
Abstract
In this work, a series of novel arylamide derivatives containing piperazine moiety were designed and synthesised as tubulin polymerisation inhibitors. Among 25 target compounds, compound 16f (MY-1121) exhibited low nanomolar IC50 values ranging from 0.089 to 0.238 μM against nine human cancer cells. Its inhibitory effects on liver cancer cells were particularly evident with IC50 values of 89.42 and 91.62 nM for SMMC-7721 and HuH-7 cells, respectively. Further mechanism studies demonstrated that compound 16f (MY-1121) could bind to the colchicine binding site of β-tubulin and directly act on β-tubulin, thus inhibiting tubulin polymerisation. Additionally, compound 16f (MY-1121) could inhibit colony forming ability, cause morphological changes, block cell cycle arrest at the G2 phase, induce cell apoptosis, and regulate the expression of cell cycle and cell apoptosis related proteins in liver cancer cells. Overall, the promising bioactivities of compound 16f (MY-1121) make the novel arylamide derivatives have the value for further development as tubulin polymerisation inhibitors with potent anticancer activities.
Introduction
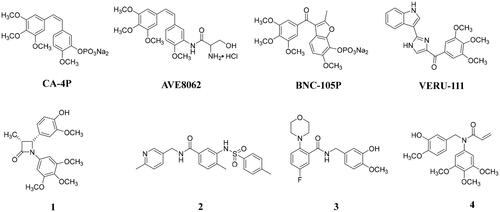
Microtubules, which are mainly composed mainly of α, β-tubulin heterodimers, serve as key component of the cytoskeletonCitation1. Given the important roles in the multiple biological functions, microtubule has been one important and successful target for the human cancer treatment. At present, a total of seven binding sites of tubulin have been identifiedCitation2,Citation3, and there are already microtubule inhibitors targeting taxane and vinca alkaloid sites approved by the FDA for clinical tumour treatment. Colchicine binding site, as one of the tubulin binding sites, is also receiving increasing attention due to their ability to overcome the shortcomings and limitations associated with taxane or vinca alkaloid binding site inhibitorsCitation4,Citation5. Although there are no FDA-approved colchicine binding site inhibitors (CBSIs) for clinical tumour treatment, several CBSIs (e.g. CA-4PCitation6, AVE8062Citation7, BNC-105pCitation8, and VERU-111Citation9; ) have been or are being studied in clinical trials due to their significant antitumor activities. Additionally, many groups also have reported potent anticancer effective CBSIs with different chemical structure typesCitation10–17, and the arylamide tubulin inhibitors have also attracted much attention because of their strong antitumor abilitiesCitation18–21. For example, compound 1Citation18, benzamide derivative 2Citation19, and N-benzylbenzamide derivative 3Citation20 could potently inhibit the polymerisation of tubulin and exhibit significant antiproliferative activities on several cancer cell lines. Recently, we also reported N-benzylarylamide 4 as a tubulin polymerisation inhibitor with potent antiproliferative activities in two-digit nanomolar IC50 valuesCitation21.
Figure 1. Structures of typical CBSIs under clinical trials and reported CBSIs.
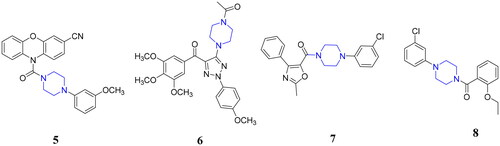
The structures of FDA approved marketed drugs generally contain nitrogen heterocycles, especially piperazines, which might help to adjust the lipid–water partition coefficient and acid–base equilibrium constant of drugsCitation22–24. In fact, piperazine groups are also frequently used to design antitumor drugsCitation25. Recently, many groups have also reported high anticancer potency tubulin polymerisation inhibitors with piperazine moietiesCitation26–35. Müller and coworkers reported a N‑heterocyclic (4-phenylpiperazin-1-yl)methanone 5 as a tubulin polymerisation inhibitor (), which was derived from the phenoxazine and phenothiazineCitation26. Compound 5 exhibited potent antiproliferative potency against a panel of cancer cell lines with a mean GI50 value of 3.3 nM and potent inhibitory effect on tubulin polymerisation by acting on the colchicine binding site. Compound 6 posing a N-acyl-piperazine moiety, as the tubulin polymerisation inhibitor, showed significant antiproliferative activities against SGC-7901, A549, and Hela cells (IC50 values of 0.084–0.221 μM)Citation31. In addition, arylpiperazinyl oxazole 7 exhibited potent anticancer effects in vitro and in vivo by the inhibition of tubulin polymerisation and excellent vascular-disrupting activityCitation32. Piperazine-based compound 8 was identified as a tubulin polymerisation inhibitor with the activity of sensitising cancer cells to apoptotic ligandsCitation35.
Figure 2. Structures of piperazine-based tubulin inhibitors.
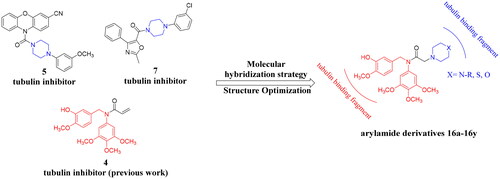
In a continuation of our interest in developing novel tubulin polymerisation inhibitorsCitation21,Citation36, we designed and synthesised a series of novel arylamide derivatives containing piperazine moiety as inhibitors of tubulin polymerisation. In this work, we assumed that linking N-benzylarylamide fragment to the piperazine moieties through the principle of molecular hybridisation strategy could afford novel arylamide derivatives containing piperazine moiety, which might bind to the colchicine binding site of β-tubulin with potent anticancer activities (). Therefore, a total of 25 target compounds were designed, synthesised and explored for antiproliferative activities against three selected tumour cell lines. Among them, the representative compound 16f (MY-1121) was identified as a tubulin polymerisation inhibitor by binding to the colchicine binding site with potent anticancer activities.
Figure 3. Design strategy of arylamide derivatives as inhibitors of tubulin polymerisation.
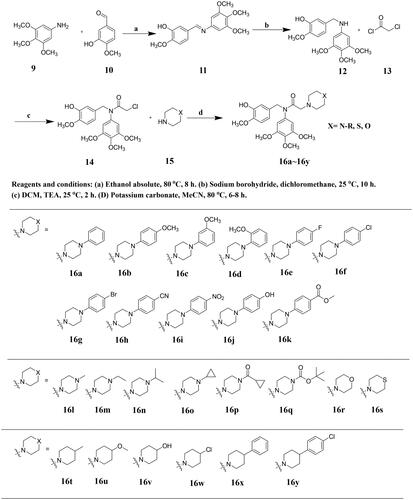
Synthesis of novel arylamide derivatives 16a–16y
The synthetic route of the designed arylamide derivatives 16a–16y is outlined in Scheme 1. A condensation reaction between commercially available 3-hydroxy-4-methoxybenzaldehyde (10) and 3,4,5-trimethoxyaniline (9) afforded the imine intermediate 11, which was further reduced with sodium borohydride (NaBH4) to get 2-methoxy-5-(((3,4,5-trimethoxyphenyl)amino)methyl)phenol (12). Then, 12 was subjected to a substitution reaction in the presence of triethylamine (TEA) to obtain 2-chloro-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (13), which were then treated with commercially available morpholine, thiomorpholine, or different substituted piperazines and piperidines to give the corresponding designed arylamide derivatives 16a–16y.
Scheme 1. Synthesis of target compounds 16a–16y.
Results and discussion
Antiproliferative activities of compounds 16a–16y against MGC-803, HCT-116, and SMMC-7721 cells
To explore the antiproliferative activities of designed arylamide derivatives 16a–16y, we determined the MTT assay to test the antiproliferative potency on three selected human cancer cells, including human liver cancer cells SMMC-7721, human gastric cancer cells MGC-803, and human colon cancer cells HCT-116. Additionally, the well-known tubulin polymerisation inhibitor colchicine was selected as the positive control. The results of the antiproliferative activities of designed arylamide derivatives 16a–16y are shown in .
Table 1. Antiproliferative activities of compounds 16a–16k and colchicine.
Table 2. Antiproliferative activities of compounds 16l–16s and colchicine.
Table 3. Antiproliferative activities of compounds 16t–16y and colchicine.
