
Abstract
Nanotechnology-based strategies can overcome the limitations of conventional cancer therapies. Hence, novel series of pyrimidine Schiff bases (4–9) were employed in the synthesis of selenium nanoparticle forms (4NPs–9NPs). All selenium nano-sized forms exerted greater inhibitions than normal-sized compounds, far exceeding 5-fluorouracil activity. Compound 4 showed effective anti-proliferative activity against MCF-7(IC50 3.14 ± 0.04 µM), HepG-2(IC50 1.07 ± 0.03 µM), and A549(IC50 1.53 ± 0.01 µM) cell lines, while its selenium nanoform 4NPs showed excellent inhibitory effects, with efficacy increased by 96.52%, 96.45%, and 93.86%, respectively. Additionally, 4NPs outperformed 4 in selectivity against the Vero cell line by 4.5-fold. Furthermore, 4NPs exhibited strong inhibition of CDK1(IC50 0.47 ± 0.3 µM) and tubulin polymerase(IC50 0.61 ± 0.04 µM), outperforming 4 and being comparable to roscovitine (IC50 0.27 ± 0.03 µM) and combretastatin-A4(IC50 0.25 ± 0.01 µM), respectively. Moreover, both 4 and 4NPs arrested the cell cycle at G0/G1 phase and significantly forced the cells towards apoptosis. Molecular docking demonstrated that 4 and 4NPs were able to inhibit CDK1 and tubulin polymerase binding sites.
Introduction
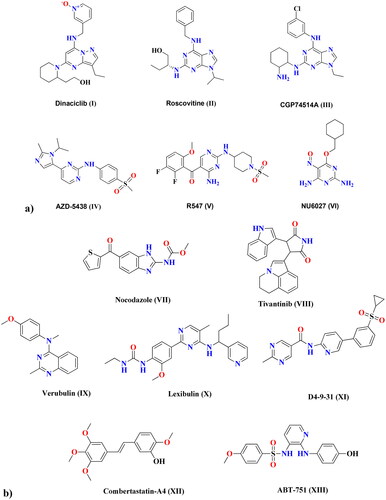
Increased mortality due to cancer is alarming this occurs on account of the ability of cancer cells to develop resistance to anti-proliferative agents in a variety of ways. Combining different drugs that target different pathways (dual inhibitors), as well as designing targeted therapies with high selectivity to cancer cells rather than healthy cells, are key to overcoming this resistance. Metal nanoparticles (NPs) can deliver chemotherapeutic-loaded NPs and are increasingly being used as alternatives to current tumour therapies, including radiotherapy, surgical therapy, and traditional chemotherapy, due to their ability to overcome many of the drawbacks of traditional treatments, such as limited water solubility, non-specific biodistribution, and low bioavailabilityCitation1. One of the most significant characteristics of cancer cells is dysregulated division, which results in aberrant cell growth. Consequently, therapeutic targets that prevent cell proliferation would be successful anti-proliferative agentsCitation2. Cell cycle regulators known as cyclin-dependent kinases (CDKs) are essential for transcription and differentiation. Catalytic cyclin subunits are responsible for the upregulation of CDK activity, while endogenous CDK inhibitors (CKIs) are responsible for their downregulation. The interaction between cyclin, CDKs, and CKIs is crucial for cell growth. Members of the CDK family play a variety of roles within cellsCitation3. In particular, CDK1 is important in the mitotic progression of the cell through the S and G2 phases, which depends on the cyclin A-CDK1 complex formationCitation4,Citation5. The development of the cyclin B-CDK1 complex is essential for the cell to proceed through mitosisCitation6. Even in the absence of the interphase CDKs (CDK2, 3, 4, and 6), it is possible for CDK1 to drive the necessary events during a cell cycle. This explains why CDK1 inhibitors are essential therapeutic targets when designing anti-proliferative agents. Structurally, CDK1 inhibitors are of various central cores, including; flavonesCitation7, benzimidazolesCitation8, and thiazolonesCitation9. Furthermore, pyrimidines represent strategic motifs in many CDK1 inhibitors, with high activity and IC50 values in the nanomolar range, including dinaciclib (I)Citation10, roscovitine (II)Citation11, and CGP74514A (III)Citation12, as examples of purines and purine isosteres. As well, AZD-5438 (IV)Citation13, R547 (V)Citation14, and NU6027 (VI)Citation15 are other examples of pyrimidines CDK1 inhibitors with potent anti-proliferative activity. shows different cyclin-dependent kinase 1 inhibitors that share structural features of a central heterocyclic parent core attached to aromatic hydrophobic moieties via different linkers. Microtubules are highly dynamic protein filaments that undergo structural changes during cell division. They assemble the mitotic spindle and carry out modifications needed to advance the cell cycle all the way to the generation of daughter cells. Microtubules are known to be attractive and promising targets for developing of anti-proliferative drugs. New therapeutic approaches are based on the understanding of the regulation and dysregulation of microtubule dynamics and their role in pathological processesCitation16. Microtubules and CDK1 are interrelated, where microtubular spindle formation is dependent on the phosphorylation/dephosphorylation of CDK1. During the interphase, the inactive form of CDK1 permits the aggregation α and β-tubulin dimer causing mitotic spindle stabilisation. Once activated at mitosis, CDK1 destabilises microtubulin by inhibiting microtubule-associated proteins, causing the disappearance of mitotic spindles in the cytoplasmCitation17. Five exogenous substances were identified as anti-microtubule drugs that act at various binding sites, including vinca alkaloidCitation18, maytansineCitation19, taxane, laulimalideCitation20, and colchicineCitation21. Laulimalide, vinca, maytansine, and taxane sites are localised on α-β tubulin heterodimer, while the colchicine site is localised on the tubulin intradimer interfaceCitation22. The colchicine binding site (CBS) has received the most attention in research. Agents that target the β-subunit of curved tubulin and prevent it from acquiring a straight structure can inhibit the assembly of microtubules by interacting at CBSCitation21. depicts various anti-microtubule and CBSI drugs. Nocodazole (VII) and tivantinib (VIII) are reported to have binary function inhibitors that act as anti-microtubules through binding at the CBS, as well as target cancer-related kinasesCitation23. Furthermore, pyrimidine-based verubulin (IX)Citation24, lexibulin (X)Citation23, and D4-9-31 (XI)Citation25 have been found to be potent colchicine binding site inhibitors (CBSIs). Structurally, well-known compounds that bind at the CBS share some common features, including two aryl systems linked via different groups. Combretastatin-A4 (XII) has two aryls linked via an alkene linker, while ABT-751 (XIII) has a sulphonamide linkerCitation26. In the case of verubulin (IX), an amino linker connects both aryl systems, while nocodazole (VII) has a ketone linkerCitation27.
Figure 1. (a) Cyclin-dependent kinase 1 (CDK1) inhibitors. (b) Anti-microtubule and CBSI drugs.
