
Abstract
A series of novel triazole derivatives containing aryl-propanamide side chains was designed and synthesised. In vitro antifungal activity studies demonstrated that most of the compounds inhibited the growth of six human pathogenic fungi. In particular, parts of phenyl-propionamide-containing compounds had excellent, broad-spectrum antifungal activity against Candida albicans SC5314, Cryptococcus neoformans 22-21, Candida glabrata 537 and Candida parapsilosis 22-20 with MIC values in the range of ≤0.125 µg/mL–4.0 µg/mL. In addition, compounds A1, A2, A6, A12 and A15 showed inhibitory activities against fluconazole-resistant Candida albicans and Candida auris. Preliminary structure-activity relationships (SARs) are also summarised. Moreover, GC-MS analysis demonstrated that A1, A3, and A9 interfered with the C. albicans ergosterol biosynthesis pathway by inhibiting Cyp51. Molecular docking studies elucidated the binding modes of A3 and A9 with Cyp51. These compounds with low haemolytic activity and favourable ADME/T properties are promising for the development of novel antifungal agents.
Introduction
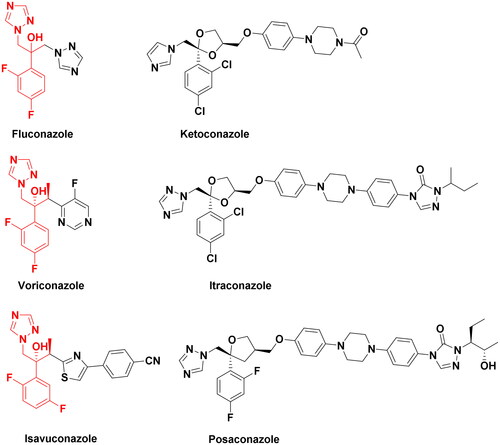
In recent years, invasive fungal infections (IFIs), such as invasive candidiasis and aspergillosis, pose an increasingly serious threat to human healthCitation1,Citation2. A retrospective study based on single-centre autopsy data from China (3,447 cases) showed that the prevalence of IFIs has been steadily increasing for decadesCitation3. The mortality rates of IFIs in China are also on the rise with estimates ranging from 36.6% to 60%Citation4. On one hand, long-term use of fluconazole has caused the widespread emergence of fluconazole resistance in Candida albicans which usually results in clinical treatment failure in settings where alternative treatment options are not available. On the other hand, COVID-19-associated fungal infections continue to increase, especially in critically ill, hospitalised, or immunocompromised patientsCitation5. Azoles, such as fluconazole, ketoconazole, voriconazole and itraconazole, etc. (), are frequently used antifungal agents in clinical practice. Azoles primarily target fungal Cyp51 (lanosterol 14α-demethylase). The pharmacophore of antifungal triazoles contains a triazole ring, a hydroxyl group and a dihalophenyl linked to a two-carbon chain as shown in . The major difference between the structures of novel triazole analogues is on the side chains attached to the pharmacophoreCitation6–8.
Figure 1. Structures of the triazole antifungal agents and novel triazole compounds.
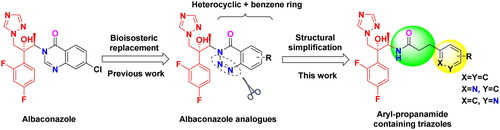
In our previous work, we designed and synthesised a series of albaconazole analogues by replacing the quinazolinone unit with its bioisostere 1,2,3-benzotriazin-4-one (). Most analogues exhibited improved in vitro antifungal activities compared to fluconazole. Inspired by the amide moiety that existed in the quinazolinone and 1,2,3-benzotriazin-4-one, we conceived that if we further simplified the N-heterocycle at the same time as keeping the amide linker with various aromatic rings, it would give novel, potent triazoles. Hence, in this study, a series of aryl-propanamide side chains containing triazole derivatives were rationally designed, synthesised and their in vitro antifungal activities were further evaluated.
Figure 2. Design strategy of target compounds.