As described in , we first explored the antiproliferative activities of compounds 16a–16k with aryl piperazine groups. As expected, most of compounds exhibited potent antiproliferative activities against these three human cancer cells with IC50 values less than 0.1 μM. Among them, the compound 16f (also named MY-1121) bearing the 1-(4-chlorophenyl)piperazine group exhibited the most potent inhibitory activities on MGC-803 (IC50 = 0.092 μM), HCT-116 (IC50 = 0.098 μM), and SMMC-7721 (IC50 = 0.088 μM) cells, which was similar to the positive control colchicine. In addition, the SMMC-7721 cells were more sensitive to compounds 16a–16k with lower IC50 values. Therefore, we discussed the structure–activity relationships based on the antiproliferative activities of compounds 16a–16k on SMMC-7721 cells. Compared with compound 16a, it was found that the substituents and their positions of the aryl piperazines showed a certain influence on the inhibitory activities. Among compounds 16b–16d, the antiproliferative activity of compound 16b with substituent in the para position of aryl piperazine was superior to that in the meta (compound 16c) and ortho (compound 16d) positions. Compared with compound 16a, the introduction of electron withdrawing groups (–CN, –OH, –F, –Cl, –Br, and –COOCH3) on the para position of benzene ring helped to improve the antiproliferative activities, while the introduction of electron donating group –OCH3 in the para, meta, or ortho position could impair the inhibitory potency.
We further explored the effects of replacement of aryl piperazine groups with alkyl piperazines, carbonyl piperazines, morpholine, or thiomorpholine on the antiproliferative activities. As shown in , the target compounds 16l–16s exhibited reduced antiproliferative activities with IC50 values ranging from 0.88 to 6.69 μM than that of compounds 16a–16k. Among them, compound 16l bearing the 1-methylpiperazine group exhibited the most potent inhibitory activities with IC50 values of 1.21 (for MGC-803), 0.88 (for HCT-116), and 1.58 μM (for SMMC-7721). Comparing the antiproliferative activities of compounds 16l–16s, it was obvious to figure out that the compounds 16l–16o bearing the alkyl piperazines were more effective in inhibiting the proliferation of cancer cells than that of compounds 16p–16s bearing the carbonyl piperazines (compounds 16p–16q), morpholine (compound 16r), or thiomorpholine (compound 16s). In addition, the extension of alkyl chains (compounds 16l–16o) reduced inhibitory activities.
In addition, we also explored the effects of replacement of piperazine groups with piperidine groups on the antiproliferative activities (). Unfortunately, when the substituent piperazine groups were replaced with the piperidine groups, the antiproliferative activities of the compounds were obviously reduced with IC50 values ranging from 5.01 to 8.16 μM, indicating that piperazine groups were more help to maintain inhibitory effects than that of the piperidine groups.
Antiproliferative activities of compound MY-1121 against other 7 cells
Given the most potent antiproliferative activities of compound MY-1121 against MGC-803, HCT-116, and SMMC-7721 cells, six additional cancer cell lines (human liver cancer cells MHCC-97H, HuH-7 and HepG2, human gastric cancer cells BGC-823 and NCI-N87, and human oesophageal cancer cells KYSE450) and one normal liver cells HL-7702 were chosen to further explore the broad-spectrum antiproliferative activities. As shown in , compound MY-1121 also exhibited potent antiproliferative activities against these selected cancer cells with IC50 values from 91.6 to 238.2 nM. Among them, human liver cancer cells HuH-7 and HepG2 were more sensitive to compound MY-1121 with IC50 values of 91.6 and 100.8 nM, respectively. Importantly, compound MY-1121 displayed more reduced antiproliferative activity against normal liver cells HL-7702.
Table 4. Antiproliferative activities of compound MY-1121 against six additional cancer cell lines.
Compound MY-1121 inhibited tubulin polymerisation by acting on the colchicine binding site of β-tubulin
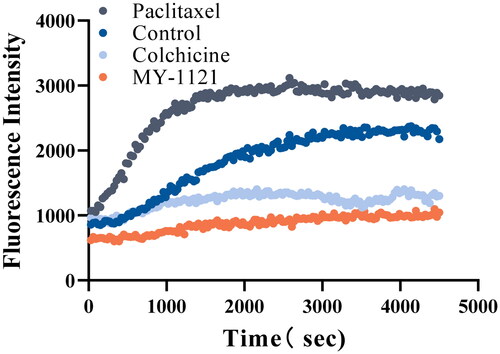
To explore whether these compounds we designed interacted with tubulin system, the most potent compound MY-1121 was selected to determine the inhibitory effect on tubulin polymerisation by the cell-free tubulin polymerisation assay. Additionally, the well-known tubulin polymerisation inhibitor colchicine and microtubule-stabilising agent paclitaxel were chosen as the reference drugs. As shown in , compound MY-1121 significantly decreased the absorbance, indicating that it could inhibit the polymerisation of tubulin, which was consistent with the effect of colchicine, and the effect was stronger than that of colchicine. While the paclitaxel obviously increased the absorbance, suggesting that it could enhance the polymerisation of tubulin. These results indicated that compound MY-1121 exhibited slightly less potent tubulin polymerisation inhibitory activity than that of colchicine.
Figure 4. Effects of compound MY-1121 on tubulin polymerisation under cell free condition. The vertical coordinate indicates the degree of tubulin polymerisation. The experiments were repeated thrice.
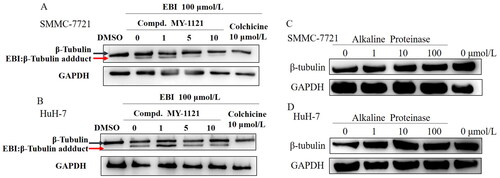
To confirm whether the compound MY-1121 could act on the colchicine binding site of β-tubulin, the EBI competitive assay and alkaline protease assay were carried out in SMMC-7721 and HuH-7 cells. In the EBI competitive assay, if the tested compound exhibit good affinity with the colchicine binding site of β-tubulin, the β-tubulin adduct band in the western blot results will be reduced. As shown in , compound MY-1121 significantly reduced the signal level of the β-tubulin adduct band in a concentration-dependent manner, which was consistent with the effect of the colchicine. These results indicated that compound MY-1121 showed good affinity with the colchicine binding site of β-tubulin. In the alkaline protease assay (), compound MY-1121 could directly bind to β-tubulin, leading to a decrease in the degradation ability of alkaline protease on β-tubulin. These results indicated that compound MY-1121 could act on the colchicine binding site of β-tubulin.
Figure 5. Compound MY-1121 bind to β-tubulin directly on colchicine binding site. (A&B). EBI competition assay, the affinity of the compound with colchicine binding site was negatively correlated with the level of the tubulin adduct band; (C&D). Alkaline protease assay. Cells were treated with Alkaline Proteinase and different concentration of compound MY-1121. The band signal of β-tubulin was negatively correlated with the ability of Alkaline Protease to hydrolyze β-tubulin. The binding of the compound to β-tubulin was able to inhibit the hydrolysis of β-tubulin by Alkaline Protease.
Inhibition of intracellular microtubules
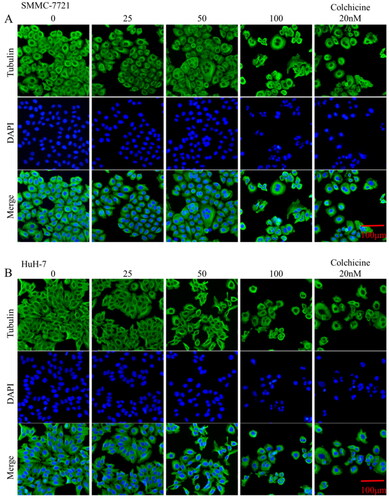
To profile the intracellular inhibitory abilities of the representative compound MY-1121 against microtubules, the immunofluorescence staining assay was performed in SMMC-7721 and HuH-7 cells. As shown in , with the increase concentrations of compound MY-1121, the proportion of tubulin (green) polymerised into filamentous structures (microtubules) gradually decreased, indicating that compound MY-1121 inhibited the ordered polymerisation of tubulin. However, the cell nucleus did not stopped replicating, resulting in a phenomenon of multiple nuclei in the high-dose group in SMMC-7721 and HuH-7 cells. Additionally, these intracellular inhibitory effects were consistent with the positive control colchicine. Taken together, these above results indicated that MY-1121 could inhibit microtubules at the cellular level.
Figure 6. Effects of compound MY-1121 on microtubule network in liver cancer cells SMMC-7721 and HuH-7. Cells were treated for 48 h. β-tubulin was stained green and cell nuclei were stained blue.
