structure-based virtual screening. Among them, HP-4 exhibited the most potent inhibitory effects (IC50 = 17.64 ± 1.45 nM) against the Hsp90 protein, which was about 7.7 times stronger than that of MPC-3100 (a positive inhibitor targeting Hsp90). In vitro cytotoxicity assay suggested that HP-4 could effectively inhibit the proliferation of a series of tumour cells, including HCT-116, HeLa, A549, A2780, DU145, HepG2 and A498. Furthermore, in vivo assay displayed that HP-4 had significant anti-tumour effects on HCT-116 cell-derived xenograft models. These data demonstrate that HP-4 could be a potential lead compound for the further investigation of anti-tumour drugs.
Introduction
Heat shock proteins (Hsps) are highly conserved in almost all organismsCitation1. As ATP-dependent molecular chaperones, Hsps are mainly responsible for the proper folding of client proteins, thereby regulating cellular homeostasisCitation2. According to gene homology and molecular weight, Hsps have been classified into small Hsps, Hsp40/60, Hsp70, Hsp90, and Hsp110Citation3. Hsp90 is one of the most abundant Hsps, which accounts for 1–2% of the total cellular protein and increases to 3–5% after exposure to external stressCitation4,Citation5. Hsp90 maintains the stability, conformation and function of many client proteins including EGFR, MET, HER2, AKT, CDK4 and cyclin DCitation6–9. Notably, most of these client proteins are overexpressed in malignant tumours and involve in the proliferation, angiogenesis, invasion and metastasis of tumour cellsCitation10. Moreover, the protein levels of Hsp90 in tumour cells are 2–10 times higher than that of their normal counterparts, suggesting its importance in tumour cell growthCitation11,Citation12. Therefore, Hsp90 is considered a promising target for cancer therapy.
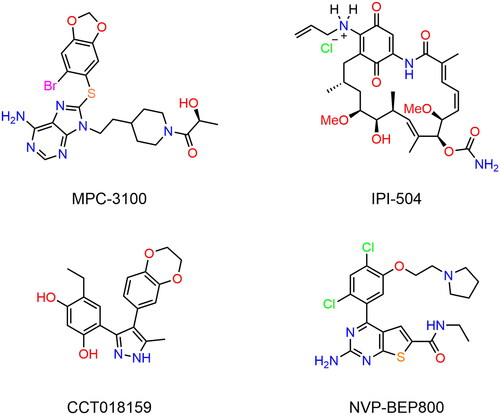
Hsp90 mainly exists as dimers in cells, and each monomer has three main domains including an N-terminal domain (NTD), a middle domain and a C-terminal domain (CTD)Citation13,Citation14. The NTD contains an ATP-binding pocket, which is essential for the folding of client proteins by binding ATPCitation15. The middle domain is crucial in client binding and the CTD is responsible for dimerisationCitation16. Many classical Hsp90 inhibitors (shown in ) have been designed to target the ATP-binding pocket of NTD, and they can block ATP hydrolysis thereby leading to the degradation of client proteinsCitation17. Kim et al. reported a purine-based Hsp90 inhibitor MPC-3100, which significantly suppressed the growth of various human tumour-induced xenografts and possessed superior pharmacokineticsCitation6. Moreover, MPC-3100 has successfully passed phase I clinical trials, yet poor solubility was a major obstacle to its further clinical developmentCitation18. IPI-504 was a water-soluble analogue of 17-AAG (an efficient Hsp90 inhibitor) and the potential anti-tumour effects of IPI-504 have been validated in preclinical experimentsCitation19,Citation20. However clinical experiments revealed that the patient developed hepatic failure and ketoacidosis while receiving IPI-504 medicationsCitation21. Sharp et al. discovered a novel 3,4-diaryl pyrazole Hsp90 inhibitor CCT018159, which possessed better potency against Hsp90 protein compared to 17-AGG with the potential advantage of water solubilityCitation22. However, the cellular inhibitory activity of CCT018159 was inferior to that of 17-AGG. NVP-BEP800 is a 2-aminothieno[2,3-d]pyrimidine Hsp90 inhibitor with nanomolar activity against a series of human tumour cell lines, yet the weak water solubility hindered its application in some contextsCitation23,Citation24. So far, there are no available Hsp90 inhibitors on the market due to their poor solubility, limited efficacy, and multiple side effectsCitation25,Citation26. Therefore, in this study, we aimed to discover novel Hsp90 inhibitors with high efficacy and broad anti-tumour activity.
Figure 1. The chemical structures of known inhibitors of Hsp90.