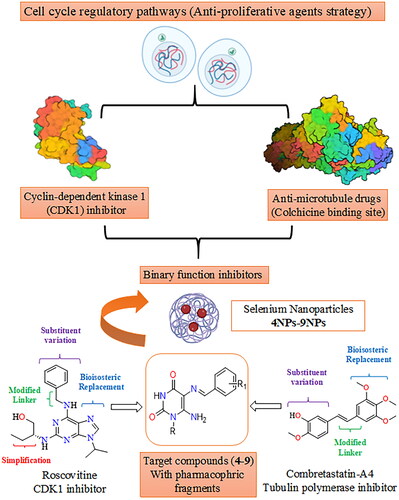
Metal nanoparticles are one of the potential next-generation anticancer therapies as a result of the advancement of nanotechnology in medical research. Selenium (Se) is an essential trace element that is required for many cellular functions via selenoprotein incorporationCitation28. The significance of selenium nanoparticles (SeNPs) originates from their bioavailability, low toxicity, interaction with proteins, and biocompatibility, particularly when compared to organic and inorganic seleniumCitation29. SeNPs were demonstrated to have potent anti-proliferative activityCitation30 due to their ability to selectively target cancerous cells while having left normal cells undamagedCitation31. They are effective in breast, prostate, and lung cancerCitation32–34. Compounds containing selenium nanoparticles are reported to regulate a variety of biological functions in cancer cells, including drug delivery, autophagy, and apoptosisCitation35. Additionally, SeNP base compounds can inhibit the growth of tumours by blocking the formation of new blood vessels that supply them with nutrientsCitation36. Furthermore, these compounds can reduce inflammation and oxidative stress, which are both associated with cancer development and progressionCitation31. On the other hand, unmodified nano-selenium is generally unstable and has a propensity to agglomerate, which significantly lowers its bioavailabilityCitation37. However, many biological macromolecules, including polysaccharides, proteins, and polyphenols, showed the ability to operate as modifiers and regulators for the coating of nano-selenium, thus being candidates for anticancer drugsCitation38,Citation39. Although there is no conclusive evidence of selenium’s ability to prevent cancer, it is essential to reorganise the status of Se and its compounds, whether they are inorganic, organic, or Se-containing nanoparticles, in order to gain a better result of Se’s potential for cancer prevention and treatmentCitation40. This work aims to design pyrimidine Schiff bases 4–9 as potential dual function CDK1 and microtubule CBS inhibitors. Structural modification of roscovitine (II) and combretastatin-A4 (XII) was achieved via bioisosteric replacement, linker modification, substituent variation, and simplification to afford two aryl systems represented by aminopyrimidine-dione and a substituted phenyl ring connected via an imino linker, as shown in , as well as their corresponding selenium nanoparticle forms (SeNPs) 4NPs–9NPs, are being synthesised as a potential alternative chemotherapy strategy. The activity of the synthesised compounds and their SeNPs as anti-proliferative agents will be tested in vitro against three cell lines, including the breast cancer cell line (MCF-7), human liver cancer cell line (HepG-2), and lung tumour cell line (A549). In the molecular modelling study, the compounds will be docked into both the enzyme CDK1 and the CBS of microtubule binding sites to compare their interaction modes and binding scores to native ligands. In support of the claim that the designed pyrimidine Schiff bases are inhibitors of both enzymes, CDK1 and tubulin polymerase CBS, the compounds with the greatest inhibitory effects will further be assessed in vitro for enzyme inhibition activity. Also, the most active compounds will further be investigated for selective cytotoxicity against Vero cell lines.
Figure 2. Design of novel pyrimidine Schiff bases and their SeNPs as dual CDK1 and tubulin polymerase inhibitors.
Results and discussion
Chemistry
The preparation of the target-designed compounds, pyrimidine Schiff bases 4–9, and their selenium nanoforms 4NPs–9NPs are outlined in Scheme 1 and , respectively.
Scheme 1. Formation of Schiff′s bases 4–9. Reagents and conditions: (i) NaNO2/AcOH/H2O, r.t, 30 min; (ii) (NH4)2S, 75 °C, 15 min and (iii) aromatic aldehydes/gl. AcOH/heated under fusion, 20 min.
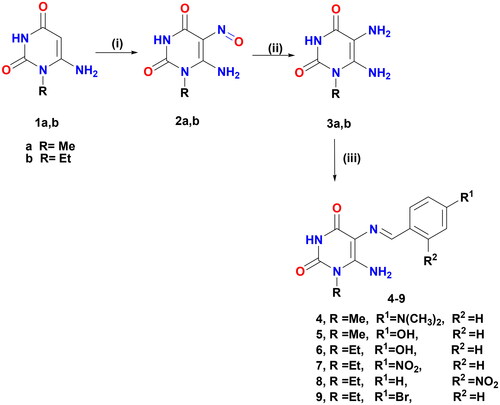
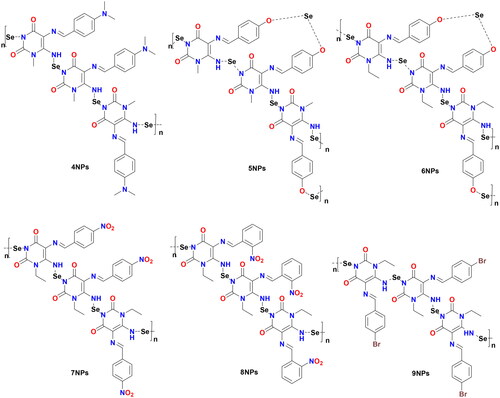
Figure 3. Chemical structures of synthesised Het-SeNPs 4NPs–9NPs.
Synthesis of pyrimidine Schiff bases 4–9
5,6-Diaminouracils (3a, b) were utilised as a starting substrate for the synthesis of Schiff’s bases. These compounds were prepared by nitrosation of 6-aminouracil (1a, b) in a stirred aqueous solution of sodium nitrite in glacial acetic acid at room temperature, affording 2a, b. The latter was then reduced by the reducing agent ammonium sulphide to furnish the starting material 3a,bCitation41–46. Pyrimidine Schiff bases 4–9 were formed under solvent-free conditions with excellent yields (84–94%) in a short time by condensation of the primary amine of 5,6-diaminouracil (3a, b) with a variety of aromatic aldehydes, including 4-(dimethylamino)benzaldehyde, 4-hydroxybenzaldehyde, 4-nitrobenzaldehyde, 4-bromobenzaldehyde, and 2-nitrobenzaldehyde. The reaction was carried out in an acidic medium, glacial acetic acid, to protonate the unstable carbinolamine intermediate and catalysed dehydration via water loss, giving the Schiff’s base products. When compared to other approaches, the current approach is considered green and eco-friendly for the production of various Schiff’s bases due to the use of accessible starting materials, solvent-free conditions, and simple product isolation, as well as high yields. These compounds were established by full spectroscopic data and elemental analysis. The 1H NMR of Schiff’s bases 4–9 was devoid of δ values of C5–NH2 and confirmed the presence of NH-3 and C6–NH2 protons in the range of δ 10.62–10.82 and 7.12–7.62 ppm, respectively, that were disappeared by D2O. Also, their 1H NMR demonstrated singlet signals ascribed to azomethine proton at δ 9.54–9.89 ppm, as well as their 13C NMR revealed a distinctive signal for azomethine carbon between δ 157.89 and 159.12 ppm. Both the 1H NMR and 13C NMR spectra clearly showed the presence of additional protons and carbon signals.
Synthesis of heterocyclic selenium nanoparticles 4NPs–9NPs in situ using pyrimidine Schiff bases (4–9):
Nanoparticles have a higher surface area to volume ratio than micro- or sub-micrometer-sized particles, which can be used to bind significant amounts of either active/targeting biomolecules (enzymes, proteins, DNA) or chemical compounds (chemotherapeutic agents). Therefore, the team’s work aimed to create new selenium nanoparticles (SeNPs) using the synthesised pyrimidine Schiff bases (4–9), which have appropriate low reducing but high stabilising properties during SeNPs preparation. The organic heterocycle derivatives reduced the Se+ cation to Se0 using ascorbic acid as a catalyst that acts as an aldehyde to form SeNPs and stabilise the nano-structure of SeNPsCitation47,Citation48. The organic compounds with reductive groups, including -OH, -NH, -NH2, and others, could reduce selenium cation. The structure of the synthesised 4NPs–9NPs is presented in . Selenium nanoparticle compounds (4NPs–9NPs) were confirmed by UV-Spectrophotometer, TEM technique, and particle size distribution.
Ultraviolet-visible (UV-Vis) spectra
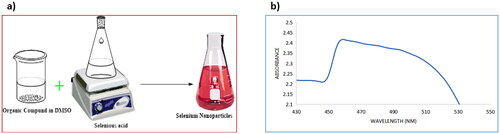
The first indication of the formation of nanoparticles was the solution turning red after adding H2SeO3 to Schiff bases (4–9) dissolved in DMSO and stirring for 1 h at 60 °C, as shown in . The formation of selenium nanoparticles 4NPs–9NPs was monitored using UV-Vis spectroscopy. The UV-Vis absorption spectrum of selenium nanoparticles is depicted in . The colloidal solution of selenium exhibits an absorption peak at 530 nm corresponding to the surface plasmon resonance peaks in the absorption spectra.
Figure 4. (a) A schematic diagram of the synthesis of selenium nanoparticles; (b) UV-Vis spectrum of selenium nanoparticles.
