
Abstract
Sulphur fluoride exchange (SuFEx) is a category of click chemistry that enables covalent linking of modular units through sulphur connective hubs. Here, we reported an efficient synthesis and in situ screening method for building a library of sulphonamides on the picomolar scale by SuFEx reaction between a sulphonyl fluoride (RSO2F) core and primary or secondary amines. This biocompatible SuFEx reaction would allow us to rapidly synthesise sulphonamide molecules, and evaluate their ChE inhibitory activity. Compound T14-A24 was identified as a reversible, competitive, and selective AChE inhibitor (Ki = 22 nM). The drug-like evaluation showed that T14-A24 had benign BBB penetration, remarkable neuroprotective effect, and safe toxicological profile. In vivo behavioural study showed that T14-A24 treatment improved the Aβ1 − 42-induced cognitive impairment, significantly prevented the effects of Aβ1 − 42 toxicity. Therefore, this SuFEx click reaction can accelerate the discovery of lead compounds.
Introduction
Over the past 20 years, click chemistry has been shown to meet the strict conditions of modular synthesis because of simple operation, material availability, efficiency, and selectivity, which concept can facilitate the construction of diverse functions by changing the building blocks and assembly orderCitation1,Citation2. Sulphur fluoride exchange (SuFEx) allows S–F to exchange with a range of nucleophilic reagents, including alcohols and amines, to form new linkages, resulting in stable S–N, S–O, and S–C covalent bondsCitation3,Citation4. The simple and robust operation of the SuFEx reaction makes this modern click reaction perfect for modular synthesis and access to different click librariesCitation5.
SuFEx connectors enable the modular construction of functional molecules through SuFExable hubs including: sulphuryl fluoride (SO2F2), ethenesulfonyl fluoride (ESF), and thionyl tetrafluoride (SOF4)Citation6,Citation7. This unique SuFEx reaction is thought to be made possible by the strict condition to stabilise the departing fluoride ion in its transit away from the strong original covalent bond to sulfurCitation8,Citation9. The S − N bonds formed in this embodiment of SuFEx reaction are common in pharmaceuticals; for example, over 150 sulphonamides can be found in the market, which include antibacterial, anti-tumor, anti-obesity, anti-thyroid, and anti-neuropathic pain activitiesCitation10,Citation11.
An approach derived from combinatorial chemistry has been attracting attention in the last two decades: microtiter plate-based synthesis coupled with in situ screening through click chemistry, which uses automation to rapidly detect biological or biochemical activityCitation12,Citation13. This approach leverages high yield organic reactions with high selectivity, starting with a set of building blocks to generate a focused library in a microplate, in which a compound is ideally formed in each wellCitation14. SuFEx click chemistry can achieve the high reactivity of sulphonyl fluorides with suitable amines. The formation of the intended sulphonamides would be efficient, with high purity and high yield.
As a proof-of-concept, fragment splicing-based molecular design would be carried out using cholinesterases (ChEs), containing a peripheral anionic site (PAS), a deep and narrow gorge, and a catalytic active site (CAS)Citation15,Citation16. Potent ChE inhibitors can be designed through a proper linker between CAS and PAS to enable the hybrid to fall into both active site cavitiesCitation17,Citation18. Here, we reported an efficient in situ screening method for building a library of sulphonamides on the picomolar scale using the SuFEx reaction between a sulphonyl fluoride (RSO2F) core and primary or secondary amines (). This biocompatible reaction condition would allow us to rapidly discover and synthesise ChE inhibitors, and evaluate a focused drug library of sulphonamide molecules.
Figure 1. Lead discovery by SuFEx click chemistry, followed by direct screening against AChE and BuChE.
Materials and methods
Chemistry
The reagents and solvents were commercially purchased and no further purification was required. Reactions were checked by thin-layer chromatography (TLC) on precoated silica gel plates (Qingdao Marine Chemical Factory, GF254). Melting points are determined on a XT4MP apparatus (Taike Corp., Beijing, China) and are not corrected. The purity (relative content) of active compounds was determined by HPLC through area normalisation method. 1H NMR and 13C NMR spectra were recorded on Bruker AV-400 or AV-600 MHz instruments using CDCl3 as solvent. High-resolution mass spectra (HRMS) were obtained on an Agilent 1260-6221 TOF mass spectrometry.
General synthetic information of title sulphonamides
To a solution of the sulphonyl fluoride T (0.1 mmol) in MeCN (3 mL) was added the amine A (1.2 equiv.) and K2CO3 (1.5 equiv.). The suspension solution was stirred at room temperature for 8 h. After the reaction was completed, the reaction mixture was poured into water (15 mL). After stirring for 10 min, the mixture was extracted with ethyl acetate (30 mL × 3), and the combined ethyl acetate layer was washed twice with saturated sodium chloride solution. The organic layer was dried over anhydrous sodium sulphate overnight and concentrated; the residue was purified by automated chromatography system to afford title sulphonamides T-A.