Nowadays, structure-based virtual screening (SBVS), including pharmacophore mapping and molecular docking, is widely used in drug discovery due to its efficiency and convenienceCitation27,Citation28. Additionally, molecular dynamics (MD) simulation is usually carried out to estimate the stability of protein-ligand systemsCitation29. In this study, we successfully identified four potent Hsp90 inhibitors from an internal database using SBVS. Then the results of MD simulations indicated the good stability of four protein-hit systems. Further in vitro assays revealed that the four hits exhibited strong inhibitory activities against the Hsp90 protein. Among them, the optimal inhibitor HP-4 significantly suppressed the proliferation of multiple human tumour cells and the progression of HCT-116 cell-derived xenograft.
Materials and methods
Cell culture
Human tumour cells including HCT-116, HeLa, A549, A2780, DU145, HepG2 and A498 cells were obtained from American Type Culture Collection (Manassas, VA, USA). HCT-116, DU145 and A2780 cells were cultured in RPMI-1640 medium. HeLa, A549, HepG2 and A498 cells were cultured in Dulbecco’s modified Eagle’s medium. All media were supplemented with 10% foetal bovine serum and 1% penicillin-streptomycin. Cell cultures were maintained in the humidified incubator with 5% CO2 at 37 °C.
Generation of pharmacophore model
According to the method previously reportedCitation30, the establishment of the pharmacophore model was carried out in the Molecular Operating Environment (MOE) program. The X-ray crystallographic structure of Hsp90 protein (PDB ID: 2XJX) was retrieved from the Protein Data Bank (PDB) with a resolution value of 1.66 Å. The Hsp90 protein was prepared by removing water, adding polar hydrogen bonds, merging non-polar hydrogen bonds, and calculating gasteiger charges using MOE. Then, the Ligand Interactions tool of MOE was used to analyse the interaction between Hsp90 active-site residues and the original ligand. Finally, based on the results of interaction analysis, pharmacophore features including hydrogen-bond donor, one aromatic centre, and hydrophobic feature were manually built using the Pharmacophore Query Editor of MOE.
Validation of pharmacophore model
To validate the selectivity of the generated Hsp90 pharmacophore model, the virtual screening of a testing database including 19 known Hsp90 inhibitors and 1110 decoy compounds downloaded from the enhanced Database of Useful Decoys (DUDe) was performed using the pharmacophore search protocol of MOECitation31,Citation32. Then MedCalc software was employed to analyse the receiver operating characteristic (ROC) curveCitation33. In ROC analysis, the area under the curve (AUC) is generally used to assess the ability of the pharmacophore model to distinguish active or inactive compounds.
Virtual screening
The virtual screening was carried out as described previouslyCitation34. First of all, an internal compound database containing 35 000 compounds was created by the combinatorial chemistry method, and the Energy Minimise algorithm in MOE was employed to generate three-dimensional (3D) structures of all compounds in the database. Then, 8750 compounds with drug-like properties from the database were filtered by the following modified Lipinski’s rules: molecular weight (MW) ≤ 600 Da, number of hydrogen-bond donors (nHD) ≤ 10, number of hydrogen-bond acceptors (nHA) ≤ 10, and log P(o/w) ≤ 6. Subsequently, the Pharmaceutical Search module of MOE was used to perform a pharmacophore-based virtual screening, and the generated Hsp90 pharmacophore model served as a 3D query. The root mean square distance (RMSD) value was used to evaluate the matching degree between each compound and the pharmacophore model. In general, a lower RMSD value suggests a better match. The 27 screened compounds with RMSD < 1 Å were then subjected to molecular docking.
Molecular docking
The crystallographic structure of Hsp90 protein (PDB ID: 2XJX) from the protein data bank was prepared by removing water, adding polar hydrogen bonds, merging non-polar hydrogen bonds, and calculating gasteiger charges using MOE. In this study, molecular docking interactions were performed using the Dock tool of MOE as described previouslyCitation34. Each selected compound was docked into the active site of Hsp90 with the triangle matcher method, and the docking score was calculated by the dG scoring function. In general, the lower docking score indicates a stronger binding affinity between the ligand and Hsp90. Finally, four compounds were selected for further study according to the docking scores.
MD Simulation
MD simulations were performed by GROMACS 2021.5 as previously reported to explore whether each Hsp90-hit system was stableCitation35. The topol file of each hit was generated by Acpype Server (www.bio2byte.be) with GAFF force field and the topol file of Hsp90 was built by GROMACS 2021.5 with AMBER99SB-ILDN force field. A cubic box of 0.9 nm was constructed with the Hsp90-hit system placed in the centre and simple point charge (SPC) water molecules were added to solvate the Hsp90-hit system. Subsequently, appropriate Na+ and Cl− were added to keep the system neutral, and the system was then subjected to a 5000-step energy minimisation with the steepest descent algorithm. NVT simulation was performed for 100 ps with a V-rescale thermostat to keep the system at 300K and later another 100 ps NPT simulation was conducted with a Parinello–Rahman barostat to maintain the system at 1 atm. Finally, each system run for 100 ns MD simulations, and the atomic coordinates of each system were recorded at every 10 ps for further analysis.