Transmission electron microscope (TEM)
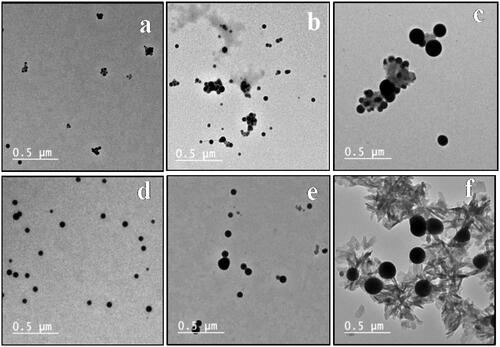
Transmission electron microscopy (TEM) confirmed the characterisation of selenium colloidal nanoparticles dispersed in an aqueous solution without the formation of aggregationsCitation49. TEM was used to examine the prepared selenium Nanoparticles, either alone or loaded onto the synthesised pyrimidine Schiff bases (4–9). The presence of formed selenium nanoparticles in the tested specimens was illustrated in . Selenium nanoparticles are found in spherical shapes with minor aggregate, and their particle sizes range from 24.98 to 60.78 nm. When selenium nanoparticles are loaded onto the investigated compounds 4–9, they spread uniformly throughout both samples, with the nanoparticles encompassing the organic heterocyclic compounds, as graphically illustrated in . The synthesised Schiff bases contributed to the prevention of agglomerates in selenium nanoparticles. It appears that the nitrogen-based compounds’ stabilising effect contributed to the existence of Se nanoparticles separated from each other with convenient distributionsCitation50,Citation51.
Figure 5. TEM of SeNPs, (a), (b), (c), (d), (e), and (f) TEM of SeNPs loaded onto 4–9, respectively.
Size distribution and zeta potential
Dynamic light scattering (DLS) measurements were used to determine the particle size of the synthesised 4NPs–9NPs. The particle size illustrated in Figure S7 in the Supplementary File has an average of 50.43 to 80.90 nm. The reason for the large particle size, which is obtained by DLS rather than TEM measurement, is due to the fact that TEM provides an image for a specific measurement range, whereas DLS provides an overall image of nanoparticles and their aggregations. In addition, DLS denotes the hydrodynamic radius of nanoparticles (hydrated particles) or coated particles in an aqueous solution, while TEM determines the dried diameter of nanoparticlesCitation52. Furthermore, the Polydispersity Index (PDI) of the SeNPs system changed in the range of 0.013 to 0.666 after interaction with heterocyclic compounds, indicating significant changes in the nanoparticle size distribution, as depicted in Figure S8 of the Supplementary File. Moreover, the zeta potential of untreated SeNPs was reported to be −30.2 mVCitation53, which increased in the range of −17.3 to +8.15 mV due to the adsorption of positively-charged heterocyclic compounds molecules onto the matrix of SeNPs (Figure S9 in the Supplementary File). This indicates that electrostatic interactions played an important role in the formation of heterocyclic derivatives-loaded nanoparticles. It is worth noting that zeta potential is a key indicator of colloidal suspension stability. As a result, the negative charge of the heterocyclic derivatives-loaded nanoparticles indicates that the whole system is stableCitation54. shows the Mean Particle Size (MPS), PDI, mean SD, and zeta potential of heterocyclic-selenium nanoparticles 4NPs–9NPs.
Table 1. Particle size and zeta potential of Het-SeNPs 4NPs–9NPs.
Biological evaluation
In vitro cytotoxicity assay
In general, as shown in , it was noticed that the selenium nano-sized pyrimidine Schiff bases (4NPs–9NPs) exhibited a more potent inhibitory effect than normal-sized pyrimidine Schiff base candidates (4–9) on human cancer cell lines, much outperforming the activity of clinically used standard 5-fluorouracil. For pyrimidine Schiff base candidates (4–9), the most effective inhibitory activity was observed for compounds 4- and 6-bearing dimethylamino and hydroxyl functionalities, respectively – when compared to other members of the series. 4NPs achieved an efficacy that exceeds its normal-sized form (4) by 98.74%, 97.20%, and 97.38% (p ≤ 0.001) when introduced into MCF-7, HepG-2, and A569 cell lines, respectively. As well, 6NPs showed a significant increase in efficiency by 96.52%, 96.45%, and 93.86% against MCF-7, HepG-2, and A569, respectively, when compared to its normal-sized form (6). Based on the aforementioned findings, candidates 4 and 6 and their nanoforms were selected for further biological estimations.
Table 2. The anti-proliferative activity of pyrimidine Schiff bases (4–9) and their nano-sized forms (4NPs–9NPs) against human cancer cell lines (IC50 µM).
To determine the selectivity of compounds 4 and 6 and their nanoforms 4NPs and 6NPs, these candidates were screened against HepG-2 and normal Vero cell lines. The selectivity of nano-sized forms of both compounds was comparable to that of 5-fluorouracil. Treatment of cell lines with 4NPs and 6NPs achieved a significant increase in selectivity by 8.78-fold and 2.24-fold, respectively, when compared with the reference drug, which was attributed to the increased bioavailability of these compounds. It was also observed that compounds 4 and 4NPs were more selective than compounds 6 and 6NPs by 2.21-fold and 3.93-fold, respectively (). Therefore, compounds 4 and 4NPs were selected for evaluating cell cycle analysis and apoptotic-inducing effect.
Table 3. The selectivity of the most potent candidates 4 and 6 in both normal and nanoformulations (4NPs and 6NPs).
In-vitro inhibitory activity of CDK1 and tubulin polymerisation CBS
It was noticed that the inhibitory activity of nanoformulation 4NPs against CDK1 was significantly increased by 4.6-fold compared to compound 4. 4NPs showed CDK1 inhibitory effect approaching that of roscovitine ( and ). Compound 4 and its nanoformulation (4NPs) exerted a colchicine binding site inhibitory (CBSI) activity as well, evidenced by IC50 values. The inhibitory effect of the nanoformulation of compound 4 was comparable to that of combretastatin-A4. Based on our findings, the incorporation of nanoparticles into the test compound maximised its efficacy as a tubulin assembly destabilising agent, thus a mitotic inhibitor ( and ).
Figure 6. The inhibitory effect of the compounds 4 and 4NPs on (a) cyclin-dependent kinase 1, (b) Tubulin polymerisation through colchicine binding site. (*) indicate to the significant differences between compound 4NPs-treated and compound 4-treated cells, where (***) indicates to p < 0.001. All experiments were performed in triplicates.
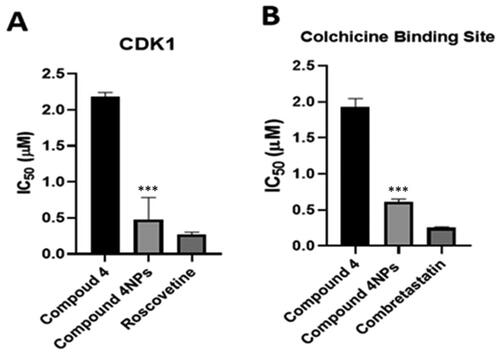
Table 4. The inhibitory activity of the most potent candidates 4 and 4NPs against both CDK1 and tubulin polymerase (CBS) compared to two reference drugs.
Flow cytometric analysis of cell-cycle distribution
Flow cytometric analysis of the cell cycle demonstrated that HepG-2 cells, which were found to be the most affected in cytotoxicity screening, were arrested at the G1 phase when treated with both 4 and nanoformulation 4NPs. This arrest was evidenced by the accumulation of cells at G0/G1 as well as a decrease in the percentage of cells in the S phase by 49.15% in comparison with untreated cells, as displayed in and . Compared to 4, the nanoformulation 4NPs caused a significant rise in the proportion of cells arrested at G0/G1, indicating enhancement of the efficiency of the test compound. shows cytometric histograms of HepG-2 cell cycle phases.
Figure 7. (a) The effect of the most potent compounds 4 and 4NPs on cell cycle phases of HepG-2. Flow cytometric histograms of HepG-2 cell cycle phases; (b) untreated cells, (c) treated with compound 4 alone, and (d) treated with compound 4NPs. All experiments were performed in triplicate.
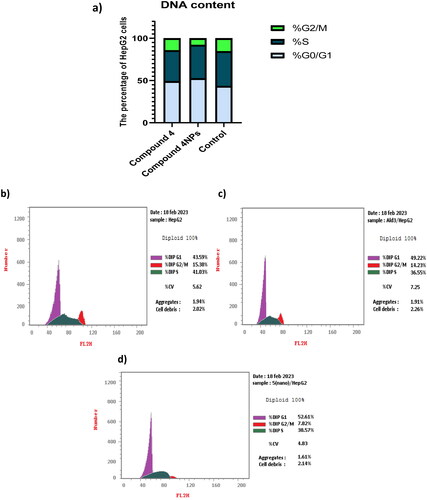
Table 5. The effect of the most potent candidates 4 and 4NPs on cell cycle phases of HepG-2.