Results and discussion
Chemistry
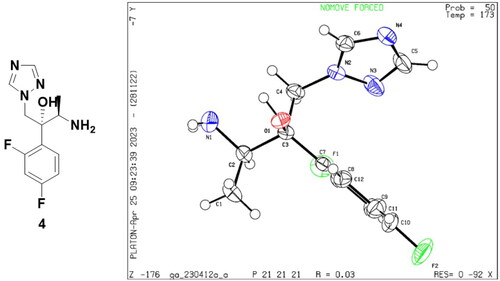
According to Scheme 1, twenty-seven novel triazole derivatives were synthesised. The key intermediate (2 R,3R)-2-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)butane-2,3-diol (1) was prepared as in our previous workCitation9. In the next step, triazolyl-diol 1 was converted into methane sulphonate and then eliminated to give epoxide 2. Epoxide 2 was reacted with NaN3 to form ring-opening azide compound 3. Next, 3 was subjected to catalytic hydrogenation to afford amine compound 4, which was followed by a condensation reaction with various commercially available substituted propionic acids to obtain target compounds A1-A27. The chiral structure of key intermediates amine 4 was clearly confirmed by X-ray crystallographic analysis. The atom labelling and thermal ellipsoids of amine 4 are shown in . It crystallised in the orthorhombic crystal system and P212121 space group with a = 8.9511(3) Å, b = 10.4014(4) Å, c = 13.8084(4) Å, α = 90°, β = 90° and δ = 90° (Table S1 in the supplementary material). Based on the results, the absolute configuration at C-2 and C-3 in amine 4 was determined to be R. All the newly synthesised compounds were characterised and confirmed by 1H NMR, 13C NMR, and LC-MS as described in Supporting Information.
Scheme 1. Synthesis of the target compounds. (a) (i) Et3N, MsCl, DCM, 0 °C, 1 h; (ii) NaOH, H2O, 0 °C, 4 h; (b) NH4Cl, NaN3, DMF, 80 °C, 10 h; (c) Pd/C, H2, MeOH, r.t., 8 h; (d) Substituted propionic acid, PyBOP, DIEA, DMF, r.t., 5 h.
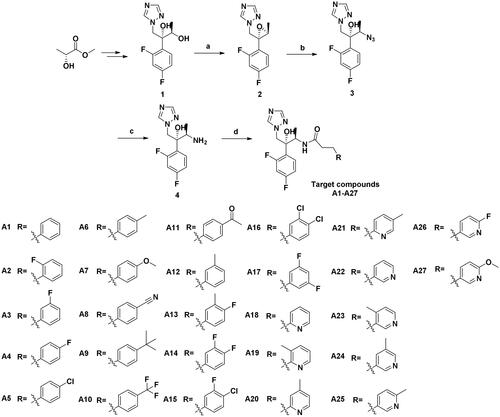
Figure 3. The single-crystal structure of amine 4.
In vitro antifungal activity and structure-activity relationships
According to the Clinical Laboratory Standards Institute (CLSI, M27-A3 and M60) protocolsCitation10,Citation11, the target compounds were evaluated for their in vitro antifungal activity by the broth microdilution method. The fungi strains used in this study were obtained from the American Type Culture Collection (ATCC) and Changhai Hospital (Shanghai, China). The MIC was defined as the minimum concentration of drugs to inhibit ≥80% growth of fungal cells compared with drug-free control. Fluconazole (FCZ) and ketoconazole (KCZ) were used as positive controls.
As shown in , the majority of the 17 phenyl-propionamide-containing compounds (A1-A17), except for A8 and A11, showed strong antifungal activity against Candida albicans SC5314 and Candida glabrata 537 with MIC values ranging from 0.125 µg/mL to 0.5 µg/mL, which were comparable to or greater than FCZ and KCZ. Good inhibitory activity was exhibited against Candida parapsilosis 22–20 and Cryptococcus neoformans 22–21 with MIC ranging from 0.5 µg/mL to 4.0 µg/mL and ≤0.125 µg/mL to 1.0 µg/mL, respectively. FCZ had no inhibitory effect (MIC > 64.0 µg/mL) on either Candida tropicalis 8915 or Aspergillus fumigatus 7544. Nine compounds (A2, A3, A10, A12–A17) were at least 2-fold more effective than FCZ with MIC 8.0 µg/mL–64.0 µg/mL. Of note, compounds A2, A3, A10, A12–A17 exhibited broad-spectrum antifungal against six fungal strains. However, compounds containing pyridinyl-propionamide (A18–A27) showed weaker activity in vitro and had a narrow spectrum compared with phenyl-propionamide compounds. For example, all of the pyridinyl-propionamide derivatives had no activity against either Candida tropicalis 8915 and Aspergillus fumigatus 7544. Similar to the above results, the introduction of a pyridine ring also resulted in the loss of antifungal activity against C. albicans, C. glabrata, C. parapsilosis and C. neoformans.