Fluorescence polarisation (FP) measurements
Hsp90 protein was obtained from Abcam (Cambridge, MA, USA). The Hsp90 fluorescence polarisation binding assay was carried out as described previouslyCitation36. Gerdanamycin (GM), a natural product that binds to the ATP pocket of Hsp90, was used to identify novel Hsp90 inhibitors based on the competition of fluorescent (BODIPY)-labelled GM with Hsp90 protein. Different concentrations of inhibitors and Hsp90 in the assay buffer were mixed in black 96-well microplates. Ten minutes later BODIPY-labelled GM was added at a final concentration of 5 nM. The binding reaction was allowed to proceed for 3 h at room temperature. Fluorescence polarisation was measured on a SpectraMax GeminiXS (Molecular Devices, Sunnyvale, CA) with excitation and emission wavelengths of 485 and 530 nm, respectively.
In vitro cytotoxicity assay
According to the experimental method reported previouslyCitation37, cells were plated on each well (5 × 104 cells) and kept overnight. Then, different concentrations of inhibitors were added to each well and incubated at 37 °C for 72 h. Thereafter, the medium was removed, and MTT was added. Cells were incubated with medium containing MTT for an additional 4 h, the supernatant was removed by centrifugation, and 150 μL of DMSO was added to each well and gently shaken for 15 min. The absorbance of the plate was recorded at 570 nm using a microplate reader.
In vivo anti-tumour activity
All experimental protocols were approved by the Animal Ethics Committee of China Pharmaceutical University. According to the experimental method reported previouslyCitation37, cultured cells of HCT-116 were washed with D-Hank’s solution and prepared as a suspension at a concentration of 106 cells/mL in culture medium. The cells were injected subcutaneously into the right side of BALB/c nude mice. Mice were randomly divided into three groups: control group, low-dose treatment group (5 mg/kg), and high-dose treatment group (10 mg/kg). Each group was treated with the intraperitoneal injection of inhibitors. The tumour was measured every three days, and the tumour volume was calculated using the formula (c × c × d)/2 (c, the smallest diameter; d, the largest diameter).
Results and discussion
Generation of pharmacophore model
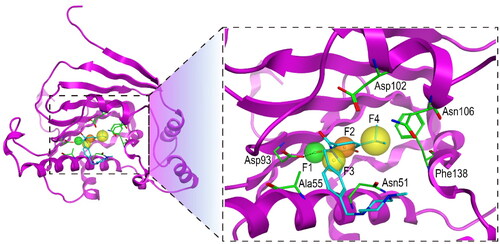
In this study, the X-ray crystallographic structure of Hsp90 protein (PDB ID: 2XJX) complex with the original ligand was retrieved from the protein data bank. After the Hsp90 protein was prepared by removing water, adding polar hydrogen bonds, merging non-polar hydrogen bonds, and calculating gasteiger charges using MOE, this protein was used to establish a Hsp90 pharmacophore model. Based on the interaction analysis of Hsp90 and the original ligand, a pharmacophore model consisting of one hydrogen-bond donor (F1), one aromatic centre (F2), and two hydrophobic features (F3 and F4) was generated (). The F1 feature overlapped the hydroxyl group of the original ligand, which formed the hydrogen-bond interaction with the carboxylate of Asp93. The hydrophobic feature F3 formed a hydrophobic interaction with Ala55. The F2 and F4 which mapped the aromatic ring and hydrophobic group of the ligand, respectively, formed hydrophobic interactions with the side chain benzene ring of Phe138. The pharmacophore features described above were used for subsequent virtual screening of Hsp90 inhibitors.
Figure 2. The pharmacophore model of Hsp90. The hydrogen-bond donor (F1) and aromatic centre (F2) were indicated by green and orange spheres, respectively. The two hydrophobic centroid features F3 and F4 were indicated by yellow spheres. The dashed black line represented hydrogen-bond interaction. Each key residue was annotated with a three-letter amino acid code.
Validation of pharmacophore model
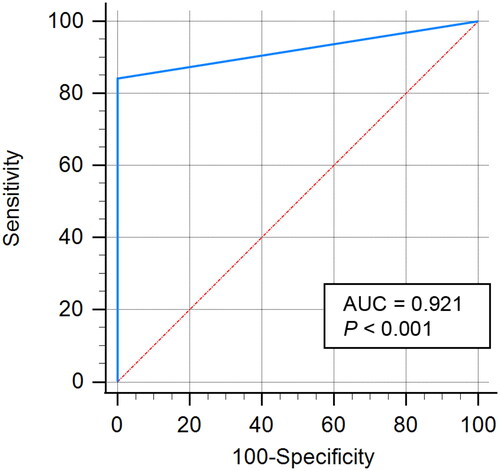
For virtual screening, the specificity and accuracy of the pharmacophore model are significant. Hence, the constructed Hsp90 pharmacophore model was validated using a testing set database consisting of 19 known inhibitors of Hsp90Citation31,Citation32 and 1110 decoy compounds from DUDE. Firstly, we combined the activity compounds with bait compounds and then conducted preliminary virtual screening using the Hsp90 pharmacophore model. The ROC curve was analysed to evaluate the quality of the Hsp90 pharmacophore modelCitation33. As shown in , the AUC value was extremely close to 1.0 (0.921), indicating the good ability of the Hsp90 pharmacophore model to differentiate true actives from decoy compounds (p < 0.001).