Cellular apoptosis analysis
Screening of HepG-2 cells using PI/Annexin V clarified that compounds 4 and 4NPs forced the cells towards apoptosis significantly. The most powerful apoptotic effect was obtained when compound 4NPs was used, as evidenced by a remarkable increase in the percentage of apoptotic cells approaching 31% when compared to that treated with compound 4. Furthermore, the test compounds 4 and 4NPs increased the percentage of necrotic cells by 5.50- and 4.14-fold, respectively, compared to untreated cells ( and ). Our findings suggested that the transformation of compound 4 to nano-sized form 4NPs increased its apoptotic-inducing potency, accordingly killing cancer cells.
Figure 8. (a) The apoptosis-inducing effect of the most potent candidates 4 and 4NPs on HepG-2. (*) indicate to the significant differences between compound 4NPs-treated and compound 4-treated cells, where (*) indicates to p < 0.05, (**) p < 0.01. All experiments were performed in triplicates. (b–d). Flow cytometric dot plot of PI/annexin V screening of HepG-2; (b) untreated cells, (c) cells treated with compound 4, and (d) cells treated with compound 4NPs. All experiments were performed in triplicate.
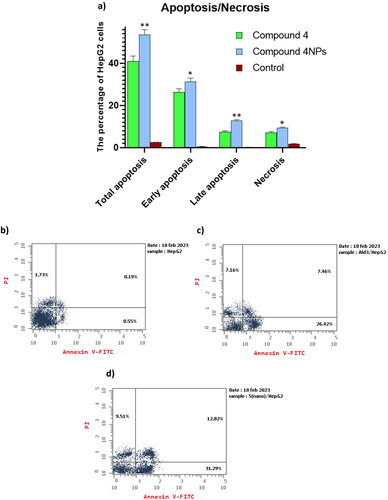
Table 6. The apoptotic effect of the most potent candidates 4 and 4NPs on HepG-2.
Molecular docking simulation
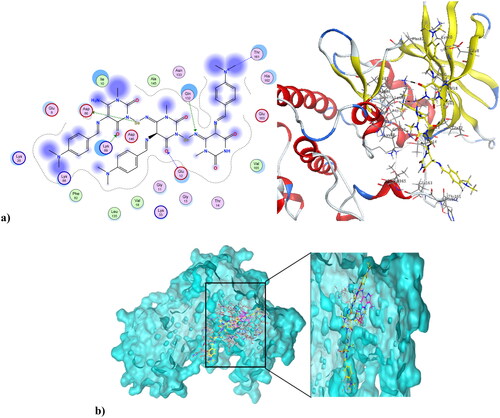
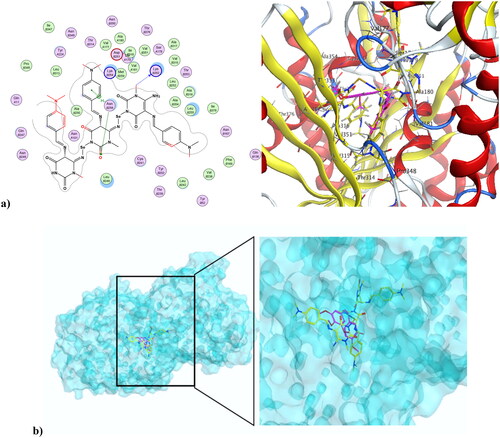
Cancer is associated with uncontrolled cell proliferation that results in most cases from dysregulation of the cell cycleCitation55. This dysregulation is often characterised by an increase in the level of cyclin-dependent kinases and their catalytic cyclin subunits or a decrease in endogenous cyclin kinase inhibitorsCitation56,Citation57. This explains why CDKs are such attractive therapeutic targets for designing potent and selective anti-proliferative agents. CDK1, like all its family, is characterised by a conserved protein kinase domain with a small N-terminal lobe and a large C-terminal fold that are connected by a hinge. Dinaciclib is an ATP-competitive CDK inhibitor that can bind to cyclin-free CDK1 [58]. As shown in Figure S10 in the Supplementary File, dinaciclib binds at the active site of CDK1 via three types of interactions. The first is an H-bond interaction formed by the NH linker at the key amino acid Leu83, while the second is a hydrophobic interaction formed by the core pyrazole moiety of dinaciclib with the two lobes at amino acids Val18, Leu135, and Ile10. The third type of interaction is achieved via stacking interaction of the pyridine moiety of dinaciclib against the key amino acid Ile10. All these pull the amino acids closer to the inside of the active site, narrowing it and resulting in the inhibitory activity of dinaciclib. The newly synthesised compound 4 was capable of binding at the CDK1 active site, exerting a docking score of −6.25 K.cal/mol compared to the docking score of −8.46 K.cal/mol of dinaciclib. depicts compound 4 at the CDK1 active site (PDB ID: 6GU6). Compound 4 formed an H-bond interaction with the key Leu83 amino acid through its carbonyl at position-4 of the pyrimidinedione ring system. It formed an additional hydrogen bond interaction at Gly81 via NH at position-3 of the pyrimidinedione. Similar to dinaciclib, it exhibited hydrophobic interaction at key amino acids Val18, Leu135, and Ile10. shows the alignment of 4 and dinaciclib at the CDK1 active site (PDB ID: 6GU6). The nano form of 4, compound 4NPs was docked into the CDK1 active site, exhibiting a docking score of −9.36 K.cal/mol. It was capable of binding at the active site, where it formed several H-bond interactions with different amino acids, including Asp86, Lys89, Gly12, Thr161, and Gln132. Also, it formed hydrophobic interactions with key amino acids such as Ile10, Lys89, Val18, Leu135, Phe82, and Gln132, as illustrated in . Additionally, the alignment of 4NPs and dinaciclib at the CDK1 active site (PDB ID: 6GU6) is displayed in .
Figure 9. (a) Compound 4 at the active site of CDK1 (PDB ID: 6GU6), and (b) alignment of compound 4 and dinaciclib at the active site of CDK1 (PDB ID: 6GU6).
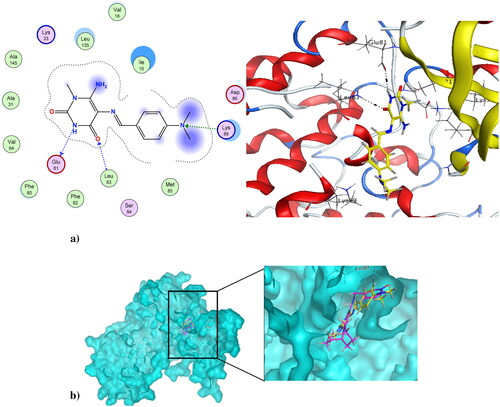
Figure 10. (a) Nano compound 4NPs at the active site of CDK1 (PDB ID: 6GU6). (b) The alignment of 4NPs and dinaciclib at the active site of CDK1 (PDB ID: 6GU6).
Microtubules play vital roles in cellular processes, where they help maintain cell shape. They participate in cellular division through the formation of the mitotic spindle. Microtubule targeting agents, also known as microtubule destabilising agents, are potent anti-proliferative agentsCitation23,Citation59,Citation60. Combretastatin-A4 is a diphenyl derivative with a high potency of disrupting the cell cycle as well as angiogenic activitiesCitation61,Citation62. Combretastatin-A4 can inhibit microtubules through binding at the colchicine binding site (CBS), resulting in disruption of cancer cell growth and powerful cytotoxic activity. The nature of combretastatin A4 interaction at the colchicine binding site (CBS) is mainly hydrophobic, with hydrophobic interactions with many key amino acids at the pocket, including Asn352, Asn349, Leu255, Leu248, Val238, Cys241, and Ala316. It forms H-bond interactions at Thr179 and Val238. Figure S11 in the Supplementary File shows combretastatin-A4 at the colchicine binding site (CBS) of microtubules (PDB ID: 5LYJ)Citation63. The newly synthesised compound 4 was capable of interacting at CBS of microtubules in a mode similar to combretastatin-A4, showing a docking score of −7.79 K.cal/mol, which was comparable to combretastatin A4’s docking score of −8.54 K.cal/mol. The pyrimidinedione of compound 4 aligned with the di-substituted phenyl ring of combretastatin A4. Compound 4 formed three H-bond interactions at Thr179, Met259, and Lys254. Also, it formed Arene-Hydrogen interactions at Lys254 and Asn258. Additionally, it showed hydrophobic interactions with a number of amino acids at the colchicine binding site, including Lys254, Asn254, and Lys352. shows compound 4 at the CBS of microtubules (PDB ID: 5LYJ), while depicts the alignment of compound 4 and combretastatin A4 at the CBS of microtubules (PDB ID: 5LYJ). On the other hand, compound 4NPs, the nano form of 4, was also capable of interacting at CBS, showing a docking score of −9.27K.cal/mol. illustrates that the nano compound 4NPs formed H-bond interactions at Lys352 and Asn258. Similar to combretastatin -A4, nano compound 4NPs formed hydrophobic interactions with many amino acids, including Leu255, Lys352, Lys254, Thr179, Leu248, Gln247, Gln11, and Ala316. shows the alignment of nano compound 4NPs and combretastatin- A4 at the CBS of microtubules (PDB ID: 5LYJ).
