
Abstract
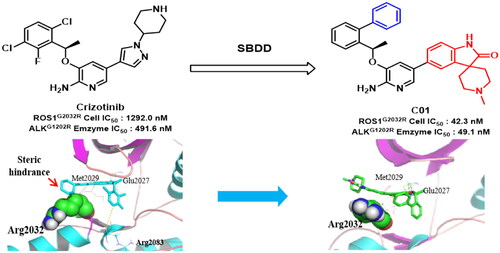
Clinical treatment by FDA-approved ROS1/ALK inhibitor Crizotinib significantly improved the therapeutic outcomes. However, the emergence of drug resistance, especially driven by acquired mutations, have become an inevitable problem and worsened the clinical effects of Crizotinib. To combat drug resistance, some novel 2-aminopyridine derivatives were designed rationally based on molecular simulation, then synthesised and subjected to biological test. The preferred spiro derivative C01 exhibited remarkable activity against CD74-ROS1G2032R cell with an IC50 value of 42.3 nM, which was about 30-fold more potent than Crizotinib. Moreover, C01 also potently inhibited enzymatic activity against clinically Crizotinib-resistant ALKG1202R, harbouring a 10-fold potency superior to Crizotinib. Furthermore, molecular dynamic disclosed that introducing the spiro group could reduce the steric hindrance with bulky side chain (Arginine) in solvent region of ROS1G2032R, which explained the sensitivity of C01 to drug-resistant mutant. These results indicated a path forward for the generation of anti Crizotinib-resistant ROS1/ALK dual inhibitors.
GRAPHICAL ABSTRACT
SAR of a series of 2-aminopyridine ROS1/ALK inhibitors was studied.
The spiro derivatives exhibited remarkable dual ROS1/ALK activity.
C01 potently inhibited Crizotinib-refractory ALKG1202R and ROS1G2032R mutants.
Molecular simulations gave a probable explanation for the activity of C01.
Highlight
Introduction
The abnormal expression of Porto-oncogene tyrosine protein kinase (ROS1) and anaplastic lymphoma kinase (ALK) have become attractive therapeutic targets since Crizotinib was granted by U.S. Food and Drug Administration (FDA) as a first-line treatment for patients with ROS1- or ALK- positive advanced non-small cell lung cancer (NSCLC)Citation1. ROS1, an orphan receptor tyrosine kinase, was initially discovered as a part of FIG-ROS1 fusion protein in glioblastoma (GBM) in 1987 and until 2007 that the first ROS1 rearrangement (SLC3A42-ROS1) in NSCLC was detectedCitation2,Citation3. Subsequent research demonstrated that CD74-ROS1 was the most frequent rearrangement, accounting for 42% patients of NSCLC harbouring ROS1 fusion partnerCitation4,Citation5. Oncogenic ROS1 fusion proteins are identified to be a driving force in approximately 1–2% of NSCLC patients, and most of them are highly sensitive to CrizotinibCitation6–9. However, Crizotinib inevitably develop drug resistance after one to two years of clinical application due to the activating genetic mutations, especially point mutations within the ROS1 kinase domain. Some ROS1 mutants associated with drug resistance, such as G2032R, D2033N, S1986Y/F, L2026M, and L1951R, have been identified in patients who relapsed on Crizotinib and other ROS1 inhibitorsCitation10–13. Among these acquired mutations, the solvent-front G2032R mutant was demonstrated to be the first-reported mutant conferring resistance to Crizotinib and it was also recognised as the most frequent and recalcitrant oneCitation14,Citation15. Molecular basic highlights that G2032R mutant confers resistance to ROS1 inhibitors through steric hindrance of drug bindingCitation14.
Similar to ROS1, ALK is prone to chromosomal rearrangements leading to transforming fusion proteins including nucleophosmin (NPM)-ALK and echinoderm microtubule associated protein like 4 (EML4)-ALK etc., which defined another molecular subtype of NSCLCCitation16,Citation17. The EML4-ALK fusion protein is demonstrated to be responsibility for about 5% of NSCLC patients and majority of them benefit from the clinical effects of CrizotinibCitation18. Compared to ROS1, ALK is more easily mutated within the kinase domain, resulting in lots of Crizotinib-refractory mutants including F1174L, G1269A, G1202R, S1206Y, L1152R, L1196M, and insertion mutant 1151TinsCitation19–22. It’s worth noting that the G1202R ALK mutant, corresponding to G2032R in ROS1 domain, is highly resistant to Crizotinib and other next-generation ALK inhibitors. Numerous ROS1/ALK inhibitors were design to overcome ROS1 and ALK mutants but few of them could potently inhibit both ROS1G2032R and ALKG1202R mutantsCitation23–26. Herein, it was necessary to design novel ROS1/ALK dual inhibitors that particularly targeted ROS1G2032R and ALKG1202R locating at the solvent-front region, to overcome the drug-resistance for cancers treated with Crizotinib.
Experimental
Chemistry
All reagents and solvents were obtained from commercial vendors and were used without further purification. All air- or moisture-sensitive reactions were conducted under nitrogen atmosphere. Yields of all reactions were not optimised. Reactions were monitored by thin-layer chromatography (TLC) on silica gel plates under an ultraviolet (UV) light at 254 nm. Flash column chromatography separations were performed on Yamazen AI-580 flash chromatography (Yamazen Co., Osaka, Japan) at 254 nm with normal phase silica gel (200 – 300 mesh, Merck Co., USA). Mass spectra (ESI-MS) were recorded on Waters ZQ4000 (Waters Co., USA). High resolution mass spectra (HRMS) were performed on a Thermo Fisher Scientific Orbitrap Fusion Tribrid (Q-OT-qIT) mass spectrometer (Thermo Fisher Scientific, Bremen, German). 1H and 13C NMR spectra were acquired on a Bruker AVIII 400 MHz and 101 MHz spectrometer (Bruker Co., Switzerland) with tetramethylsilane (TMS) as the internal reference. The chemical shift (δ) was given in ppm and the coupling constant (J) was given in Hz. Melting points were obtained on X-4 Melting-point Apparatus with Microscope (Yuhua Instrument Co., Ltd., Henan, China) and the results were uncorrected. The purity of the desired compound was measured by ultra-high performance liquid chromatography (UPLC), using an ACQUITY-UPLC BEH C18 column (1.7 μm, 2.1*50mm) and eluting with a mixture of solvents MeOH (A) and 0.1% formic acid aqueous (B) from VA:VB = 15:85 to 50:50. Peaks were detected at 254 nm with a flow rate of 0.25 ml/min.
General procedure A for the preparation of compounds 2a–2c
To a solution of acetyl benzene derivative (20 mmol) in THF (30 ml), (-)-diisopinocampheyl chloroborane (6.4 g, 20 mmol) in THF (20 ml) was added at −40 °C dropwise to keep the temperature below −27 °C, the reaction mixture was stirred at −40 °C for 3 h. After additional reaction in a period of 18 h at room temperature, the resulting mixture was concentrated and 50 ml TBME was added, followed by diethanolamine (4.2 g, 40 mmol) in a quick stir at 60 °C. After 1 h of reaction, the mixture was filtered through a pad of silica gel using additional TBME (30 ml) as an eluent. The combined organic layer was concentrated and purified by flash chromatography eluting with 1–5% ethyl acetate in petroleum ether, to provide the desired product as colourless oil.
(S)-1-phenylethanol (2a)
Compound 1a (2.4 g, 20.0 mmol) was subjected to asymmetric reduction according to the general procedure A to give compound 2a (1.7 g, yield: 72%). 1H NMR (400 MHz, DMSO) δ 7.37–7.27 (m, 4H), 7.22 (t, J = 7.0 Hz, 1H), 5.13 (d, J = 4.0 Hz, 1H), 4.76–4.68 (m, 1H), 1.33 (d, J = 6.8 Hz, 3H), ESI-MS m/z: 123.1 [M + H]+.
(S)-1–(2-fluorophenyl)ethanol (2b)
Compound 1b (2.7 g, 20.0 mmol) was subjected to asymmetric reduction according to the general procedure A to obtain compound 2b (2.1 g, yield: 75%). 1H NMR (400 MHz, DMSO) δ 7.54 (t, J = 7.6 Hz, 1H), 7.28 (dd, J = 14.4, 7.2 Hz, 1H), 7.19 (t, J = 7.4 Hz, 1H), 7.11 (dd, J = 6.8, 8.4 Hz, 1H), 5.28 (d, J = 4.4 Hz, 1H), 5.04–4.95 (m, 1H), 1.34 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 141.5 [M + H]+.
(S)-1–(2-bromophenyl)ethanol (2c)
Compound 1c (10.0 g, 50.0 mmol) was subjected to asymmetric reduction according to the general procedure A to give compound 2c (6.8 g, yield: 68%). 1H NMR (400 MHz, DMSO) δ 7.54 (s, 1H), 7.41 (d, J = 8.0 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 7.28 (t, J = 7.6 Hz, 1H), 5.29 (d, J = 4.4 Hz, 1H), 4.77–4.66 (m, 1H), 1.32 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 201.0/203.0 [M + H]+.
General procedure B for the preparation of compounds 3a–3c
To a solution of phenylethanol derivative in dry THF (20 ml), 2-nitropyridin-3-ol and PPh3 (1.5 equivalents) were added under the N2 atmosphere. After stirring 1 h at 0 °C, diisopropyl azodiformate (1.5 equivalents) was added and the reaction mixture was stirred at room temperature under N2 atmosphere for 6 h. The mixture was concentrated, diluted with water (30 ml) and extracted with ethyl acetate (30 ml). The combined organic layer was dried over MgSO4, concentrated and purified by flash chromatography eluting with 1–10% ethyl acetate in petroleum ether, to get the target compound.
(R)-2-nitro-3–(1-phenylethoxy)pyridine (3a)
Compound 2a (1.7 g, 14.4 mmol) was reacted with 2-nitropyridin-3-ol (2.2 g, 15.8 mmol) according to the general procedure B to obtain compound 3a (2.5 g, yield: 71%). 1H NMR (400 MHz, MeOD) δ 7.96 (d, J = 4.4 Hz, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.48 (dd, J = 8.8, 4.8 Hz, 1H), 7.42 (d, J = 7.2 Hz, 2H), 7.37 (t, J = 7.4 Hz, 2H), 7.30 (t, J = 7.2 Hz, 1H), 5.68 (q, J = 6.4 Hz, 1H), 1.66 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 245.5 [M + H]+.
(R)-3–(1-(2-fluorophenyl)ethoxy)-2-nitropyridine (3b)
Compound 2b (2.1 g, 15.0 mmol) was reacted with 2-nitropyridin-3-ol (2.2 g, 16.5 mmol) according to the general procedure B give compound 3b (2.4 g, yield: 62%). 1H NMR (400 MHz, DMSO) δ 8.09 (d, J = 4.4 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.68 (dd, J = 8.4, 4.4 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.41–7.35 (m, 1H), 7.28–7.21 (m, 2H), 6.04 (q, J = 6.3 Hz, 1H), 1.63 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 263.7 [M + H]+.
(R)-3–(1-(2-bromophenyl)ethoxy)-2-nitropyridine (3c)
Compound 2c (6.8 g, 34.0 mmol) was reacted with 2-nitropyridin-3-ol (5.3 g, 37.5 mmol) according to the general procedure B to obtain compound 3c (7.0 g, yield: 65%). 1H NMR (400 MHz, MeOD) δ 7.96 (d, J = 4.4 Hz, 1H), 7.63 (d, J = 8.8 Hz, 1H), 7.48 (dd, J = 8.8, 4.8 Hz, 1H), 7.42 (d, J = 7.2 Hz, 2H), 7.37 (t, J = 7.4 Hz, 1H), 7.30 (t, J = 7.2 Hz, 1H), 5.68 (q, J = 6.4 Hz, 1H), 1.66 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 323.5/325.5 [M + H]+.
General procedure C for the preparation of compounds 4a–4b and 7a–7h
To a solution of nitro derivative, ethanol (15 ml), acetic acid (3 ml), and H2O (1 ml), Fe (4.0 equivalents) was added in a quick stir. Refluxed the reaction for 2 h and the mixture was concentrated, diluted with water (30 ml) and extracted with ethyl acetate (30 ml). The combined organic layer was dried over MgSO4, concentrated and purified by flash chromatography eluting with 10–40% ethyl acetate in petroleum ether, to get the target compound.
(R)-3–(1-phenylethoxy)pyridin-2-amine (4a)
Compound 3a (2.5 g, 10.2 mmol) was reduced according to the general procedure C to obtain compound 4a (1.7 g, 80% yield). 1H NMR (400 MHz, MeOD) δ 7.49 (d, J = 1.6 Hz, 1H), 7.39 (dt, J = 18.8, 7.2 Hz, 5H), 7.30 (t, J = 7.0 Hz, 1H), 6.97 (d, J = 1.6 Hz, 1H), 5.45 (q, J = 6.4 Hz, 1H), 1.68 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 215.3 [M + H]+.
(R)-3–(1-(2-fluorophenyl)ethoxy)pyridin-2-amine (4b)
Compound 3b (2.4 g, 9.3 mmol) was reduced according to the general procedure C to give compound 4b (1.8 g, 82% yield). 1H NMR (400 MHz, DMSO) δ 7.54 (t, J = 7.6 Hz, 1H), 7.46 (d, J = 4.8 Hz, 1H), 7.37–7.30 (m, 1H), 7.20 (dd, J = 12.0, 4.8 Hz, 2H), 6.85 (d, J = 8.0 Hz, 1H), 6.38 (dd, J = 8.0, 5.2 Hz, 1H), 5.75 (s, 2H), 5.69 (q, J = 6.4 Hz, 1H), 1.61 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 233.1 [M + H]+.