N-(4-Fluorophenethyl)-2–(1-(2-methoxyphenyl)-1H-1,2,3-triazol-4-yl)ethane-1-sulphonamide (T2-A7)
Light white oil, yield 77%; 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1H), 7.74 (dd, J = 7.8, 1.7 Hz, 1H), 7.44 (ddd, J = 8.7, 7.7, 1.7 Hz, 1H), 7.27 (s, 1H), 7.16–7.08 (m, 4H), 7.04–6.91 (m, 2H), 3.86 (s, 3H), 3.47 (dd, J = 8.4, 6.6 Hz, 2H), 3.34–3.22 (m, 4H), 2.81 (t, J = 7.0 Hz, 2H), 1.26 (d, J = 5.1 Hz, 4H). HRMS (ESI) m/z [M + H]+: 405.1391 calcd for C19H22FN4O3S: 405.1383.
N-(2–(5-chloro-1H-indol-3-yl)ethyl)-2–(1-(2-methoxy-4-nitrophenyl)-1H-1,2,3-triazol-4-yl)ethane-1-sulphonamide (T11-A21)
Light white oil, yield 85%; 1H NMR (400 MHz, CDCl3) δ 8.26 (s, 1H), 7.69 (s, 1H), 7.52 (d, J = 2.0 Hz, 1H), 7.30 (s, 2H), 7.23 (d, J = 8.6 Hz, 1H), 7.13 (d, J = 2.4 Hz, 1H), 7.10 (dd, J = 8.6, 2.0 Hz, 1H), 7.07 (s, 1H), 3.43 (q, J = 6.4 Hz, 2H), 3.33 (dd, J = 9.1, 6.2 Hz, 2H), 3.14 (dd, J = 9.0, 6.3 Hz, 2H), 2.98 (t, J = 6.5 Hz, 2H), 2.40 (s, 6H). HRMS (ESI) m/z [M – H]+: 458.1417, calcd for C22H25ClN5O2S: 458.1408.
6,7-Dimethoxy-2-((2–(1-(2-methoxy-4-nitrophenyl)-1H-1,2,3-triazol-4-yl)ethyl)sulphonyl)-1,2,3,4-tetrahydroisoquinoline (T11-A23)
Light yellow oil, yield 81%; 1H NMR (500 MHz, CDCl3) δ 7.80 (s, 1H), 7.28 (s, 2H), 7.06 (s, 1H), 6.56 (dd, J = 250.3, 28.3 Hz, 2H), 4.42 (s, 2H), 3.84 (s, 6H), 3.58 (t, J = 5.9 Hz, 2H), 3.45 (dd, J = 8.7, 6.5 Hz, 2H), 3.31 (dd, J = 8.9, 6.4 Hz, 2H), 2.86 (t, J = 5.9 Hz, 2H), 2.39 (s, 6H). HRMS (ESI) m/z [M + Na]+: 479.1723, calcd for C23H28N4O4SNa: 479.1715.
3–(4-(2-((6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)sulphonyl)ethyl)-1H-1,2,3-triazol-1-yl)quinoline (T14-A23)
Light white oil, yield 84%; 1H NMR (500 MHz, CDCl3) δ 9.29 (d, J = 2.6 Hz, 1H), 8.43 (d, J = 2.6 Hz, 1H), 8.20 (d, J = 8.5 Hz, 1H), 7.93 (dd, J = 8.3, 1.4 Hz, 1H), 7.82 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.68 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H), 6.56 (d, J = 21.2 Hz, 2H), 4.44 (s, 2H), 3.82 (d, J = 10.1 Hz, 6H), 3.60 (t, J = 5.9 Hz, 2H), 3.49 (t, J = 7.4 Hz, 2H), 3.38 (t, J = 7.5 Hz, 2H), 2.86 (t, J = 6.0 Hz, 2H). HRMS (ESI) m/z [M + H]+: 480.1700, calcd for C24H26N5O4S: 480.1693.
(2-(5-Methoxy-1H-indol-3-yl)ethyl)-2–(1-(quinolin-3-yl)-1H-1,2,3-triazol-4-yl)ethane-1-sulphonamide (T14-A24)
Light yellow oil, yield 75%; 1H NMR (500 MHz, CDCl3) δ 9.30 (d, J = 2.7 Hz, 1H), 8.45 (d, J = 2.6 Hz, 1H), 8.21 (d, J = 8.5 Hz, 1H), 8.01–7.92 (m, 2H), 7.85 (d, J = 10.0 Hz, 2H), 7.69 (t, J = 7.4 Hz, 1H), 7.53 (d, J = 9.3 Hz, 1H), 7.21 (d, J = 8.9 Hz, 1H), 7.06 (dd, J = 25.9, 2.4 Hz, 2H), 6.83 (dd, J = 8.8, 2.4 Hz, 1H), 3.85 (s, 3H), 3.48 (q, J = 6.4 Hz, 2H), 3.39 (t, J = 7.6 Hz, 2H), 3.19 (t, J = 7.7 Hz, 2H), 3.03 (t, J = 6.3 Hz, 2H). HRMS (ESI) m/z [M – H]+: 477.1709, calcd for C24H25N6O3S: 477.1700.