Binding model of compound MY-1121 with tubulin
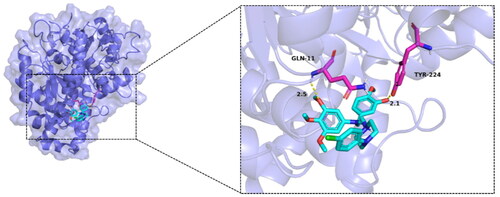
Given the compound MY-1121 was identified as a novel tubulin polymerisation inhibitor, a molecular docking was performed to study the possible binding mode of compound MY-1121 with tubulin (PDB: 1SA0). We analysed the interactions between protein and ligand (), all functional residues were identified and classified according to their interactions. There were multiple groups of residues used to form interactions between receptor protein and ligand, such as the hydrogens bond formed by GLN11 and TYR224 of TUBA1D and ligand. With these interaction forces, the binding energy of protein–ligand complex was −6.6 kcal/mol, which was a good performance and might help to exhibit potent activities.
Figure 7. Molecular docking results of compound MY-1121 with tubulin (PDB: 1AS0). The hydrogen bond, ionic interactions, and hydrophobic interactions are depicted as yellow, magentas, and green dashed lines, respectively.
Compound MY-1121 inhibited cell proliferation
As the executor of cell proliferation, when the polymerisation of tubulin is inhibited, cell proliferation is also inhibited. Therefore, after verifying the inhibitory effects of compound MY-1121 on tubulin polymerisation, we further explored the effects of compound MY-1121 on the proliferation of liver cancer cells SMMC-7721 and HuH-7.
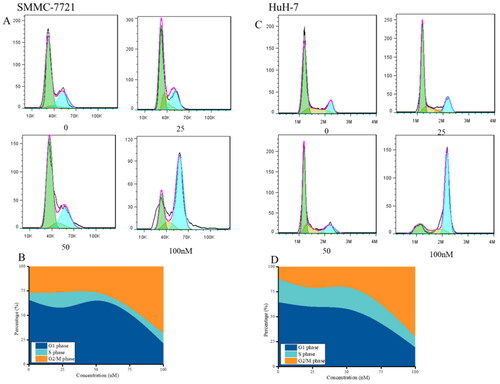
The results in suggested that compound MY-1121 caused strong arresting effects on the cell cycle in SMMC-7721 and HuH-7 cells, and the percentage of G2/M phase cells in two liver cancer cells increased by 40% (SMMC-7721 cells) and 57% (HuH-7 cells), respectively, indicating that compound MY-1121 arrested SMMC-7721 and HuH-7 cell cycle at the G2 phase.
Figure 8. Effects of compound MY-1121 on cell cycle distribution. Liver cancer cells SMMC-7721 (A, B) and HuH-7 (C, D) were treated for 48 h. The cells were stained with propidium iodide and analysed via flow cytometry to measure the cell cycle profile.
Considering that the colony forming ability of cells is a manifestation of cell proliferation activity, we further investigated the effects of compound MY-1121 on the colony forming ability of HuH-7 cells by the colony formation assay. As shown in , when cells were treated with compound MY-1121 for seven days, the ability of cells to format colonies was concentration-dependently inhibited, and the number of colonies was also significantly reduced in a concentration-dependent manner.
Figure 9. Effects of compound MY-1121 on colony formatting ability. Liver cancer cells SMMC-7721 (A) and HuH-7 (B) were treated for seven days.

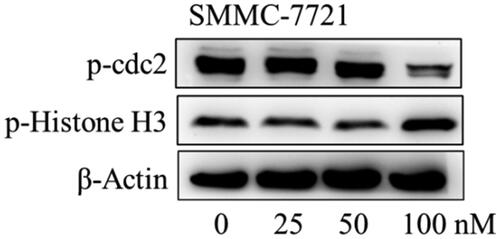
The effects of compound MY-1121 on the expression of cell cycle regulatory proteins were further tested by Western blotting assay. At the protein levels, compound MY-1121 induced down-regulation on the expression of G2 phase-related protein p-cdc2, and up-regulation on the expression of M phase marker protein p-histone H3 ().
Figure 10. The effects of compound MY-1121 on cell cycle related proteins in SMMC-7721 cell were conducted via Western blotting assay. Cells were treated with indicated concentrations of compound MY-1121 for 48 h.
In summary, compound MY-1121 could arrest liver cancer cells SMMC-7721 and HuH-7 cells in the G2/M phase and regulate the expression of related proteins.
Effects of compound MY-1121 on morphology changes
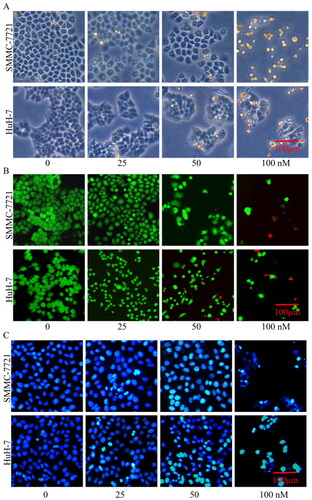
Effects of compound MY-1121 on morphology changes in SMMC-7721 and HuH-7 cells were further explored. As shown in , under the compound MY-1121 effects, the SMMC-7721 and HuH-7 cells became smaller and rounder (upper panel in ), the cell density decreased and the dead cells (red) increased (middle panel in ), and the nuclei became brighter and concentrated (lower panel in ).
Figure 11. Effects of compound MY-1121 on morphology changes of liver cancer cells. Liver cancer cells were treated for 48 h. (A) Cell morphology in the bright field; (B) number of live cells (green) to dead cells (red); and (C) cell nuclei changes.
Compound MY-1121 induced apoptosis in liver cancer cells SMMC-7721 and HuH-7
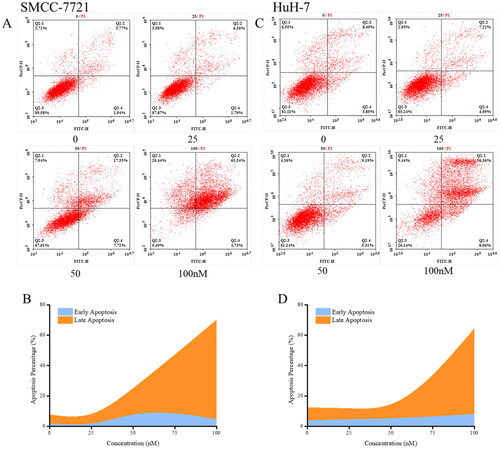
Previous studies have shown that tumour cell cycle arrest and inhibition of proliferation have the potential to trigger the development of apoptosis. Therefore, the effects of compound MY-1121 on apoptosis of liver cancer cells SMMC-7721 and HuH-7 were examined. The changes in cell morphology after the treatment of compound MY-1121 suggested that it could induce apoptosis in liver cancer cells SMMC-7721 and HuH-7 (). The annexin-V/PI assay further validated the concentration-dependent induction of apoptosis by compound MY-1121 in SMMC-7721 and HuH-7 cells (). As shown in , after 48 h of treatment with compound MY-1121, the apoptosis rates of SMMC-7721 and HuH-7 cells were both close to 70%, indicating that compound MY-1121 exhibited a clear apoptosis-inducing effect.
Figure 12. Cell apoptosis induced by compound MY-1121. Liver cancer cell SMMC-7721 (A, B) and HuH-7 (C, D) were treated indicated concentrations of compound MY-1121 for 48 h.
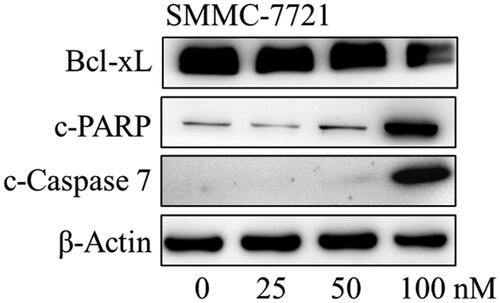
Considering that compound MY-1121 could induce apoptosis in SMMC-7721 and HuH-7 cells, further detection of the effects on apoptosis-related proteins were conducted by the Western blotting assay. As shown in , it was found that compound MY-1121 caused a decrease in the expression of anti-apoptotic protein Bcl-xL, and an increase in the expression of apoptosis executor cleaved caspase-7, as well as its cleavage substrate cleaved-PARP. These above results indicated that compound MY-1121 effectively induced apoptosis and regulated the expression of apoptosis related proteins in liver cancer cells SMMC-7721 and HuH-7.
