
Abstract
A chromone-peptidyl hybrids series was synthesised and rationally repurposed towards identification of potential antileishmanial hits against visceral leishmaniasis. Three hybrids 7c, 7n, and 7h showed potential IC50 values (9.8, 10, and 12 µM, respectively) which were comparable to erufosine IC50 (9.8 µM) but lower potency than miltefosine IC50 (3.5 µM). Preliminary assessment of cytotoxicity using human THP-1 cells presented chromone-peptidyl hybrids 7c and 7n as non-cytotoxic up to 100 µM while erufosine and miltefosine had CC50 of 19.4 µM and >40 µM, respectively. In silico studies pinpointed the N-p-methoxyphenethyl substituent at the peptidyl moiety together with the oxygen-based substituted functions of the phenyl ring of the chromone moiety as crucial players in binding to LdCALP. Together, these findings present chromone-peptidyl hybrids 7c and 7n as potential and anticipated non-cytotoxic antileishmanial hit compounds for possible development of potential antileishmanial agents against visceral leishmaniasis.
Graphical Abstract
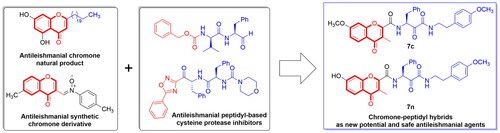
Introduction
About 1 billion humans are inhabitants in tropical and sub-tropical regions endemic to pathogens causative for neglected tropical diseases (NTDs). Unfortunately, the majority of populations in these regions involve underdeveloped poor communities and hence, development of therapeutics is largely neglected by pharmaceutical companiesCitation1. NTDs epidemiology is complicated and often influenced by different environmental factors. Many NTDs have complex life cycles, animal reservoirs, and vectors for transmission. These and other reasons make it difficult to control them for public health.
World Health Organisation (WHO) has referred to leishmaniasis as an eminent NTD. Clinical manifestation of leishmaniasis could be: (1) cutaneous leishmaniasis (CL) characterised by skin lesions; (2) mucocutaneous leishmaniasis (MCL) distinguished by lesions on the skin and mouth; or (3) visceral leishmaniasis (VL), which encompasses high fever, anaemia, hepatosplenomegaly, and immune deficiencyCitation2. WHO estimates that up to 90 000 new cases of VL and up to 1 million new cases of CL worldwide per annumCitation3. Untreated VL could be a deadly NTDCitation3. Current available VL treatments include amphotericin B, paromomycin, miltefosine, fluconazole, ketoconazole, pentamidine or pentavalent antimonial saltsCitation4–9. Unfortunately, all have limitations and/or toxicities. The safe, effective, and affordable antileishmanial drug free from drawbacks of current treatments has not been achieved yetCitation10–12.
Economically, drug-repurposing offers a potential opportunity for urgent drug development while cutting-off initial development costs. The cost of classical development of a new chemical moiety into an effective drug may require several hundred million dollars and takes up to ten yearsCitation13–20. This makes the search for new anti-leishmaniasis medications, which primarily affects poor countries, economically unviable procedure for pharmaceutical corporationsCitation21,Citation22. In several outcomes, the repurposing rational circumvented such issues and offered hits that can then be used as springboardsCitation23,Citation24. In fact, several antileishmanial drugs were obtained from such strategy. Therefore, this strategy might be viewed as a successful approach for the development of antileishmanial agents.
Calpains are a group of mammalian proteolytic proteins which are calcium-dependent cysteine proteases that are involved in several cellular functions. It was discovered that Leishmania has a calpain-like protein (CALP) whose inhibition was found to be connected to induction of Leishmania’s cell deathCitation25. Fortunately, MDL28170 () which was originally developed as an inhibitor of mammalian calpain cysteine proteases was found to show potential antileishmanial activity. MDL28170 showed a dose-dependent antiproliferative activity against L. amazonensis promastigotesCitation26,Citation27. Furthermore, another cysteine protease inhibitor derivative 2 () was discovered to possess potential antileishmanial activityCitation28. Consequently, repurposing cysteine protease inhibitors, particularly, calpain inhibitors was suggested as an interesting rational to generate new antileishmanial lead compoundsCitation26,Citation27,Citation29,Citation30.
Figure 1. Chemical structures of cysteine protease inhibitors with potential antileishmanial activity and the design rational of chromone-peptidyl hybrids as new antileishmanial agents.

Oxidative stress and production of reactive oxygen species have important roles in several diseasesCitation31–34. Consequently, antioxidants targeting oxidative stress might be helpful agents against such diseasesCitation35. Interestingly, several antileishmanial molecules were reported with antioxidant activityCitation36–38. Accordingly, chemical entities combining antioxidant activity with other antileishmanial mechanisms might be interesting candidates for investigation.
Materials and methods
Chemistry
Compounds were prepared as detailed in the supplementary information and reported in literatureCitation39.