Methods for library construction
In 96-well plate, to a dimethyl sulfoxide (DMSO) solution of the sulphonyl fluoride derivative (20 µL, 4 mM, final conc. 1 mM) was added amine library in DMSO (20 µL, 6 mM, final conc. 1.5 mM) and PBS buffer (pH 7.4, with 1,8-diazabicyclo[5.4.0]undec-7-ene, 40 µL, 6 mM, final conc. 1.5 mM) and the reaction was shaken at 37 °C overnight. The library solution was diluted 50-fold into the buffer (1% DMSO final concentration) and a twofold serial dilution was prepared for the measurement of inhibition. The plate was centrifuged, sealed, and incubated at 37 °C with a humidifier overnight. For the 96-well format, the amine library was tested in phosphate buffered saline (PBS) + DMSO (without difluoride) alone.
EeAChE and eqBuChE inhibition assays
AChE of electric eel and BuChE of horse serum were determined according to Ellman’s method. The absorbance was detected at 410 nm using a Biotek Synergy HTX Multi-Mode reader. Inhibition curves were used to calculate IC50 values.
Kinetic studies of eeAChE inhibition
The same test setup was used for kinetic studies with six concentrations of substrate (0.1–1 mM) and four concentrations of inhibitor (0–0.32 µM). The effect of different doses of compound T14-A24 on the catalytic activity of AChE (0–0.72 U/mL) at 37 °C was also investigated.
hAChE and hBuChE inhibition assays
The compounds were dissolved in DMSO and diluted with ethanol to five concentrations of 200, 100, 10, 1, and 0.1 μM. Briefly, 40 μL of buffer, 10 μL of different concentrations of the compounds for testing, and 10 μL of human AChE (hAChE)/ human BuChE (hBuChE) were added to a 96-well plate. DTNB (Citation20 μL) was added after 10 min of incubation at 37 °C and incubated for another 10 min. In the end, 20 μL of ACh or BuCh was added and the absorbance was measured at 412 nm. IC50 was calculated by SPSS17.0.
Molecular docking study
The crystal structure of hAChE (PDB ID: 4EY4) was used as a template, and the structure was optimised using Discovery Studio Client v20.1.0 (DS). In addition, the Full Minimization function under the DS Small Molecule module was used to prepare the ligands. The Prepare Protein function under the Macromolecules module is used to prepare the protein receptor, which mainly includes removal of water molecules, hydrogenation for the protein, supplementation of non-intact amino acid residues, etc. Then, using CDOCKER, the title compound was docked to the active site of the protein. The optimal docking pose is selected based on the software’s comprehensive score of receptor–ligand interactions, which is -CDOCKER_INTERACTION_ENERGY.
MTT assays
In vitro, the cytotoxic effects of compounds on L02 cells and hepG2 cells, the neuroprotective effects on PC12 cells were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) using donepezil as positive controls.
PAMPA-BBB Penetration Assay
Commercial drugs and test compounds were used in the assay, and the concentrations of the test chemicals in the donor and recipient wells were quantified using a UV plate spectrometer (PerkinElmer VICTOR Nivo, Finland).
Animal studies
Male mice, 18–24 g, 6–8 weeks were used. Forty male mice were randomly assigned into five groups: (i) blank control group; (ii) saline + vehicle; sham-operation group; (iii) oligomerized Aβ1–42 peptide + vehicle; model group; (iv) oligomerized Aβ1–42 peptide + donepezil (15 mg/kg, po),donepezil group, (v) oligomerized Aβ1–42 peptide + T14-A24 (10 mg/kg, po), T14-A24 group.
Morris water maze (MWM) was used to test cognitive function and mice were randomly selected for behavioural testing. Recorded the number of times the target area was crossed, the time in the target NW quadrant, the total distance, the distance in the NW quadrant, the time into the NW quadrant, and the average speed, and compared across experimental groups. All mice were sacrificed after completion of the behavioural study and brains were collected to assess the total Aβ1-42 content using a mouse ELISA kit.
Statistical analysis
Data are reported as mean ± SEM of at least three independent experiments and data analysis was performed with GraphPad Prism 8 software.
Results and discussion
Fragment screening on ChEs activity (T1–T17)
Recently, new aliphatic sulphonyl fluorides containing a triazole moiety were efficiently synthesised through the region-selective cycloaddition reaction of but-3-ene-1,3-disulfonyl difluoride and organic azidesCitation19. Primary screening of compounds T1–T17 (Scheme 1) was performed with equine BuChE (eqBuChE) and electrophorus electricus AChE (eeAChE) using modified Ellman’s methodCitation20. As shown in Supplementary Table S1 and , all of compounds T1–T17 exhibited inhibitory activity against AChE and BuChE. The structure–activity relationships (SAR) showed: (i) most compounds displayed better inhibitory activity against BuChE than against AChE, amongst them, compounds T2, T3, T11, T12, and T14–T17 had almost same inhibitory activity against AChE as BuChE; (ii) halogenated aryl at 1-position of the triazole ring showed potent BuChE inhibition, for example, T4 (4-F, 69.0%), T6 (3-Cl, 71.5%), T8 (4-Br, 82.4%), T9 (3,4-diCl, 88.8%); (iii) benzyl at 1-position of the triazole ring decreased ChE inhibitory activity, for example, T1 (47.9%, 53.6%) > T15 (26.5%, 30.6%).