Figure 11. (a) Compound 4 at the CBS of microtubules (PDB ID: 5LYJ). (b) The alignment of compound 4 and combretastatin-A4 at the CBS of microtubules (PDB ID: 5LYJ).
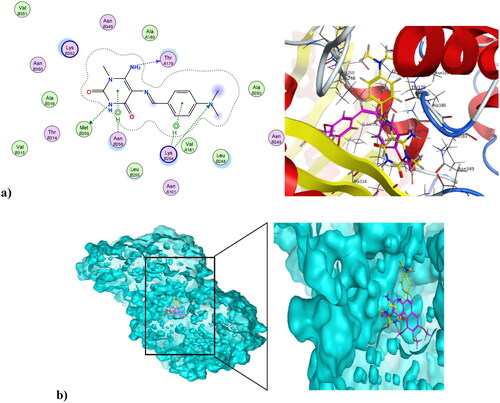
Figure 12. (a) Nano compound 4NPs at the CBS of microtubules (PDB ID: 5LYJ). (b) The alignment of nano compound 4NPs and combretastatin-A4 at the CBS of microtubules (PDB ID: 5LYJ).
Conclusion
Selenium-based nanoparticles are believed to improve tumour targeting of chemotherapeutic agents, resulting in more effective tumour inhibition. Thus, the pyrimidine Schiff bases (4–9) were synthesised under solvent-free conditions by condensation of 5,6-diaminouracil (3a,b) with various aromatic aldehydes and characterised using spectroscopic methods and elemental analyses. These synthesised compounds were formulated in selenium nanoforms (4NPs–9NPs), which were confirmed by UV-Spectrophotometer, TEM technique, and particle size distribution. All the normal-sized compounds and Se-nanoforms were tested in vitro against MCF-7, HepG-2, and A569 cell lines to determine their anti-proliferative activities. Interestingly, all selenium nano-sized forms had a greater inhibitory effect than normal-sized compounds, much outperforming the activity of clinically used standard 5-FU. Compound 4 demonstrated effective anti-proliferative activity against MCF-7 (IC50 3.14 ± 0.04 µM), HepG-2 (IC50 1.07 ± 0.03 µM), and A549 (IC50 1.53 ± 0.01 µM), while its selenium nanoform 4NPs showed an excellent inhibitory effect, with efficacy increased by 96.52%, 96.45%, and 93.86%, respectively, against the three cell lines when compared to 4. Additionally, compound 4 showed a pronounced selectivity comparable to 5-fluorouracil against Vero cell lines, whereas 4NPs demonstrated a five-fold increase in selectivity compared to 4 and a nine-fold increase over 5-FU. Furthermore, 4 exhibited IC50 values of 2.18 ± 0.06 µM and 1.92 ± 0.12 µM against CDK1 and tubulin polymerase at the colchicine binding site (CBS), respectively. While 4NPs strongly inhibited both CDK1 with an IC50 of 0.47 ± 0.3 µM, comparable to roscovitine (IC50 0.27 ± 0.03 µM), and tubulin polymerase CBS with an IC50 of 0.61 ± 0.04 µM, comparable to combretastatin-A4 (IC50 0.25 ± 0.01 µM). Moreover, compound 4 stopped the cell cycle at G0/G1 phase and forced the cells towards apoptosis, whereas its nanoform 4NPs caused a significant rise in the proportion of cells arrested at G0/G1 and its apoptotic-inducing potency, indicating an improvement in the test compound’s efficiency. In the molecular docking simulation, 4 and 4NPs were able to bind to CDK1 and CBS binding sites in inhibitory modes. These findings imply that pyrimidine Schiff base in selenium nanoform 4NPs is a promising scaffold with a possible anti-proliferative candidate for further research.
Materials and methods
Chemistry
Melting points (°C) were measured using the Stuart melting point apparatus (SMP 30) and are uncorrected. Pre-coated (0.25 mm) silica gel plates (Merck 60 F254, Germany) were used to monitor reactions, and spots were visualised using a UV lamp (254 nm). Ethyl acetate: toluene (1:1) and chloroform: methanol (9:1) were used as elution systems. NMR spectra were recorded in (DMSO) at 1H NMR (400 MHz) and 13C NMR (100 MHz) on a Bruker NMR spectrometer (δ ppm), Zagazig university using TMS as an internal standard. Mass spectra were done on the direct inlet part of the mass analyser in a Thermo Scientific GCMS model ISQ at Al-Azhar University’s Regional Centre for Mycology and Biotechnology (RCMB), Egypt. The UV-Vis (Shimadzu spectrophotometer) was used to monitor the selenium nanoparticles formation. The UV-Vis spectra were taken between 400 and 700 nm. High-Resolution Transmission Electron Microscopy (HRTEM) JEOL (JEM-2100 TEM) was used to obtain the shape and size of the SeNPs. A drop of colloidal solution was placed on a 400 mesh Carbon-coated Copper grid and evaporated in the air at room temperature to prepare samples for TEM measurements. The average diameter, size distribution, and zeta potential of samples were measured using a particle size analyser (Nano-ZS, Malvern Instruments Ltd., UK). For measuring Size distribution and zeta potential, the sample was sonicated for 30–60 min just before assessment.
Materials
The starting compounds, 6-aminouracil 1a,b, nitroso derivatives 2a,b, 5,6-diaminouracil derivatives 3a,b were prepared according to the reported methodCitation41–46. Aldrich Chemicals Co., USA, as well as commercial sources, provided all of the chemicals and reagents used.
General method for the preparation of (E)-6-amino-1-alkyl-5-(arylideneamino)pyrimidine-2,4(1H,3H)-diones (4–9)
A mixture of 5,6-diaminouracils 3a,b (1.75 mmol) and different appropriate aromatic aldehydes (1.75 mmol) was heated under fusion in the presence of glacial acetic acid (0.7 ml) for 20 min. The formed residue was collected by methanol, filtered, washed with methanol, recrystallized from MeOH-DMF (4:1), and then dried in the oven to afford the desired Schiff′s bases 4–9 in excellent yields.
(E)-6-amino-5-((4-(dimethylamino)benzylidene)amino)-1-methylpyrimidine-2,4(1H,3H)-dione (4)
Yellow solid, Yield: 88%; m.p.= 294–296 °C; 1H NMR (400 MHz, DMSO) δ 10.62 (s, 1H, NH uracil), 9.54 (s, 1H, CH arylidene), 7.68 (d, J = 8.8 Hz, 2H, Ar–H), 7.12 (s, 2H, NH2), 6.71 (d, J = 8.8 Hz, 2H, Ar–H), 3.31 (s, 3H, CH3), 2.96 (s, 6H, 2CH3). 13C NMR (100 MHz, DMSO) δ 157.98, 153.95, 151.08, 150.61, 149.25, 128.49, 126.70, 111.70, 99.27, 39.89, 29.27 ppm. MS: m/z (rel. int. %) = 287 (M+, 11), 281 (59), 262 (51), 234 (53), 228 (42), 215 ,(61) 209 (66), 201 (52), 197 (75), 189 (73), 169 (100), 162 (61), 125 (80), 104 (78). Anal. Calcd (%) for C14H17N5O2 (287.32): C, 58.52; H, 5.96; N, 24.38; Found: C, 58.69; H, 6.12 N, 24.17.