General procedure D for the preparation of compounds 5a–5b and 8a–8h
The amino derivative was dissolved in acetonitrile (15 ml), N-Bromosuccinimide (1.05 equivalents) was added slowly to mixture at 0 °C. After reacted 30 min in an ice bath, the resulting mixture was concentrated and purified by flash chromatography eluting with 5–15% ethyl acetate in petroleum ether to provide the target compound.
(R)-5-bromo-3–(1-phenylethoxy)pyridin-2-amine (5a)
Compound 4a (1.7 g, 8.1 mmol) was reacted with N-Bromosuccinimide (1.5 g, 8.6 mmol) according to the general procedure D to obtain compound 5a (1.3 g, yield: 56%). 1H NMR (400 MHz, MeOD) δ 7.49 (d, J = 1.6 Hz, 1H), 7.39 (dt, J = 18.8, 7.2 Hz, 4H), 7.30 (t, J = 7.0 Hz, 1H), 6.97 (d, J = 1.6 Hz, 1H), 5.45 (q, J = 6.4 Hz, 1H), 1.68 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 293.5/295.5[M + H]+.
(R)-5-bromo-3–(1-(2-fluorophenyl)ethoxy)pyridin-2-amine (5b)
Compound 4b (1.8 g, 7.6 mmol) was reacted with N-Bromosuccinimide (1.4 g, 8.0 mmol) according to the general procedure D to obtain compound 5b (1.4 g, yield: 60%). 1H NMR (400 MHz, MeOD) δ 7.52 (d, J = 2.0 Hz, 1H), 7.46 (t, J = 7.0 Hz, 1H), 7.35 (dd, J = 14.8, 7.2 Hz, 1H), 7.23–7.11 (m, 2H), 6.95 (d, J = 1.6 Hz, 1H), 5.73 (q, J = 6.4 Hz, 1H), 1.71 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 311.5/313.5 [M + H]+.
General procedure E for the preparation of compounds 6a–6h and A01–C07
To a solution of bromide derivative and corresponding boric acid ester (1.2 equivalents) in DMF (10 ml) and H2O (1.2 ml), K2CO3 (2.5 equivalents) was added, followed by Pd(PPh3)4 (0.05 equivalents) under N2 atmosphere. The reaction was stirred at 85 °C under N2 atmosphere for 8 h. The resulting mixture was cooled to room temperature, diluted with water (30 ml), and extracted with ethyl acetate (50 ml). The combined organic layer was washed with saturated solution of NaCl (50 ml), dried over MgSO4, concentrated and purified by flash chromatography eluting with 1–10% methanol in dichloromethane to give the desired compound.
(R)-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)-2-nitropyridine (6a)
Compound 3c (2.0 g, 6.2 mmol) was reacted with 1-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (1.5 g, 7.4 mmol) according to the general procedure E to obtain compound 6a (1.4 g, yield: 70%). 1H NMR (400 MHz, MeOD) δ 7.12–7.07 (m, 1H), 7.01 (s, 1H), 6.83 (s, 2H), 6.74 (d, J = 4.8 Hz, 3H), 6.60–6.50 (m, 4H), 6.26 (d, J = 8.8 Hz, 1H), 4.96 (q, J = 6.3 Hz, 1H), 0.93 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 325.1 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)-2-nitropyridine (6b)
Compound 3c (2.0 g, 6.2 mmol) was reacted with 4,4,5,5-tetramethyl-2-phenyl-1,3,2-dioxaborolane (1.5 g, 7.4 mmol) according to the general procedure E to obtain compound 6b (1.5 g, yield: 78%). 1H NMR (400 MHz, DMSO) δ 8.02 (dd, J = 4.4, 0.8 Hz, 1H), 7.58–7.54 (m, 1H), 7.52 (dt, J = 6.8, 2.4 Hz, 2H), 7.49 (s, 1H), 7.48–7.41 (m, 2H), 7.40–7.36 (m, 3H), 7.25 (dd, J = 7.6, 1.6 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 5.53 (q, J = 6.3 Hz, 1H), 1.61 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 321.5 [M + H]+.
(R)-3–(1-(4'-fluoro-[1,1'-biphenyl]-2-yl)ethoxy)-2-nitropyridine (6c)
Compound 3c (0.50 g, 1.5 mmol) was reacted with 4,4,5,5-tetramethyl-2-phenyl-1,3,2-dioxaborolane (0.41 g, 1.8 mmol) according to the general procedure E to obtain compound 6c (0.40 g, yield: 75%). 1H NMR (400 MHz, DMSO) δ 8.03 (d, J = 4.4 Hz, 1H), 7.57 (dd, J = 8.4, 4.4 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.48–7.37 (m, 4H), 7.35–7.29 (m, 3H), 7.23 (dd, J = 10.4, 7.6 Hz, 2H), 5.54 (q, J = 6.3 Hz, 1H), 1.60 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 339.5 [M + H]+.
(R)-3–(1-(4'-methoxy-[1,1'-biphenyl]-2-yl)ethoxy)-2-nitropyridine (6d)
Compound 3c (0.50 g, 1.5 mmol) was reacted with 2–(4-methoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (0.43 g, 1.8 mmol) according to the general procedure E to obtain compound 6d (0.45 g, yield: 82%). 1H NMR (400 MHz, DMSO) δ 8.01 (dd, J = 4.4, 1.2 Hz, 1H), 7.55 (dd, J = 8.8, 4.8 Hz, 1H), 7.50 (dd, J = 7.6, 1.2 Hz, 1H), 7.43–7.34 (m,2H), 7.32–7.29 (m, 2H), 7.22 (dd, J = 7.6, 1.2 Hz, 1H), 7.14 (dd, J = 8.8, 1.2 Hz, 1H), 7.08–7.04 (m, 2H), 5.55 (q, J = 6.3 Hz, 1H), 4.03 (q, J = 7.1 Hz, 1H) 3.83 (s, 3H), 1.62 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 351.4 [M + H]+.
(R)-3–(1-(4'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)-2-nitropyridine (6e)
Compound 3c (0.50 g, 1.5 mmol) was reacted with 4,4,5,5-tetramethyl-2-(p-tolyl)-1,3,2-dioxaborolane (0.40 g, 1.8 mmol) according to the general procedure E to obtain compound 6e (0.44 g, yield: 85%). 1H NMR (400 MHz, DMSO) δ 8.01 (dd, J = 4.4, 1.2 Hz, 1H), 7.56 (dd, J = 8.4, 4.4 Hz, 1H), 7.51 (dd, J = 7.6, 1.6 Hz, 1H), 7.45–7.35 (m, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.28–7.25 (m, 2H), 7.22 (dd, J = 7.6, 1.6 Hz, 1H), 7.14 (dd, J = 8.8, 1.2 Hz, 1H), 5.53 (q, J = 6.3 Hz, 1H), 2.39 (s, 3H), 1.61 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 335.3 [M + H]+.
(R)-3–(1-(3'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)-2-nitropyridine (6f)
Compound 3c (0.50 g, 1.5 mmol) was reacted with 4,4,5,5-tetramethyl-2-(m-tolyl)-1,3,2-dioxaborolane (0.40 g, 1.8 mmol) according to the general procedure E to obtain compound 6f (0.43 g, yield: 83%). 1H NMR (400 MHz, DMSO) δ 8.02 (dd, J = 4.4, 0.8 Hz, 1H), 7.57 (dd, J = 7.6, 1.2 Hz, 1H), 7.54 (dd, J = 7.7, 1.3 Hz, 1H), 7.47–7.35 (m, 3H), 7.26–7.18 (m, 3H), 7.15 (d, J = 8.4 Hz, 2H), 5.54 (q, J = 6.3 Hz, 1H), 2.35 (s, 3H), 1.61 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 335.3 [M + H]+.
(R)-3–(1-(4'-ethoxy-[1,1'-biphenyl]-2-yl)ethoxy)-2-nitropyridine (6g)
Compound 3c (0.50 g, 1.5 mmol) was reacted with 2–(4-ethoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (0.46 g, 1.8 mmol) according to the general procedure E to obtain compound 6g (0.41 g, yield: 73%). 1H NMR (400 MHz, DMSO) δ 8.01 (d, J = 4.4 Hz, 1H), 7.55 (dd, J = 8.8, 4.8 Hz, 1H), 7.52–7.48 (m, 1H), 7.44–7.34 (m, 2H), 7.29 (d, J = 8.8 Hz, 2H), 7.22 (dd, J = 7.6, 1.2 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 7.04 (d, J = 8.4 Hz, 2H), 5.55 (q, J = 6.3 Hz, 1H), 4.10 (q, J = 6.9 Hz, 2H),1.62 (d, J = 6.0 Hz, 3H),1.37(t, J = 7.0 Hz, 3H), ESI-MS m/z: 365.5 [M + H]+.
(R)-2-nitro-3–(1-(4'-(trifluoromethoxy)-[1,1'-biphenyl]-2-yl)ethoxy)pyridine (6h)
Compound 3c (0.50 g, 1.5 mmol) was reacted with 4,4,5,5-tetramethyl-2–(4-(trifluoromethoxy)phenyl)-1,3,2-dioxaborolane (0.55 g, 1.8 mmol) according to the general procedure E to obtain compound 6h (0.48 g, yield: 70%). 1H NMR (400 MHz, DMSO) δ 8.03 (dd, J = 4.8, 1.2 Hz, 1H), 7.71–7.67 (m, 1H), 7.59–7.54 (m, 2H), 7.49–7.42 (m, 5H), 7.28–7.25 (m, 1H), 5.56 (q, J = 6.3 Hz, 1H), 1.61 (dd, J = 6.4, 2.0 Hz, 4H), ESI-MS m/z: 405.5 [M + H]+.
(R)-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)pyridin-2-amine (7a)
Compound 6a (1.4 g, 4.3 mmol) was reduced according to the general procedure C to obtain compound 7a (1.1 g, 85% yield). 1H NMR (400 MHz, DMSO) δ 7.75 (d, J = 8.0 Hz, 1H), 7.55 (s, 1H), 7.50 (t, J = 7.6 Hz, 1H), 7.43–7.36 (m, 2H), 7.30 (d, J = 7.6 Hz, 1H), 6.37–6.30 (m, 3H), 5.70 (s, 2H), 5.21 (q, J = 6.3 Hz, 1H), 3.57 (s, 3H), 1.54 (d, J = 6.0 Hz, 3H), ESI-MS m/z: 295.1 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (7b)
Compound 6b (1.5 g, 4.8 mmol) was reduced according to the general procedure C to obtain compound 7b (1.2 g, 88% yield). 1H NMR (400 MHz, DMSO) δ 8.02 (dd, J = 4.4, 0.8 Hz, 1H), 7.58–7.54 (m, 3H), 7.52 (dt, J = 6.8, 2.4 Hz, 2H), 7.49 (s, 1H), 7.48–7.41 (m, 2H), 7.40–7.36 (m, 3H), 7.25 (dd, J = 7.6, 1.6 Hz, 1H), 7.16 (d, J = 7.6 Hz, 1H), 5.53 (q, J = 6.3 Hz, 1H), 1.61 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 291.5 [M + H]+.
(R)-3–(1-(4'-fluoro-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (7c)
Compound 6c (0.40 g, 1.2 mmol) was reduced according to the general procedure C to obtain compound 7c (0.29 g, 80% yield). 1H NMR (400 MHz, DMSO) δ 7.67 (dd, J = 8.0, 1.2 Hz, 1H), 7.44–7.29 (m, 7H), 7.19 (dd, J = 7.6, 1.2 Hz, 1H), 6.32–6.24 (m, 2H), 5.67 (s, 2H), 5.20 (q, J = 6.3 Hz, 1H), 1.59 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 309.5 [M + H]+.
(R)-3–(1-(4'-methoxy-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (7d)
Compound 6d (0.45 g, 1.3 mmol) was reduced according to the general procedure C to obtain compound 7d (0.32 g, 81% yield). 1H NMR (400 MHz, DMSO) δ 7.64 (dd, J = 7.6, 1.2 Hz, 1H), 7.40–7.36 (m, 2H), 7.35–7.31 (m, 1H), 7.29–7.26 (m, 2H), 7.17 (dd, J = 7.6, 1.2 Hz, 1H), 7.09–7.04 (m, 2H), 6.26–6.24 (m, 2H), 5.66 (s, 2H), 5.24 (q, J = 6.3 Hz, 1H), 3.83 (s, 3H), 1.61 (d, J = 6.0 Hz, 3H), ESI-MS m/z: 321.4 [M + H]+.
(R)-3–(1-(4'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (7e)
Compound 6e (0.44 g, 1.3 mmol) was reduced according to the general procedure C to obtain compound 7e (0.37 g, 91% yield). 1H NMR (400 MHz, DMSO) δ 7.65 (dd, J = 8.0, 1.6 Hz, 1H), 7.40–7.35 (m, 2H), 7.35–7.31 (m, 3H), 7.23 (d, J = 8.0 Hz, 2H), 7.17 (dd, J = 7.6, 1.2 Hz, 1H), 6.26–6.24 (m, 2H), 5.66 (s, 2H), 5.22 (q, J = 6.3 Hz, 1H), 2.40 (s, 3H), 1.60 (d, J = 6.3 Hz, 3H), ESI-MS m/z: 305.7 [M + H]+.