Figure 13. The effects of compound MY-1121 on apoptosis-related proteins in SMMC-7721 cell were conducted via Western blotting assay. Cells were treated indicated concentrations of compound MY-1121 for 48 h.
Conclusions
In summary, a series of novel arylamide derivatives containing piperazine moiety as inhibitors of tubulin polymerisation have been designed, synthesised, and evaluated. The representative compound MY-1121 exhibited low nanomolar IC50 values ranging from 0.089 to 0.238 μM against nine cancer cells. Its inhibitory effects on liver cancer cells were particularly evident, with IC50 values of 89.42 and 91.62 nM for SMMC-7721 and HuH-7 cells, respectively. Further research found that compound MY-1121 could bind to the colchicine binding site of β-tubulin and directly act on β-tubulin, thus inhibiting tubulin polymerisation both in liver cancer cells and cell-free conditions. The mechanism studies demonstrated compound MY-1121 could cause cell cycle arrest at G2 phase, induce cell apoptosis, and regulate the expression of cell cycle and cell apoptosis related proteins in liver cancer cells. In addition, compound MY-1121 could also inhibit the colony forming ability of SMMC-7721 cells and cause morphological changes in MMC-7721 and HuH-7 cells. Therefore, novel arylamide derivatives containing piperazine moiety could serve as effective tubulin polymerisation inhibitors, which caused inhibition of cell proliferation and induction of apoptosis in liver cancer cells.
Experimental
General
All the commercially available materials (Aladdin Co., Ltd., Shanghai, China, Macklin Co., Ltd., Shanghai, China, or Innochem Co., Ltd., Beijing, China), solvents (Aladdin Co., Ltd., Shanghai, China, Macklin Co., Ltd., Shanghai, China, or Innochem Co., Ltd., Beijing, China), antibodies (Cell Signaling Technology, Beverly, MA), and cancer cells (Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai, China) were purchased from commercial companies. NMR and HRMS spectra were recorded using the Bruker av400 and Bruker Micromass (Billerica, MA), respectively. The purity of compounds was determined by reversed phase C18 high performance liquid chromatography with UV detection at 254 nm and 25 °C.
Synthesis of target compounds 16a–16y
The synthesis methods of compounds 11, 12, and 14 refer to our previous collaboration and were provided in the supporting informationCitation21.
To a solution of 14 (1.0 mmol, 1.0 equiv.) in anhydrous acetonitrile (20 mL), commercially available morpholine, thiomorpholine, or different substituted piperazines and piperidines (1.0 mmol, 1.0 equiv.), and K2CO3 (1.0 mmol, 0.5 equiv.) were added, and then the mixture was stirred for 6–8 h at 80 °C. Upon completion, the resultant mixture was concentrated to get the white crude product, which was further purified by chromatography on silica gel (PE:EA = 1:1) to afford corresponding designed arylamide derivatives 16a–16y.
N-(3-Hydroxy-4-methoxybenzyl)-2-(4-phenylpiperazin-1-yl)-N-(3,4,5-trimethoxyphenyl) acetamide (16a)
White powder; yield: 58.2%; m.p. 118–119 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.94 (s, 1H), 7.20 (t, J = 7.8 Hz, 2H), 6.90 (d, J = 8.1 Hz, 2H), 6.84–6.69 (m, 3H), 6.57 (d, J = 8.0 Hz, 1H), 6.43 (s, 2H), 4.68 (s, 2H), 3.72 (s, 3H), 3.68 (s, 6H), 3.66 (s, 3H), 3.06 (s, 6H), 2.55 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 169.05, 153.25, 151.47, 147.30, 146.77, 137.78, 137.18, 130.82, 129.37, 119.75, 119.24, 116.19, 115.80, 112.41, 106.38, 60.55, 59.42, 56.42, 56.10, 52.86, 52.03, 48.69. HRMS: calcd. C29H36N3O6 [M + H]+, 522.2599, found: 522.2607.
N-(3-Hydroxy-4-methoxybenzyl)-2-(4-(4-methoxyphenyl)piperazin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16b)
White powder; yield: 62.1%; m.p. 108–109 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 6.88–6.76 (m, 5H), 6.70 (d, J = 1.8 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.43 (s, 2H), 4.68 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.04 (s, 2H), 2.99–2.89 (m, 4H), 2.60–2.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.98, 153.33, 153.24, 147.30, 146.79, 145.87, 137.79, 137.17, 130.84, 119.73, 117.76, 116.22, 114.71, 112.42, 106.39, 60.54, 59.45, 56.42, 56.10, 55.64, 52.97, 52.04, 50.06. HRMS: calcd. C30H38N3O7 [M + H]+, 522.2704, found: 522.2710.
N-(3-Hydroxy-4-methoxybenzyl)-2-(4-(3-methoxyphenyl)piperazin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16c)
White powder; yield: 49.7%.; m.p. 112–113 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.08 (t, J = 8.2 Hz, 1H), 6.81 (d, J = 8.2 Hz, 1H), 6.70 (d, J = 1.7 Hz, 1H), 6.58–6.32 (m, 6H), 4.67 (s, 2H), 3.72 (s, 3H), 3.70 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.11–2.99 (m, 6H), 2.55–2.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.52, 160.17, 152.75, 152.35, 146.81, 146.31, 137.32, 136.73, 130.37, 129.51, 119.23, 115.74, 111.99, 107.97, 105.94, 104.06, 101.41, 60.00, 58.94, 55.99, 55.59, 54.78, 52.34, 51.56, 48.18. HRMS: calcd. C30H38N3O7 [M + H]+, 522.2704, found: 522.2714.
N-(3-Hydroxy-4-methoxybenzyl)-2-(4-(2-methoxyphenyl)piperazin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16d)
White powder; yield: 59.2%; m.p. 121–122 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 6.95–6.79 (m, 5H), 6.70 (d, J = 1.6 Hz, 1H), 6.57 (d, J = 7.8 Hz, 1H), 6.43 (s, 2H), 4.68 (s, 2H), 3.75 (s, 3H), 3.72 (s, 3H), 3.68 (s, 6H), 3.65 (s, 3H), 3.05 (s, 2H), 2.94–2.82 (m, 4H), 2.61–2.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 169.07, 153.24, 152.44, 147.29, 146.77, 141.70, 137.81, 137.16, 137.13, 130.83, 122.86, 121.32, 119.73, 118.37, 116.18, 112.42, 106.38, 60.56, 59.52, 56.43, 56.10, 55.78, 53.10, 52.02, 50.51. HRMS: calcd. C30H38N3O7 [M + H]+, 522.2704, found: 522.2709.
2-(4-(4-Fluorophenyl)piperazin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16e)
White powder; yield: 49.7%; m.p. 124–125 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.05–6.98 (m, 2H), 6.91 (dd, J = 9.1, 4.6 Hz, 2H), 6.81 (d, J = 8.2 Hz, 1H), 6.70 (d, J = 1.8 Hz, 1H), 6.57 (d, J = 5.8 Hz, 1H), 6.41 (d, J = 12.4 Hz, 2H), 4.67 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.05 (s, 2H), 3.02–2.96 (m, 4H), 2.58–2.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.52, 157.09, 154.76, 152.75, 147.89 (d, J = 1.9 Hz), 146.81, 146.31, 137.01 (d, J = 58.8 Hz), 130.37, 119.23, 116.97, 115.28, 115.06, 111.96, 105.94, 60.00, 58.89, 55.98, 55.90, 55.67, 52.34, 51.55, 49.00. HRMS: calcd. C29H35FN3O6 [M + H]+, 540.2504, found: 540.2511.
2-(4-(4-Chlorophenyl)piperazin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16f, MY-1121)
White powder; yield: 62.1%; m.p. 132–133 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.21 (d, J = 9.0 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 6.81 (d, J = 8.2 Hz, 1H), 6.70 (d, J = 1.7 Hz, 1H), 6.57 (d, J = 7.5 Hz, 1H), 6.43 (s, 2H), 4.68 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.12–3.02 (m, 6H), 2.57–2.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.99, 153.25, 150.26, 147.30, 146.76, 137.75, 137.16, 130.80, 129.05, 122.72, 119.74, 117.25, 116.20, 112.41, 106.38, 60.55, 59.33, 56.42, 56.10, 52.66, 52.05, 48.50. HRMS: calcd. C29H35ClN3O6 [M + H]+, 556.2209, found: 556.2211.