Table 1. In vitro antifungal activity of the target compounds against tested fungi (MIC, μg/mL).
Preliminary structure-activity relationships (SARs) show a correlation between in vitro antifungal activities and chemical structures. Overall, the phenyl-propanamide containing triazoles inhibited fungi better than the corresponding pyridinyl analogues. Moreover, the antifungal effect of meta-F-substituted compound (A3) on the terminal phenyl is better than that of the ortho and para-F-substituted (A2 and A4) against C. tropicalis and A. fumigatus. Interestingly, among the para-substituted compounds (A4–A11), almost all compounds had no activity against C. tropicalis, except for A10. Furthermore, compounds with electron-withdrawing groups in para- position of cyan (A8), trifluoromethyl (A10), and acetyl group (A11) resulted decreased activity against C. albicans and C. parapsilosis. Replacement of meta-F with CH3 yielded A12 which had a similar potency to A3 against tested fungi. The introduction of another F or Cl atom to the terminal phenyl, whilst keeping the meta-substituted position, yielded the di-substituted compounds A13–A17. These displayed excellent and broader-spectrum in vitro activity against all of the tested strains. For example, A13–A17 had the best MIC values (≤ 0.125 µg/mL) against C. albicans and C. glabrata. When compared with 2-pyridinyl containing series A18–A21 (H or different CH3 substituted position) and related 3-pyridinyl analogues A22-A25, A18–A21 had relatively greater activity against C. glabrata, C. parapsilosis and C. neoformans, and was even more efficacious against C. albicans.
Evaluation of MIC against FCZ-resistant strains
The alarming impact of drug resistance amongst Candida spp. is an emerging problem globallyCitation12. Rising rates of azole-resistant Candida spp. are associated with an increase in treatment failures. Candida auris is characterised by a high level of multi-drug resistance. It has been reported in more than 45 countries and has caused serious hospital outbreaks with crude mortality rates as high as 72%Citation13,Citation14. Therefore, we further evaluated the in vitro antifungal activity of our compounds against fluconazole-resistant strains by the microdilution method assay.
As shown in , the MIC values of FCZ against C. albicans 901 and C. auris 918 and 919 were greater than 256.0 µg/mL, indicating that these isolates were highly FCZ-resistant. These triazole derivatives exhibited moderate inhibitory effects against C. albicans 901. A1 and A5 showed the best activity with MIC of 1.0 µg/mL against C. alb 901. A1, A2, A6, A12 and A15 demonstrated inhibitory effects against the two C. auris strains with MIC of 32.0 µg/mL or 64.0 µg/mL. The above results demonstrated that with appropriate structural modifications on the triazole compound side chains, the problem of drug resistance could be overcome.
Table 2. In vitro antifungal activity of the potent compounds against FCZ-resistant C. albicans and C. auris isolates measured by MIC (μg/mL).
GC-MS analysis of sterol composition in C. albicans
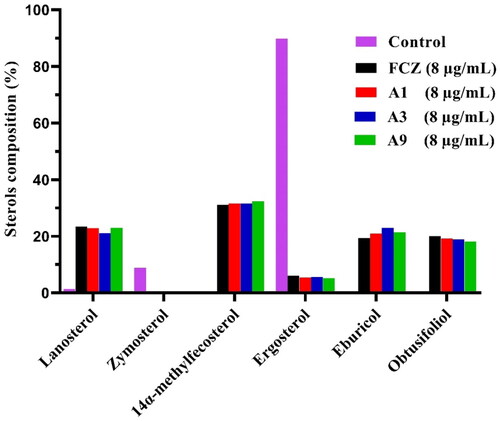
Gas chromatography-mass spectrometry (GC-MS) was used to examine the composition of sterols in fungal cells as a preliminary method to verify whether our target compounds inhibit Cyp51Citation15. Therefore, compounds A1, A3, A9 and FCZ at 8 µg/mL were used to treat C. albicans SC5314, and their sterol profiles were analysed as shown in . As the major fungal sterol, ergosterol accounted for 89.85% of the total sterols in the control group, zymosterol accounted for 8.80%, in addition to several other sterols. When treated with either FCZ, A1, A3, or A9 the ergosterol content of the fungal cells decreased to 6.08%, 5.44%, 5.63% and 5.17%, respectively. In contrast, the content of other sterols in the bypass pathway increased. For instance, treatment with A1 resulted in the following: the proportion of lanosterol, 14α-methylfecosterol, eburicol and obtusifoliol increased to 23.42%, 31.11%, 19.29% and 20.09%, respectively. Our target compounds had a consistent effect and the same mechanism of action as FCZ by inhibiting the production of ergosterol on Cyp51.