Scheme 1. Chemical structures of compounds T1–T17.
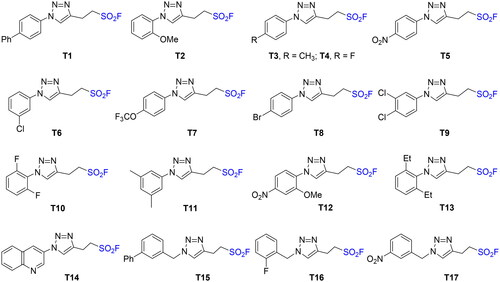
Figure 2. Potency and selectivity profile of inhibitory activity of compounds T1–T17 (A) and amines A1–A24 (B) against AChE and BuChE.
Fragment splicing has been used to design and optimise lead compounds towards new skeleton with target bioactivityCitation21. Herein, a series of novel aliphatic sulphonyl fluorides containing a triazole unit were identified as fragments to find potent ChE inhibitors by fragment splicing and coupling strategy. To balance inhibitory activity between AChE and BuChE, we selected compounds T2, T3, T11, and T14 to perform next fragment splicing-based molecular design.
Binding mode analysis on fragment sulphonyl fluorides
ChE inhibitors are typically used to maintain the level of acetylcholine activityCitation22. The PAS of AChE is centred on Tyr72, Asp74, Tyr124, and Trp 286 as the coreCitation23. The equally critical CAS at the bottom of the binding pocket consists of an enzymatic catalytic triad, Ser203, Glu334, His447, and residues including Asp285, Phe330, and Trp86 at the centerCitation24,Citation25. BuChE is highly similar to AChE, sharing 65% homologous amino acid sequenceCitation26. Their active sites both include CAS, choline-binding pocket, and PAS. The catalytic triad in hBuChE consists of three conserved residues: Ser198, His438, Glu325, which in hAChE are Ser203, His447, Glu334Citation27.
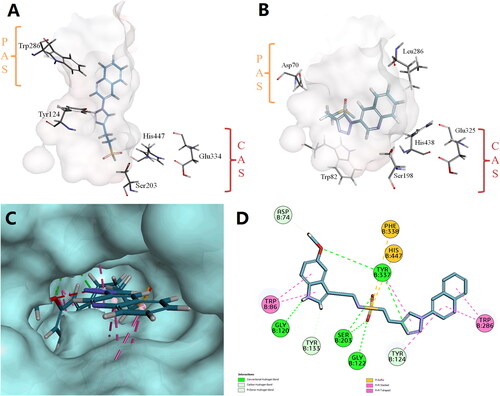
Compound T14 had dual AChE and BuChE inhibition. As shown in , compound T14 was well inserted into the deep CAS active pocket of hAChE and hBuChE. Thus, the sulphonyl fluoride hub as a fragment linker could permit further structural modifications to penetrate the PAS at the entrance of the gorge. The designed molecules may increase the interaction by binding to multiple sites of ChE, including PAS as well as CAS active pockets. This will help to carry out fragment splicing-based molecular design.
Figure 3. Binding orientation of T14 to the active site cavities of (A) hAChE (PDB ID: 4EY4) and (B) hBuChE (PDB ID: 6QAA). Top view of docking result (C) and 2-D (D) plots of T14-A24 onto hAChE (PDB: 4EY4).
Optimisation of microtiter plate-based SuFEx reaction
Microtiter plate-based chemistry has been developed as a new strategy for rapid identification and optimisation of enzyme inhibitorsCitation28,Citation29. Compounds T2, T3, T11, and T14 were selected for fragment splicing-based molecular design. In this work, we would develop a one-step, overnight SuFEx click reaction to carry out small-scale high throughput screening. The sulphonyl fluoride hubs were used to generate the S − N bonds as a fragment linker in this embodiment of SuFEx reaction with appropriate nucleophile aminesCitation30,Citation31. According to the structural characteristics of ChE inhibitors, the amine fragments were selected to be commercially available or synthetically (Supplementary Scheme S1). Then, the SuFEx hubs in T2, T3, T11, and T14 were coupled by SuFEx reaction with a panel of 24 amines to produce 96 analogs in biocompatible solvent (DMSO/PBS, 50/50) and overnight incubation at 37 °C. This will increase the chance of success during fragment-to-lead optimisation stage.
In situ screening of target sulphonamides
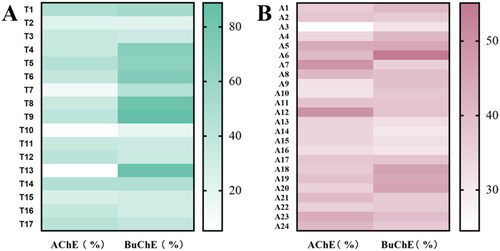
The reaction products were directly screened for eqBuChE and eeAChE inhibitory activity using modified Ellman’s methodCitation20. Scatter plots of the screening results showed that the compounds had better AChE inhibitory activity, and quinolyl triazolyl sulfonylfluoride (T14) was more potent than phenyl triazolyl sulfonylfluorides (T2, T3, and T11) in and . As shown in Supplementary Table S2, 96 sulphonamides were obtained from SuFEx click reaction. In addition, ChE inhibition of amine molecules alone was evaluated as displayed in , with no appreciable effect on the assay (Data are shown in Supplementary Table S3). According to potency, lipophilicity, and molecular weight, we selected a number of molecules and resynthesised them on a milligram scale (Scheme 2), and their potency in the original screen was fully confirmed ( and Supplementary Table S4). Some of sulphonamide products showed improved ChE inhibitory activity, amongst them, compound T14-A24 had the most potent AChE activity (IC50 = 6 nM for AChE).