Figure 3. ROC curve of the generated Hsp90 pharmacophore model.
Virtual screening
showed the screening strategy of Hsp90 inhibitors in this study. First, the modified Lipinski’s rules were employed to filter drug-like compounds from the database, and 8750 compounds were retrieved. Then, the 8750 filtered compounds were subjected to the pharmacophore-based virtual screening, and the constructed Hsp90 pharmacophore model was used as a 3D query. In this step, 27 compounds with RMSD values lower than 1 Å were screened. Subsequently, a docking-based virtual screening was performed to predict the binding affinities between 27 compounds and the Hsp90 active site, and a classical Hsp90 inhibitor MPC-3100 was chosen as the positive control. Since the docking score of MPC-3100 was −13.31 kcal/mol, the value of −13.31 was used as the threshold for selecting candidate inhibitors of Hsp90. Finally, four top compounds (termed as HPs 1–4) with a docking score lower than −13.31 kcal/mol were chosen for further study ( and ).
Figure 4. The protocol for virtual screening and biological evaluation of Hsp90 inhibitors.
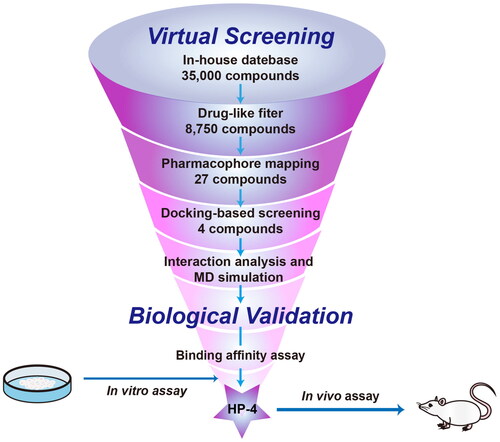
Figure 5. The chemical structures of HPs 1–4.
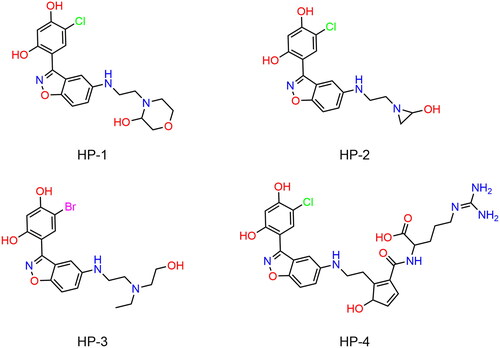
Table 1. The drug-like properties, docking sores and biological data of HPs 1–4 and MPC-3100.
Interaction analysis
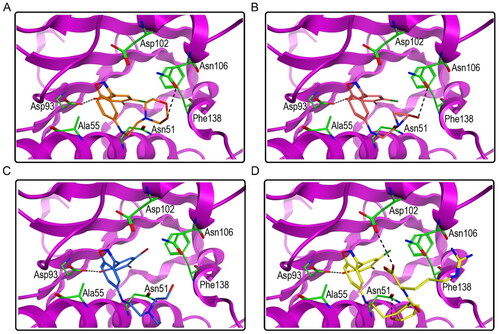
A molecular docking study was further performed to predict the binding poses of HPs 1–4 to the Hsp90. showed the interactions between HPs 1–4 and Hsp90. We found that all of the HPs 1–4 created hydrogen-bond interactions with Asp93 and the benzene ring of them formed hydrophobic interactions with Ala55, which matched well with the generated pharmacophore features F1 and F3. Moreover, all of the HPs 1–4 formed hydrophobic interactions with Phe138, which corresponded to the pharmacophore features F2 and F4. Notably, both HP-1 and HP-2 formed an extra hydrogen bond with Asp 106 while HP-4 created an additional hydrogen bond with Asp 102 and Asn 51, respectively. These extra hydrogen bonds may assist in stabilising the inhibitor in the active site of Hsp90. Overall, these results indicate that HPs 1–4 can interact with critical residues of Hsp90 and could be potent Hsp90 inhibitors.
Figure 6. The docking modes of HPs 1–4 with key residues of Hsp90. (A) HP-1; (B) HP-2; (C) HP-3; (D) HP-4. The dashed black line represents hydrogen-bond interaction.