(R)-3–(1-(3'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (7f)
Compound 6f (0.43 g, 1.3 mmol) was reduced according to the general procedure C to obtain compound 7f (0.33 g, 85% yield). 1H NMR (400 MHz, DMSO) δ 7.67 (dd, J = 7.6, 1.2 Hz, 1H), 7.40 (dd, J = 7.2, 1.6 Hz, 3H), 7.33 (m, J = 7.6, 1.6 Hz, 1H), 7.26 (d, J = 7.6 Hz, 1H), 7.18 (dd, J = 7.6, 1.6 Hz, 1H), 7.12 (d, J = 8.8 Hz, 2H), 6.32–6.24 (m, 2H), 5.65 (s, 2H), 5.21 (q, J = 6.4 Hz, 1H), 2.38 (s, 3H), 1.60 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 305.3 [M + H]+.
(R)-3–(1-(4'-ethoxy-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (7g)
Compound 6g (0.41 g, 1.2 mmol) was reduced according to the general procedure C to obtain compound 7g (0.26 g, 68% yield). 1H NMR (400 MHz, DMSO) δ 7.64 (d, J = 7.6 Hz, 1H), 7.40–7.34 (m, 2H), 7.33–7.28 (m, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.17 (dd, J = 7.6, 1.2 Hz, 1H), 7.05 (d, J = 8.8 Hz, 2H), 6.26–6.24 (m, 2H), 5.67 (s, 2H), 5.24 (q, J = 6.3 Hz, 1H), 4.10 (q, J = 6.9 Hz, 2H), 1.61 (d, J = 6.4 Hz, 3H), 1.37 (t, J = 6.8 Hz, 3H), ESI-MS m/z: 335.5 [M + H]+.
(R)-3–(1-(4'-(trifluoromethoxy)-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (7h)
Compound 6h (0.48 g, 1.1 mmol) was reduced according to the general procedure C to obtain compound 7h (0.27 g, 65% yield). 1H NMR (400 MHz, DMSO) δ 7.69 (dd, J = 8.0, 1.2 Hz, 1H), 7.48 (s, 3H), 7.44–7.34 (m, 3H), 7.22 (dd, J = 7.6, 1.6 Hz, 1H), 6.28–6.26 (m, 1H), 5.80 (s, 1H), 5.66 (s, 2H), 5.19 (q, J = 6.3 Hz, 1H), 1.59 (d, J = 6.3 Hz, 3H), ESI-MS m/z: 375.5 [M + H]+.
(R)-5-bromo-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)pyridin-2-amine (8a)
Compound 7a (1.1 g, 3.7 mmol) was reacted with N-Bromosuccinimide (0.69 g, 3.9 mmol) according to the general procedure D to obtain compound 8a (0.83 g, yield: 60%). 1H NMR (400 MHz, DMSO) δ 7.74 (d, J = 7.6 Hz, 1H), 7.59 (s, 1H), 7.53–7.47 (m, 2H), 7.42 (t, J = 7.4 Hz, 1H), 7.33 (d, J = 7.6 Hz, 1H), 6.36 (s, 2H), 6.03 (s, 2H), 5.22 (q, J = 6.1 Hz, 1H), 2.57 (s, 3H), 1.60 (d, J = 6.0 Hz, 3H), ESI-MS m/z: 373.0/375.0 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)-5-bromopyridin-2-amine (8b)
Compound 7b (1.2 g, 4.2 mmol) was reacted with N-Bromosuccinimide (0.78 g, 4.4 mmol) according to the general procedure D to obtain compound 8b (0.96 g, yield: 62%). 1H NMR (400 MHz, DMSO) δ 7.69 (dd, J = 7.6, 1.2 Hz, 1H), 7.56–7.53 (m, 2H), 7.52–7.49 (m, 1H), 7.42 (dd, J = 5.6, 2.0 Hz, 1H), 7.39 (dd, J = 6.2, 1.6 Hz, 1H), 7.35 (dd, J = 6.8, 1.6 Hz, 2H), 7.23 (dd, J = 7.2, 1.2 Hz, 1H), 6.20 (d, J = 2.0 Hz, 1H), 6.00 (s, 2H), 5.16 (q, J = 6.3 Hz, 1H), 1.71 (d, J = 6.0 Hz, 3H), ESI-MS m/z: 369.0/371.0 [M + H]+.
(R)-5-bromo-3–(1-(4'-fluoro-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (8c)
Compound 7c (0.29 g, 0.90 mmol) was reacted with N-Bromosuccinimide (0.18 g, 1.0 mmol) according to the general procedure D to obtain compound 8c (0.19 g, yield: 54%). 1H NMR (400 MHz, DMSO) δ 7.70 (dd, J = 8.0, 16 Hz, 1H), 7.44 (d, J = 2.0 Hz, 1H), 7.41 (dd, J = 7.6, 1.2 Hz, 1H), 7.37 (d, J = 7.2 Hz, 5H), 7.23 (dd, J = 7.6, 1.2 Hz, 1H), 6.21 (d, J = 2.0 Hz, 1H), 6.00 (s, 2H), 5.18 (q, J = 6.3 Hz, 1H), 1.70 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 387.0/389.0 [M + H]+.
(R)-5-bromo-3–(1-(4'-methoxy-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (8d)
Compound 7d (0.32 g, 1.0 mmol) was reacted with N-Bromosuccinimide (0.20 g, 1.1 mmol) according to the general procedure D to obtain compound 8d (0.24 g, yield: 60%). 1H NMR (400 MHz, DMSO) δ 7.65 (dd, J = 7.6, 1.2 Hz, 1H), 7.43 (d, J = 2.0 Hz, 1H), 7.36 (m, 2H), 7.29–7.26 (m, 2H), 7.21 (dd, J = 7.2, 1.2 Hz, 1H), 7.12–7.08 (m, 2H), 6.20 (d, J = 2.0 Hz, 1H), 6.01 (s, 2H), 5.20 (q, J = 6.3 Hz, 1H), 3.84 (s, 3H), 1.71 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 399.4/401.4 [M + H]+.
(R)-5-bromo-3–(1-(4'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (8e)
Compound 7e (0.37 g, 1.2 mmol) was reacted with N-Bromosuccinimide (0.23 g, 1.3 mmol) according to the general procedure D to obtain compound 8e (0.28 g, yield: 61%). 1H NMR (400 MHz, DMSO) δ 7.65 (dd, J = 8.0, 1.6 Hz, 1H), 7.40–7.35 (m, 1H), 7.35–7.31 (m, 3H), 7.23 (d, J = 8.0 Hz, 2H), 7.17 (dd, J = 7.6, 1.2 Hz, 1H), 6.26–6.24 (m, 2H), 5.66 (s, 2H), 5.22 (q, J = 6.3 Hz, 1H), 2.40 (s, 3H), 1.60 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 383.4/385.4 [M + H]+.
(R)-5-bromo-3–(1-(3'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (8f)
Compound 7f (0.33 g, 1.1 mmol) was reacted with N-Bromosuccinimide (0.21 g, 1.2 mmol) according to the general procedure D to obtain compound 8f (0.29 g, yield: 70%). 1H NMR (400 MHz, DMSO) δ 7.68 (d, J = 7.6 Hz, 1H), 7.45–7.38 (m, 3H), 7.35 (m, J = 7.6, 1.6 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 7.22 (dd, J = 7.6, 1.2 Hz, 1H), 7.13 (d, J = 8.0 Hz, 2H), 6.23 (d, J = 1.6 Hz, 1H), 6.00 (s, 2H), 5.15 (q, J = 6.3 Hz, 1H), 2.42 (s, 3H), 1.71 (d, J = 6.0 Hz, 3H), ESI-MS m/z: 383.4/385.4 [M + H]+.
(R)-5-bromo-3–(1-(4'-ethoxy-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (8g)
Compound 7g (0.26 g, 0.8 mmol) was reacted with N-Bromosuccinimide (0.16 g, 0.9 mmol) according to the general procedure D to obtain compound 8g (0.18 g, yield: 55%). 1H NMR (400 MHz, DMSO) δ 7.65 (d, J = 7.2 Hz, 1H), 7.43 (d, J = 1.6 Hz, 1H), 7.40–7.31 (m, 3H), 7.26 (d, J = 8.8 Hz, 2H), 7.21 (d, J = 7.6 Hz, 1H), 7.08 (d, J = 8.4 Hz, 2H), 6.20 (d, J = 2.0 Hz, 1H), 6.06 (s, 1H), 5.20 (q, J = 6.3 Hz, 1H), 4.12 (q, J = 7.1 Hz, 2H), 1.71 (d, J = 6.4 Hz, 3H), 1.38 (t, J = 6.8 Hz, 3H), ESI-MS m/z: 413.5/415.5 [M + H]+.
(R)-5-bromo-3–(1-(4'-(trifluoromethoxy)-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-2-amine (8h)
Compound 7h (0.27 g, 0.7 mmol) was reacted with N-Bromosuccinimide (0.15 g, 0.8 mmol) according to the general procedure D to obtain compound 8h (0.17 g, yield: 53%). 1H NMR (400 MHz, DMSO) δ 7.72 (dd, J = 8.0, 1.2 Hz, 1H), 7.55–7.50 (m, 2H), 7.48–7.43 (m, 3H), 7.42–7.36 (m, 1H), 7.29–7.22 (m, 1H), 6.22 (d, J = 2.0 Hz, 1H), 6.12 (s, 1H), 5.98 (s, 2H), 5.19 (q, J = 6.3 Hz, 1H), 1.70 (d, J = 6.4 Hz, 3H), ESI-MS m/z: 453.4/455.4 [M + H]+.
(R)-5–(1-methyl-1H-pyrazol-5-yl)-3–(1-phenylethoxy)pyridin-2-amine (A01)
Compound 5a (0.32 g, 1.1 mmol) was reacted with 1-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.27 g, 1.3 mmol) according to the general procedure E to obtain A01 (0.24 g, yield: 75%). 1H NMR (400 MHz, MeOD) δ 7.52 (d, J = 1.2 Hz, 1H), 7.43–7.38 (m, 3H), 7.36–7.32 (m, 2H), 7.28–7.23 (m, 1H), 6.86 (d, J = 2.0 Hz, 1H), 6.15 (d, J = 2.0 Hz, 1H), 5.48 (q, J = 6.4 Hz, 1H), 3.45 (s, 3H), 1.69 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 151.29, 142.14, 141.22, 137.85, 137.13, 128.45, 127.52, 125.34, 118.51, 105.06, 76.38, 35.65, 23.31. ESI-MS m/z: 295.1 [M + H]+.
(R)-5–(1-methyl-1H-pyrazol-4-yl)-3–(1-phenylethoxy)pyridin-2-amine (A02)
Compound 5a (0.32 g, 1.1 mmol) was reacted with 1-methyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.27 g, 1.3 mmol) according to the general procedure E to obtain A02 (0.23 g, yield: 72%). 1H NMR (400 MHz, MeOD) δ 7.68 (s, 1H), 7.64 (s, 1H), 7.54 (s, 1H), 7.45 (d, J = 7.6 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.26 (t, J = 7.4 Hz, 1H), 7.14 (s, 1H), 7.06 (d, J = 12.4 Hz, 2H), 5.53 (q, J = 6.3 Hz, 1H), 3.86 (s, 3H), 1.69 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 142.29, 135.31, 128.31, 127.45, 127.08, 126.81, 125.36, 120.16, 116.82, 116.63, 76.22, 37.62, 37.42, 23.06. ESI-MS m/z: 295.1 [M + H]+.
(R)-5–(1-ethyl-1H-pyrazol-4-yl)-3–(1-phenylethoxy)pyridin-2-amine (A03)
Compound 5a (0.32 g, 1.1 mmol) was reacted with 1-ethyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.29 g, 1.3 mmol) according to the general procedure E to obtain A03 (0.23 g, yield: 69%). 1H NMR (400 MHz, MeOD) δ 7.76 (s, 1H), 7.65 (s, 1H), 7.56 (s,1H), 7.46 (d, J = 7.6 Hz, 2H), 7.37 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.2 Hz, 1H), 7.07 (s, 1H), 5.54 (q, J = 6.4 Hz, 1H), 4.16 (q, J = 7.3 Hz, 2H), 1.70 (d, J = 6.4 Hz, 3H), 1.45 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 149.63, 142.31, 135.23, 133.45, 128.30, 127.45, 125.37, 125.31, 119.91, 116.67, 76.22, 46.42, 23.03, 14.49. ESI-MS m/z: 309.5 [M + H]+.
(R)-3–(1-phenylethoxy)-5–(1-propyl-1H-pyrazol-4-yl)pyridin-2-amine (A04)
Compound 5a (0.32 g, 1.1 mmol) was reacted with 1-propyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.31 g, 1.3 mmol) according to the general procedure E to obtain A04 (0.21 g, yield: 60%). 1H NMR (400 MHz, MeOD) δ 7.75 (s, 1H), 7.56 (s, 1H), 7.46 (d, J = 7.6 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.4 Hz, 1H), 7.18 (s, 1H), 7.08 (d, J = 8.0 Hz, 1H), 5.54 (q, J = 6.4 Hz, 1H), 4.08 (t, J = 7.0 Hz, 1H), 3.96 (t, J = 7.0 Hz, 1H), 1.90–1.78 (m, 2H), 1.70 (d, J = 6.4 Hz, 3H), 0.89 (dd, J = 15.8, 7.6 Hz, 5H). 13C NMR (101 MHz, MeOD) δ 142.32, 135.22, 128.30, 127.45, 126.92, 126.01, 125.37, 116.64, 115.96, 76.23, 53.34, 53.15, 23.32, 23.28, 23.03, 9.83. ESI-MS m/z: 323.2 [M + H]+.