Scheme 2. Chemical structures of purified compounds in milligram-scale.
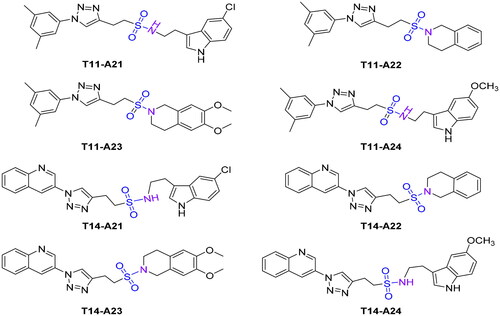
Figure 4. SuFEx-enabled high-throughput ChE screening. Scatter plot of % inhibition rates of reaction crudes at 20 µM.

Kinetic study of eeAChE inhibition
To investigate the type of inhibition of AChE, an enzyme kinetic study of the active compound T14-A24 was carried outCitation32,Citation33. As shown in , the curves of enzyme activity with enzyme concentration (0, 0.045, 0.090, 0.18, 0.36, and 0.72 U/mL) in the presence of different concentrations of T14-A24, the slope of the straight lines decreases with increasing inhibitor concentration, indicating that compound T14-A24 is an inhibitory substance, moreover, these straight lines are intersected to one point, indicating that this inhibition is reversible. As shown in , the slope of the Lineweaver–Burk plot increases (Km increases) but the intercept remains unchanged (Vmax is unaffected) as the concentration of compound T14-A24 increases, indicating that this inhibitor does not affect the maximum rate of the reaction, and it can be concluded that this reversible inhibition is competitive. Furthermore, in the Lineweaver–Burk secondary plot, the dissociation constant Ki for compound T14-A24 was estimated to be 22 nM.
Figure 5. A: Relationship between eqBuChE inhibition and various concentrations of T14-A24; (B) Lineweaver–Burk plots of eqBuChE inhibition kinetics of T14-A24.
Inhibition of hAChE and hBuChE
To determine the potency and selectivity of compounds for the human enzymes, ChE inhibitory activity was detected on hAChE and hBuChE. As shown in , compounds T11-A22, T11-A24, T14-A22, and T14-A24 showed stronger inhibitory effect on hAChE (IC50 = 2.32, 4.36, 2.18 and 1.35 μM for hAChE, respectively), while they hardly inhibited hBuChE even at 20 μM. Hence, compounds T11-A22, T11-A24, T14-A22, and T14-A24 were found as highly selective hAChE inhibitors.
Table 1. Inhibitory activity on hAChE and hBuChETable Footnotea.
Molecular docking of T14-A24
As shown in and , the mode of interaction of receptor–ligand binding is visualised by the CDOCKER molecular docking module of Discovery Studio 2020. Compound T14-A24 was well inserted into the binding groove of hAChE by the following actions: (i) π–π interactions between the benzene ring and Trp86, Tyr124, and Trp286; (ii) π–S interactions between sulphur on the sulphonyl group and Phe338, His447; (iii) hydrogen bonding interactions between oxygen on the sulphonyl group and Gly122, Ser203, between the benzene ring and Tyr124, Gly120 and between the hydroxyl group on the benzene ring and Asp74.
Cytotoxicity assays
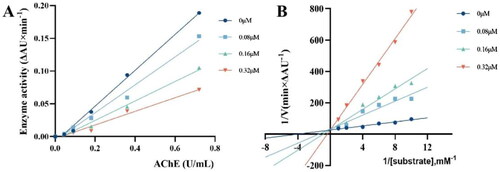
To investigate the safety, the cytotoxicity of compounds with better activity against HepG2 and L02 was measured with MTTCitation34,Citation35. As shown in Supplementary Figure S1, most of the compounds have more than 80% cell activity for HepG2 and L02 cells at 25 μM. Therefore compound T14-A24 was selected as the representative compound. As shown in , the cell survival rate of compound T14-A24 did not change significantly at concentrations of 10 μM and 25 μM, and when the concentration of T14-A24 was increased to 50 μM, the survival rate of HepG2 and L02 cells were maintained at 71.0% and 80.1%, respectively. The results suggested that the target molecule T14-A24 has a broad therapeutic safety for L02 cells and HepG2 cells.
Figure 6. Cytotoxic effects of T14-A24 on (A) L02 cells and (B) HepG2 cells. C: Neuroprotective effect of T14-A24 against H2O2-induced PC12 neurons injury.