(E)-6-amino-5-((4-hydroxybenzylidene)amino)-1-methylpyrimidine-2,4(1H,3H)-dione (5)
Yellow solid, Yield: 84%; m.p.= 259–260 °C; 1H NMR (400 MHz, DMSO) δ 10.65 (s, 1H, NH uracil), 9.57 (s, 1H, CH arylidene), 9.30 (s, 1H, OH), 7.70 (d, J = 8.6 Hz, 2H, Ar–H), 7.18 (s, 2H, NH2), 6.78 (d, J = 8.6 Hz, 2H, Ar–H), 3.32 (s, 3H, CH3).13C NMR (100 MHz, DMSO) δ 158.76, 158.01, 154.32, 150.07, 149.30, 130.04, 128.88, 115.36, 99.08, 29.34 ppm. MS: m/z (rel. int. %) = 260 (M+, 38), 248 (60), 245 (60), 190 (48), 184 (96), 180 (46), 173 (56), 159 (72), 154 (80), 138 (50), 82 (48), 70 (100). Anal. Calcd (%) for C12H12N4O3 (260.25): C, 55.38; H, 4.65; N, 21.53; Found: C, 55.67; H, 4.82; N, 21.74.
(E)-6-amino-1-ethyl-5-((4-hydroxybenzylidene)amino)pyrimidine-2,4(1H,3H)-dione (6)
Yellow solid, Yield: 85%; m.p.= 274–275 °C; 1H NMR (400 MHz, DMSO) δ 10.64 (s, 1H, NH uracil), 9.71 (s, 1H, CH arylidene), 9.57 (s, 1H, OH), 7.70 (d, J = 8.4 Hz, 2H, Ar–H), 7.23 (s, 2H, NH2), 6.79 (d, J = 8.4 Hz, 2H, Ar–H), 3.94 (q, J = 6.8 Hz, 2H, CH2), 1.15 (t, J = 6.8 Hz, 3H, CH3) ppm.13C NMR (100 MHz, DMSO) δ 159.12, 158.41, 153.79, 151.01, 149.45, 130.31, 129.31, 115.86, 99.43, 37.58, 13.36 ppm. MS: m/z (rel. int. %) = 274 (M+, 31), 258 (100), 251 (97), 245 (50), 224 (58), 201 (46), 152 (78), 99 (52). Anal. Calcd (%) for C13H14N4O3 (274.28): C, 56.93; H, 5.15; N, 20.43; Found: C, 57.04; H, 5.37; N, 20.69.
(E)-6-amino-1-ethyl-5-((4-nitrobenzylidene)amino)pyrimidine-2,4(1H,3H)-dione (7)
Orange solid, Yield: 94%; m.p.>300 °C; 1H NMR (400 MHz, DMSO) δ 10.82 (s, 1H, NH uracil), 9.77 (s, 1H, CH arylidene), 8.20 (d, J = 9.0 Hz, 2H, Ar–H), 8.15 (d, J = 9.0 Hz, 2H, Ar–H), 7.62 (s, 2H, NH2), 3.97 (q, J = 7.0 Hz, 2H, CH2), 1.17 (t, J = 7.0 Hz, 3H, CH3) ppm.13C NMR (100 MHz, DMSO) δ 157.94, 154.66, 148.91, 146.97, 145.92, 145.01, 127.87, 123.73, 99.58, 37.24, 13.00. ppm. MS: m/z (rel. int. %) = 303 (M+, 49), 289 (76), 282 (58), 267 (48), 249 (50), 244 (55), 225 (74), 213 (62), 197 (40), 189 (78), 177 (76), 172 (100), 147 (68), 113 (56), 107 (70). Anal. Calcd (%) for C13H13N5O4 (303.28): C, 51.49; H, 4.32; N, 23.09; Found: C, 51.72; H, 4.50; N, 23.21.
(E)-6-amino-1-ethyl-5-((2-nitrobenzylidene)amino)pyrimidine-2,4(1H,3H)-dione (8)
Orange solid, Yield: 92%; m.p.>300 °C; 1H NMR (400 MHz, DMSO) δ 10.75 (s, 1H, NH uracil), 9.89 (s, 1H, CH arylidene), 8.40 (d, J = 7.6 Hz, 1H, Ar–H), 7.87 (d, J = 7.6 Hz, 1H, Ar–H), 7.69 (t, J = 7.6 Hz, 1H, Ar–H), 7.59–7.51 (m, 1H, Ar–H), 7.45 (s, 2H, NH2), 3.95 (q, J = 6.8 Hz, 2H, CH2), 1.16 (t, J = 6.8 Hz, 3H, CH3) ppm.13C NMR (100 MHz, DMSO) δ 157.89, 154.57, 148.88, 148.56, 142.58, 132.49, 131.59, 129.25, 128.58, 123.69, 99.44, 37.21, 12.96 ppm. MS: m/z (rel. int. %) = 303 (M+, 32), 216 (30), 169 (47), 118 (38), 97 (44), 80 (45), 80 (45), 77 (59), 57 (100). Anal. Calcd (%) for C13H13N5O4 (303.28): C, 51.49; H, 4.32; N, 23.09; Found: C, 51.74; H, 4.51; N, 23.28.
(E)-6-amino-5-((4-bromobenzylidene)amino)-1-ethylpyrimidine-2,4(1H,3H)-dione (9)
Yield: 92%; m.p. >300 °C; 1H NMR (400 MHz, DMSO) δ 10.53 (s, 1H, NH), 9.64 (s, 1H, CH arylidene), 7.85 (d, J = 8.5 Hz, 2H, Ar–H), 7.56 (d, J = 8.5 Hz, 2H, Ar–H), 7.42 (s, 2H, NH2), 3.95 (q, J = 6.9 Hz, 2H, CH2), 1.15 (t, J = 6.9 Hz, 3H, CH3). 13C NMR (100 MHz, DMSO) δ 157.96, 153.89, 150.50, 148.82, 137.92, 131.99, 131.34, 129.07, 128.25, 122.06, 98.88, 37.05, 13.02. ppm. MS: m/z (rel. int. %) = 339 (M + 2, 43), 337 (M+, 37), 313 (35), 247 (39), 238 (65), 192 (66), 145 (42), 131 (46), 123 (57), 93 (100), 77 (54); Anal. Calcd (%) for C13H13BrN4O2 (337.18): C, 46.31; H, 3.89; 70; N, 16.62. Found: C, 46.49; H, 4.07; 70; N, 16.87.
Synthesis of in-situ selenium nanoparticles (SeNPs) 4NPs-9NPs using synthesised pyrimidine Schiff bases 4–9
In a conical flask, selenious acid (H2SeO3, 0.013 gm, 0.01 mmol) was dissolved in deionised water (80 ml). After heating the selenious acid solution to 60 °C, compounds 4–9 (0.01 mmol) in DMSO (10 ml) were added. 200 µL of 40 mM ascorbic acid was added as a catalyst after 1 h of continuous stirring at 60 °C, and the ruby red SeNPs were suspended. UV-Spectrophotometer, particle size distribution, and TEM were used to confirm and characterise the formation of selenium nanoparticles.
Biological evaluation
In vitro cytotoxicity assay
The cytotoxic activity of all synthesised compounds, both normal and nano-sized forms, was evaluated against three cancer cell lines, breast cancer (MCF-7), hepatocellular carcinoma (HepG-2), and non-small cell lung cancer (A549), while the normal Vero cell line was performed for the promising compounds 4, 6 and their nano-sized 4NPs, 6NPs, using the MTT assay according to the literatureCitation64. The cell lines were obtained from ATCC (American Type Culture Collection). 5-Fluorouracil was used as a control. It is worth noting that freshly prepared nanoparticle-based compounds were used in each of the conducted experiments. The protocol of the MTT assay was illustrated in detail in the Supplementary File.
In vitro enzymes inhibition assay
Estimation of the activity of CDK1
The most active compound 4 and its selenium nanoform 4NPs were evaluated for their CDK1 inhibition activity. Anti-CDK1/cyclin B1 assay was performed in vitro according to the manufacturer’s instructionsCitation4 as described in the Supplementary File.
Determination of colchicine–tubulin binding capacity
Additionally, the colchicine–tubulin binding site inhibition activity of the test compounds 4 and 4NPs was investigated using the reported method, as described in the Supplementary FileCitation65.
Flow cytometric analysis of cell-cycle distribution
Cell cycle flow cytometry was performed using propidium iodide (PI) staining (ab139418_Propidium Iodide Flow Cytometry Kit/BD)Citation66. IC50 of both 4 and 4NPs was introduced to HepG-2 cells and then incubated for 48 h. The cells were then fixed in 70% ethanol for 12 h at 4 °C. After that, the cells were washed with cold PBS, incubated with 100 μL RNase A at 37 °C for 30 min, and stained with 400 μL PI for another 30 min at room temperature in the dark. Flowing software was used to analyse the data after measuring the stained cells with an Epics XL-MCL™ Flow Cytometer (Beckman Coulter).