(R)-3–(1-(2-fluorophenyl)ethoxy)-5–(1-methyl-1H-pyrrol-2-yl)pyridin-2-amine (A05)
Compound 5b (0.35 g, 1.1 mmol) was reacted with 1-methyl-2–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole (0.27 g, 1.3 mmol) according to the general procedure E to obtain A05 (0.22 g, yield: 65%). 1H NMR (400 MHz, MeOD) δ 7.77 (s, 1H), 7.71 (s, 1H), 7.56 (s, 1H), 7.50 (dd, J = 15.4, 7.8 Hz, 1H), 7.36 (dd, J = 14.2, 6.9 Hz, 1H), 7.20–7.10 (m, 4H), 7.03 (d, J = 6.8 Hz, 2H), 5.83 (q, J = 6.4 Hz, 1H), 3.88 (s, 3H), 1.74 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 151.01, 140.21, 137.25, 129.30, 129.22, 126.63, 126.59, 124.44, 124.40, 118.05, 115.03, 114.82, 112.46, 69.68, 69.65, 21.59. ESI-MS m/z: 312.5 [M + H]+.
(R)-3–(1-(2-fluorophenyl)ethoxy)-5–(1-methyl-1H-pyrazol-4-yl)pyridin-2-amine(A06)
Compound 5b (0.35 g, 1.1 mmol) was reacted with 1-methyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.27 g, 1.3 mmol) according to the general procedure E to obtain A06 (0.26 g, yield: 75%). UPLC purity: 95.65%. 1H NMR (400 MHz, MeOD) δ 7.71 (s, 1H), 7.67 (s, 1H), 7.56 (s, 1H), 7.50 (dd, J = 15.6, 8.0 Hz, 1H), 7.32 (dd, J = 14.0, 6.8 Hz, 1H), 7.20–7.10 (m, 1H), 7.06 (d, J = 6.8 Hz, 2H), 5.83 (q, J = 6.3 Hz, 1H), 3.88 (s, 1H), 1.73 (d, J = 6.4 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 135.26, 133.99, 129.47, 129.39, 127.07, 126.88, 126.84, 126.80, 124.52, 124.49, 120.11, 118.38, 116.81, 116.05, 115.04, 114.82, 69.69, 69.66, 37.60, 37.43, 21.42. ESI-MS m/z: 313.3 [M + H]+.
(R)-5–(1-ethyl-1H-pyrazol-4-yl)-3–(1-(2-fluorophenyl)ethoxy)pyridin-2-amine (A07)
Compound 5b (0.35 g, 1.1 mmol) was reacted with 1-ethyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.29 g, 1.3 mmol) according to the general procedure E to obtain A07 (0.22 g, yield: 62%). 1H NMR (400 MHz, MeOD) δ 7.78 (s, 1H), 7.68 (s, 1H), 7.58 (s, 1H), 7.52 (t, J = 7.4 Hz, 1H), 7.32 (dd, J = 13.6, 6.4 Hz, 1H), 7.21–7.11 (m,2H), 7.08 (s, 1H), 5.84 (q, J = 6.4 Hz, 1H), 4.18 (q, J = 7.3 Hz, 2H), 1.74 (d, J = 6.4 Hz, 3H), 1.46 (t, J = 7.4 Hz, 3H), 1.40 (t, J = 7.2 Hz, 2H). 13C NMR (101 MHz, MeOD) δ 149.63, 140.24, 135.18, 133.88, 129.47, 129.39, 126.94, 126.89, 126.85, 125.30, 124.52, 124.49, 119.84, 118.49, 116.10, 115.23, 115.04, 114.82, 69.72, 69.69, 46.55, 46.44, 29.30, 29.00, 21.40, 14.49, 14.44. ESI-MS m/z: 327.3 [M + H]+.
(R)-3–(1-(2-fluorophenyl)ethoxy)-5–(1-propyl-1H-pyrazol-4-yl)pyridin-2-amine(A08)
Compound 5b (0.35 g, 1.1 mmol) was reacted with 1-propyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.31 g, 1.3 mmol) according to the general procedure E to obtain A08 (0.20 g, yield: 55%). 1H NMR (400 MHz, MeOD) δ 7.76 (s, 1H), 7.68 (s, 1H), 7.58 (s, 1H), 7.51 (t, J = 7.2 Hz, 1H), 7.31 (dd, J = 14.8, 7.6 Hz, 1H), 7.20–7.04 (m, 4H), 5.83 (q, J = 6.4 Hz, 1H), 4.09 (t, J = 7.0 Hz, 2H), 1.87 (q, J = 7.2, 2H), 1.73 (d, J = 6.4 Hz, 3H), 0.94–0.87 (m, 4H). 13C NMR (101 MHz, MeOD) δ 149.65, 140.23, 135.16, 134.01, 129.47, 129.39, 126.92, 126.90, 126.86, 125.99, 124.53, 124.49, 119.76, 118.47, 116.05, 115.96, 115.03, 114.81, 69.70, 69.68, 53.34, 53.17, 23.32, 23.29, 21.58, 21.42, 9.84. ESI-MS m/z: 341.7 [M + H]+.
(R)-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)-5–(1-methyl-1H-pyrrol-2-yl)pyridin-2-amine (B01)
Compound 8a (0.16 g, 0.43 mmol) was reacted with 1-methyl-2–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole (0.11 g, 0.52 mmol) according to the general procedure E to obtain B01 (0.12 g, yield: 72%). 1H NMR (400 MHz, DMSO) δ 7.79 (d, J = 7.6 Hz, 1H), 7.55–7.53 (m, 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.40 (m, J = 7.6, 1.2 Hz, 1H), 7.28 (dd, J = 7.6, 0.8 Hz, 1H), 6.75 (t, J = 2.2,1H), 6.29 (d, J = 1.6 Hz, 2H), 5.99 (t, J = 3.2,1H), 5.84–5.81 (m, 3H), 5.28 (q, J = 6.3 Hz, 1H), 3.29 (d, J = 6.8 Hz, 6H), 1.61 (d, J = 6.0 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 150.78, 141.84, 139.74, 138.58, 138.29, 131.37, 130.57, 130.15, 128.98, 128.17, 126.66, 123.58, 118.67, 107.79, 107.61, 107.49, 72.42, 36.33, 34.53, 23.61. ESI-MS m/z: 374.2 [M + H]+.
(R)-5–(1-methyl-1H-pyrazol-5-yl)-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)pyridin-2-amine (B02)
Compound 8a (0.16 g, 0.43 mmol) was reacted with 1-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.11 g, 0.52 mmol) according to the general procedure E to obtain B02 (0.13 g, yield: 78%). 1H NMR (400 MHz, DMSO) δ 7.81 (d, J = 8.0 Hz, 1H), 7.62–7.47 (m, 4H), 7.43–7.36 (m, 2H), 7.28 (d, J = 7.6 Hz, 1H), 6.37 (s, 1H), 6.28 (d, J = 1.2 Hz, 1H), 6.11–6.07 (m, 3H), 5.32 (q, J = 6.3 Hz, 1H), 3.55 (s, 3H), 3.36 (s, 2H), 1.62 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 151.61, 140.90, 140.09, 138.37, 137.15, 135.53, 131.55, 130.06, 129.02, 128.19, 126.89, 119.92, 116.61, 107.59, 72.74, 38.97, 36.94, 25.36, 24.33. ESI-MS m/z: 375.3 [M + H]+.
(R)-5–(1-methyl-1H-pyrazol-4-yl)-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)pyridin-2-amine (B03)
Compound 8a (0.16 g, 0.43 mmol) was reacted with 1-methyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.11 g, 0.52 mmol) according to the general procedure E to obtain B03 (0.11 g, yield: 68%). 1H NMR (400 MHz, DMSO) δ 7.85 (d, J = 7.6 Hz, 1H), 7.71 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 1.6 Hz, 1H), 7.47–7.53 (m,2H), 7.40 (t, J = 7.4 Hz, 1H), 7.29 (d, J = 7.6 Hz, 1H), 6.53 (s, 1H), 6.33 (d, J = 1.6 Hz, 1H), 5.69 (s, 2H), 5.37 (q, J = 6.3 Hz, 1H), 3.84 (s, 3H), 3.44 (s, 3H), 1.58 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 150.61, 141.90, 140.09, 138.37, 136.15, 135.53, 130.55, 130.06, 129.02, 128.19, 126.89, 119.92, 116.61, 107.59, 72.74, 38.97, 36.94, 25.36, 23.33. ESI-MS m/z: 375.3 [M + H]+.
(R)-5–(1-ethyl-1H-pyrazol-4-yl)-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)pyridin-2-amine (B04)
Compound 8a (0.16 g, 0.43 mmol) was reacted with 1-ethyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.12 g, 0.52 mmol) according to the general procedure E to obtain B04 (0.12 g, yield: 74%). 1H NMR (400 MHz, CDCl3) δ 7.73 (s, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.63 (s, 1H), 7.50 (m, J = 7.4 Hz, 2H), 7.45–7.34 (m, 2H), 7.24 (d, J = 7.6 Hz, 1H), 6.56 (s, 1H), 6.27 (s, 1H), 5.42 (q, J = 12.1, 5.9 Hz, 1H), 4.85 (s, 2H), 4.22 (q, J = 7.2 Hz, 2H), 3.56 (s, 3H), 1.64 (d, J = 6.1 Hz, 3H), 1.55 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 141.91, 138.37, 136.19, 136.02, 135.39, 130.53, 130.05, 129.03, 128.95, 128.84, 128.18, 126.90, 125.71, 116.55, 107.63, 73.94, 72.66, 46.71, 46.55, 36.91, 25.36, 23.36, 15.87, 15.81. ESI-MS m/z: 389.6 [M + H]+.
(R)-3–(1-(2–(1-methyl-1H-pyrazol-5-yl)phenyl)ethoxy)-5–(1-propyl-1H-pyrazol-4-yl)pyridin-2-amine (B05)
Compound 8a (0.16 g, 0.43 mmol) was reacted with 1-propyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.12 g, 0.52 mmol) according to the general procedure E to obtain B05 (0.10 g, yield: 60%). 1H NMR (400 MHz, CDCl3) δ 7.84–7.65 (m, 2H), 7.62 (s, 1H), 7.50 (s, 2H), 7.45–7.32 (m, 2H), 7.23 (d, J = 7.4 Hz, 1H), 6.57 (s, 1H), 6.27 (s, 1H), 5.54–5.31 (q, 1H), 4.88 (s, 2H), 4.11 (t, J = 6.7 Hz, 2H), 3.57 (s, 3H), 1.93 (q, J = 14.1, 7.0 Hz, 2H), 1.63 (d, J = 5.7 Hz, 3H), 0.96 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 149.04, 141.34, 140.23, 140.13, 138.53, 136.01, 135.00, 130.44, 130.01, 128.62, 127.95, 126.11, 125.39, 119.52, 119.30, 116.95, 107.17, 73.12, 53.97, 36.85, 23.66, 23.34, 11.14. ESI-MS m/z: 403.5 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)-5–(1-methyl-1H-pyrrol-2-yl)pyridin-2-amine (B06)
Compound 8b (0.16 g, 0.43 mmol) was reacted with 1-methyl-2–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrrole (0.12 g, 0.52 mmol) according to the general procedure E to obtain B06 (0.12 g, yield: 72%). 1H NMR (400 MHz, DMSO) δ 7.71 (dd, J = 8.0, 1.2 Hz, 1H), 7.45–7.39 (m, 5H), 7.34 (m, J = 7.6, 1.2 Hz, 1H), 7.23–7.17 (m, 3H), 6.76–6.73 (m, 1H), 6.22 (d, J = 2.0 Hz, 1H), 6.02 (dd, J = 3.6, 2.8 Hz, 1H), 5.83–5.76 (m, 3H), 5.24 (q, J = 6.3 Hz, 1H), 3.25 (s, 3H), 1.68 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 150.55, 140.66, 140.24, 139.93, 138.87, 138.10, 131.43, 129.94, 129.42, 128.81, 128.62, 128.08, 127.90, 126.21, 123.55, 118.40, 117.68, 107.78, 107.40, 72.28, 34.63, 24.07. ESI-MS m/z: 370.3 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)-5–(1-methyl-1H-pyrazol-5-yl)pyridin-2-amine (B07)
Compound 8b (0.16 g, 0.43 mmol) was reacted with 1-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.11 g, 0.52 mmol) according to the general procedure E to obtain B07 (0.10 g, yield: 61%). 1H NMR (400 MHz, DMSO) δ 7.73 (dd, J = 8.0, 1.2 Hz, 1H), 7.53 (d, J = 2.0 Hz, 1H), 7.44–7.38 (m, 5H), 7.34 (m, J = 7.6, 1.2 Hz, 1H), 7.21–7.16 (m, 3H), 6.22 (d, J = 1.6 Hz, 1H), 6.08–6.02 (m, 3H), 5.28 (q, J = 6.3 Hz, 1H), 3.49 (s, 3H), 1.70 (d, J = 6.0 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 151.72, 140.73, 140.20, 139.72, 138.88, 138.57, 138.13, 129.91, 129.42, 128.83, 128.64, 128.18, 127.93, 126.30, 117.78, 114.48, 105.42, 73.94, 72.34, 37.32, 25.36, 23.95. ESI-MS m/z: 371.3 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)-5–(1-methyl-1H-pyrazol-4-yl)pyridin-2-amine (B08)
Compound 8b (0.16 g, 0.43 mmol) was reacted with 1-methyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.11 g, 0.52 mmol) according to the general procedure E to obtain B08 (0.12 g, yield: 76%). UPLC purity: 95.29%. 1H NMR (400 MHz, DMSO) δ 7.75 (dd, J = 8.0, 1.2 Hz, 1H), 7.63 (d, J = 1.6 Hz, 1H), 7.60 (s, 1H), 7.56–7.50 (m, 3H), 7.41 (m, J = 7.6, 1.6 Hz, 1H), 7.38–7.31 (m, 4H), 7.21 (dd, J = 7.6, 1.2 Hz, 1H), 6.29 (d, J = 2.0 Hz, 1H), 5.70 (s, 2H), 5.34 (q, J = 6.3 Hz, 1H), 3.85 (s, 3H), 1.70 (d, J = 6.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 150.31, 140.57, 140.31, 139.99, 139.49, 135.56, 135.44, 129.94, 129.61, 128.95, 128.57, 128.11, 128.05, 126.69, 126.30, 119.95, 117.45, 115.34, 72.04, 38.99, 24.23. ESI-MS m/z: 371.4 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)-5–(1-ethyl-1H-pyrazol-4-yl)pyridin-2-amine (B09)
Compound 8b (0.16 g, 0.43 mmol) was reacted with 1-ethyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.12 g, 0.52 mmol) according to the general procedure E to obtain B09 (0.11 g, yield: 66%). 1H NMR (400 MHz, DMSO) δ 7.75 (dd, J = 8.0, 1.2 Hz, 1H), 7.68–7.64 (m, 2H), 7.56–7.49 (m, 3H), 7.41 (dt, J = 7.6, 1.2 Hz, 2H), 7.34 (ddd, J = 8.0, 4.0, 2.4 Hz, 3H), 7.21 (dd, J = 7.6, 1.2 Hz, 1H), 6.31 (d, J = 1.6 Hz, 1H), 5.71 (s, 2H), 5.34 (q, J = 6.3 Hz, 1H), 4.13 (q, J = 7.3 Hz, 2H), 1.70 (d, J = 6.0 Hz, 3H), 1.40 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 150.29, 140.56, 140.33, 140.00, 139.49, 135.57, 135.30, 129.96, 129.58, 128.93, 128.57, 128.10, 128.04, 126.28, 125.19, 119.71, 117.56, 115.37, 72.08, 46.72, 24.25, 15.88. ESI-MS m/z: 385.3 [M + H]+.