Neuroprotective study
The protection of compound T14-A24 against radical damage was appraised by measuring the protective capacity of the compound against H2O2 injuryCitation36,Citation37. T14-A24 showed a dose-dependent protective effect against H2O2-induced damage in PC12 cells (). When the concentration was 25 μM, T14-A24 exhibited a slightly stronger neuroprotective effect than the positive control, donepezil, with a cell viability of 69.3%. When the concentration was decreased to 5 μM, the cell viability dropped to 55.9%. It indicated that T14-A24 can arrest the H2O2-generated free hydroxyl radicals.
PAMPA-BBB Penetration Assay
Parallel Artificial Membrane Permeation Assay (PAMPA) is an in vitro model of passive diffusion used to predict the blood-brain barrier (BBB) permeabilityCitation38,Citation39. To validate the experimental procedure, we have selected six commercially available drugs with reported values. The relationship plot between the experimental and bibliographic data showed a good linear correlation, Pe (exp.) = 0.9092 Pe (bibl.) + 4.1235 (R2 = 0.9309) (Supplementary Figure S2). T14-A24 was measured according to this procedure and the results in show that the compounds tested had permeability values above 4 × 10−6 cm/s, indicating a potential BBB permeability for T14-A24.
Table 2. Results of the PAMPA-BBB assay for six commercial drugs used in the experimental procedure validation and compounds.
Behavioural studies
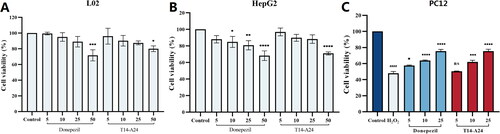
In vivo acute toxicity assessment has proven that T14-A24 has a high in vivo safety profile (Supplementary Figure S3). Then in order to investigate the anti-AD effect of compound T14-A24, we established a model of cognitive impairment induced by Aβ1–42Citation40,Citation41. There was no significant variation in body weight growth between the two groups of mice during the experiment described in , indicating that the compounds have a high in vivo safety profile. As shown in , the learning and memory abilities of the AD model group, mice injected with Aβ intra-cerebroventricularly, were much lower than those of the control group. As shown in ), the compound group, on the one hand, substantially reduced the time to find the platform compared to the model group. On the other hand, T14-A24 shortens the route to the platform and enhances the selectivity of the destination quadrant compared to the donepezil group. This suggests that both T14-A24 and donepezil significantly improved Aβ1–42-induced cognitive dysfunction. The results of the test with mouse Aβ1–42 ELISA kit are shown in , the total level of Aβ1–42 was clearly increased in the icv Aβ1–42 group compared with the control or sham group, indicating successful modelling. Meanwhile, Aβ1–42 was remarkably decreased in mice treated with the positive drug or compound, indicating that the compound can reduce the amount of amyloid peptide in the brain by inhibiting the level of AChE, thus exerting a neuroprotective effect, resulting in a substantial improvement of AD symptoms in mice.
Figure 7. Effects of T14-A24 and donepezil on oligomeric Aβ1–42-induced damage experiments in the MWM task. (A) Protocol followed for in vivo experiments. icv: intraventricular injection; po: orally; MWM: Morris water maze. (B) Daily body weight of mice in different groups during treatment. (C) Learning curves of the escape latencies during the acquisition phase of different groups. (D) Average footprints of mice in MWM on the last day of the study. (E) The time in the target quadrant during the acquisition phase of different groups. (F) The number of times the platform was crossed during the acquisition phase of different groups.
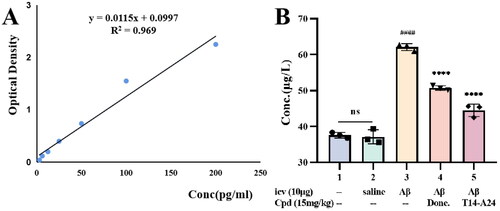
Figure 8. The Aβ1 − 42 total amount was quantified by using a mouse Aβ1 − 42 ELISA kit. (A) Standard curve; (B) Aβ1 − 42 total amount in mice brains of different groups. Calculate brain tissue Aβ1–42 content according to linear regression equation, data are presented as mean ± SEM (n = 8; ####p < 0.0001 (vs. control group), ***p < 0.001, ****p < 0.0001 vs. Aβ1–42 peptide model group).
Conclusion
In summary, as a method for the rapid synthesis of functional molecules, SuFEx click chemistry was used to develop a workflow that allows for in situ synthesis of sulphonamide inhibitors based on SuFEx reaction for high-throughput screening of their cholinesterase activity. This biocompatible SuFEx reaction made us rapidly discover sulphonamide molecule T14-A24 as a reversible, competitive, and selective AChE inhibitor (Ki = 22 nM). In addition, T14-A24 demonstrated exceptional neuroprotective efficacy and moderate BBB penetration potential. In vitro and in vivo safety experiments exhibited that T14-A24 had good neurological and hepatic safety. Subsequent in vivo behavioural studies showed that T14-A24 therapy improved the Aβ1 − 42-induced cognitive impairment, greatly reduced Aβ1 − 42 toxicity, and nearly recovered cognitive function. Furthermore, the evaluation of the Aβ1 − 42 total amount confirmed its anti-amyloidogenic effects better than the positive donepezil. In conclusion, T14-A24 described here represented a potent and selective AChE inhibitor and had a potential lead for the treatments of Alzheimer’s disease. More importantly, SuFEx-enabled lead discovery can accelerate the development of robust biological probes and drug candidates.