2-(4-(4-Bromophenyl)piperazin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16g)
White powder; yield: 58.7%; m.p. 119–120 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.32 (d, J = 8.9 Hz, 2H), 6.83 (dd, J = 19.1, 8.6 Hz, 3H), 6.70 (d, J = 1.5 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.42 (s, 2H), 4.67 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.33 (s, 2H), 3.07–3.04 (m, 4H), 2.57–2.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.50, 152.76, 150.11, 146.81, 146.31, 137.29, 136.73, 131.43, 130.36, 119.23, 117.20, 115.75, 111.95, 109.84, 105.93, 60.00, 58.86, 55.99, 55.90, 55.59, 52.14, 47.91. HRMS: calcd. C29H35BrN3O6 [M + H]+, 600.1704, found: 600.1696.
2-(4-(4-Cyanophenyl)piperazin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16h)
White powder; yield: 67.1%; m.p. 142–142 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.57 (t, J = 9.2 Hz, 2H), 6.99 (d, J = 9.0 Hz, 2H), 6.81 (d, J = 8.2 Hz, 1H), 6.69 (d, J = 1.6 Hz, 1H), 6.56 (d, J = 7.9 Hz, 1H), 6.42 (s, 2H), 4.67 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.31–3.23 (m, 4H), 3.06 (s, 2H), 2.57–2.52 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.46, 153.12, 152.78, 146.82, 146.31, 137.22, 133.23, 130.33, 119.98, 119.25, 115.73, 114.01, 113.96, 111.96, 105.93, 98.16, 60.08, 58.69, 55.99, 55.91, 55.68, 51.88, 46.44. HRMS: calcd. C30H35N4O6 [M + H]+, 547.2551, found: 547.2554.
N-(3-hydroxy-4-methoxybenzyl)-2-(4-(4-nitrophenyl)piperazin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16i)
White powder; yield: 67.1%; m.p. 118–119 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.04 (d, J = 9.3 Hz, 2H), 7.00 (d, J = 9.3 Hz, 2H), 6.81 (d, J = 8.2 Hz, 1H), 6.70 (s, 1H), 6.57 (d, J = 7.6 Hz, 1H), 6.42 (s, 2H), 4.67 (s, 2H), 3.72 (s, 3H), 3.68 (s, 6H), 3.65 (s, 3H), 3.43–3.38 (m, 4H), 3.08 (s, 2H), 2.63–2.53 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.94, 155.16, 153.29, 147.30, 146.76, 137.68, 137.65, 137.27, 130.77, 126.18, 119.76, 116.20, 113.07, 112.41, 106.38, 60.55, 59.02, 56.44, 56.10, 52.29, 52.05, 46.89. HRMS: calcd. C29H35N4O8 [M + H]+, 567.2449, found: 567.2453.
N-(3-hydroxy-4-methoxybenzyl)-2-(4-(4-hydroxyphenyl)piperazin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16j)
White powder; yield: 59.7%; m.p. 122–123 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.90 (s, 1H), 8.78 (s, 1H), 6.81 (d, J = 8.2 Hz, 1H), 6.72 (dd, J = 17.6, 5.4 Hz, 3H), 6.63 (d, J = 8.9 Hz, 2H), 6.56 (d, J = 6.5 Hz, 1H), 6.42 (s, 2H), 4.67 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.03 (s, 2H), 2.95–2.82 (m, 4H), 2.54–2.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.57, 152.73, 150.81, 146.81, 146.31, 144.18, 137.35, 136.70, 130.38, 124.50, 119.20, 117.64, 115.40, 111.99, 105.94, 60.09, 60.00, 55.98, 55.90, 52.58, 51.54, 49.98. HRMS: calcd. C29H36N3O7 [M + H]+, 538.2548, found: 538.2559.
Methyl 4-(4-(2-((3-hydroxy-4-methoxybenzyl)(3,4,5-trimethoxyphenyl)amino)-2-oxoethyl)piperazin-1-yl)benzoate (16k)
White powder; yield: 59.7%; m.p. 131–132 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 7.77 (d, J = 8.9 Hz, 2H), 6.95 (d, J = 9.0 Hz, 2H), 6.81 (d, J = 8.2 Hz, 1H), 6.70 (d, J = 1.7 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.43 (s, 2H), 4.67 (s, 2H), 3.77 (s, 3H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.29–3.19 (m, 4H), 3.06 (s, 2H), 2.61–2.52 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.95, 166.61, 154.33, 153.27, 147.29, 146.77, 137.73, 137.11, 131.16, 130.80, 119.74, 118.51, 116.19, 113.77, 112.36, 106.33, 60.53, 59.22, 56.41, 56.07, 52.51, 52.02, 51.92, 47.15. HRMS: calcd. C31H38N3O8 [M + H]+, 580.2653, found: 580.2653.
N-(3-hydroxy-4-methoxybenzyl)-2-(4-methylpiperazin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16l)
White powder; yield: 48.2%; m.p. 127–128 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.68 (d, J = 1.7 Hz, 1H), 6.55 (d, J = 7.7 Hz, 1H), 6.40 (s, 2H), 4.65 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 2.96 (s, 2H), 2.42–2.32 (m, 4H), 2.30–2.16 (m, 4H), 2.11 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.12, 153.19, 147.27, 146.75, 137.85, 137.10, 130.83, 119.68, 116.16, 112.39, 106.35, 60.54, 59.58, 56.40, 56.10, 55.15, 52.76, 51.99, 46.20. HRMS: calcd. C24H34N3O6 [M + H]+, 460.2442, found: 460.2452.
2-(4-Ethylpiperazin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16m)
White powder; yield: 47.8%; m.p. 122–123 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.68 (d, J = 1.9 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 6.40 (s, 2H), 4.66 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 2.96 (s, 2H), 2.41–2.31 (m, 5H), 2.30–2.21 (m, 5H), 0.95 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.13, 153.19, 147.27, 146.77, 137.88, 137.10, 130.85, 119.68, 116.17, 112.40, 106.34, 60.54, 59.63, 56.40, 56.10, 52.92, 52.83, 52.10, 52.00, 12.40. HRMS: calcd. C25H36N3O6 [M + H]+, 474.2559, found: 474.2607.
N-(3-hydroxy-4-methoxybenzyl)-2-(4-isopropylpiperazin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16n)
White powder; yield: 67.1%; m.p. 126–127 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.68 (d, J = 1.8 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 6.40 (s, 2H), 4.66 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 2.97 (s, 2H), 2.64–2.56 (m, 1H), 2.42–2.33 (m, 8H), 0.95 (s, 3H), 0.93 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 168.62, 152.69, 146.79, 146.30, 137.42, 136.65, 130.39, 119.18, 115.74, 111.97, 105.86, 60.06, 59.20, 55.95, 55.58, 53.73, 52.61, 51.54, 47.88, 18.04. HRMS: calcd. C26H38N3O6 [M + H]+, 488.2755, found: 488.2758.
2-(4-Cyclopropylpiperazin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16o)
White powder; yield: 59.5%; m.p. 134–135 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (s, 1H), 6.57 (d, J = 8.2 Hz, 1H), 6.45 (d, J = 1.7 Hz, 1H), 6.32 (d, J = 8.0 Hz, 1H), 6.16 (s, 2H), 4.42 (s, 2H), 3.48 (s, 3H), 3.44 (s, 6H), 3.41 (s, 3H), 2.72 (s, 2H), 2.26–2.17 (m, 4H), 2.16–2.02 (m, 4H), 1.35–1.27 (m, 1H), 0.12 (q, J = 6.0, 4.1 Hz, 2H), 0.00 (q, J = 2.5 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 168.64, 152.70, 146.79, 146.29, 137.38, 136.67, 130.38, 119.19, 115.68, 111.94, 105.92, 60.08, 59.12, 55.96, 55.58, 52.73, 52.38, 51.51, 37.97, 5.50. HRMS: calcd. C26H36N3O6 [M + H]+, 486.2599, found: 486.2608.
2-(4-(Cyclopropanecarbonyl)piperazin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16p)
White powder; yield: 61.3%; m.p. 124–125 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 6.81 (d, J = 8.1 Hz, 1H), 6.68 (s, 1H), 6.56 (d, J = 4.0 Hz, 1H), 6.44 (d, J = 30.2 Hz, 2H), 4.66 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.62–3.36 (m, 4H), 3.04 (s, 2H), 2.49–2.27 (m, 4H), 1.99–1.86 (m, 1H), 0.74–0.61 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 170.85, 169.44, 168.47, 152.78, 146.82, 146.31, 137.20, 130.32, 119.28, 115.75, 111.99, 105.91, 60.08, 59.97, 56.00, 55.91, 55.67, 55.59, 51.55, 10.17, 6.86. HRMS: calcd. C27H36N3O7 [M + H]+, 514.2548, found: 514.2553.