(R)-3–(1-([1,1'-biphenyl]-2-yl)ethoxy)-5–(1-propyl-1H-pyrazol-4-yl)pyridin-2-amine (B10)
Compound 8b (0.16 g, 0.43 mmol) was reacted with 1-propyl-4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (0.12 g, 0.52 mmol) according to the general procedure E to obtain B10 (0.10 g, yield: 58%). 1H NMR (400 MHz, CDCl3) δ 7.68 (s, 1H), 7.62 (d, J = 7.3 Hz, 1H), 7.56–7.43 (m, 5H), 7.37 (m, J = 18.7 Hz, 4H), 7.31–7.23 (m, 1H), 6.46 (s, 1H), 5.41 (q, J = 6.1 Hz, 1H), 4.81 (s, 2H), 4.11 (t, J = 6.8 Hz, 2H), 2.13–1.86 (m, 2H), 1.76 (d, J = 6.0 Hz, 3H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 148.79, 140.36, 140.33, 140.22, 139.29, 136.16, 134.70, 129.96, 129.30, 128.48, 128.34, 127.75, 127.61, 125.45, 125.18, 119.80, 119.22, 116.24, 72.66, 53.97, 24.31, 23.74, 11.15. ESI-MS m/z: 399.5 [M + H]+.
(R)-5–(5-(1-([1,1'-biphenyl]-2-yl)ethoxy)-6-aminopyridin-3-yl)-1'-methylspiro[indoline-3,4'-piperidin]-2-one (C01)
Compound 8b (0.16 g, 0.43 mmol) was reacted with 1′-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)spiro[indoline-3,4′-piperidin]-2-one (0.18 g, 0.52 mmol) according to the general procedure E to obtain C01 (0.12 g, yield: 56%). UPLC purity: 94.34%. 1H NMR (400 MHz, MeOD) δ 7.67 (d, J = 7.7 Hz, 1H), 7.62–7.56 (m, 1H), 7.49–7.36 (m, 4H), 7.33 (t, J = 7.3 Hz, 1H), 7.30–7.25 (m, 2H), 7.24–7.16 (m, 1H), 6.87 (d, J = 7.8 Hz, 1H), 6.80 (s, 1H), 6.53 (s, 1H), 5.41 (dd, J = 12.0, 5.8 Hz, 1H), 3.54–3.41 (m, 2H), 3.24–3.13 (m, 2H), 2.78 (s, 3H), 2.18–2.04 (m, 4H), 1.79 (d, J = 6.1 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 181.70, 150.54, 141.34, 140.75, 140.24, 140.07, 139.21, 138.75, 135.10, 132.03, 129.42, 129.07, 128.08, 127.96, 127.46, 127.25, 126.12, 125.24, 123.02, 120.28, 116.98, 107.62, 72.63, 49.73, 43.62, 31.15, 29.24, 22.88. ESI-MS m/z: 505.4 [M + H]+.
(R)-5–(6-amino-5–(1-(4'-fluoro-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-3-yl)-1'-methylspiro[indoline-3,4'-piperidin]-2-one (C02)
Compound 8c (0.16 g, 0.41 mmol) was reacted with 1′-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)spiro [indoline-3,4′-piperidin]-2-one (0.17 g, 0.49 mmol) according to the general procedure E to obtain C02 (0.10 g, yield: 48%). UPLC purity: 95.16%. 1H NMR (400 MHz, MeOD) δ 7.68 (dd, J = 7.8, 0.9 Hz, 1H), 7.60 (d, J = 1.7 Hz, 1H), 7.40 (ddd, J = 10.4, 8.8, 4.7 Hz, 2H), 7.30 (m, J = 7.5, 1.2 Hz, 1H), 7.27–7.20 (m, 2H), 7.17 (dd, J = 7.6, 1.2 Hz, 1H), 7.14–7.07 (m, 2H), 6.84–6.79 (m, 2H), 6.50 (d, J = 1.8 Hz, 1H), 5.41 (q, J = 6.3 Hz, 1H), 3.17–3.07 (m, 2H), 2.91–2.80 (m, 2H), 2.54 (s, 3H), 2.07–1.89 (m, 4H), 1.77 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 182.33 (s), 162.24 (d, J = 245.8 Hz), 150.55 (s), 141.34 (s), 140.01 (s), 139.68 (s), 139.31 (s), 138.39 (s), 136.36 (d, J = 3.4 Hz), 135.25 (s), 132.94 (s), 130.90 (d, J = 8.1 Hz), 129.56 (s), 128.12 (s), 127.56 (s), 126.18 (s), 125.47 (s), 123.40 (s), 120.02 (s), 117.32 (s), 114.83 (d, J = 21.6 Hz), 107.39 (s), 72.58 (s), 49.91 (s), 44.59 (s), 44.11 (s), 32.00 (s), 22.64 (s). ESI-MS m/z: 523.6 [M + H]+.
(R)-5–(6-amino-5–(1-(4'-methoxy-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-3-yl)-1'-methylspiro[indoline-3,4'-piperidin]-2-one (C03)
Compound 8c (0.20 g, 0.50 mmol) was reacted with 1′-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)spiro [indoline-3,4′-piperidin]-2-one (0.20 g, 0.60 mmol) according to the general procedure E to obtain C03 (0.13 g, yield: 50%). UPLC purity: 92.70%. 1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.42–7.30 (m, 3H), 7.24 (d, J = 8.4 Hz, 3H), 7.00 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 7.8 Hz, 1H), 6.79 (s, 1H), 6.56 (s, 1H), 5.49 (q, J = 6.3 Hz, 1H), 4.98 (s, 2H), 3.92 (s, 3H), 3.32–3.17 (m, 2H), 3.05–2.92 (m, 2H), 2.64 (s, 3H), 2.39–2.22 (m, 2H), 2.08–1.96 (m, 2H), 1.77 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 182.30, 158.94, 150.05, 140.97, 140.31, 140.06, 139.25, 138.24, 135.55, 132.83, 132.56, 130.49, 130.14, 128.05, 127.80, 126.66, 125.52, 123.59, 119.99, 116.72, 113.96, 107.73, 72.87, 55.45, 50.21, 45.48, 43.98, 32.17, 24.03. ESI-MS m/z: 535.4 [M + H]+.
(R)-5–(6-amino-5–(1-(4'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-3-yl)-1'-methylspiro[indoline-3,4'-piperidin]-2-one (C04)
Compound 8c (0.20 g, 0.52 mmol) was reacted with 1′-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)spiro [indoline-3,4′-piperidin]-2-one (0.21 g, 0.62 mmol) according to the general procedure E to obtain C04 (0.15 g, yield: 57%). UPLC purity: 96.99%. 1H NMR (400 MHz, MeOD) δ 7.65 (dd, J = 7.8, 1.2 Hz, 1H), 7.59 (s, 1H), 7.45–7.39 (m, 1H), 7.36 (dd, J = 7.6, 1.4 Hz, 1H), 7.31 (m, J = 7.5, 1.4 Hz, 1H), 7.25–7.17 (m, 3H), 7.14 (d, J = 8.1 Hz, 2H), 6.88 (dd, J = 7.8, 1.6 Hz, 1H), 6.84 (d, J = 1.4 Hz, 1H), 6.53 (d, J = 1.8 Hz, 1H), 5.43 (q, J = 6.3 Hz, 1H), 3.89–3.76 (m, 2H), 3.58–3.43 (m, 2H), 3.01 (s, 3H), 2.40 (s, 3H), 2.38–2.26 (m, 2H), 2.20–2.07 (m, 2H), 1.78 (d, J = 6.3 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 181.03, 150.51, 141.42, 140.79, 140.18, 139.19, 139.05, 137.25, 137.13, 134.71, 131.17, 129.49, 128.93, 128.68, 127.78, 127.44, 126.05, 125.19, 122.71, 120.52, 117.25, 107.76, 72.80, 66.67, 49.55, 42.66, 30.27, 22.77, 19.82. ESI-MS m/z: 519.4 [M + H]+.
(R)-5–(6-amino-5–(1-(3'-methyl-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-3-yl)-1'-methylspiro[indoline-3,4'-piperidin]-2-one (C05)
Compound 8c (0.21 g, 0.55 mmol) was reacted with 1′-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)spiro [indoline-3,4′-piperidin]-2-one (0.23 g, 0.66 mmol) according to the general procedure E to obtain C05 (0.13 g, yield: 47%). UPLC purity: 99.12%. 1H NMR (400 MHz, DMSO) δ 7.72 (dd, J = 7.7, 0.8 Hz, 1H), 7.63 (s, 1H), 7.49–7.30 (m, 4H), 7.27–7.16 (m, 2H), 7.16–7.05 (m, 2H), 6.75 (dd, J = 7.7, 1.2 Hz, 1H), 6.70 (s, 1H), 6.36 (d, J = 1.5 Hz, 1H), 5.92 (s, 2H), 5.32 (q, J = 6.2 Hz, 1H), 3.08–2.88 (m, 2H), 2.84–2.60 (m, 2H), 2.47 (s, 3H), 2.22 (s, 3H), 1.96–1.85 (m, 2H), 1.85–1.75 (m, 2H), 1.73 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 181.42, 151.28, 142.20, 140.76, 140.07, 139.89, 139.47, 138.43, 138.19, 136.87, 132.97, 130.06, 129.94, 128.63, 128.53, 128.09, 127.11, 126.70, 126.12, 125.06, 124.47, 119.37, 116.13, 107.36, 72.18, 50.20, 45.58, 44.12, 32.20, 24.34, 21.29. ESI-MS m/z: 519.6 [M + H]+.
(R)-5–(6-amino-5–(1-(4'-fluoro-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-3-yl)-1'-methylspiro[indoline-3,4'-piperidin]-2-one (C06)
Compound 8c (0.15 g, 0.36 mmol) was reacted with 1′-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)spiro [indoline-3,4′-piperidin]-2-one (0.15 g, 0.43 mmol) according to the general procedure E to obtain C06 (0.09 g, yield: 45%). UPLC purity: 99.78%. 1H NMR (400 MHz, MeOD) δ 7.66 (d, J = 7.7 Hz, 1H), 7.58 (d, J = 1.5 Hz, 1H), 7.42 (d, J = 7.7 Hz, 1H), 7.35 (m, J = 7.6, 1.1 Hz, 1H), 7.29 (m, J = 7.5, 1.2 Hz, 1H), 7.22–7.11 (m, 3H), 6.92 (d, J = 8.6 Hz, 2H), 6.84 (dd, J = 7.7, 1.2 Hz, 1H), 6.79 (d, J = 1.2 Hz, 1H), 6.51 (d, J = 1.6 Hz, 1H), 5.47 (q, J = 6.3 Hz, 1H), 4.07 (q, J = 7.0 Hz, 2H), 3.31–3.24 (m, 2H), 3.05–2.98 (m, 2H), 2.66 (s, 3H), 2.12–1.98 (m, 4H), 1.78 (d, J = 6.3 Hz, 3H), 1.42 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 181.97, 158.45, 150.52, 141.30, 140.54, 140.13, 139.31, 138.59, 135.07, 132.43, 132.28, 130.20, 129.58, 127.59, 127.42, 126.19, 125.31, 123.18, 120.18, 117.21, 114.01, 107.55, 72.59, 63.20, 49.86, 44.13, 43.68, 31.62, 22.74, 13.77. ESI-MS m/z: 549.6 [M + H]+.