MD Simulations
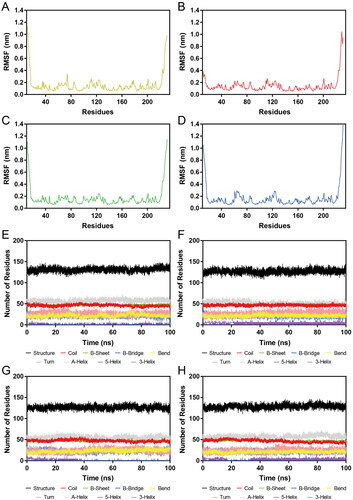
Subsequently, HPs 1–4-Hsp90 complexes were subjected to 100 ns MD simulations for the evaluation of the stability of HPs 1–4 at the active site of Hsp90 evolved with time. The RMSD was calculated to determine the stability of the system during the whole MD simulations. It was observed that the RMSDs of HP-1-Hsp90 and HP-2-Hsp90 were relatively stable at about 0.45 nm, the RMSD of HP-3-Hsp90 complex was relatively stable at about 0.40 nm, and the RMSD of HP-4-Hsp90 complex was relatively stable at about 0.30 nm (). Moreover, we also measured the RMSDs of HPs 1–4. displayed the RMSD plots for HPs 1–4 during the simulation, respectively. We found that though some fluctuations were observed, the RMSDs of HPs 1–4 finally tended to equilibrium at about 0.16 nm, 0.18 nm, 0.20 nm, and 0.36 nm, respectively. These data suggest that HPs 1–4-Hsp90 complexes were fairly stable during the whole simulations and HPs 1–4 could bind stably with Hsp90. Furthermore, the mobility of backbone Cα atoms of each Hsp90 residue was depicted using the root mean square fluctuations (RMSF) values to evaluate the flexibility of HPs 1–4-Hsp90 complexes systems. A smaller RMSF value indicates less residue flexibility and the lower fluctuations suggest better protein stability. showed that the key residues Ala55, Asp93, and Phe138 in the Hsp90 active site of four complexes systems were all not too flexible in the whole simulations with RMSFs less than 0.10 nm, reflecting that HPs 1–4 could interact strongly with these crucial residues. Based on the above RMSD and RMSF analysis, we can conclude that HPs 1–4 could stably bind to the active pocket of the Hps90. Finally, DSSP analysis was carried out to evaluate whether the secondary structure of the Hsp90 protein was changed during the whole MD simulations. As shown in , there were no obvious changes in structural elements, suggesting the structural stability of Hsp90.
Figure 7. RMSD of atoms during MD simulations. (A, B) RMSDs of HP-1-Hps90 complexes and HP-1, respectively; (C, D) RMSDs of HP-2-Hps90 complexes and HP-2, respectively; (E, F) RMSDs of HP-3-Hps90 complexes and HP-3, respectively; (G, H) RMSDs of HP-4-Hps90 complexes and HP-4, respectively;.
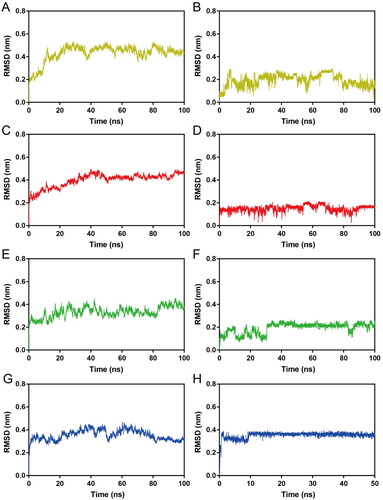
Figure 8. (A–D) RMSF of Cα atoms of Hsp90 residues in HPs 1–4-Hsp90 complexes, respectively. The colour code is HP-1-Hsp90 (yellow), HP-2-Hsp90 (red), HP-3-Hsp90 (green), and HP-4-Hsp90 (bule). (E–H) Secondary structure of Hsp90 in HPs 1–4-Hsp90 complexes, respectively.
Inhibitory activities of HPs 1–4 on Hsp90
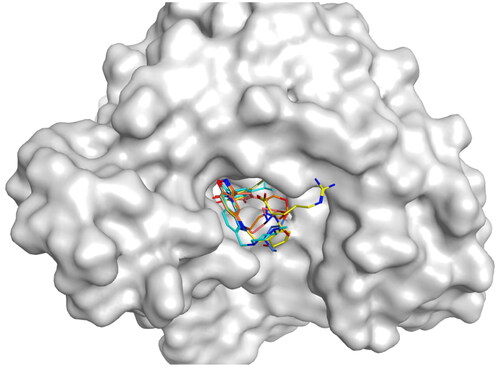
Previous studies have confirmed that GM binds to the ATP binding site of the N-terminal domain (NTD) of Hsp90, and GM dose-dependently inhibits the ATPase activity of Hsp90Citation38. Hence, we conducted a fluorescent (BODIPY)-labelled GM-FP experiment to verify whether HPs 1–4 could competitively bind to the ATP-binding pocket of the NTD of Hsp90. The Hsp90 ATP inhibitor MPC-3100 served as a positive control. As shown in , all four compounds could competitively inhibit the ATP binding site activity of GM to Hsp90 with IC50 values ranging from 17.64 to 61.69 nM, which were all smaller than 100 nM. Among them, HP-4 exhibited the most potent inhibitory effects with the IC50 value of 17.64 ± 1.45 nM. Furthermore, we measured the inhibitory effects of MPC-3100 against Hsp90 protein with the IC50 value of 136.16 ± 4.27 nM. By comparison, the inhibitory activity of HP-4 on Hsp90 was stronger than that of MPC-3100 by about 7.7-fold. In addition, to further verify that HPs 1–4 targeted the ATP binding site, a docking study was conducted. As shown in , the original ligand and all HPs 1–4 were able to bind to the ATP-binding pocket of NTD of Hsp90.