Biological evaluations
Evaluation of antileishmanial activity and safety
Standard evaluation protocols were adopted evaluation as presented in the supplementary information and reported in literatureCitation14,Citation40,Citation41.
In silico study
The amino acid sequence of CALP of L. donovani was obtained from https://www.uniprot.org (UniProt sequence ID: E9BTE3) and applied to construct homology model of LdCALP. SWISS-MODEL and ProMod3 were utilised in template search and alignment. Validation and refinement of the generated model were performed with the co-crystallized calpain inhibitor (PDB ID: 1TL9). Docking simulations were performed utilising the model and the results were visualised via DS visualiser.
Results and discussion
Design and repurposing rational
MDL28170 (1) and compound 2 structure might be consisted of two parts; a left aryl moiety which is connected to a right peptidyl moiety (). The carbonyl-based formyl or ketone functions at the peptidyl moieties of MDL28170 (1) and compound 2, respectively are responsible for the cysteine protease inhibitory activity of these compounds. Meanwhile, chromone is an interesting privileged aryl moiety scaffold with known potential antileishmanial activityCitation42. Among reported antileishmanial chromones, 2-henicosyl-5,7-dihydroxy-4H-chromen-4-one (3, ) is a chromone-based natural product isolated from Bomarea setacea and bears a long alkyl chain at position 2Citation43. Noteworthy, 2-henicosyl-5,7-dihydroxy-4H-chromen-4-one (3, ) has antioxidant activity and protection against infection-associated excessive stress has been claimed important in leishmaniasisCitation43. The long alkyl chain might suggest that antileishmanial activity could be sterically tolerant to large substituents at position 2. In addition, chromone derivative 4 having p-tolyl-nitrone substituent at position 3 was also found to possess potential antileishmanial activityCitation44. In fact, the literature reported scare efforts to develop chromone hybrids as antileishmanial agents such as chromone-quinoline hybrids 5 and chromone-triclosan hybrids 6 ()Citation45,Citation46.
Hybridisation is a drug design strategy recognised capable of generating potential bioactive compoundsCitation47–51. Considering the above-mentioned data, we postulated designed chromone-peptidyl hybrid structures 7 as possible novel antileishmanial hit compounds. As shown in , the design involved replacement of the aryl moiety of MDL28170 and compound 2 by the privileged chromone moiety. As structural information gained from antileishmanial natural product 3 suggested that position 2 is sterically tolerant to large substituents, the peptidyl moiety was placed at this position and its N-terminal was coupled to the chromone ring through amide moiety while the p-tolyl-nitrone moiety at position 3 of compound 4 was replaced by the smaller methyl moiety to maintain the presence of only one large substituent at the right side. As the phenyl ring of the chromone moiety of compound 3 has hydroxyl substituents, diverse oxygen-based substituted functions and patterns were introduced at the phenyl ring of the chromone moiety in the designed compounds to explore possible relationships between structure and activity. Simultaneously, the peptidyl moiety was designed based on the privileged α-ketoamide function in the peptidyl moiety as a carbonyl-based function known for inhibition of calpain cysteine protease. In addition, the C-terminal of the peptidyl moiety was converted into a primary amide or one of proposed diverse secondary amides to explore possible structure activity relationship. Interestingly, we found that the targeted compounds according to the proposed design for the new antileishmanial agents conform to the structure of some compounds that have been previously investigated as agents against anti-neurodegenerative diseases eliciting inhibition of human calpain and antioxidant activity. Hence, we have synthesised these compounds and evaluated their antileishmanial activity. In this lieu, the current effort might be considered a rational repurposing.
Chemistry
Towards a concise synthesis of targeted chromone-peptidyl hybrids, the synthesis of target compounds (7a–q) was conducted in two or three linear steps as depicted in Scheme 1. The intermediate β-amino-α-hydroxy amides (10a–l) were obtained through a coupling reaction between the appropriate chromone-2-carboxylic acid derivative (8a–e)Citation39 and the appropriate β-amino-α-hydroxy amide (9a–c), using 1-ethyl-3–(3-dimethylaminopropyl)carbodiimide.HCl (EDC)/1-hydroxybenzotriazole (HOBt) coupling system. Subsequent oxidation of (10a–l) under Dess-Martin periodinane (DMP) conditionsCitation52 afforded the corresponding chromone-peptidyl hybrids (7a–l) in good yields. Concerning the chromone-peptidyl hybrids (7m–q) carrying free hydroxyl group(s) on the phenyl ring, their synthesis was achieved through acidic deprotection of the methoxymethoxy groups using methanolic HCl.
Scheme 1. Synthesis of target chromone-peptidyl hybrids (7a–q). Reagents and conditions: (i) EDC, HOBt, DMF, stir 0 °C-rt, 2h; (ii) Dess-Martin periodinane, DMF, 0 °C-rt, 10% Na2S2O3 solution, 2h (7a–l); (iii) 1% methanolic HCl, 70 °C, 2 h (7m–q).