Cellular apoptosis analysis
The apoptotic potency of compounds 4 and 4NPs was investigated using an Annexin-V assayCitation67 and biparametric cytofluorimetric analysis. The liver HepG-2 cells have been subjected to treatment with an IC50 of both 4 and 4NPs for 48 h before being trypsinized, centrifuged, and rinsed twice with PBS before being suspended in 500 μL of binding buffer and double stained for 15 min with 5 μL Annexin V-FITC and 5 μL PI in the dark at room temperature. Stained cells have been counted and analysed with an Epics XL-MCL™ Flow Cytometer and Flowing software.
Molecular docking simulation
Molecular docking has been performed using the Molecular Operating Environment (MOE) software, according to the literatureCitation68. First, the MOE builder was used to build ligands in 3D conformation via 3D protonation and assigning partial charges, followed by energy minimisation using the MMFF94x force field. The biological targets needed for the docking study were obtained from the RCSB-Protein Data Bank (PDB ID: 6GU6)Citation58; cyclin-dependent kinase 1 (CDK1) in complex with dinaciclib and combretastatin-A4 at the colchicine binding site of microtubules (PDB ID: 5LYJ)Citation63. The proteins were prepared in a four-step preparation protocol, including 3D protonation and auto-correction for atoms types, connections, and assigning charges. Finally, the active sites were defined with the help of MOE Alpha Site Finder. Dummy atoms have been generated for the alpha spheres and used as polar-hydrophobic descriptors of the active sites of both 6GU6 and 5LYJ. The MOE-induced fit docking protocol was adopted. For validation, re-docking of the native ligands against both the 6GU6 and 5LYJ active sites was performed, and the cut-off of root-mean-square deviation (RMSD) values were < 2.0 Å. The interaction of ligands at the active sites was assessed via bond rotation inside the active site using the triangle matcher approach and London dG scoring function as first scoring function. Five poses out of 30 were retained after using GBVI/WSA dG force field-based scoring function as a second scoring function. Poses that had a lower RMSD and a higher S-value were recorded.
Consent form
Authors accept the final version submitted to the journal.
Supplemental Material
Download PDF (5.7 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Xu J-J, Zhang W-C, Guo Y-W, Chen X-Y, Zhang Y-N. Metal nanoparticles as a promising technology in targeted cancer treatment. Drug Deliv. 2022;29(1):664–678.
- Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, Zheng Q, Cai C. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res. 2021;11(5):1913.
- Yang B, Quan Y, Zhao W, Ji Y, Yang X, Li J, Li Y, Liu X, Wang Y, Li Y, et al. Design, synthesis and biological evaluation of 2-((4-sulfamoylphenyl) amino)-pyrrolo [2,3-d] pyrimidine derivatives as CDK inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):2169282.
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130–146.
- Wu L, Qu X. Cancer biomarker detection: recent achievements and challenges. Chem Soc Rev. 2015;44(10):2963–2997.
- Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17(2):93–115.
- Zhang XH, Hsiang J, Rosen ST. Flavopiridol (Alvocidib), a cyclin-dependent kinases (CDKs) inhibitor, found synergy effects with niclosamide in cutaneous t-cell lymphoma. J Clin Haematol. 2021;2(2):48.
- Cho YS, Angove H, Brain C, Chen CH-T, Cheng H, Cheng R, Chopra R, Chung K, Congreve M, Dagostin C, et al. Fragment-based discovery of 7-azabenzimidazoles as potent, highly selective, and orally active CDK4/6 inhibitors. ACS Med Chem Lett. 2012;3(6):445–449.
- Freeman-Cook KD, Hoffman RL, Behenna DC, Boras B, Carelli J, Diehl W, Ferre RA, He Y-A, Hui A, Huang B, et al. Discovery of PF-06873600, a CDK2/4/6 inhibitor for the treatment of cancer. J Med Chem. 2021;64(13):9056–9077.
- Saqub H, Proetsch-Gugerbauer H, Bezrookove V, Nosrati M, Vaquero EM, de Semir D, Ice RJ, McAllister S, Soroceanu L, Kashani-Sabet M, et al. Dinaciclib, a cyclin-dependent kinase inhibitor, suppresses cholangiocarcinoma growth by targeting CDK2/5/9. Sci Rep. 2020;10(1):1–13.
- Cicenas J, Kalyan K, Sorokinas A, Stankunas E, Levy J, Meskinyte I, Stankevicius V, Kaupinis A, Valius M. Roscovitine in cancer and other diseases. Ann Transl Med. 2015;3(10):135–147.
- Dai Y, Dent P, Grant S. Induction of apoptosis in human leukemia cells by the CDK1 inhibitor. Cell Cycle. 2002;1(2):128–137.
- Tadesse S, Zhu G, Mekonnen LB, Lenjisa JL, Yu M, Brown MP, Wang S. A novel series of N-(pyridin-2-yl)-4-(thiazol-5-yl) pyrimidin-2-amines as highly potent CDK4/6 inhibitors. Future Med Chem. 2017;9(13):1495–1506.
- Sorf A, Novotna E, Hofman J, Morell A, Staud F, Wsol V, Ceckova M. Cyclin-dependent kinase inhibitors AZD5438 and R547 show potential for enhancing efficacy of daunorubicin-based anticancer therapy: interaction with carbonyl-reducing enzymes and ABC transporters. Biochem Pharmacol. 2019;163:290–298.
- Mesguiche V, Parsons RJ, Arris CE, Bentley J, Boyle FT, Curtin NJ, Davies TG, Endicott JA, Gibson AE, Golding BT, et al. 4-Alkoxy-2, 6-diaminopyrimidine derivatives: inhibitors of cyclin dependent kinases 1 and 2. Bioorg Med Chem Lett. 2003;13(2):217–222.
- Lafanechère L. The microtubule cytoskeleton: an old validated target for novel therapeutic drugs. Front Pharmacol. 2022;13:3888.
- Serpico AF, Febbraro F, Pisauro C, Grieco D. Compartmentalized control of Cdk1 drives mitotic spindle assembly. Cell Reports. 2022;38(4):110305.
- Gigant B, Wang C, Ravelli RBG, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature. 2005;435(7041):519–522.
- Prota AE, Bargsten K, Diaz JF, Marsh M, Cuevas C, Liniger M, Neuhaus C, Andreu JM, Altmann K-H, Steinmetz MO, et al. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc Natl Acad Sci U S A. 2014;111(38):13817–13821.
- Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann K-H, Miller JH, Díaz JF, Steinmetz MO. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew Chem Int Ed Engl. 2014;53(6):1621–1625.
- Ravelli RBG, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428(6979):198–202.
- Dorléans A, Gigant B, Ravelli RBG, Mailliet P, Mikol V, Knossow M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proc Natl Acad Sci U S A. 2009;106(33):13775–13779.
- Wang Y, Zhang H, Gigant B, Yu Y, Wu Y, Chen X, Lai Q, Yang Z, Chen Q, Yang J, et al. Structures of a diverse set of colchicine binding site inhibitors in complex with tubulin provide a rationale for drug discovery. FEBS J. 2016;283(1):102–111.
- Chamberlain MC, Grimm S, Phuphanich S, Recht L, Zhu JZ, Kim L, Rosenfeld S, Fadul CE, Brain Tumor Investigational Consortium (BTIC). A phase 2 trial of verubulin for recurrent glioblastoma: a prospective study by the Brain Tumor Investigational Consortium (BTIC). J Neurooncol. 2014;118(2):335–343.
- Siddiqui-Jain A, Hoj JP, Cescon DW, Hansen MD. Pharmacology and in vivo efficacy of pyridine-pyrimidine amides that inhibit microtubule polymerization. Bioorg Med Chem Lett. 2018;28(5):934–941.
- Lu Y, Chen J, Xiao M, Li W, Miller DD. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res. 2012;29(11):2943–2971.
- McLoughlin EC, O’Boyle NM. Colchicine-binding site inhibitors from chemistry to clinic: a review. Pharmaceuticals. 2020;13(1):8.
- Ferro C, Florindo HF, Santos HA. Selenium nanoparticles for biomedical applications: from development and characterization to therapeutics. Adv Healthcare Mater. 2021;10(16):2100598.
- Ikram M, Javed B, Raja NI, Mashwani Z-u-R. Biomedical potential of plant-based selenium nanoparticles: a comprehensive review on therapeutic and mechanistic aspects. Int J Nanomed. 2021;16:249–268.
- Singh KR, Nayak V, Singh J, Singh AK, Singh RP. Potentialities of bioinspired metal and metal oxide nanoparticles in biomedical sciences. RSC Adv. 2021;11(40):24722–24746.