Figure 9. The docking modes of HPs 1–4 and the original ligand bind to the ATP-binding pocket of NTD of Hsp90.
In vitro cytotoxicity evaluation
In view of the high potential inhibitory activities of HPs 1–4 on Hsp90 protein, a typical MTT method was used to assess their cytotoxicity against human colon tumour cells HCT-116. MPC-3100 was chosen as a positive control. As shown in , the IC50 value of MPC-3100 inhibiting the growth of HCT-116 cells was 779.59 nM. Moreover, HPs 1–4 significantly suppressed the proliferation of HCT-116 cells with IC50 values ranging from 148.52 nM to 559.37 nM, which were all smaller than that of MPC-3100. Among them, HP-4 exhibited the best anti-tumour activity, which is about 5 times stronger than that of MPC-3100. Subsequently, we evaluated the inhibitory effects of HP-4 on other human tumour cells. As shown in , HP-4 exhibited good broad-spectrum anti-tumour effects on a series of tumour cells including HeLa, A549, A2780, DU145, HepG2, and A498.
Figure 10. The cytotoxicity of MPC-3100 and HPs 1–4 against human colon cancer HCT-116 cells.
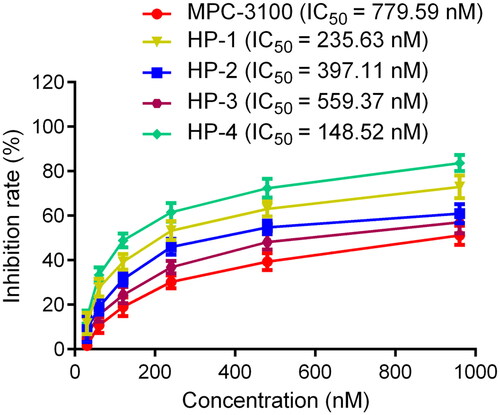
Table 2. Biochemical profile of HP-4.
In vivo anti-tumour effects of HP-4
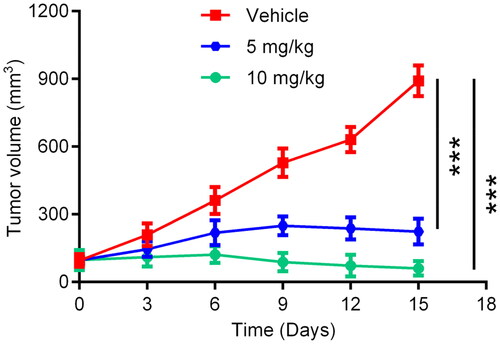
In order to further investigate the anti-tumour efficacy of HP-4 in vivo, HCT-116 cell-derived xenograft models were established in BALB/c nude mice. The HCT-116 tumour-bearing mice were divided into three groups including HP-4 (5 mg/kg), HP-4 (10 mg/kg) and the vehicle control group. As shown in , the tumour volume of mice after the administration of HP-4 was remarkably reduced, and HP-4 could significantly inhibit the growth of HCT-116 tumour compared with the vehicle control. These results indicate that HP-4 exhibited strong therapeutic effects on HCT-116 tumour-bearing mice.
Figure 11. In vivo anti-tumour effects of HP-4. Data are represented as mean ± SD, n = 5. ***p < 0.001.
Conclusion
Since the importance of Hsp90 in the proliferation of tumour cells, it has been a very promising target in the development of anti-tumour drugs. However, no Hsp90 inhibitors have been available on the market so far. In order to discover novel and efficient Hsp90 inhibitors, we successfully identified four compounds using a combined virtual screening strategy. In vitro test results showed that the four compounds not only had strong inhibitory activities on Hsp90 protein but also exhibited significant inhibitory effects on the proliferation of human tumour cells. Among them, the optimal inhibitor HP-4 could significantly inhibit the tumour growth of HCT-116 tumour-bearing mice. In general, HP-4 could be a promising lead compound targeting Hsp90. Structural optimisation of HP-4 will be further carried out in our laboratory for the development of novel Hsp90 drugs.