In vitro evaluations
Evaluation of antileishmanial activity
In vitro L. donovani promastigotes-based evaluation model
The activity of the synthesised target compounds against the promastigote stage of L. donovani was performed using an established in vitro promastigotes-based modelCitation14. For each tested compound, two different concentrations (50 and 25 µM) were employed. The highest 50 µM concentration was used to exclude almost inactive compounds, while the 25 µM concentration was used to indicate the most potent compounds. Miltefosine and erufosine served as reference standard drugs utilising a resazurin-based assayCitation53. The results are presented in .
Table 1. % Growth inhibition of L. donovani promastigotes elicited by chromone peptidyl hybrids (7a–q).
Analysis of the results showed that among hybrids 7a–c having 7-methoxy-substituted chromone moiety the N-p-methoxyphenethyl derivative 7c and the primary amide derivative 7a were active while the N-benzyl derivative 7b elicited a poor activity. Switching from 7-methoxy to the larger substituent 7-methoxymethoxy-substituted chromone moiety did not improve the activity of the N-benzyl derivative 7d. Replacement of the 7-methoxy-substituted chromone moiety by 6,7-dimethoxy-substituted chromone moiety showed that the N-p-methoxyphenethyl derivative 7h maintains a high potential activity at both tested concentrations while the primary amide derivative 7f lost the activity and the N-benzyl derivative 7 g still have poor activity. Increasing the size of the substituent at position 6 of the chromone moiety by employing 6-methoxymethoxy-7-methoxy-substituted chromone moiety resulted in enhancing the activity of the N-benzyl derivative 7i while preserving the activity of N-p-methoxyphenethyl derivative 7j. However, increasing the size of substituents at both positions 6 and 7 of the chromone moiety by using 6,7-di(methoxymethoxy)-substituted chromone moiety was detrimental to the activity of the N-p-methoxyphenethyl derivative 7 l but preserved the activity of the N-benzyl derivative 7k. Investigating the activity of hybrids having 7-hydroxy-substituted chromone moiety 7m and 7n in relation to hybrids 7b and 7c having 7-methoxy-substituted chromone show no significant difference in activity of N-benzyl or N-p-methoxyphenethyl derivatives indicating minimum impact of such molecular modification of the chromone moiety on the activity. Meanwhile, both N-benzyl and N-p-methoxyphenethyl derivatives 7o and 7p possessing the 6-hydroxy-7-methoxy-substituted chromone moiety showed equivalent inhibition values. However, the activity of 6,7-dihydroxy-substituted chromone moiety-based N-benzyl derivative 7q was poor. Noteworthy, the poorly active 6,7-dihydroxy-substituted chromone derivative 7q was reported to have a potential antioxidant activityCitation39. Meanwhile the phenolic hydroxy moiety responsible for the antioxidant activity was lacking from several highly active compounds such as 7h and 7c. These findings suggest that the antioxidant activity, despite might be a contributor to the activity of some compounds such as 7n, it is not the major contributor to the activity of other tested compounds. On the other side, the N-substituent might be the most influential on activity with the N-p-methoxyphenethyl derivatives are the most active.
In vitro potency evaluation against L. donovani promastigotes
Seven hybrid compounds (7a, 7c, 7h, 7j, 7n, 7o and 7p) elicited the best inhibitory activity at the tested two doses and, hence, were selected for further potency evaluation in comparison with miltefosine and erufosine standard drugs to assess their potency as potential hit compounds. This evaluation was conducted adopting the more rigorous dose-dependent activity evaluation model to unveil the potency of the hybrids that showed inhibition by more than 80% in the previous double concentrations assay. The lower doses used for the generation of the dose-response curve would distinguish between potent and less potent compounds. The determined IC50 values for inhibition of the growth of L. donovani were illustrated in . While the primary amide derivative 7a and the N-benzyl derivative 7o were impotent, the three N-p-methoxyphenethyl derivatives 7c, 7h, and 7n were potent showing IC50 of 9.8, 12, and 10 µM, respectively which is very close to the potency of erufosine. It might be deduced that these three hybrids based on 7-methoxychromone, 6,7-dimethoxychromone and 7-hydroxychromone moieties might be potential hit compounds. Meanwhile, N-p-methoxyphenethyl derived hybrids 7j, and 7p based on 6-methoxymethoxy-7-methoxychromone and 6-hydroxy-7-methoxychromone moieties were much less potent (). These findings suggest that derivatives 7c, 7h, and 7n might serve as potential hit compounds for further development.
Table 2. Determined IC50 values against L. donovani promastigotes for the most active chromone peptidyl hybrids.