- Chen G, Yang F, Fan S, Jin H, Liao K, Li X, Liu G-B, Liang J, Zhang J, Xu J-F, et al. Immunomodulatory roles of selenium nanoparticles: novel arts for potential immunotherapy strategy development. Front Immunol. 2022;13:3939.
- Liao G, Tang J, Wang D, Zuo H, Zhang Q, Liu Y, Xiong H. Selenium nanoparticles (SeNPs) have potent antitumor activity against prostate cancer cells through the upregulation of miR-16. World J Surg Onc. 2020;18(1):11.
- Tian J, Wei X, Zhang W, Xu A. Effects of selenium nanoparticles combined with radiotherapy on lung cancer cells. Front Bioeng Biotechnol. 2020;8:598997.
- Al-Otaibi AM, Al-Gebaly AS, Almeer R, Albasher G, Al-Qahtani WS, Abdel Moneim AE. Potential of green-synthesized selenium nanoparticles using apigenin in human breast cancer MCF-7 cells. Environ Sci Pollut Res Int. 2022;29(31):47539–47548.
- Nayak V, Singh KR, Singh AK, Singh RP. Potentialities of selenium nanoparticles in biomedical science. New J Chem. 2021;45(6):2849–2878.
- Ehudin MA, Golla U, Trivedi D, Potlakayala SD, Rudrabhatla SV, Desai D, Dovat S, Claxton D, Sharma A. Therapeutic benefits of selenium in hematological malignancies. Int J Mol Sci. 2022;23(14):7972.
- Kumar A, Prasad KS. Role of nano-selenium in health and environment. J Biotechnol. 2021;325:152–163.
- Alavi M, Rai M, Martinez F, Kahrizi D, Khan H, Rose Alencar de Menezes I, Douglas Melo Coutinho H, Costa J. The efficiency of metal, metal oxide, and metalloid nanoparticles against cancer cells and bacterial pathogens: different mechanisms of action. Cell Mol Biomed Rep. 2022;2(1):10–21.
- Kuršvietienė L, Mongirdienė A, Bernatonienė J, Šulinskienė J, Stanevičienė I. Selenium anticancer properties and impact on cellular redox status. Antioxidants. 2020;9(1):80.
- Nogueira CW, Barbosa NV, Rocha JB. Toxicology and pharmacology of synthetic organoselenium compounds: an update. Arch Toxicol. 2021;95(4):1179–1226.
- El-Kalyoubi S, Agili F, Adel I, Tantawy MA. Novel uracil derivatives depicted potential anticancer agents: in vitro, molecular docking, and ADME study. Arab J Chem. 2022;15(4):103669.
- El-Kalyoubi S, Agili F, Zordok WA, El-Sayed ASA. Synthesis, in silico prediction and in vitro evaluation of antimicrobial activity, DFT calculation and theoretical investigation of novel xanthines and uracil containing imidazolone derivatives. Int J Mol Sci. 2021;22(20):10979.
- El-Kalyoubi S, Agili F. Synthesis, in silico prediction and in vitro evaluation of antitumor activities of novel pyrido [2, 3-d] pyrimidine, xanthine and lumazine derivatives. Molecules. 2020;25(21):5205.
- El-Kalyoubi SA, Taher ES, Ibrahim TS, El-Behairy MF, Al-Mahmoudy AMM. Uracil as a Zn-binding bioisostere of the allergic benzenesulfonamide in the design of quinoline–uracil hybrids as anticancer carbonic anhydrase inhibitors. Pharmaceuticals. 2022;15(5):494.
- El-Kalyoubi S, El-Sebaey SA, Rashad AM, Al-Ghulikah HA, Ghorab MM, Elfeky SM. Synthesis, DFT calculations, and anti-proliferative evaluation of pyrimidine and selenadiazolopyrimidine derivatives as dual Topoisomerase II and HSP90 inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):2198163.
- El-Kalyoubi S, Elbaramawi SS, Zordok WA, Malebari AM, Safo MK, Ibrahim TS, Taher ES. Design and synthesis of uracil/thiouracil based quinoline scaffolds as topoisomerases I&II inhibitors for chemotherapy: a new hybrid navigator with DFT calculation. Bioorg Chem. 2023;136:106560.
- Kapur M, Soni K, Kohli K. Green synthesis of selenium nanoparticles from broccoli, characterization, application and toxicity. Adv Tech Biol Med. 2017;05(01):2379–1764.
- Wadhwani SA, Gorain M, Banerjee P, Shedbalkar UU, Singh R, Kundu GC, Chopade BA. Green synthesis of selenium nanoparticles using Acinetobacter sp. SW30: optimization, characterization and its anticancer activity in breast cancer cells. Int J Nanomed. 2017;12:6841–6855.
- Ranjitha V, Ravishankar V. Extracellular synthesis of selenium nanoparticles from an actinomycetes streptomyces griseoruber and evaluation of its cytotoxicity on HT-29 cell line. Pharm Nanotechnol. 2018;6(1):61–68.
- Khalil A, Hassan ML, Ward AA. Novel nanofibrillated cellulose/polyvinylpyrrolidone/silver nanoparticles films with electrical conductivity properties. Carbohydr Polym. 2017;157:503–511.
- Khalil AM, Georgiadou V, Guerrouache M, Mahouche-Chergui S, Dendrinou-Samara C, Chehimi MM, Carbonnier B. Gold-decorated polymeric monoliths: in-situ vs ex-situ immobilization strategies and flow through catalytic applications towards nitrophenols reduction. Polymer. 2015;77:218–226.
- Fan W, Yan W, Xu Z, Ni H. Formation mechanism of monodisperse, low molecular weight chitosan nanoparticles by ionic gelation technique. Colloids Surf B Biointerfaces. 2012;90:21–27.
- Iqbal MS, Abbas K, Qadir MI. Synthesis, characterization and evaluation of biological properties of selenium nanoparticles from Solanum lycopersicum. Arab J Chem. 2022;15(7):103901.
- Ashrafpour S, Moghadam TT. Interaction of silver nanoparticles with lysozyme: functional and structural investigations. Surf Interfaces. 2018;10:216–221.
- Barnum KJ, O’Connell MJ. Cell cycle regulation by checkpoints. Cell Cycle Control Mech Protocols. 2014;1170:29–40.
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166.
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development. 2013;140(15):3079–3093.
- Wood DJ, Korolchuk S, Tatum NJ, Wang L-Z, Endicott JA, Noble MEM, Martin MP. Differences in the conformational energy landscape of CDK1 and CDK2 suggest a mechanism for achieving selective CDK inhibition. Cell Chem Biol. 2019;26(1):121.e5–130.e5.
- Kavallaris M. Erratum: microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10(4):309–309.
- Hammond JW, Cai D, Verhey KJ. Tubulin modifications and their cellular functions. Curr Opin Cell Biol. 2008;20(1):71–76.
- Nam N-H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr Med Chem. 2003;10(17):1697–1722.
- Bhattacharyya B, Panda D, Gupta S, Banerjee M. Anti‐mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28(1):155–183.
- Gaspari R, Prota AE, Bargsten K, Cavalli A, Steinmetz MO. Structural basis of cis-and trans-combretastatin binding to tubulin. Chem. 2017;2(1):102–113.
- van de Loosdrecht AA, Beelen RH, Ossenkoppele GJ, Broekhoven MG, Langenhuijsen MM. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J Immunol Methods. 1994;174(1-2):311–320.
- Tahir SK, Kovar P, Rosenberg SH, Ng SC. Rapid colchicine competition-binding scintillation proximity assay using biotin-labeled tubulin. Biotechniques. 2000;29(1):156–160.
- El-Zoghbi MS, El-Sebaey SA, Al-Ghulikah HA, Sobh EA. Design, synthesis, docking, and anticancer evaluations of new thiazolo [3, 2-a] pyrimidines as topoisomerase II inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):2175209.
- Al-Ghulikah HA, El-Sebaey SA, Bass AKA, El-Zoghbi MS. New pyrimidine-5-carbonitriles as COX-2 inhibitors: design, synthesis, anticancer screening, molecular docking, and in silico ADME profile studies. Molecules. 2022;27(21):7485.
- Elfeky SM, Almehmadi SJ, Tawfik SS. Synthesis, in-silico, and in-vitro study of novel chloro methylquinazolinones as PI3K-δ inhibitors, cytotoxic agents. Arab J Chem. 2022;15(2):103614.