(R)-5–(6-amino-5–(1-(4'-(trifluoromethoxy)-[1,1'-biphenyl]-2-yl)ethoxy)pyridin-3-yl)-1'-methylspiro[indoline-3,4'-piperidin]-2-one (C07)
Compound 8c (0.16 g, 0.35 mmol) was reacted with 1′-methyl-5–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) spiro[indoline-3,4′-piperidin]-2-one (0.15 g, 0.42 mmol) according to the general procedure E to obtain C07 (0.10 g, yield: 48%). UPLC purity: 95.46%.1H NMR (400 MHz, MeOD) δ 7.72 (d, J = 7.7 Hz, 1H), 7.61 (s, 1H), 7.47–7.40 (m, 2H), 7.35 (d, J = 7.1 Hz, 3H), 7.32–7.25 (m, 2H), 7.22 (d, J = 7.7 Hz, 1H), 6.85 (s, 2H), 6.54 (s, 1H), 5.42 (q, J = 11.6, 5.4 Hz, 1H), 3.73–3.46 (m, 2H), 3.31–3.23 (m, 2H), 2.86 (s, 3H), 2.26–2.15 (m, 2H), 2.14–2.05 (m, 2H), 1.79 (d, J = 5.4 Hz, 3H). 13C NMR (101 MHz, MeOD) δ 181.39, 150.72, 148.47, 141.46, 140.01, 139.38, 139.32, 139.24, 138.87, 135.33, 131.72, 130.78, 129.49, 128.39, 127.66, 125.99, 125.61, 122.95, 121.75, 120.50, 120.21, 117.44, 107.54, 72.75, 49.65, 43.26, 30.81, 29.25, 22.46. ESI-MS m/z: 589.6 [M + H]+.
Kinase inhibitory activity
Kinase inhibitory detection for ROS1, ALKWT, ALKG1202R, and ALKL1196M (Carna Biosciences, lot: 10CBS-0385B for ROS1; 13CBS-0537B for ALK) were performed with the HTRF KinEASE-TK assay from Cisbio according to the manufacturer’s instructions. The dosage of enzymes in each reaction well was set to 0.012 ng of ROS1 (200 ng/ul), 0.25 ng of ALK (including ALKWT, ALKG1202R, and ALKL1196M) (200 ng/ul). Enzymatic assay was carried out as has been previously described. The IC50 values of target compounds were calculated using nonlinear regression with normalised dose-response fit employing Prism GraphPad software.
Antiproliferative MTT assay
The antiproliferative activity of target compounds were investigated for two human cancer cell lines HCC-78 and NCI-H3122 by the standard MTT assay, with Crizotinib as the positive control. Both cell lines were purchased from Cell Bank of China Science Academy (Shanghai, China). All solvents were purchased from Gibco (Gibco BRL, Grand Island, NY, USA). The cell lines were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium supplement with 10% foetal bovine serum (FBS). Approximately 2 × 103 cells, suspended in medium, were plated into each well of a 96-well plate and grown at 37 °C in a humidified atmosphere with 5% CO2 for 24 h. The following day, various concentrations of tested compounds were added to the culture medium and incubated for 72 h. Fresh MTT was added to each well at the terminal concentration of 5 mg/ml in phosphate buffered saline (PBS), and incubated with cells at 37 °C for 4 h. The formazan crystals in each well were dissolved in 100 μl DMSO, and the absorbency at 570 nm was measured with an enzyme-linked immunosorbent assay plate reader. All target compounds were tested three times in each of the cell lines and the IC50 values were calculated using Prism GraphPad software.
Antiproliferative CCK-8 assay
The antiproliferative activity of compound B08 and C01–C03 was evaluated against CD74-ROS1G2032R and CD74-ROS1L2026M expressing Ba/F3 cell lines by the standard CCK-8 assay, with Crizotinib as the positive control. Both cell lines were constructed by lentivirus transfection. All solvents were purchased from Gibco (Gibco BRL, Grand (RPMI) 1640 medium supplement with 10% foetal bovine serum (FBS). Approximately 1 × 104 cells, suspended in medium, were plated into each well of a 96-well plate and grown for 4 h. Next, different concentrations of tested compounds were added to the culture medium and incubated for 72 h. Then CCK-8 was added to each well (10 μl/100 μl), and incubated with cells at 37 °C for 1 h. Then, the absorbency at 450 nm was measured with an enzyme-linked immunosorbent assay plate reader. All target compounds were tested three times in each of the cell lines and the IC50 values were calculated using Prism GraphPad software.
Western blotting
Western blotting analysis was performed as described. Approximately 2.0 × 106 Ba/F3 cells were plated into each well of a six-well plate and grown for 24 h. Next, different concentrations of Crizotinib and C01 were added to the culture medium for 8h. Then the cells were digested and centrifuged with trypsin and lysed with RIPA lysis buffer, centrifuged at 12000 rpm at 4 °C for 30 min, later collected the supernatant. Equal amount of protein was separated by 15% SDS-PAGE gel and were transferred to polyvinylidene difluoride (PVDF) membranes. After incubated overnight at 4 °C with the respective primary antibodies (1: 1000), a horse-radish peroxidase (HRP)-conjugated secondary antibody (1: 2000) was sequentially added. After adding clarity Western ECL substrate, use the multifunctional imaging analysis system (protein Simple-FluorChem R) to visualise protein strip.
Apoptosis assay
NCI-H3122 and HCC-78 cells were seeded in six-well plates at 1.5 × 105 cells per well for 24 h, then treated with different concentrations of Crizotinib or C01 for 48 h. Floating and attached cells were harvested by washing with phosphate-buffered saline (PBS), exposuring to trypsin and centrifuging at 3000 rpm for 10 min. The cells were resuspended in 1 ml PBS and centrifuged again. Apoptosis assay was measured with the use of an Annexin-V-FITC and PI Staining Kit (BestBio). After centrifugation, the PBS was removed, and the cell pellet was carefully resuspended in 400 μl Annexin-V-FITC binding solution. Annexin-V-FITC Staining solution and PI Staining solution were added and incubated for 15 min at 4 °C under the condition of protection from light. Then the samples were analysed for fluorescence with a flow cytometer (FACSCalibur, BD Biosciences).
Molecular modelling
Molecular docking study was performed on Glide module of Schrödinger suite (New York, NY)Citation27. The crystal structures of ROS1 and ALK in complex with Crizotinib (PDB ID: 3ZBF and 2XP2, respectively) were obtained from protein data bank (PDB) (http://www.rcsb.org/)Citation14,Citation28. As the template using the crystal structure of the wide-type model, the Schrödinger 11.1 software was performed on constructing the G2032R mutant by replacing the specific residue Gly2032 with Arg2032 in ROS1 and the G1202R mutant by replacing the specific residue Gly1202 with Arg1202 in ALK. The proteins were preprocessed and minimised using Protein Preparation wizard module. Hydrogen atoms were added and crystallographic waters were removed, missing side chains or missing loops were filled using prime module. A restrained partial minimisation was subsequently accomplished with the default root mean square deviation (RMSD) value of 0.3 Å using Optimised Potentials for Liquid Simulations (OPLS) 2005 force field. The receptor grid box was generated at the centre of cocrystalised inhibitors using Receptor grid generation module under the default parameter values. After the ligands were prepared with the Ligprep module, they were docked into the proteins using extra precision mode (XP) and the best docking configurations based on the XP Glide scores (G-scores) were selected as the most probable binding conformation.
The MD simulations were performed by using Amber12 programCitation29. The Glide XP results of C01 binding to the ROS1-G2032R mutant were used as initial structures in two simulation systems. Geometry optimisation and the electrostatic potential calculations for C01 was carried out at the HF/6-31G* level of the Gaussian 09 suiteCitation30. The atomic partial charges were computed by the restrained electrostatic potential (RESP)Citation31 fitting method. AMBER 99SB force fieldsCitation32 and General AMBER Force Field (GAFF)Citation33 were employed to generate the parameters of protein and C01, respectively. Each system was placed in the TIP3P water box with a 10 Å buffer size, and neutralised by adding Na+, then the leap module was performed to generate parameters and coordinate files. The system of Crizotinib binding ROS1-G2032R was built in the same way as C01. Three consecutive minimisation stages were performed to relax all systems prior to the MD simulation: (i) the protein and ligand were restrained by a 5.0 kcal·mol−1·Å − 2 harmonic constraint potential and only water molecules and ions were minimised with 8000 steps of steepest descent minimisation and 7000 steps of conjugate gradient minimisation; (ii) the protein backbone and ligand atoms were constrained with a force constant 5.0 kcal mol−1 Å − 2 and other atoms were minimised by 5000 steps of steepest descent minimisation and subsequent 5000 steps of conjugate gradient minimisation; (iii) all atoms were relaxed through respective 5000 steps of steepest descent method and conjugate gradient method. The systems were then heated from 0 to 300 K, followed by 500 ps density equilibration. After that, 20 ns MD simulations were performed using the PMEMD module of AMBER 12 program in the NPT condition. The particle mesh Ewald (PME)Citation34 algorithm was used to describe electrostatic interactions in all calculations. A cut-off distance of 10 Å was set for electrostatic and van der Waals interactions. The SHAKECitation35 algorithm was assigned to constrain all bonds involving hydrogen atoms. The representative structure of each system was produced by cluster analysis and visualised by PyMOL. The binding-free energies (ΔGbind) were calculated by using MM-GBSA and MM-PBSACitation36,Citation37 procedure in AMBER12.
Results and discussion
Rational design based on molecular modelling strategies
Owing to highly homologous amino acid sequences at ATP binding site, ROS1 and ALK share similar sensitivity to a number of inhibitors. 2-Aminopyridine nucleus was recognised as the specific structure of the ROS1/ALK dual inhibitors, lots of compounds harbouring 2-aminopyridine nucleus structure, such as Crizotinib, PF-06469015, Lorlatinib and 2e (), showed superior ROS1/ALK inhibitory potencyCitation28,Citation38,Citation39. Among these compounds, the macrocycle derivative Lorlatinib and our previously reported agent 2e retained persistent potency to variety of drug-resistant mutants including ROS1-G2032R, with cellular IC50 values as 203 nM and 105 nM, respectivelyCitation40–42. Furthermore, Lorlatinib exhibited robust cellular phosphorylation ALK activity against G1202R mutant with an IC50 value as 77 nMCitation43. The success of Lorlatinib and 2e confer the potential for 2-aminopyridine analogues to overcome ROS1G2032R, ALKG1202R, and other Crizotinib-resistant mutants. To further improve the sensitivity profile against both wild-type and drug-resistant mutants, we identify 2-aminopyridine structure as the basic skeleton of ROS1/ALK dual inhibitors and carry out a series of optimizations under the guidance of molecular modelling ().
Figure 1. Structures of representative ROS1/ALK dual inhibitors.
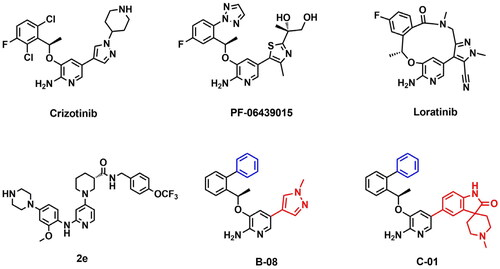
Figure 2. Design strategies of target compounds based on cocrystal structures of Crizotinib with ROS1WT (PDB 3ZBF) and ALKWT (PDB 2XP2). (A) Docking structure of Crizotinib binding to apo-G2032R ROS1 receptor. (B) Docking structure of Crizotinib binding to apo-G1202R ALK receptor. (C) 2D structure of Crizotinib. (D) Docking posture of Crizotinib binding to ROS1 receptor. (E) Design strategies for novel 2-aminopyridine analogues as ROS1/ALK dual inhibitors. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article).
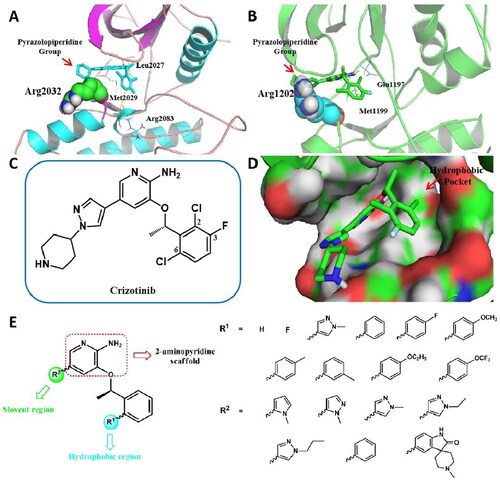
To elucidate the molecular mechanism of ROS1G2032R- and ALKG1202R -mediated resistance, molecular docking simulations on Crizotinib with G2032R-ROS1 and G1202R-ALK were conducted, by incorporating Arg2032 and Arg1202 mutants respectively (. As the docking results showed, in ROS1 () or ALK () kinase domain, the steric hindrance between pyrazolopiperidine group and bulky side chain of Arginine conferred resistance to Crizotinib. Thus, we introduced various tiny substituted pyrazoles and aromatic groups at the solvent-front region (R2 group) to increase binding affinities with ROS1G2032R and ALKG1202R mutants. In addition, we previously disclosed a promising c-MET/ALK inhibitor SMU-B, which harboured a spiro structure and exhibited superior enzymatic IC50 values against ROS1 (1.48 nM, Figure S1) and ALK (<0.5 nM)Citation44. The successful design of SMU-B demonstrated the promise of novel spiro compounds to improve the inhibitory activities against ROS1 and ALK. Moreover, we hypothesised that introducing the spiro groups could keep the rigid of desired compounds and thus reduce the steric hindrance at the solvent region. Therefore, we also introduced the preferred spiro group at the solvent region to generate the target compounds ().