Tert-butyl 4-(2-((3-hydroxy-4-methoxybenzyl)(3,4,5-trimethoxyphenyl)amino)-2-oxoethyl)piperazine-1-carboxylate (16q)
White powder; yield: 56.3%; m.p. 129–130 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (d, J = 8.5 Hz, 1H), 6.81 (t, J = 8.3 Hz, 1H), 6.68 (d, J = 7.6 Hz, 1H), 6.56 (s, 1H), 6.40 (d, J = 8.5 Hz, 2H), 4.65 (s, 2H), 3.71 (s, 3H), 3.66 (s, 6H), 3.64 (s, 3H), 3.30–3.20 (m, 4H), 3.02 (s, 2H), 2.43–2.30 (m, 4H), 1.40 (s, 9H). 13C NMR (100 MHz, DMSO-d6) δ 168.54, 153.54, 153.28, 147.79, 147.30, 146.79, 137.70, 130.81, 119.76, 116.23, 112.43, 106.37, 60.52, 59.13, 56.44, 56.11, 52.91, 52.45, 46.20, 41.37, 21.61. HRMS: calcd. C28H40N3O8 [M + H]+, 546.2810, found: 546.2819.
N-(3-hydroxy-4-methoxybenzyl)-2-morpholino-N-(3,4,5-trimethoxyphenyl) acetamide (16r)
White powder; yield: 58.8%; m.p. 132–133 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 6.87–6.51 (m, 3H), 6.41 (s, 2H), 4.66 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.57–3.43 (m, 4H), 2.99 (s, 2H), 2.47–2.30 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.88, 153.26, 147.29, 146.79, 137.75, 137.15, 130.83, 119.73, 116.20, 112.38, 106.34, 66.69, 60.52, 59.70, 56.40, 56.07, 53.36, 52.01. HRMS: calcd. C23H31N2O7 [M + H]+, 447.2126, found: 447.2132.
N-(3-hydroxy-4-methoxybenzyl)-2-morpholino-N-(3,4,5-trimethoxyphenyl) acetamide (16s)
White powder; yield: 65.1%; m.p. 127–128 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.67 (d, J = 1.8 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 6.39 (s, 2H), 4.65 (s, 2H), 4.03 (q, J = 7.1 Hz, 1H), 3.85 (t, J = 13.4 Hz, 1H), 3.71 (s, 3H), 3.67 (s, 6H), 3.64 (s, 3H), 3.05 (s, 2H), 2.72–2.60 (m, 4H), 2.51–2.49 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 169.26, 153.24, 147.27, 146.75, 137.80, 137.10, 130.81, 119.72, 116.18, 112.37, 106.32, 60.53, 60.29, 56.42, 56.08, 54.37, 52.00, 27.76. HRMS: calcd. C23H31N2O6S [M + H]+, 463.1897, found: 463.1897.
N-(3-hydroxy-4-methoxybenzyl)-2-(4-methylpiperidin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16t)
White powder; yield: 68.2%; m.p. 141–131 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.91 (s, 1H), 6.86–6.36 (m, 5H), 4.65 (s, 2H), 3.72 (s, 3H), 3.66 (s, 6H), 3.64 (s, 3H), 2.94 (s, 2H), 2.67 (d, J = 10.1 Hz, 2H), 1.96 (t, J = 10.8 Hz, 2H), 1.48 (d, J = 11.7 Hz, 2H), 1.28–1.16 (m, 1H), 1.02 (dd, J = 21.6, 10.8 Hz, 2H), 0.84 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 169.35, 153.16, 147.25, 146.75, 137.98, 137.04, 130.89, 119.67, 116.16, 112.36, 106.31, 60.52, 60.11, 56.37, 56.07, 53.49, 51.99, 34.43, 30.36, 22.33. HRMS: calcd. C25H35N2O6 [M + H]+, 459.2490, found: 459.2496.
N-(3-hydroxy-4-methoxybenzyl)-2-(4-methoxypiperidin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16u)
White powder; yield: 48.3%; m.p. 131–132 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.92 (s, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.68 (d, J = 1.3 Hz, 1H), 6.55 (d, J = 7.9 Hz, 1H), 6.40 (s, 2H), 4.65 (s, 2H), 3.72 (s, 3H), 3.66 (s, 6H), 3.65 (s, 3H), 3.19 (s, 3H), 3.02 (d, J = 46.4 Hz, 3H), 2.66–2.53 (m, 2H), 2.13 (t, J = 9.7 Hz, 2H), 1.74 (d, J = 9.8 Hz, 2H), 1.31 (d, J = 9.4 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 169.30, 153.20, 147.27, 146.78, 137.93, 137.09, 130.89, 119.68, 116.18, 112.38, 106.33, 75.96, 60.52, 59.62, 56.38, 56.08, 55.20, 51.99, 50.87, 31.20. HRMS: calcd. C25H35N2O7 [M + H]+, 475.2439, found: 475.2446.
N-(3-hydroxy-4-methoxybenzyl)-2-(4-hydroxypiperidin-1-yl)-N-(3,4,5-trimethoxyphenyl)acetamide (16v)
White powder; yield: 55.7%; m.p. 146–147 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (s, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.69 (s, 1H), 6.55 (d, J = 7.8 Hz, 1H), 6.39 (s, 2H), 4.66 (s, 2H), 4.48 (s, 1H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 2.95 (s, 2H), 2.61 (d, J = 9.5 Hz, 2H), 2.09 (t, J = 9.8 Hz, 2H), 1.63 (d, J = 9.9 Hz, 2H), 1.30 (d, J = 9.5 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 168.86, 152.69, 146.78, 146.30, 137.44, 136.66, 130.43, 119.18, 115.69, 111.94, 105.91, 66.06, 60.07, 59.22, 55.95, 55.57, 51.52, 50.74, 34.43. HRMS: calcd. C24H33N2O7 [M + H]+, 461.2282, found: 461.2282.
2-(4-Chloropiperidin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16w)
White powder; yield: 58.2%; m.p. 135–136 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (s, 1H), 6.80 (d, J = 8.2 Hz, 1H), 6.68 (d, J = 1.4 Hz, 1H), 6.55 (d, J = 7.8 Hz, 1H), 6.40 (s, 2H), 4.66 (s, 2H), 4.12 (s, 1H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.01 (s, 2H), 2.64 (s, 2H), 2.29 (t, J = 9.1 Hz, 2H), 1.96 (d, J = 10.2 Hz, 2H), 1.66 (d, J = 9.0 Hz, 2H). 13C NMR δ 168.69, 152.74, 146.80, 146.31, 137.35, 136.71, 130.37, 119.21, 115.73, 111.97, 105.89, 60.07, 58.81, 57.89, 55.89, 55.58, 51.55, 50.47, 35.41. HRMS: calcd. C24H32ClN2O6 [M + H]+, 479.1943, found: 479.1944.
2-(4-Chloropiperidin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16x)
White powder; yield: 61.7%; m.p. 126–126 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.23 (dt, J = 13.2, 6.9 Hz, 5H), 6.81 (d, J = 8.2 Hz, 1H), 6.71 (s, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.43 (s, 2H), 4.68 (s, 2H), 3.72 (s, 3H), 3.67 (s, 6H), 3.65 (s, 3H), 3.04 (s, 2H), 2.83 (d, J = 9.7 Hz, 2H), 2.40 (t, J = 11.9 Hz, 1H), 2.14 (t, J = 10.7 Hz, 2H), 1.69–1.50 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.92, 152.73, 146.79, 146.31, 146.25, 137.59, 136.64, 130.45, 128.30, 126.57, 125.93, 119.22, 115.72, 111.96, 105.85, 60.00, 59.60, 55.98, 55.67, 53.34, 51.58, 41.42, 33.09. HRMS: calcd. C30H37N2O6 [M + H]+, 521.2646, found: 521.2654.