Figure 4. GC-MS analysis of sterols in C. albicans cells. The fungal strain was treated with DMSO (Control), FCZ, compounds A1, A3 or A9 at 8 μg/mL for 8 h.
Evaluation of hemolysis
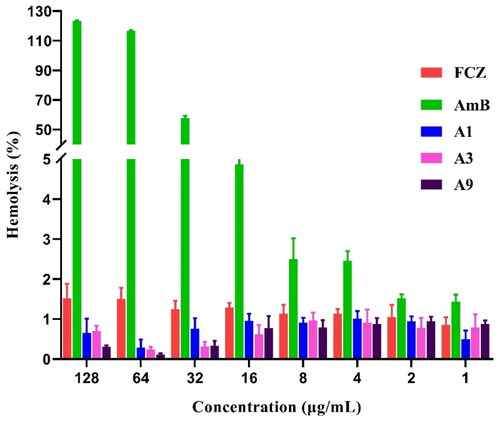
Haemolysis rate is an important indicator of drug safetyCitation16. We performed rabbit haemolysis experiments to evaluate the safety of our synthesised compounds. At a concentration of 16 µg/mL, AmB haemolysis averaged 4.87%, while FCZ, A1, A3 and A9 haemolysis averaged 1.30%, 0.95%, 0.61%, and 0.78%, respectively, demonstrating minimal effect to the rabbit red cells (). When AmB was given at 32 µg/mL, it caused significant haemolysis (57.70%), and at 64 µg/mL, it resulted in 100% haemolysis. In contrast, even at the highest tested concentration (256 µg/mL), FCZ, A1, A3 and A9 only induced 1.52%, 0.64%, 0.71% and 0.31% haemolysis, respectively, indicating that they did not cause significant haemolysis and that our compounds have a similar safety profile as FCZ in rabbit red blood cells.
Figure 5. Haemolytic effect of A1, A3 and A9 against rabbit red blood cells at different indicated concentrations.
Molecular docking study
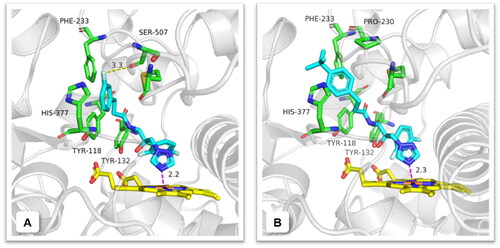
To investigate the binding mode of target compounds, a molecular docking study was carried out using the Surflex-Dock module in the SYBYL-X 2.0. Representative compounds A3 and A9 (MIC ≤0.125 µg/mL against C. albicans) were docked into C. albicans Cyp51 (PDB ID: 5TZ1) active site. As depicted in , the N(4) atom of the 1,2,4-triazole formed a coordination bond with the haem Fe-atom, and the hydroxyl group and difluorophenyl group were considered as forming nonbonding interactions like Tyr132. The phenyl-propanamide side chain stretched into the Cyp51 channel to form hydrophobic and van der Waals interactions with surrounding residues such as Tyr118, Phe233 and His377. Notably, the 3-fluorine atom on the terminal phenyl of compound A3 was found to form an additional halogen bond with Ser507 in the distance of 3.3 Å, while compound A9 (), shared a similar binding mode with A3, mainly interacted with surrounding residues by hydrophobic and van der Waals. In addition, the docking of two selected compounds A3 and A9 with proper fitting into the binding site exhibited binding free energies (ΔGb) of −169.50 and −142.78 kcal/mol, respectively, corresponding to the reference drug fluconazole (ΔGb: −123.85 kcal/mol). This result was correlated to their antifungal potency against C. albicans.