With respect to the “head” benzyloxy group, the C6-substituent of the phenyl points to a hydrophobic pocket, which allows larger substituent groups to be imported (). In order to make possible bindings with amino acids around the hydrophobic region and improve the ROS1/ALK inhibitory activity, variety of pyrazolyl and phenyl derived groups are subsequently introduced at R1 region (). Another benefit of the introduction of aromatic groups into the hydrophobic pocket would be the structural balance of the target compound, which helps to alleviate steric hindrance at the solvent region on the other side. Moreover, according to previous studies, the removal of halogens from benzyloxy group was beneficial to improving potency, lipophilic efficiency, and in vitro clearance, thus providing better likelihood for improving pharmaceutical propertiesCitation38.
On the basis of the structural analysis above, a series of novel 2-aminopyridine derivatives were designed and synthesised rationally and the target compounds were subjected to anti-proliferative evaluations and enzymatic assays against ROS1, ALK as well as c-MET. Furthermore, the binding modes were disclosed by docking and molecular dynamics (MD) simulation, which confirmed the design strategy and structure-activity relationships (SARs).
Chemistry
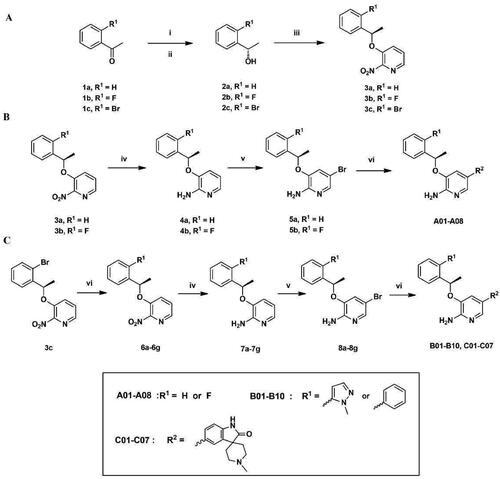
The target compounds were prepared according to the synthetic routes disclosed in . At first, acetophenone derivatives (1a–1c) were subjected to asymmetric reduction in the present of (-)-diisopinocampheyl chloroborane (DIP-Cl) to obtain the compounds 2a–2c. Mitsunobu reaction of 2a–2c with 2-nitropyridin-3-ol led to key intermediates 3a–3c (). The intermediates 3a and 3b were then subjected to Fe-catalysed reduction to give compounds 4a–4b, following by brominating on 2-aminopyridine scaffold in the present of N-Bromosuccinimine (NBS) to afford compounds 5a–5b. Suzuki coupling of 5a–5b with variety of borate ester substituents finally got the target compounds A01–A08 (). In addition, various aromatic substituents were introduced on intermediate 4c in Suzuki coupling conditions to give compounds 6a–6g. After a series of reactions including Fe-catalysed reduction, brominating and Suzuki coupling, compounds 6a–6g were finally developed to the target compounds B01–B10 and C01–C07 (). The chemical structures of these compounds were confirmed by 1H NMR, 13C NMR and ESI-MS.
Figure 3. (A–C) General synthetic routes of 2-aminopyridine derivatives. Reagents and conditions: (i) (-)-DIP-Cl, THF, −35 °C, 3 h, rt, 18 h; (ii) diethanolamine, TBME, 60 °C, reflux, 1 h, rt, 2 h; (iii) DIAD, PPh3, 2-nitropyridin-3-ol, dry THF, 0 °C, 8 h; (iv) Fe, AcOH, EtOH, 85 °C, reflux, 2 h; (v) NBS, MeCN, 0 °C, 30 min; (vi) potassium carbonate, Pd(PPh3)4, DOX, H2O, 85 °C, 8 h.
Kinase inhibitory activities and SARs study
The target compounds as well as the positive control Crizotinib were evaluated for inhibitory activities against ROS1, ALK, and c-MET enzymes using homogeneous time resolved fluorescence (HTRF) assay and the dose-response curve was carried out to determine the half maximal inhibitory concentration (IC50) values. As a general trend, most of the desired compounds showed moderate to excellent inhibitory potency against ROS1 and ALK enzymes, with IC50 values from 963.9 nM to 4.6 nM, which confirmed the rational design of 2-aminopyridine derivatives as dual ROS1/ALK inhibitors. In contrast, the sensitivity of all the target compounds against c-MET enzyme was relatively weak, with inhibition rate at 1 μM less than 70% ().
Table 1. The enzymatic and anti-proliferative activities of target compounds A01–A08.
Table 2. The enzymatic and anti-proliferative activities of target compounds B01–B10.
Table 3. The enzymatic and anti-proliferative activities of target compounds C01–C07.
In the first round of optimisation, we removed the halogen substitutions at the “head” benzyloxy group and replaced the R2 moiety with different pyrazole derived substituents, leading to analogues A01–A04. These inhibitors exhibited relatively poor activities against ROS1 and ALK enzymes. Further optimisation introduced fluorine atom at R1 position, thus got the target compounds A05–A08. Compared to A01–A04, the enzymatic activities of F-substituted derivatives A05–A08 had no obvious improvement. Analysis of these compounds found that prolonged the carbon chain on the pyrazole group resulted in a loss of enzymatic activity. For example, compound A04 with a propyl substituent displayed approximate 10-fold decrease in ROS1 potency compared to A02 with a methyl substituent (963.9 nM vs. 99.5 nM).
The first round of structural optimisation highlighted that the H or F atom replacement at R1 position was ineffective to improve the enzymatic activity. Molecular modelling indicated that the R1 position resided in a relatively roomy hydrophobic region, which allowed larger substituents. Aiming to make possible bindings with amino acids around the hydrophobic region and improve the ROS1 and ALK inhibitory activities, 1-methyl-1H-pyrazol-5-yl was subsequently introduced and generated inhibitors B01–B05. The results showed that these inhibitors harboured comparable potency with F-substituted A05–A08, with IC50 values ranging from 374.3 nM to 80.5 nM for ROS1 enzyme and from 1165.0 nM to 286.0 nM for ALK enzyme. Furthermore, by replacing the 1-methyl-1H-pyrazol-5-yl with a more hydrophobic phenyl at R1 position, compounds B06–B10 exhibited significant improvement of enzymatic activities. For example, the ROS1 and ALK IC50 values of compound B08 were 37.4 nM and 67.4 nM, which were about 10-fold and 4-fold more potent than that of compound B03 (374.3 nM for ROS1 and 286.0 nM for ALK).
This round of structural modification confirmed the importance of phenyl ring at R1 position. Next work was conducted by keeping phenyl ring at R1 position and introducing a novel spiro fragment to replace pyrazole substituent at R2 position, thus yielded compounds C01. As expected, C01 displayed superior ROS1 and ALK enzymatic activities with IC50 values of 4.7 nM and 25.6 nM, respectively. To further explore the influence of derived group on phenyl ring at R1 position, various functionalities including fluoro, methoxyl, methyl, ethyoxyl, trifluoromethoxy, and trifluoromethyl were imported. The introduction of fluoro got compound C02, which also showed excellent activities against ROS1 and ALK (4.6 nM and 30.1 nM, respectively), similarly to that of C01. It was observed that C01 and C02 were more potent than Crizotinib in ROS1 potency, while were comparable to Crizotinib in ALK potency. However, other derived compounds (C03–C07) showed decreased activities, and the larger derived group led to the less active potency, indicating that the volume of hydrophobic pocket was limited.
In general, the enzymatic assays highlighted the significance of 2-aminopyridine as basic scaffold of dual ROS1/ALK inhibitors; while hydrophobic phenyl or 4-fluorophenyl at R1 position and novel spiro group at R2 position were the preferred combination. Moreover, the target compound demonstrated excellent selectivity profile for ROS1 and ALK over c-MET, which was not seen in the enzymatic assays of Crizotinib and SMU-BCitation28,Citation44.
In vitro anti-proliferative activity
All target compounds as well as Crizotinib were investigated for anti-proliferative activities against HCC-78 cell line harbouring SLC34A2-ROS1 fusion and NCI-H3122 cell line harbouring EML4-ALK fusion by the standard MTT assay. The results were illuminated as IC50 values and summarised in . As the results showed, the tendency of cellular potency was consistent with that of enzymatic potency for most target compounds. For example, H-or F-substituted compounds A01–A08, which were recognised as weaker inhibitors in enzymatic assay, exhibited moderated anti-proliferative activities against HCC-78 and NCI-H3122 cell lines with IC50 values over 10.0 μM. Modification of R1 position with substituted pyrazol group (B01–B05) further diminished cellular potency, especially for ALK-expressing H3122 cell. Similar to enzymatic assay, introducing a hydrophobic phenyl at R1 position (B06–B10) led to an obvious increase in both HCC-78 and NCI-H3122 cellular activities. Compound B08 exhibited potent anti-proliferative activities against HCC-78 and NCI-H3122 cells with IC50 values of 4.6 μM and 5.7 μM, which turned out to be similar to that of Crizotinib (4.5 μM and 4.1 μM, respectively). Additionally, the spiro analogues C01–C07, the most potent inhibitors in enzymatic assay, also showed the best anti-proliferative activities with IC50 values ranging from 5.6 μM to 1.4 μM. The optimal inhibitors in enzymatic and cellular assays were subjected to further evaluation of anti-mutation activity.
Impressive activities against Crizotinib-resistant ROS1G2032R and ALKG1202R mutants
In order to evaluate the sensitivity towards different ROS1 or ALK mutants, the representative inhibitors, including B08 and C01–C03, were assayed for their anti-resistant activities against Ba/F3 cells and biological enzymes.
As shown in , the anti-proliferative activity of compound C01 and C02 against CD74-ROS1WT cell was comparable to Crizotinib. With regard to CD74-ROS1L2026M cell, the selected inhibitor C02 showed potent activity with an IC50 value of 21.3 nM, slightly better than 31.5 nM of Crizotinib (). Moreover, compared to Crizotinib, all the selected inhibitors exhibited better anti-proliferative activity against CD74-ROS1G2032R cell, an acknowledge Crizotinib-refractory mutant within hydrophobic region (). In particular, the optimal compound C01 showed superior ROS1G2032R cellular activity with an IC50 value of 42.3 nM, which was about 30-fold more potent than Crizotinib (). ALKG1202R is a hydrophobic mutant similar to ROS1G2032R, which also confers resistance to Crizotinib and most of listed ALK inhibitors. In enzymatic assay, the spiro derivatives C01 and C02 potently inhibited ALKG1202R mutant with IC50 values of 49.1 nM and 275.5 nM, superior to 491.6 nM of Crizotinib (). Whereas, all the selected compounds displayed diminished enzymatic activity against another ALKL1196M mutant compared to Crizotinib, which was not consistent with their potency in EML4-ALKL1196M cell (). In general, these results demonstrated that the representative inhibitors, especially the spiro analogue C01, remained persistent potency to Crizotinib-resistant ROS1G2032R and ALKG1202R mutants, which confirmed our previous design strategy.
Figure 4. In vitro activities against Crizotinib-resistant ROS1G2032R and ALKG1202R mutants. Dose–response curves for proliferation of Ba/F3 CD74-ROS1WT cell (A), Ba/F3 CD74-ROS1L2026M cell (B), Ba/F3 CD74-ROS1G2032R cell (C), Ba/F3 EML4-ALKWT cell (D) and Ba/F3 EML4-ALKL1196M cell (E) after 72 h exposure to B08, C01–C03, and Crizotinib. (F) Enzyme-based activities of B08, C01, C02, and Crizotinib against ALKG1202R mutant. All data are the average of n ≥ 2 ± standard deviation.
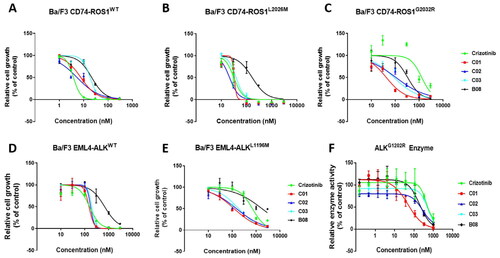
Table 4. Enzymatic and cellular activities to ROS1 and ALK mutants.
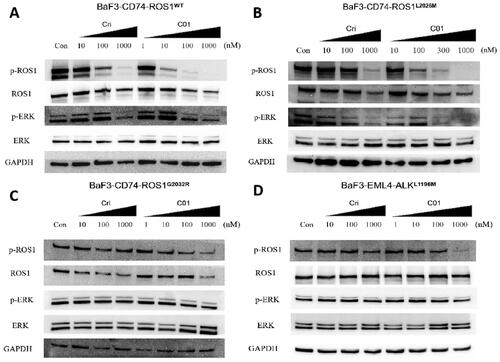
Intracellular phosphorylation effects of C01 against ROS1 and ALK
The effects of C01 on the phosphorylation of ROS1 and ALK and downstream signalling pathway were conducted on Ba/F3 cells. Consistent with results from cell proliferation assays, treatment of CD74-ROS1WT and CD74-ROS1L2026M cell with C01 and Crizotinib led to attenuated phosphorylation of ROS1 and its key downstream molecule ERK in a dose-dependent manner (). Furthermore, although significant inhibition of ROS1 phosphorylation occurred in a dose-dependent manner in CD74-ROS1G2032R cell treated with C01, the positive Crizotinib did not reduce ROS1 phosphorylation at concentration up to 1000 nM (), of which was also seen in EML4-ALKL1196M cell (). These findings demonstrated that the optimal compound C01 exhibited persistent potency to Crizotinib-refractory ROS1G2032R and ALKL1196M mutants in cellular activity.