2-(4-Chloropiperidin-1-yl)-N-(3-hydroxy-4-methoxybenzyl)-N-(3,4,5-trimethoxyphenyl)acetamide (16y)
White powder; yield: 67.7%; m.p. 151–152 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (s, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 6.81 (d, J = 8.2 Hz, 1H), 6.71 (s, 1H), 6.57 (d, J = 8.1 Hz, 1H), 6.43 (s, 2H), 4.69 (s, 2H), 3.72 (s, 3H), 3.68 (s, 6H), 3.66 (s, 3H), 3.04 (s, 2H), 2.83 (d, J = 9.7 Hz, 2H), 2.42 (t, J = 11.9 Hz, 1H), 2.15 (t, J = 10.8 Hz, 2H), 1.56 (dt, J = 21.9, 11.6 Hz, 4H). 13C NMR (100 MHz, DMSO-d6) δ 168.87, 152.74, 146.80, 146.32, 145.21, 137.55, 136.66, 130.45, 128.48, 128.46, 128.20, 119.21, 115.74, 111.97, 105.87, 59.99, 59.48, 55.89, 55.58, 53.19, 51.57, 40.73, 32.94. HRMS: calcd. C30H36ClN2O6 [M + H]+, 555.2256, found: 555.2260.
Cell culture
All the cells were obtained from the ATCC (Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai, China). The cells were cultured in the RPMI-1640 medium supplemented with 10% foetal bovine serum (FBS), 100 units/mL penicillin, and 10 mg/mL penicillin streptomycin. All the cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C.
In vitro tubulin polymerisation assay
Blank solvent was used as the blank control group, the test compound was used as the experimental group, colchicine was used as the positive drug group, and paclitaxel was used as the negative drug group. The temperature of the fluorescence microplate reader was set to 37 °C, and the above drug mixture was added to each well in a 96-well plate. Fluorescence microplate reader parameters were set with an excitation wavelength of 360 nm and emission wavelength of 450 nm, and the fluorescence intensity was detected every 1 min for a total of 60 times. The reaction system for tubulin polymerisation was prepared. After preheating, the culture plate was removed and the prepared tubulin polymerisation reaction system was quickly added to each well. The plate was then placed in the microplate reader, and the program was started to begin measuring. Data were collected and a curve was drawn.
EBI competitive assay
Different liver cancer cells in logarithmic growth phase were treated with blank solvent, different concentrations of compound, and positive control drugs for 2 h. EBI was added to each group to make the final concentration 100 μM, and incubated for 2 h. A blank control group without EBI treatment was also set up. Cells from each group were collected, and total protein was extracted. Western blot was used to detect the tubulin adduct band. The signal of the tubulin adduct band is negatively correlated with the affinity of the compound and the colchicine binding site of β-tubulin.
Alkaline protease assay
Tumour cells were collected and lysate. Different concentrations of compounds were used to treat the lysate, with a blank solvent group set up, and incubated at 4 °C for 1 h. Proteins in the lysates were hydrolysed by adding subtilisin at a ratio of 1:500, incubated at room temperature for 15 min, and 1 mM PMSF was added to terminate the protein hydrolysis. A control group without hydrolysis was also set up. Western blot was used to detect changes in the level of the target protein.
Molecular docking study
The X-ray crystal structures of TUBA1D (PDB: 1SA0) were retrieved from the Protein Data Bank. The protonation state of all the compounds was set at pH = 7.4, and the compounds were expanded to 3D structures using Open Babel. AutoDock Tools (ADT3) were applied to prepare and parametrise the receptor protein and ligands. The docking grid documents were generated by AutoGrid of sitemap, and AutoDock Vina (1.2.0) was used for docking simulation. The optimal pose was selected to analysis interaction. Finally, the protein–ligand interaction figure was generated by PyMOL. The TUBA1D protein is represented as a slate cartoon model, ligand is shown as a cyan stick, and their binding sites are shown as magentas stick structures. Nonpolar hydrogen atoms are omitted. The hydrogen bond, ionic interactions, and hydrophobic interactions are depicted as yellow, magentas, and green dashed lines, respectively.
Calcein-AM/PI staining
Different liver cancer cells in the logarithmic growth phase were treated with blank solvent, different concentrations of compounds for 48 h. Calcein-AM and PI dyes were added to 1× assay buffer to prepare a double staining solution. After washing the liver cancer cells, they were incubated with the double staining solution for 0.5 h. The results were observed and recorded under a fluorescence microscope.
DAPI staining
Liver cancer cells SMMC-7721 and HuH-7 were treated with drugs at different concentrations of compound for 48 h. After incubation, the cells were washed and fixed in a 4% paraformaldehyde solution at room temperature for 10 min. The paraformaldehyde solution was removed, and the cells were washed again. DAPI staining solution was added to the cells and incubated in the dark at room temperature for 30 min. After incubation, the DAPI staining solution was removed and the cells were washed again. The cells were then observed using a fluorescence microscope to visualise the nuclei and images were recorded.
Immunofluorescence staining
Cells were seeded in 24-well cell culture plates and then treated with vehicle control 0.1% DMSO, different concentrations of compound MY-1121 or colchicine for 6 h. After the treatment, cells were fixed with 4% paraformaldehyde, and then infiltrated for three times with PBS containing 0.1% Triton X-100. Five percent BSA was added into each well before blocking for 30 min at 25 °C. After that, cells were incubated with anti-β-tubulin at 4 °C overnight. Then, we discarded the anti-β-tubulin, added PBS to each well for washing three times, added a fluorescent secondary antibody, and incubated it at room temperature under the dark condition for 1 h. Next, we discarded the fluorescent secondary antibody, washed wells with PBS for three times, and then each well was added DAPI (2 μg/mL) and further incubated at room temperature under the dark condition for 25 min. Finally, the fluorescence microscope was used to visualise the cells.
Western blotting assay
Cells treated with compound MY-1121 were collected, and then lysed to obtain prepared protein samples. The prepared protein samples were denatured and separated by SDS-PAGE, and then transferred to the PVDF membrane. The membrane was sealed with skim milk (5%), and then incubated at 4 °C overnight with the first antibody conjugate. After the incubation, the membrane was washed with TBST/TBS for three times (10 min each time), and then was incubated with the second antibody for another 1.5 h. The target proteins were analysed after washing ECL Ultrasensitive Luminol and Ultrasensitive Imaging Analyzer.
Supplemental Material
Download PDF (2.3 MB)Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. U2004123 and 82273782 for Sai-Yang Zhang).
Disclosure statement
The authors declared that there was no conflict of interest about this paper.
Correction Statement
This article was originally published with errors, which have now been corrected in the online version. Please see Correction (http://dx.doi.org/10.1080/14756366.2023.2255447)
Additional information
Funding
References
- Mühlethaler T, Gioia D, Prota AE, Sharpe ME, Cavalli A, Steinmetz MO. Comprehensive analysis of binding sites in tubulin. Angew Chem Int Ed Engl. 2021;60(24):13331–13342.
- Shuai W, Wang G, Zhang Y, Bu F, Zhang S, Miller DD, Li W, Ouyang L, Wang Y. Recent progress on tubulin inhibitors with dual targeting capabilities for cancer therapy. J Med Chem. 2021;64(12):7963–7990.
- Yang J, Yu Y, Li Y, Yan W, Ye H, Niu L, Tang M, Wang Z, Yang Z, Pei H, et al. Cevipabulin–tubulin complex reveals a novel agent binding site on α-tubulin with tubulin degradation effect. Sci Adv. 2021;7:eabg4168.
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010;10(3):194–204.
- Mosca L, Ilari A, Fazi F, Assaraf YG, Colotti G. Taxanes in cancer treatment: activity, chemoresistance and its overcoming. Drug Resist Updat. 2021;54:100742.
- Nathan P, Zweifel M, Padhani AR, Koh DM, Ng M, Collins DJ, Harris A, Carden C, Smythe J, Fisher N, et al. Phase I trial of combretastatin A4 phosphate (CA4P) in combination with bevacizumab in patients with advanced cancer. Clin Cancer Res. 2012;18(12):3428–3439.
- Eskens FA, Tresca P, Tosi D, Van Doorn L, Fontaine H, Van der Gaast A, Veyrat-Follet C, Oprea C, Hospitel M, Dieras V. A phase I pharmacokinetic study of the vascular disrupting agent ombrabulin (AVE8062) and docetaxel in advanced solid tumours. Br J Cancer. 2014;110(9):2170–2177.
- Pal S, Azad A, Bhatia S, Drabkin H, Costello B, Sarantopoulos J, Kanesvaran R, Lauer R, Starodub A, Hauke R, et al. A phase I/II trial of BNC105P with everolimus in metastatic renal cell carcinoma. Clin Cancer Res. 2015;21(15):3420–3427.