In vitro preliminary safety assessment
Towards preliminary assessment of safety of the promising N-p-methoxyphenethyl derivatives 7c, 7h, and 7n, which emerged as potential inhibitors of leishmania in above assessments, the human monocytic THP-1 cell line was employed. A hit compound should not trigger cytotoxicity of the human cell to be a safe compound. Evaluation was conducted in comparison with the standard drugs miltefosine and erufosine. As displayed in , the results showed that among the tested derivatives, hybrids 7c and 7n lacking substituents at position 6 of the chromone moiety were not toxic up to 100 µM concentrations and thus much safer than both miltefosine and erufosine. Meanwhile, hybrid 7h, whose chromone moiety have methoxy substituent at position 6 showed a CC50 value of 29.1 µM indicating that it was safer than erufosine but more toxic than miltefosine. Accordingly, it might be concluded that hybrids 7c and 7n could serve as non-toxic potential antileishmanial hit compounds for further development.
Table 3. In vitro evaluation of cytotoxicity on THP-1 cell of compounds 7c, 7h, and 7n.
In silico simulation study
A molecular docking study was conducted to get molecular insights of potential hit compounds. Since no 3D-structure is known for calpain-like cysteine peptidase of L. donovani (LdCALP), homology modelling was performed to establish a valid 3D-model that might be recruited for further in silico studies which were performed in comparison with reference compound MDL28170.
Homology modelling
Multiple sequence alignment of CALP sequences of L. donovani, L. infantum, L. mexicana, T. cruzi, and T. brucei (UniProt sequences ID: E9BTE3, A4ICQ6, E9ASJ8, V5D9W4, and A0A3L6KUN6, respectively) revealed that CALP is highly conserved in Leishmania species but not in Trypanosoma species (Supplementary Figure S1) which might be in accordance with literatureCitation54. To construct a 3D model, a template was searched for LdCALP (UniProt sequence ID: E9BTE3) using HHblits of SWISS-MODEL which suggested the calpain protease (PDB ID: 1TL9) as a best fit with 25% sequence identity (). Applying these results, a homology model was built with ProMod3Citation55–57. Estimation of the quality of the built model, Ramachandran plot was addressed which showed that more than 95% of the amino acid residues were in the favoured regions (). This result suggested that the established LdCALP homology model had acceptable quality and might be utilised for further in silico studies.
Figure 2. Sequence alignment and Ramachandran plot of the generated homology model of CALP of L. donovani (A) Sequence alignment against calpain I protease core (PDB code: 1TL9); (B) Ramachandran plot of amino acid residues of the generated homology model indicated that most of them are in the favoured regions.

Molecular docking study
Molecular modelling study was performed to predict the possible binding modes of the three promising compounds (7c, 7h, and 7n) having highest in vitro potency against L. donovani promastigotes with LdCALP after refinement with brief dynamic simulation with co-crystallized inhibitor leupeptin (PDB code: 1TL9). As illustrated in , the binding site is in a cleft between an α-helices-rich domain and a β-sheets-rich domain. Docking was performed employing MDL28170 (1) as reference ligand.
Figure 3. Position of active site in three-dimensional protein structure showing co-crystallized inhibitor leupeptin in the active site.

The results showed that compounds 7c, 7h, and 7n as well as the reference ligand docked successfully into the substrate pocket of LdCALP (. While docking predicted the reference MDL28170 (1) to possess an energy score of −7.73 Kcal/mol, compounds 7c, 7h, and 7n had slightly better scores of −8.18, −8.64 and −8.07 Kcal/mol, respectively. Beyond docking scores, investigation of the binding modes of the docked structures revealed that they could establish a network of favoured interactions with amino acid residues in the binding pocket. The reference molecule, MDL28170 (1), formed two hydrogen bond donor interactions by its amide and aldehyde moieties with Leu609 and Ser691, respectively. In addition, favourable π-π, π-sulfur, π-alkyl, and carbon-hydrogen interactions were established with Ala662, Val674, Gly689, Thr690, Cys607, and His678 (). Whereas compound 7c formed favourable hydrogen bonding via its carbonyl group of the right peptidyl moiety with Leu609 in addition to two carbon-hydrogen interactions between its two other carbonyls and Gly689 and Ser691 (). Compound 7c was also capable of forming another carbon-hydrogen interaction between the alkyl side chain of 4-methoxyphenethyl moiety and Gly510. Moreover, it was predicted that 7c could establish π-π, π-sigma, π-alkyl, and alkyl interactions with His678, Cys607, Ala662, and Ala675. On the other hand, compound 7h was predicted to establish a network of favourable binding interactions including a π-sulfur interaction between its 4-methoxyphenethyl moiety and Cys607 (). In addition, it was predicted that 7h would form two favourable hydrogen bonds with Arg687 and Gly689 as well as two carbon-hydrogen interactions with Gly689 and Thr669. Furthermore, various π-sigma, alkyl, and π-alkyl interactions were predicted to form between 7h and Thr669, Thr610, Ala662, Ala679, Val672, Val674, and Trp712. Concerning compound (7n), it was predicted to form hydrogen bond between the 4-methoxyphenethyl moiety and His793 in addition to another hydrogen bond between its NH group and Ser608 (). Moreover, a carbon-hydrogen interaction was formed between 4-methoxyphenethyl moiety and Ser691. Furthermore, π-sigma, π-π, alkyl, and π-alkyl interactions were established with Ser691, His678, Cys607, Ala611, Ala672, Ala675, and Phe659. These predicted binding modes of the investigated compounds provided insights about the structural features that might have a role in binding with the active site of LdCALP and contribute to increased potency of these compounds. It was observed that 4-methoxyphenethyl moiety substitution at the terminal amide together with the oxygen-based substituted functions at the phenyl ring of the privileged chromone moiety in addition to the peptide moiety contributed positively to the binding of the investigated compounds with the active site of LdCALP.