Supplemental Material
Download PDF (2.5 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Liu Z, Li J, Li S, Li G, Sharpless KB, Wu P. SuFEx click chemistry enabled late-stage drug functionalization. J Am Chem Soc. 2018;140(8):2919–2925.
- Marra A, Dong J, Ma T, Giuntini S, Crescenzo E, Cerofolini L, Martinucci M, Luchinat C, Fragai M, Nativi C, et al. Protein glycosylation through sulfur fluoride exchange (SuFEx) chemistry: the key role of a fluorosulfate thiolactoside. Chemistry. 2018;24(71):18981–18987.
- Qin H-L, Zheng Q, Bare GAL, Wu P, Sharpless KB. A heck-matsuda process for the synthesis of beta-arylethenesulfonyl fluorides: selectively addressable bis-electrophiles for SuFEx click chemistry. Angew Chem Int Ed Engl. 2016;55(45):14155–14158.
- Li S, Wu P, Moses JE, Sharpless KB. Multidimensional SuFEx click chemistry: sequential sulfur(VI) fluoride exchange connections of diverse modules launched from an SOF4 hub. Angew Chem Int Ed Engl. 2017;56(11):2903–2908.
- Zhao C, Rakesh KP, Ravidar L, Fang W-Y, Qin H-L. Pharmaceutical and medicinal significance of sulfur (SVI)-containing motifs for drug discovery: a critical review. Eur J Med Chem. 2019;162:679–734.
- Oakdale JS, Kwisnek L, Fokin VV. Selective and orthogonal post-polymerization modification using sulfur(VI) fluoride exchange (SuFEx) and copper-catalyzed azide–alkyne cycloaddition (CuAAC) reactions. Macromolecules. 2016;49(12):4473–4479.
- Dong J, Krasnova L, Finn MG, Sharpless KB. Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew Chem Int Ed Engl. 2014;53(36):9430–9448.
- Schimler SD, Cismesia MA, Hanley PS, Froese RDJ, Jansma MJ, Bland DC, Sanford MS. Nucleophilic deoxyfluorination of phenols via aryl fluorosulfonate intermediates. J Am Chem Soc. 2017;139(4):1452–1455.
- Zelli R, Tommasone S, Dumy P, Marra A, Dondoni A. A click ligation based on SuFEx for the metal-free synthesis of sugar and iminosugar clusters. Eur J Org Chem. 2016;2016(30):5102–5116.
- Supuran CT. Carbonic anhydrase inhibition and the management of hypoxic tumors. Metabolites. 2017;7:48.
- Capasso C, Supuran CT. Sulfa and trimethoprim-like drugs – antimetabolites acting as carbonic anhydrase, dihydropteroate synthase and dihydrofolate reductase inhibitors. J Enzyme Inhib Med Chem. 2014;29(3):379–387.
- Brik A, Lin Y-C, Elder J, Wong C-H. A quick diversity-oriented amide-forming reaction to optimize P-subsite residues of HIV protease inhibitors. Chem Biol. 2002;9(8):891–896.
- Zhang Z, Zhang S-L, Wu C, Li H-H, Zha L, Shi J, Liu X, Qin H-L, Tang W. Sulfur-fluoride exchange (SuFEx)-enabled lead discovery of AChE inhibitors by fragment linking strategies. Eur J Med Chem. 2023;257:115502.
- Mamidyala SK, Finn MG. In situ click chemistry: probing the binding landscapes of biological molecules. Chem Soc Rev. 2010;39(4):1252–1261.
- Davies P, Maloney A. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2(8000):1403.
- Masson P, Froment MT, Bartels CF, Lockridge O. Asp 7O in the peripheral anionic site of human butyrylcholinesterase. Eur J Biochem. 1996;235(1–2):36–48.
- Li Q, He S, Chen Y, Feng F, Qu W, Sun H. Donepezil based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur J Med Chem. 2018;158:463–477.
- Jiang X, Zhang Z, Zuo J, Wu C, Zha L, Xu Y, Wang S, Shi J, Liu X-H, Zhang J, et al. Novel cannabidiol-carbamate hybrids as selective BuChE inhibitors: Docking-based fragment reassembly for the development of potential therapeutic agents against Alzheimer’s disease. Eur J Med Chem. 2021; 223:113735.
- Shadrack WL, Qin HL, Tang HL. Transition metal-free regioselective synthesis of triazolyl aliphatic sulfonyl fluorides. Tetrahedron. 2021;98:132425.
- Wu C, Zhang G, Zhang Z-W, Jiang X, Zhang Z, Li H, Qin H-L, Tang W. Structure–activity relationship, in vitro and in vivo evaluation of novel dienyl sulphonyl fluorides as selective BuChE inhibitors for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem. 2021;36(1):1860–1873.
- Fu Y, Zhang D, Kang T, Guo Y-Y, Chen W-G, Gao S, Ye F. Fragment splicing-based design, synthesis and safener activity of novel substituted phenyl oxazole derivatives. Bioorg Med Chem Lett. 2019;29(4):570–576.