Figure 6. Predicted binding mode of compounds A3 and A9 (blue) in the active site of CYP51. Yellow dashed lines represent the halogen bond interactions and magenta dashed lines represent the coordination bond. The image was generated using PyMol.
Theoretical evaluation of ADME/T properties
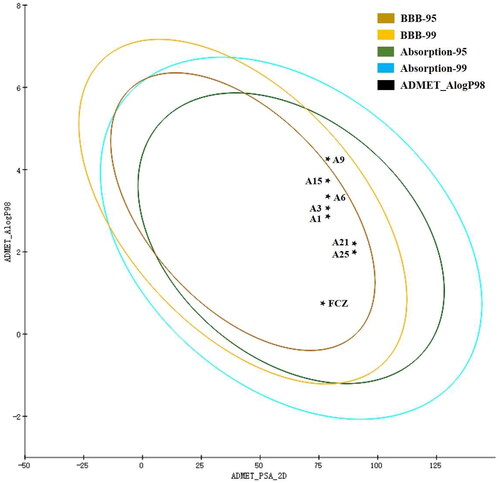
ADMET properties refer to the absorption, distribution, metabolism, excretion and toxicity properties of the molecule in the organism. ADMET properties prove useful in improving the success rate of drug developmentCitation17,Citation18. Hence, to further investigate our synthesised triazole drugs, compounds A1, A3, A6, A9, A15, A21 and A25 were predicted for in silico DS-ADMET and DS-TOPKAT modules. As shown in , nearly all compounds are positioned in the 95% and 99% confidence ellipses for the blood-brain barrier (BBB) and human intestinal absorption (HIA), indicating that these compounds have proper aqueous liposolubility and might enter the blood through HIA to exert their antifungal effect in vivo, as well as penetrate through BBB to treat fungal infections of the nervous system.
Figure 7. The evaluation egg-chart of PSA and Alog P of tested compounds. The 95% and 99% confidence limit ellipses of blood-brain barrier and intestinal absorption models.
The detailed predicated data of ADME/T were summarised in Table S2. Similar to FCZ, these triazole derivatives had proper absorption or permeation with PSA < 140, −2.0 ≤ A logP ≤ 7.0. For toxicity evaluation, these compounds were non-mutagenic, noncarcinogenic, and non-irritating. Although part of the compounds showed a certain degree of skin sensitisation, ocular irritancy or development toxicity potential (DTP) like FCZ, there were still subtle differences between the compounds. For example, A15 had the characteristics of weak skin sensitisation, A3 showed mild ocular irritancy, and A6, A21 and A25 had non-toxic DTP, which reflected the different kinds of substituents and may influence the drug safety profile.
Conclusion
In summary, based on our previous work, we simplified the quinazoline ring of albaconazole to obtain a series of novel triazoles containing aryl-propanamide side chains. In vitro, antifungal activity of the target compounds demonstrated that parts of them exhibited potent and broad-spectrum antifungal activity against tested fungi. SARs study indicated that phenyl-propanamide moiety was good for exerting antifungal activity than pyridinyl. Moreover, compounds A1, A2, A6, A12 and A15 not only had excellent antifungal activity against C. albicans and C. glabrata with MIC ≤ 0.125 µg/mL but also exhibited inhibitory activity against FCZ-resistant strains including C.auris isolates. GC-MS mechanism studies showed that selected compounds A1, A3, and A9 influenced the ergosterol biosynthesis in a manner similar to that of fluconazole via Cyp51 inhibition. In addition, A1, A3 and A9 were further evaluated for their ability to induce haemolysis of rabbit red cells. A1, A3 and A9 induced a similar level of haemolysis to that of FCZ. Molecular docking studies showed that the propanamide moiety side chains extended into the hydrophobic pocket of the CYP51 channel and m-F substituted on the terminal phenyl could form halogen interactions with Ser507. Overall, in silico evaluation of ADME/T properties of our compounds suggested that these novel triazole analogues are promising drugs. Further structure optimisation and pharmacokinetic evaluation work are currently being undertaken.