Ethical statement
All experimental protocols were reviewed and approved by the Animal Ethics Committee of China Pharmaceutical University.
Author contributions
YX, YZ and SZ performed experiments and acquired data. YX, YZ and SZ collected experimental samples. MMN and YZ performed statistical analysis. JL, YZ, ZX and LY analysed data and wrote the paper. All authors had edited and approved the final manuscript.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Additional information
Funding
References
- Hu C, Yang J, Qi Z, Wu H, Wang B, Zou F, Mei H, Liu J, Wang W, Liu Q, et al. Heat shock proteins: biological functions, pathological roles, and therapeutic opportunities. MedComm. 2022;3(3):e161.
- Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31(3):164–172.
- Wang L, Xu XL, Jiang ZY, You QD. Modulation of protein fate decision by small molecules: targeting molecular chaperone machinery. Acta Pharm Sin B. 2020;10(10):1904–1925.
- Dhamad AE, Zhou ZQ, Zhou JH, Du YC. Systematic proteomic identification of the heat shock proteins (Hsp) that interact with estrogen receptor alpha (ERα) and biochemical characterization of the ERα-Hsp70 interaction. PLoS One. 2016;11(8):e0160312.
- Bickel D, Gohlke H. C-terminal modulators of heat shock protein of 90kDa (HSP90): state of development and modes of action. Bioorg Med Chem. 2019;27(21):115080.
- Kim S-H, Bajji A, Tangallapally R, Markovitz B, Trovato R, Shenderovich M, Baichwal V, Bartel P, Cimbora D, McKinnon R, et al. Discovery of (2S)-1-[4-(2-{6-amino-8-[(6-bromo-1,3-benzodioxol-5-yl)sulfanyl]-9H-purin-9-yl}ethyl)piperidin-1-yl]-2-hydroxypropan-1-one (MPC-3100), a purine-based Hsp90 inhibitor. J Med Chem. 2012;55(17):7480–7501.
- Samant RS, Clarke PA, Workman P. The expanding proteome of the molecular chaperone HSP90. Cell Cycle. 2012;11(7):1301–1308.
- Prince T, Neckers L. A network of its own: the unique interactome of the Hsp90 cochaperone, Sba1/p23. Mol Cell. 2011;43(2):159–160.
- Echeverria PC, Bernthaler A, Dupuis P, Mayer B, Picard D. An interaction network predicted from public data as a discovery tool: application to the Hsp90 molecular chaperone machine. PLoS One. 2011;6(10):e26044.
- Wagner AJ, Chugh R, Rosen LS, Morgan JA, George S, Gordon M, Dunbar J, Normant E, Grayzel D, Demetri GD, et al. A phase I study of the HSP90 inhibitor retaspimycin hydrochloride (IPI-504) in patients with gastrointestinal stromal tumors or soft-tissue sarcomas. Clin Cancer Res. 2013;19(21):6020–6029.
- Mori M, Hitora T, Nakamura O, Yamagami Y, Horie R, Nishimura H, Yamamoto T. Hsp90 inhibitor induces autophagy and apoptosis in osteosarcoma cells. Int J Oncol. 2015;46(1):47–54.
- Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan C-G, Sun D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7(1):162–170.
- Patel HJ, Modi S, Chiosis G, Taldone T. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin Drug Discov. 2011;6(5):559–587.
- Mak OW, Sharma N, Reynisson J, Leung IKH. Discovery of novel Hsp90 C-terminal domain inhibitors that disrupt co-chaperone binding. Bioorg Med Chem Lett. 2021;38:127857.
- Verba KA, Wang RY-R, Arakawa A, Liu Y, Shirouzu M, Yokoyama S, Agard DA. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science. 2016;352(6293):1542–1547.
- Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BSJ. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem Biol. 2011;6(8):800–807.
- Kim HH, Hyun JS, Choi J, Choi KE, Jee JG, Park SJ. Structural ensemble-based docking simulation and biophysical studies discovered new inhibitors of Hsp90 N-terminal domain. Sci Rep. 2018;8(1):368.
- Kim S-H, Tangallapally R, Kim IC, Trovato R, Parker D, Patton JS, Reeves L, Bradford C, Wettstein D, Baichwal V, et al. Discovery of an L-alanine ester prodrug of the Hsp90 inhibitor, MPC-3100. Bioorg Med Chem Lett. 2015;25(22):5254–5257.
- Sequist LV, Gettinger S, Senzer NN, Martins RG, Jänne PA, Lilenbaum R, Gray JE, Iafrate AJ, Katayama R, Hafeez N, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28(33):4953–4960.
- Normant E, Paez G, West KA, Lim AR, Slocum KL, Tunkey C, McDougall J, Wylie AA, Robison K, Caliri K, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30(22):2581–2586.
- Wang H, Lu MJ, Yao MQ, Zhu W. Effects of treatment with an Hsp90 inhibitor in tumors based on 15 phase II clinical trials. Mol Clin Oncol. 2016;5(3):326–334.