- Akıncıoğlu H, Gülçin İ. Potent acetylcholinesterase inhibitors: potential drugs for Alzheimer’s disease. Mini Rev Med Chem. 2020;20(8):703–715.
- Walczak-Nowicka ŁJ, Herbet M. Acetylcholinesterase inhibitors in the treatment of neurodegenerative diseases and the role of acetylcholinesterase in their pathogenesis. Int J Mol Sci. 2021;22:9290.
- de Almeida JSFD, Dolezal R, Krejcar O, Kuca K, Musilek K, Jun D, França TCC. Molecular modeling studies on the interactions of aflatoxin B1 and its metabolites with human acetylcholinesterase. II. Interactions with the catalytic anionic site (CAS). Toxins (Basel). 2018;10:389.
- Ordentlich A, Barak D, Kronman C, Flashner Y, Leitner M, Segall Y, Ariel N, Cohen S, Velan B, Shafferman A, et al. Dissection of the human acetylcholinesterase active center determinants of substrate specificity. Identification of residues constituting the anionic site, the hydrophobic site, and the acyl pocket. J Biol Chem. 1993;268(23):17083–17095.
- Joubert J, Kapp E. Discovery of 9-phenylacridinediones as highly selective butyrylcholinesterase inhibitors through structure-based virtual screening. Bioorg Med Chem Lett. 2020;30(9):127075.
- de Andrade P, Mantoani SP, Gonçalves Nunes PS, Magadán CR, Pérez C, Xavier DJ, Hojo ETS, Campillo NE, Martínez A, Carvalho I, et al. Highly potent and selective aryl-1,2,3-triazolyl benzylpiperidine inhibitors toward butyrylcholinesterase in Alzheimer’s disease. Bioorg Med Chem. 2019;27(6):931–943.
- Lee SG, Chmielewski J. Rapid synthesis and in situ screening of potent HIV-1 protease dimerization inhibitors. Chem Biol. 2006;13(4):421–426.
- Wu C-Y, Chang C-F, Chen JS-Y, Wong C-H, Lin C-H. Rapid diversity-oriented synthesis in microtiter plates for in situ screening: discovery of potent and selective alpha-fucosidase inhibitors. Angew Chem Int Ed Engl. 2003;42(38):4661–4664.
- Liang F-S, Brik A, Lin Y-C, Elder JH, Wong C-H. Epoxide opening in water and screening in situ for rapid discovery of enzyme inhibitors in microtiter plates. Bioorg Med Chem. 2006;14(4):1058–1062.
- Jahnke W, Erlanson DA, de Esch IJP, Johnson CN, Mortenson PN, Ochi Y, Urushima T. Fragment-to-lead medicinal chemistry publications in 2019. J Med Chem. 2020;63(24):15494–15507.
- Nagar S, Argikar UA, Tweedie DJ. Enzyme kinetics in drug metabolism: fundamentals and applications. Methods Mol Biol. 2014;1113:1–6.
- Larik FA, Shah MS, Saeed A, Shah HS, Channar PA, Bolte M, Iqbal J. New cholinesterase inhibitors for Alzheimer’s disease: structure activity relationship, kinetics and molecular docking studies of 1-butanoyl-3-arylthiourea derivatives. Int J Biol Macromol. 2018;116:144–150.
- Park JW, Ha YM, Moon K-m, Kim S-r, Jeong HO, Park YJ, Lee HJ, Park JY, Song YM, Chun P, et al. De novo tyrosinase inhibitor: 4-(6,7-dihydro-5H-indeno[5,6-d]thiazol-2-yl)-benzene-1,3-diol (MHY1556). Bioorg Med Chem Lett. 2013;23(14):4172–4176.
- Tada H, Shiho O, Kuroshima K, Koyama M, Tsukamoto K. An improved colorimetric assay for interleukin 2. J Immunol Methods. 1986;93(2):157–165.
- Liu J, Zhang L, Liu D, Li B, Zhang M. Neuroprotective effects of extracts from the radix curcuma aromatica on H2O2-induced damage in PC12 cells. Comb Chem High Throughput Screen. 2018;21(8):571–582.
- Hajialyani M, Hosein Farzaei M, Echeverría J, et al. Hesperidin as a neuroprotective agent: a review of animal and clinical evidence. Molecules. 2019;24:648.
- Di L, Kerns EH, Bezar IF, Petusky SL, Huang Y. Comparison of blood–brain barrier permeability assays: in situ brain perfusion, MDR1-MDCKII and PAMPA-BBB. J Pharm Sci. 2009;98(6):1980–1991.
- Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem. 2003;38(3):223–232.
- Takeda S, Hashimoto T, Roe AD, Hori Y, Spires-Jones TL, Hyman BT. Brain interstitial oligomeric amyloid increases with age and is resistant to clearance from brain in a mouse model of Alzheimer’s disease. FASEB J. 2013;27(8):3239–3248.
- Hampel H, Mesulam M-M, Cuello AC, Farlow MR, Giacobini E, Grossberg GT, Khachaturian AS, Vergallo A, Cavedo E, Snyder PJ, et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain. 2018;141(7):1917–1933.