Figure 4. Predicted binding modes of compounds 7c, 7h, and 7n as well as the reference ligand MDL28170 with LdCALP: (A) Calculated docking pose of the reference compound MDL28170 (1) in 3D (left) and 2D view (right) into the substrate pocket of LdCALP; (B) Calculated docking pose of compound 7c in 3D (left) and 2D view (right) into the substrate pocket of LdCALP; (C) Calculated docking pose of compound 7h in 3D (left) and 2D view (right) into the substrate pocket of LdCALP; (D) Calculated docking pose of compound 7n in 3D (left) and 2D view (right) into the substrate pocket of LdCALP.

Conclusion
Rational repurposing of designed chromone-peptidyl hybrids successfully afforded two potentially active and possibly non-cytotoxic antileishmanial hit compounds 7c and 7n. Initially, a rationally designed chromone-peptidyl hybrids was performed that suggested structures previously reported in literature towards development of neurodegenerative diseases. The targeted compounds were concisely resynthesized in two or three linear synthetic steps and investigated for repurposing employing as model of L. donovani promastigotes. The results suggested that the antioxidant activity is not the major activity mediator despite, it is possible to be partially involved in the activity of some compounds. Meanwhile, analysis of structure-activity relationship suggested an influential role for the substituents at the terminal amide moiety of the peptidyl moiety. The three compounds 7c, 7n, and 7h that showed comparable potencies to erufosine drug were subjected to preliminary safety assessment which suggested that compounds 7c and 7n bearing no substituents at position 6 of the chromone moiety could be non-toxic to human cells. Towards getting insights into molecular interactions that might assist future direction, in silico study that was performed that pointed to the N-p-methoxyphenethyl substituent in the peptidyl moiety together with the oxygen-based substituted functions at the phenyl ring of the privileged chromone moiety as crucial influencers establishing networks of favourable binding interactions with the binding site of LdCALP. This suggested that these moieties are key structural features for the future development of the investigated compounds. As multiple sequence alignment showed that CALPs are highly conserved in Leishmania species, it might be anticipated that tested compounds might have pan-antileishmanial activity. However, this has to be evaluated in a future study. Together, these findings present chromone-peptidyl hybrid compounds 7c and 7n as antileishmanial hit compounds for further development of potential antileishmanial agents.
Supplemental Material
Download PDF (480.6 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Joshi G, Quadir SS, Yadav KS. Road map to the treatment of neglected tropical diseases: nanocarriers interventions. J Control Release. 2021;339:51–74.
- Bilgic-Temel A, Murrell DF, Uzun S. Cutaneous leishmaniasis: a neglected disfiguring disease for women. Int J Womens Dermatol. 2019;5(3):158–165.
- Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, Boer Md. Leishmaniasis Worldwide and Global Estimates of Its Incidence. PLoS One. 2012;7(5):e35671.
- Tuon FF, Dantas LR, de Souza RM, Ribeiro VST, Amato VS. Liposomal drug delivery systems for the treatment of leishmaniasis. Parasitol Res. 2022;121(11):3073–3082.
- Pinheiro AC, de Souza MVN. Current leishmaniasis drug discovery. RSC Med Chem. 2022;13(9):1029–1043.
- Madusanka RK, Silva H, Karunaweera ND. Treatment of Cutaneous Leishmaniasis and insights into species-specific responses: a narrative review. Infect Dis Ther. 2022;11(2):695–711.
- de Santana NS, de Oliveira de Siqueira LB, Santos-Oliveira R, dos Santos Matos AP, Ricci-Júnior E. Nanoparticles for the treatment of visceral leishmaniasis: review. J Nanoparticle Res. 2023;25:24.
- Khabsa J, Jain S, El-Harakeh A, Rizkallah C, Pandey DK, Manaye N, Honein-AbouHaidar G, Halleux C, Dagne DA, Akl EA. Stakeholders’ views and perspectives on treatments of visceral leishmaniasis and their outcomes in HIV-coinfected patients in East Africa and South-East Asia: a mixed methods study. PLoS Negl Trop Dis. 2022;16(8):e0010624.
- Kumari S, Kumar V, Tiwari RK, Ravidas V, Pandey K, Kumar A. Amphotericin B: a drug of choice for Visceral Leishmaniasis. Acta Trop. 2022;235:106661.
- Yeshaw Y, Tsegaye AT, Nigatu SG. Incidence of mortality and its predictors among adult Visceral Leishmaniasis patients at the university of Gondar hospital: a retrospective cohort study. Infect Drug Resist. 2020;13:881–891.