- Sharp SY, Boxall K, Rowlands M, Prodromou C, Roe SM, Maloney A, Powers M, Clarke PA, Box G, Sanderson S, et al. In vitro biological characterization of a novel, yynthetic diaryl pyrazole resorcinol class of heat shock protein 90 inhibitors. Cancer Res. 2007;67(5):2206–2216.
- Massey AJ, Schoepfer J, Brough PA, Brueggen J, Chène P, Drysdale MJ, Pfaar U, Radimerski T, Ruetz S, Schweitzer A, et al. Preclinical antitumor activity of the orally available heat shock protein 90 inhibitor NVP-BEP800. Mol Cancer Ther. 2010;9(4):906–919.
- Chen WG, Sin SH, Wen KW, Damania B, Dittmer DP. Hsp90 inhibitors are efficacious against Kaposi Sarcoma by enhancing the degradation of the essential viral gene LANA, of the viral co-receptor EphA2 as well as other client proteins. PLoS Pathog. 2012;8(11):e1003048.
- Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823(3):742–755.
- Li ZN, Luo Y. HSP90 inhibitors and cancer: prospects for use in targeted therapies (Review). Oncol Rep. 2022;49(1):6.
- Yang D‐S, Yang Y‐H, Zhou Y, Yu L‐L, Wang R‐H, Di B, Niu M‐M. A redox-triggered bispecific supramolecular nano-medicine based on peptide self-assembly for high-efficacy and low-toxic cancer therapy. Adv Funct Mater. 2020;30(4):1904969.
- Moreira BP, Batista ICA, Tavares NC, Armstrong T, Gava SG, Torres GP, Mourão MM, Falcone FH. Docking-based virtual screening enables prioritizing protein kinase inhibitors with in vitro phenotypic activity against schistosoma mansoni. Front Cell Infect Microbiol. 2022;12:913301.
- Zinovjev K, van der Kamp MW. Enlighten2: molecular dynamics simulations of protein-ligand systems made accessible. Bioinformatics. 2020;36(20):5104–5106.
- Wang YT, Zhang H, Li JD, Niu M-M, Zhou Y, Qu YQ. Discovery of potent and noncovalent KRASG12D inhibitors: structure-based virtual screening and biological evaluation. Front Pharmacol. 2022;13:1094887.
- Uno T, Kawai Y, Yamashita S, Oshiumi H, Yoshimura C, Mizutani T, Suzuki T, Chong KT, Shigeno K, Ohkubo M, et al. Discovery of 3-Ethyl-4-(3-isopropyl-4-(4-(1-methyl-1 H-pyrazol-4-yl)-1 H-imidazol-1-yl)-1 H-pyrazolo[3,4-b]pyridin-1-yl)benzamide (TAS-116) as a potent, selective, and orally available HSP90 inhibitor. J Med Chem. 2019;62(2):531–551.
- Llauger L, He H, Kim J, Aguirre J, Rosen N, Peters U, Davies P, Chiosis G. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90. J Med Chem. 2005;48(8):2892–2905.
- Molla M, Aljahdali M, Sumon M, Asseri A, Altayb H, Islam M, Alsaiari A, Opo F, Jahan N, Ahammad F, et al. Integrative ligand-based pharmacophore modeling, virtual screening, and molecular docking simulation approaches identified potential lead compounds against pancreatic cancer by targeting FAK1. Pharmaceuticals. 2023;16(1):120.
- Zhou YJ, Tang S, Chen TT, Niu M-M. Structure-based pharmacophore modeling, virtual screening, molecular docking and biological evaluation for identification of Potential Poly (ADP-Ribose) Polymerase-1 (PARP-1) inhibitors. Molecules. 2019;24(23):4258.
- Hu C, Guo T, Zou Y, Gao J, Gao Y, Niu M, Xia Y, Shen X, Li J. Discovery of dual S-RBD/NRP1-targeting peptides: structure-based virtual screening, synthesis, biological evaluation, and molecular dynamics simulation studies. J Enzyme Inhib Med Chem. 2023;38(1):2212327.
- Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, Chiosis G. Development of a fluorescence polarization assay for the molecular chaperone Hsp90. J Biomol Screen. 2004;9(5):375–381.
- Zheng L, Ren R, Sun X, Zou Y, Shi Y, Di B, Niu M-M. Discovery of a dual tubulin and Poly(ADP-Ribose) polymerase-1 inhibitor by structure-based pharmacophore modeling, virtual screening, molecular docking, and biological evaluation. J Med Chem. 2021;64(21):15702–15715.
- Franke J, Eichner S, Zeilinger C, Kirschning A. Targeting heat-shock-protein 90 (Hsp90) by natural products: geldanamycin, a show case in cancer therapy. Nat Prod Rep. 2013;30(10):1299–1323.