- Markowski MC, Tutrone R, Pieczonka C, Barnette KG, Getzenberg RH, Rodriguez D, Steiner MS, Saltzstein DR, Eisenberger MA, Antonarakis ES. A phase Ib/II study of sabizabulin, a novel oral cytoskeleton disruptor, in men with metastatic castration-resistant prostate cancer with progression on an androgen receptor-targeting agent. Clin Cancer Res. 2022;28(13):2789–2795.
- Peng X, Ren Y, Pan W, Liu J, Chen J. Discovery of novel acridane-based tubulin polymerization inhibitors with anticancer and potential immunomodulatory effects. J Med Chem. 2023;66(1):627–640.
- Puxeddu M, Wu J, Bai R, D’Ambrosio M, Nalli M, Coluccia A, Manetto S, Ciogli A, Masci D, Urbani A, et al. Induction of ferroptosis in glioblastoma and ovarian cancers by a new pyrrole tubulin assembly inhibitor. J Med Chem. 2022;65(23):15805–15818.,
- Ji T, Jian X, Chen L, Zeng W, Huo X, Li M, Chen P, Zhang Y, You W, Zhao P. Discovery of novel 6-p-tolyl-3-(3,4,5-trimethoxybenzyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivative as a potent tubulin inhibitor with promising in vivo antitumor activity. Eur J Med Chem. 2023;256:115437.
- Barreca M, Spanò V, Rocca R, Bivacqua R, Abel A-C, Maruca A, Montalbano A, Raimondi MV, Tarantelli C, Gaudio E, et al. Development of [1,2]oxazoloisoindoles tubulin polymerization inhibitors: further chemical modifications and potential therapeutic effects against lymphomas. Eur J Med Chem. 2022;243:114744.
- Wang G, Liu W, Fan M, He M, Li Y, Peng Z. Design, synthesis and biological evaluation of novel thiazole-naphthalene derivatives as potential anticancer agents and tubulin polymerisation inhibitors. J Enzyme Inhib Med Chem. 2021;36(1):1693–1701.
- Liu R, Zhang S, Huang M, Guo Z, Li L, Li M, Wu L, Guan Q, Zhang W. Design, synthesis and bioevaluation of 2,7-diaryl-pyrazolo[1,5-a]pyrimidines as tubulin polymerization inhibitors. Bioorg Chem. 2021;115:105220.
- Song J, Wang SY, Wang X, Jia MQ, Tian XY, Fu XJ, Jin CY, Zhang SY. Discovery of a novel coumarin-dihydroquinoxalone derivative MY-673 as a tubulin polymerization inhibitor capable of inhibiting the ERK pathway with potent anti-gastric cancer activities. Bioorg Chem. 2023;137:106580.
- Wang Y, Sun M, Wang Y, Qin J, Zhang Y, Pang Y, Yao Y, Yang H, Duan Y. Discovery of novel tubulin/HDAC dual-targeting inhibitors with strong antitumor and antiangiogenic potency. Eur J Med Chem. 2021;225:113790.
- Zhou P, Liu Y, Zhou L, Zhu K, Feng K, Zhang H, Liang Y, Jiang H, Luo C, Liu M, et al. Potent antitumor activities and structure basis of the chiral β-lactam bridged analogue of combretastatin A-4 binding to tubulin. J Med Chem. 2016;59(22):10329–10334.
- Lin S, Du T, Zhang J, Wu D, Tian H, Zhang K, Jiang L, Lu D, Sheng L, Li Y, et al. Optimization of benzamide derivatives as potent and orally active tubulin inhibitors targeting the colchicine binding site. J Med Chem. 2022;65(24):16372–16391.
- Zhu H, Li W, Shuai W, Liu Y, Yang L, Tan Y, Zheng T, Yao H, Xu J, Zhu Z, et al. Discovery of novel N-benzylbenzamide derivatives as tubulin polymerization inhibitors with potent antitumor activities. Eur J Med Chem. 2021;216:113316.
- Song J, Wang SH, Song CH, Zhang WX, Zhu JX, Tian XY, Fu XJ, Xu Y, Jin CY, Zhang SY. Discovery of N-benzylarylamide derivatives as novel tubulin polymerization inhibitors capable of activating the Hippo pathway. Eur J Med Chem. 2022;240:114583.
- Yuan S, Wang DS, Liu H, Zhang SN, Yang WG, Lv M, Zhou YX, Zhang SY, Song J, Liu HM. New drug approvals for 2021: synthesis and clinical applications. Eur J Med Chem. 2023;245(Pt 1):114898.
- Yuan S, Luo YQ, Zuo JH, Liu H, Li F, Yu B. New drug approvals for 2020: synthesis and clinical applications. Eur J Med Chem. 2021;215:113284.
- Lang DK, Kaur R, Arora R, Saini B, Arora S. Nitrogen-containing heterocycles as anticancer agents: an overview. Anticancer Agents Med Chem. 2020;20(18):2150–2168.
- Sharma A, Wakode S, Fayaz F, Khasimbi S, Pottoo FH, Kaur A. An overview of piperazine scaffold as promising nucleus for different therapeutic targets. Curr Pharm Des. 2020;26(35):4373–4385.
- Prinz H, Ridder A-K, Vogel K, Böhm KJ, Ivanov I, Ghasemi JB, Aghaee E, Müller K, Heterocyclic N. (4-Phenylpiperazin-1-yl)methanones derived from phenoxazine and phenothiazine as highly potent inhibitors of tubulin polymerization. J Med Chem. 2017;60(2):749–766.
- Wang L, Zheng Y, Li D, Yang J, Lei L, Yan W, Zheng W, Tang M, Shi M, Zhang R, et al. Design, synthesis, and bioactivity evaluation of dual-target inhibitors of tubulin and Src kinase guided by crystal structure. J Med Chem. 2021;64(12):8127–8141.
- Povedano JM, Rallabandi R, Bai X, Ye X, Liou J, Chen H, Kim J, Xie Y, Posner B, Rice L, et al. A multipronged approach establishes covalent modification of β-tubulin as the mode of action of benzamide anti-cancer toxins. J Med Chem. 2020;63(22):14054–14066.
- Yuan X-Y, Song C-H, Liu X-J, Wang X, Jia M-Q, Wang W, Liu W-B, Fu X-J, Jin C-Y, Song J, et al. Discovery of novel N-benzylarylamide-dithiocarbamate based derivatives as dual inhibitors of tubulin polymerization and LSD1 that inhibit gastric cancers. Eur J Med Chem. 2023;252:115281.
- Sun Y-X, Song J, Kong L-J, Sha B-B, Tian X-Y, Liu X-J, Hu T, Chen P, Zhang S-Y. Design, synthesis and evaluation of novel bis-substituted aromatic amide dithiocarbamate derivatives as colchicine site tubulin polymerization inhibitors with potent anticancer activities. Eur J Med Chem. 2022;229:114069.
- Huang L, Liu M, Man S, Ma D, Feng D, Sun Z, Guan Q, Zuo D, Wu Y, Zhang W, et al. Design, synthesis and bio-evaluation of novel 2-aryl-4-(3,4,5-trimethoxy-benzoyl)-5-substituted-1,2,3-triazoles as the tubulin polymerization inhibitors. Eur J Med Chem. 2020;186:111846.
- Choi MJ, No ES, Thorat DA, Jang JW, Yang H, Lee J, Choo H, Kim SJ, Lee CS, Ko SY, et al. Synthesis and biological evaluation of aryloxazole derivatives as antimitotic and vascular-disrupting agents for cancer therapy. J Med Chem. 2013;56(22):9008–9018.
- Liu J, Liu C, Zhang X, Yu L, Gong X, Wang P. Anticancer sulfonamide hybrids that inhibit bladder cancer cells growth and migration as tubulin polymerisation inhibitors. J Enzyme Inhib Med Chem. 2019;34(1):1380–1387.
- Jaiswal S, Parida SK, Murarka S, Singh P. Development of S-aryl dithiocarbamate derived novel antiproliferative compound exhibiting tubulin bundling. Bioorg Med Chem. 2022;68:116874.
- Chopra A, Anderson A, Giardina C. Novel piperazine-based compounds inhibit microtubule dynamics and sensitize colon cancer cells to tumor necrosis factor-induced apoptosis. J Biol Chem. 2014;289(5):2978–2991.
- Song J, Guan YF, Liu WB, Song CH, Tian XY, Zhu T, Fu XJ, Qi YQ, Zhang SY. Discovery of novel coumarin-indole derivatives as tubulin polymerization inhibitors with potent anti-gastric cancer activities. Eur J Med Chem. 2022;238:114467.