- Scarpini S, Dondi A, Totaro C, Biagi C, Melchionda F, Zama D, Pierantoni L, Gennari M, Campagna C, Prete A, et al. Visceral Leishmaniasis: epidemiology, diagnosis, and treatment regimens in different geographical areas with a focus on pediatrics. Microorganisms. 2022;10(10):1887.
- Wijnant G-J, Dumetz F, Dirkx L, Bulté D, Cuypers B, Van Bocxlaer K, Hendrickx S. Tackling drug resistance and other causes of treatment failure in Leishmaniasis. Front Trop Dis. 2022;3:837460.
- Jourdan J-P, Bureau R, Rochais C, Dallemagne P. Drug repositioning: a brief overview. J Pharm Pharmacol. 2020;72(9):1145–1151.
- Hassan AHE, Phan T-N, Choi Y, Moon S, No JH, Lee YS. Design, rational repurposing, synthesis, in vitro evaluation, homology modeling and in silico study of Sulfuretin Analogs as Potential Antileishmanial hit compounds. Pharmaceuticals. 2022;15(9):1058.
- Farag AK, Hassan AHE, Ahn BS, Park KD, Roh EJ. Reprofiling of pyrimidine-based DAPK1/CSF1R dual inhibitors: identification of 2,5-diamino-4-pyrimidinol derivatives as novel potential anticancer lead compounds. J Enzyme Inhib Med Chem. 2020;35(1):311–324.
- Braga SS. Multi-target drugs active against leishmaniasis: a paradigm of drug repurposing. Eur J Med Chem. 2019;183:111660.
- Rai P, Arya H, Saha S, Kumar D, Bhatt TK. Drug repurposing based novel anti-leishmanial drug screening using in-silico and in-vitro approaches. J Biomol Struct Dyn. 2022;40(21):10812–10820.
- Tabrez S, Rahman F, Ali R, Muhammad F, Alshehri BM, Alaidarous MA, Banawas S, Dukhyil AAB, Rub A. Repurposing of FDA-approved drugs as inhibitors of sterol C-24 methyltransferase of Leishmania donovani to fight against leishmaniasis. Drug Dev Res. 2021;82(8):1154–1161.
- Farag AK, Hassan AHE, Chung K-S, Lee J-H, Gil H-S, Lee K-T, Roh EJ. Diarylurea derivatives comprising 2,4-diarylpyrimidines: discovery of novel potential anticancer agents via combined failed-ligands repurposing and molecular hybridization approaches. Bioorg Chem. 2020;103:104121.
- Hassan AHE, Yoo SY, Lee KW, Yoon YM, Ryu HW, Jeong Y, Shin J-S, Kang S-Y, Kim S-Y, Lee H-H, et al. Repurposing mosloflavone/5,6,7-trimethoxyflavone-resveratrol hybrids: discovery of novel p38-α MAPK inhibitors as potent interceptors of macrophage-dependent production of proinflammatory mediators. Eur J Med Chem. 2019;180:253–267.
- Berenstein AJ, Magariños MP, Chernomoretz A, Agüero F. A multilayer network approach for guiding drug repositioning in neglected diseases. PLoS Negl Trop Dis. 2016;10(1):e0004300.
- Andrade-Neto VV, Cunha-Junior EF, Faioes VdS, Martins TP, Silva RL, Leon LL, Torres-Santos EC. Leishmaniasis treatment: update of possibilities for drug repurposing. Front Biosci. 2018;23:967–996.
- Cutinho PF, Shankar RC, Anand A, Roy J, Mehta CH, Nayak UY, Murahari M. Hit identification and drug repositioning of potential non-nucleoside reverse transcriptase inhibitors by structure-based approach using computational tools (part II). J Biomol Struct Dyn. 2020;38(13):3772–3789.
- Ferreira Letícia T, Rodrigues J, Cassiano Gustavo C, Tavella Tatyana A, Tomaz Kaira Cristina P, Baia-da-Silva Djane C, Souza Macejane F, Lima Marilia Nunes do N, Mottin M, Almeida Ludimila D, et al. Computational chemogenomics drug repositioning strategy enables the discovery of epirubicin as a new repurposed hit for Plasmodium falciparum and P. vivax. Antimicrob Agents Chemother. 2020;64(9):e02041-02019.
- Casanova M, Gonzalez IJ, Sprissler C, Zalila H, Dacher M, Basmaciyan L, Späth GF, Azas N, Fasel N. Implication of different domains of the Leishmania major metacaspase in cell death and autophagy. Cell Death Dis. 2015;6(10):e1933.
- d’Avila-Levy CM, Marinho FA, Santos LO, Martins JL, Santos ALS, Branquinha MH. Antileishmanial activity of MDL 28170, a potent calpain inhibitor. Int J Antimicrob Agents. 2006;28(2):138–142.