Figure 5. Inhibition of cellular phosphorylation of ROS1, ALK, and ERK by C01 in CD74-ROS1WT (A), CD74-ROS1L2026M (B), CD74-ROS1G2032R (C), and EML4-ALKL1196M (D) cells investigated by western blot analysis, with Crizotinib as the positive control. Cells were treated with Crizotinib or C01 at the indicated concentrations for 4 h and the levels of protein were evaluated by western blot analysis of cell lysates using specific antibodies.
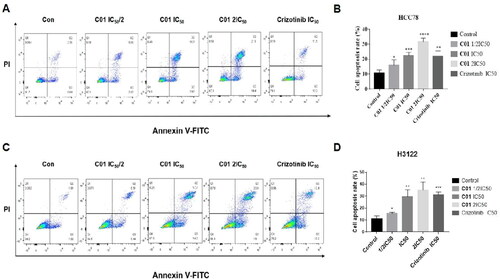
C01 induced apoptosis of HCC-78 and NCI-H3122 cells
To investigate whether target inhibitors induced apoptosis of HCC-78 and NCI-H3122 cells, the optimal compound C01 and positive control Crizotinib were determined by Annexin-V and Propidium iodide staining followed by flow cytometry. As shown in promoted apoptosis of HCC-78 cell in a dose-dependent manner, the percentage of apoptotic cell was significantly increased from 15.9% (2.0 μM, IC50/2) to 31.7% (7.8 μM, 2 IC50), compared with the vehicle group of 10.7%. Moreover, the percentage of apoptotic HCC-78 cell in C01 treatment group at 3.9 μM concentration (1 IC50) was superior to that of Crizotinib treatment group at 4.5 μM concentration (1 IC50). Likewise, the percentage of apoptotic NCI-H3122 cell observed in C01 (1.5 μM, IC50/2), C01 (3.0 μM, 1 IC50), and C01 (6.0 μM, 2 IC50) treatment groups were 15.61%, 29.47% and 35.13%, respectively, which were superior to the vehicle group (11.24%) and appeared in an obvious dose-dependent manner.
Figure 6. (A) Flow cytometric analysis of apoptosis in HCC-78 cell line treated with C01 in the concentrations of 0 (control), 2.0 μM, 3.9 μM, and 7.8 μM. (B) Graphical representation of total percentage of apoptosis induced by C01 in HCC-78 cell line. (C) Flow cytometric analysis of apoptosis in NCI-H3122 cell line treated with C01 in the concentrations of 0 (control), 1.5 μM, 3.0 μM and 6.0 μM. (D) Graphical representation of total percentage of apoptosis induced by C01 in NCI-H3122 cell line. (*p < 0.05; **p < 0.01; ***p < 0.001 vs control, n = 3).
Molecular dynamics
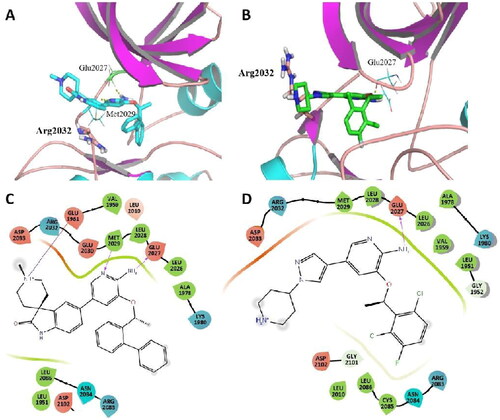
In order to investigate the increased potency of C01 against ROS1G2032R mutant compared to Crizotinib, molecular dynamics (MD) were performed to explain the possible binding conformation of selected compounds. As shown in , when C01 was bound to ROS1G2032R, the larger spiro group had an obvious shift away from Arg2032, avoiding the steric hindrance in the solvent region. Consequently, C01 kept the main contact with the ROS1G2032R in hinge region, two H-bonds were formed between amino pyridine scaffold and key amino acid residues, the NH from amino group made H-bond to Glu2027 and the N from pyridine group made hydrogen bond to Met2029, respectively. However, while Crizotinib was bound, the H-bond with Met2029 in hinge region disappeared due to the conformational change resulted from steric hindrance between pyrazolopiperidine group and bulky side chain of Arg2032 (). These MD results gave the reasonable explanation for higher potency of C01 against ROS1G2032R compared to Crizotinib.
Figure 7. Representative structures from MD simulation of C01 (A) and Crizotinib (B) in the active site of ROS1G2032R. (ROS1 in coloured cartoon, ligands, and residues in stick mode. Yellow dots represent hydrogen bonds). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.) (C) 2D diagram of the interaction between C01 and the binding site of ROS1G2032R. (D) 2D diagram of the interaction between Crizotinib and the binding site of ROS1G2032R.
Additionally, the binding-free energies of C01 and Crizotinib were also calculated by using MM-PBSA (molecular mechanics Poisson–Boltzmann surface area) and MM-GBSA (molecular mechanics Generalized-Born surface area) programs in AMBER. As Table S1 shown, MM-PBSA and MM-GBSA calculations demonstrated that C01 showed lower ΔGbind when interacting with ROS1-G2032R mutant compared to Crizotinib, which were in good agreement with the biological data. According to the energy individual components of the binding-free energies, the favourable contributors to ligand binding were van der Waals (vdW) terms, electrostatic and polar solvation energies, whereas nonpolar salvation and entropy terms oppose binding. The ΔGprep (GB) and ΔGprep (PB) also indicated that C01 could bind with ROS1-G2032R mutant more strongly, which was consistent with experimental data.
Conclusion
In summary, a series of novel 2-aminopyridine derivatives were designed, synthesised and optimised for therapeutic potency against ROS1 and ALK kinases as well as their principal mutants ROS1G2032R and ALKG1202R. Bioactivity evaluation demonstrated that most of target inhibitors showed more remarkable antitumour activity than Crizotinib against human cancer cell lines including HCC-78 and NCI-H3122. Moreover, the representative compound C01 exhibited robust Ba/F3 cellular activity against Crizotinib-refractory ROS1G2032R mutant with an IC50 value of 42.3 nM, which was about 30-fold more potent than Crizotinib. In enzymatic assay, C01 potently inhibited enzymatic activity against Crizotinib-resistant ALKG1202R, harbouring a 10-fold potency superior to Crizotinib (IC50: 49.1 nM vs. 491.6 nM). Flow cytometric analysis indicated that C01 could induce apoptosis of HCC-78 and NCI-H3122 cell lines at the concentration of 3.9 μM and 3.0 μM, respectively. Molecular dynamic disclosed that C01 could dock into the active site of ROS1G2032R and kept the main contact in hinge region, which gave a probable explanation of anti-drug-resistant mutants. These results indicated the potential of novel 2-aminopyridine derivatives for further development as potent anti-tumour agents.
Supplemental Material
Download PDF (3.2 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Dickran K, Blumenthal GM, Luo L, He K, Ingrid F, Steven L, Richard P. Benefit-risk summary of Crizotinib for the treatment of patients with ROS1 alteration-positive, metastatic non-small cell lung cancer. Oncologist. 2016;21(8):974–980.
- Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci USA. 1987;84(24):9270–9274.
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203.
- Jun HJ, Johnson H, Bronson RT, de Feraudy S, White F, Charest A. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012;72(15):3764–3774.
- Neel DS, Allegakoen DV, Olivas V, Mayekar MK, Hemmati G, Chatterjee N, Blakely CM, McCoach CE, Rotow JK, Le A, et al. Differential subcellular localization regulates oncogenic signaling by ROS1 kinase fusion proteins. Cancer Res. 2019;79(3):546–556.
- Bergethon K, Shaw AT, Ou S-HI, Katayama R, Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang R, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–870.
- Cooper WA, Lam DC, O'Toole SA, Minna JD. Molecular biology of lung cancer. J Thorac Dis. 2013;5(Suppl 5):S479–S490.
- Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res. 2013;19(15):4040–4045.
- Gainor JF, Shaw AT. Novel targets in non-small cell lung cancer: ROS1 and RET fusions. Oncologist. 2013;18(7):865–875.
- Drilon A, Somwar R, Wagner JP, Vellore NA, Eide CA, Zabriskie MS, Arcila ME, Hechtman JF, Wang L, Smith RS, et al. A novel Crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin Cancer Res. 2016;22(10):2351–2358.
- McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, Estrada-Bernal A, Davies KD, Merrick DT, Bunn PA Jr, et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin Cancer Res. 2018;24(14):3334–3347.
- Facchinetti F, Loriot Y, Kuo MS, Mahjoubi L, Lacroix L, Planchard D, Besse B, Farace F, Auger N, Remon J, et al. Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clin Cancer Res. 2016;22(24):5983–5991.
- Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17(11):637–658.
- Awad MM, Katayama R, McTigue M, Liu W, Deng YL, Brooun A, Friboulet L, Huang D, Falk MD, Timofeevski S, et al. Acquired resistance to Crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368(25):2395–2401.
- Lin JJ, Choudhury NJ, Yoda S, Zhu VW, Johnson TW, Sakhtemani R, Dagogo-Jack I, Digumarthy SR, Lee C, Do A, et al. Spectrum of mechanisms of resistance to Crizotinib and Lorlatinib in ROS1 fusion-positive lung cancer. Clin Cancer Res. 2021;27(10):2899–2909.
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284.
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S-i, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566.
- Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008;99(12):2349–2355.
- Mazot P, Cazes A, Boutterin MC, Figueiredo A, Raynal V, Combaret V, Hallberg B, Palmer RH, Delattre O, Janoueix-Lerosey I, et al. The constitutive activity of the ALK mutated at positions F1174 or R1275 impairs receptor trafficking. Oncogene. 2011;30(17):2017–2025.
- Heuckmann JM, Holzel M, Sos ML, Heynck S, Balke-Want H, Koker M, Peifer M, Weiss J, Lovly CM, Grutter C, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17(23):7394–7401.
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired Crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra17.
- Ai X, Shen S, Shen L, Lu S. An interaction map of small-molecule kinase inhibitors with anaplastic lymphoma kinase (ALK) mutants in ALK-positive non-small cell lung cancer. Biochimie. 2015;112:111–120.
- Shen J, Meng Y, Wang K, Gao M, Du J, Wang J, Li Z, Zuo D, Wu Y. EML4-ALK G1202R mutation induces EMT and confers resistance to ceritinib in NSCLC cells via activation of STAT3/Slug signaling. Cell Signal. 2022;92:110264.
- Shaw AT, Gandhi L, Gadgeel S, Riely GJ, Cetnar J, West H, Camidge DR, Socinski MA, Chiappori A, Mekhail T, et al. Alectinib in ALK-positive, Crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol. 2016;17(2):234–242.
- Fontana D, Ceccon M, Gambacorti-Passerini C, Mologni L. Activity of second-generation ALK inhibitors against Crizotinib-resistant mutants in an NPM-ALK model compared to EML4-ALK. Cancer Med. 2015;4(7):953–965.
- Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K, Singh M, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–1133.
- Glide, Schrödinger LLC. New York, NY; 2017.
- Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung P-P, Pairish M, Jia L, Meng J, Funk L, Botrous I, et al. Structure based drug design of Crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–6363.
- Case DA, Darden TA, Cheatham TE III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, et al. AMBER 12. San Francisco (CA): University of California, San Francisco; 2012.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. Gaussian 09. Wallingford (CT): Gaussian, Inc.; 2009.
- Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97(40):10269–10280.
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78(8):1950–1958.
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174.
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N·log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092.
- Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Chem. 1997;23:327–341.
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33(12):889–897.
- Rastelli G, Rio Degliesposti DA, Sgobba G. M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J Comput Chem. 2010;31:797–810.
- Huang Q, Johnson TW, Bailey S, Brooun A, Bunker KD, Burke BJ, Collins MR, Cook AS, Cui JJ, Dack KN, et al. Design of potent and selective inhibitors to overcome clinical anaplastic lymphoma kinase mutations resistant to Crizotinib. J Med Chem. 2014;57(4):1170–1187.
- Basit S, Ashraf Z, Lee K, Latif M. First macrocyclic 3(rd)-generation ALK inhibitor for treatment of ALK/ROS1 cancer: clinical and designing strategy update of Lorlatinib. Eur J Med Chem. 2017;134:348–356.
- Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, McTigue M, Deng YL, Liu W, Brooun A, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking Crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci USA. 2015;112(11):3493–3498.
- Chong CR, Bahcall M, Capelletti M, Kosaka T, Ercan D, Sim T, Sholl LM, Nishino M, Johnson BE, Gray NS, et al. Identification of existing drugs that effectively target NTRK1 and ROS1 rearrangements in lung cancer. Clin Cancer Res. 2017;23(1):204–213.
- Liu S, Jiang Y, Yan R, Li Z, Wan S, Zhang T, Wu X, Hou J, Zhu Z, Tian Y, et al. Design, synthesis and biological evaluations of 2-amino-4-(1-piperidine) pyridine derivatives as novel anti Crizotinib-resistant ALK/ROS1 dual inhibitors. Eur J Med Chem. 2019;179:358–375.
- Johnson TW, Richardson PF, Bailey S, Brooun A, Burke BJ, Collins MR, Cui JJ, Deal JG, Deng YL, Dinh D, et al. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J Med Chem. 2014;57(11):4720–4744.
- Li J, Wu N, Tian Y, Zhang J, Wu S. Aminopyridyl/pyrazinyl spiro[indoline-3,4'-piperidine]-2-ones as highly selective and efficacious c-Met/ALK inhibitors. ACS Med Chem Lett. 2013;4(8):806–810.