- Marinho FA, Gonçalves KCS, Oliveira SSC, Gonçalves DS, Matteoli FP, Seabra SH, Oliveira ACS, Bellio M, Oliveira SS, Souto-Padrón T, et al. The Calpain Inhibitor MDL28170 induces the expression of apoptotic markers in Leishmania amazonensis Promastigotes. PLoS One. 2014;9(1):e87659.
- Steert K, Berg M, Mottram JC, Westrop GD, Coombs GH, Cos P, Maes L, Joossens J, Van der Veken P, Haemers A, et al. α-Ketoheterocycles as inhibitors of Leishmania mexicana Cysteine protease CPB. ChemMedChem. 2010;5(10):1734–1748.
- Marinho FA, Sangenito LS, Oliveira SSC, De Arruda LB, D'Ávila-Levy CM, Santos ALS, Branquinha MH. The potent cell permeable calpain inhibitor MDL28170 affects the interaction of Leishmania amazonensis with macrophages and shows anti-amastigote activity. Parasitol Int. 2017;66(5):579–583.
- Ennes-Vidal V, Menna-Barreto RFS, Branquinha MH, Dos Santos ALS, D'Avila-Levy CM. Why calpain inhibitors are interesting leading compounds to search for new therapeutic options to treat leishmaniasis? Parasitology. 2017;144(2):117–123.
- Hassan AHE, Kim HJ, Park K, Choi Y, Moon S, Lee CH, Kim YJ, Cho SB, Gee MS, Lee D, et al. Synthesis and biological evaluation of O6-Aminoalkyl-Hispidol analogs as multifunctional monoamine oxidase-B inhibitors towards management of neurodegenerative diseases. Antioxidants. 2023;12(5):1033.
- Lee H-H, Shin J-S, Chung K-S, Kim J-M, Jung S-H, Yoo H-S, Hassan AHE, Lee JK, Inn K-S, Lee S, et al. 3′,4′-Dihydroxyflavone mitigates inflammatory responses by inhibiting LPS and TLR4/MD2 interaction. Phytomedicine. 2023;109:154553.
- Hassan AHE, Kim HJ, Gee MS, Park J-H, Jeon HR, Lee CJ, Choi Y, Moon S, Lee D, Lee JK, et al. Positional scanning of natural product hispidol’s ring-B: discovery of highly selective human monoamine oxidase-B inhibitor analogues downregulating neuroinflammation for management of neurodegenerative diseases. J Enzyme Inhib Med Chem. 2022;37(1):768–780.
- Gil H-S, Lee J-H, Farag AK, Hassan AHE, Chung K-S, Choi J-H, Roh E-J, Lee K-T. AKF-D52, a synthetic Phenoxypyrimidine-urea derivative, triggers extrinsic/intrinsic apoptosis and cytoprotective autophagy in human non-small cell lung cancer cells. Cancers. 2021;13(22):5849.
- Forman HJ, Zhang H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat Rev Drug Discov. 2021;20(9):689–709.
- Miranda-Sapla MM, Tomiotto-Pellissier F, Assolini JP, Carloto ACM, Bortoleti BTdS, Gonçalves MD, Tavares ER, Rodrigues JHdS, Simão ANC, Yamauchi LM, et al. trans-Chalcone modulates Leishmania amazonensis infection in vitro by Nrf2 overexpression affecting iron availability. Eur J Pharmacol. 2019;853:275–288.
- Tomiotto-Pellissier F, Alves DR, Miranda-Sapla MM, de Morais SM, Assolini JP, da Silva Bortoleti BT, Gonçalves MD, Cataneo AHD, Kian D, Madeira TB, et al. Caryocar coriaceum extracts exert leishmanicidal effect acting in promastigote forms by apoptosis-like mechanism and intracellular amastigotes by Nrf2/HO-1/ferritin dependent response and iron depletion: Leishmanicidal effect of Caryocar coriaceum leaf exracts. Biomed Pharmacother. 2018;98:662–672.
- Cataneo AHD, Tomiotto-Pellissier F, Miranda-Sapla MM, Assolini JP, Panis C, Kian D, Yamauchi LM, Colado Simão AN, Casagrande R, Pinge-Filho P, et al. Quercetin promotes antipromastigote effect by increasing the ROS production and anti-amastigote by upregulating Nrf2/HO-1 expression, affecting iron availability. Biomed Pharmacother. 2019;113:108745.
- Kim SH, Lee YH, Jung SY, Kim HJ, Jin C, Lee YS. Synthesis of chromone carboxamide derivatives with antioxidative and calpain inhibitory properties. Eur J Med Chem. 2011;46(5):1721–1728.
- Hassan AHE, Phan T-N, Yoon S, Lee CJ, Jeon HR, Kim S-H, No JH, Lee YS. Pyrrolidine-based 3-deoxysphingosylphosphorylcholine analogs as possible candidates against neglected tropical diseases (NTDs): identification of hit compounds towards development of potential treatment of Leishmania donovani. J Enzyme Inhib Med Chem. 2021;36(1):1922–1930.
- Phan T-N, Baek K-H, Lee N, Byun SY, Shum D, No JH. In vitro and in vivo activity of mTOR kinase and PI3K inhibitors Against Leishmania donovani and Trypanosoma brucei. Molecules. 2020;25(8):1980.
- Silva CFM, Pinto DCGA, Fernandes PA, Silva AMS. Evolution of chromone-like compounds as potential antileishmanial agents, through the 21st century. Expert Opin Drug Discov. 2020;15(12):1425–1439.
- Cardona-Galeano W, Yepes AF, Quintero-Saumeth J, Robledo SM, Alzate F, Rojano B. A biologically active chromone from Bomarea setacea (alstroemeriaceae): Leishmanicidal, antioxidant and multilevel computational studies. ChemistrySelect. 2022;7(45):e202203852.
- Mallick S, Dutta A, Ghosh J, Maiti S, Mandal AK, Banerjee R, Bandyopadhyay C, Pal C. protective therapy with Novel chromone derivative against Leishmania donovani infection induces Th1 response in vivo. Chemotherapy. 2011;57(5):388–393.
- Otero E, Vergara S, Robledo SM, Cardona W, Carda M, Vélez ID, Rojas C, Otálvaro F. Synthesis, Leishmanicidal and cytotoxic activity of Triclosan-Chalcone, Triclosan-chromone and Triclosan-coumarin hybrids. Molecules. 2014;19(9):13251–13266.
- Coa JC, García E, Carda M, Agut R, Vélez ID, Muñoz JA, Yepes LM, Robledo SM, Cardona WI. Synthesis, leishmanicidal, trypanocidal and cytotoxic activities of quinoline-chalcone and quinoline-chromone hybrids. Med Chem Res. 2017;26(7):1405–1414.
- Kim S-Y, Hassan AHE, Chung K-S, Kim S-Y, Han H-S, Lee H-H, Jung S-H, Lee K-Y, Shin J-S, Jang E, et al. Mosloflavone-resveratrol hybrid TMS-HDMF-5z exhibits potent in vitro and in vivo anti-inflammatory effects through NF-κB, AP-1, and JAK/STAT inactivation. Front Pharmacol. 2022;13:857789.
- Hong JY, Chung K-S, Shin J-S, Lee J-H, Gil H-S, Lee H-H, Choi E, Choi J-H, Hassan AHE, Lee YS, et al. The anti-proliferative activity of the Hybrid TMS-TMF-4f compound against human cervical cancer involves apoptosis mediated by STAT3 inactivation. Cancers. 2019;11(12):1927.
- Hassan AHE, Choi E, Yoon YM, Lee KW, Yoo SY, Cho MC, Yang JS, Kim HI, Hong JY, Shin J-S, et al. Natural products hybrids: 3,5,4′-Trimethoxystilbene-5,6,7-trimethoxyflavone chimeric analogs as potential cytotoxic agents against diverse human cancer cells. Eur J Med Chem. 2019;161:559–580.
- Alam MM, Hassan AHE, Lee KW, Cho MC, Yang JS, Song J, Min KH, Hong J, Kim D-H, Lee YS. Design, synthesis and cytotoxicity of chimeric erlotinib-alkylphospholipid hybrids. Bioorg Chem. 2019;84:51–62.
- Alam MM, Hassan AHE, Kwon YH, Lee HJ, Kim NY, Min KH, Lee S-Y, Kim D-H, Lee YS. Design, synthesis and evaluation of alkylphosphocholine-gefitinib conjugates as multitarget anticancer agents. Arch Pharm Res. 2018;41(1):35–45.
- Lee KY, Seob Lee K, Jin C, Lee YS. Design and synthesis of calpain inhibitory 6-pyridone 2-carboxamide derivatives. Eur J Med Chem. 2009;44(3):1331–1334.
- Kulshrestha A, Bhandari V, Mukhopadhyay R, Ramesh V, Sundar S, Maes L, Dujardin JC, Roy S, Salotra P. Validation of a simple resazurin-based promastigote assay for the routine monitoring of miltefosine susceptibility in clinical isolates of Leishmania donovani. Parasitol Res. 2013;112(2):825–828.
- Branquinha MH, Marinho FA, Sangenito LS, Oliveira SSC, Goncalves KC, Ennes-Vidal V, d‘Avila-Levy CM, Santos ALS. Calpains: potential targets for alternative chemotherapeutic intervention against human pathogenic Trypanosomatids. Curr Med Chem. 2013;20(25):3174–3185.
- Studer G, Tauriello G, Bienert S, Biasini M, Johner N, Schwede T. ProMod3 – a versatile homology modelling toolbox. PLoS Comput Biol. 2021;17(1):e1008667.
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303.
- Bienert S, Waterhouse A, de Beer TAP, Tauriello G, Studer G, Bordoli L, Schwede T. The SWISS-MODEL repository – new features and functionality. Nucleic Acids Res. 2017;45(D1):D313–D319.