Experiment section
Synthesis
The solvents and reagents were purchased from Bidepharm (Shanghai, China) or Aladdin (Shanghai, China) without further purification unless otherwise indicated. The column chromatography was performed on silica gel (200–300 mesh). 1H and 13C NMR spectra were recorded in DMSO-d6 using a Bruker AC-300/400P spectrometer, and TMS as a standard. Chemical shifts (δ values) and coupling constants (J values) are measured in ppm and Hz, respectively. ESI mass spectra were performed using an Agilent Technologies 6120 quadrupole LC-MS.
General procedure for the synthesis of 1-(((2 R,3S)-2–(2,4-difluorophenyl)-3-methyloxiran-2-yl)methyl)-1H-1,2,4-triazole (2)
Compound 1 (10.0 mmol) was dissolved in DCM (15 ml), and Et3N (11.0 mmol) was added at 0 °C, and then methanesulfonyl chloride (MsCl) (11.0 mmol) was slowly added. The mixture was stirred at room temperature for 1 h. After the reaction was complete, 3 M sodium hydroxide solution (approximately 6 ml) was poured into the mixture, then stirred at 0 °C for 4 h, and the reaction was monitored by TLC. After the reaction had finished (monitored by TLC), the organic phase was washed with saturated sodium chloride aqueous solution (3 × 20 ml), dried with anhydrous sodium sulphate and evaporated under vacuum to obtain the crude product of compound 2, which was recrystallized under the condition of ethyl acetate/petroleum ether as a white solid (yield 60%).
General procedure for the synthesis of (2 R,3R)-3-azido-2–(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)butan-2-ol (3)
Compound 2 (24.0 mmol) was dissolved in DMF (20 ml) followed by the addition of NaN3 (28.8 mmol) and NH4Cl (36.0 mmol). The reaction mixture was stirred at 80 °C for 10 h. The reaction was monitored by TLC. After the reaction was completed, the reaction mixture was cooled to room temperature, poured into ice water, and then stirred overnight to produce a large amount of white solid. The crude compound 3 was obtained by filtration and dried, which was directly used in the next step without further purification.
General procedure for the synthesis of (2 R,3R)-3-amino-2–(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)butan-2-ol (4)
Pd/C was slowly added to a solution of compound 3 in MeOH (20 ml). The mixture was stirred overnight in a hydrogen atmosphere at room temperature. The reaction was monitored by TLC. After the reaction was completed, Pd/C was filtered and the organic solvent was evaporated under reduced pressure to obtain compound 4 as a white solid (yield 68%).
General procedure for the synthesis of target compounds (A1–A27)
DIEA (2 mmol) and PyBOP (1.1 mmol) were added to a solution of compound 4 (1.0 mmol) and various substituted aromatic carboxylic acid (1.0 mmol) in DMF (5 ml) at room temperature. The mixture was stirred for 2 h. The reaction was monitored by TLC. After the reaction had finished, the mixture was poured into water, and then the mixture was extracted with ethyl acetate (2 × 10 ml). After washing with brine (2 × 10 ml) and drying over anhydrous Na2SO4, the organic phase was evaporated in a vacuum. The crude product was purified by silica gel column chromatography using ethyl acetate/petroleum ether (1:1) as the eluent to give target compounds A1–A27.
Biological activity
Strains and culture conditions
All fungal strains used in this study were obtained from the American Type Culture Collection (ATCC) and Changhai Hospital (Shanghai, China). All strains were cultivated by shaking (200 rpm) at 30 °C in YEPD medium (1% yeast extract, 2% peptone, and 2% dextrose) to an exponential growth stage. Ketoconazole (KCZ), fluconazole (FCZ) and amphotericin B (AmB) were purchased from Sigma and dissolved in dimethyl sulfoxide (DMSO) at a concentration of 6.4 mg/mL as stock solutions.
Broth microdilution assay
Fungal cells at the exponential growth stage were collected and resuspended in RPMI 1640 medium, then diluted to 1 × 103 cells/mL. The suspension of fungal cells was inoculated into 96-well plates. Antifungal compounds were serially added and diluted in a 2-fold series to a final concentration range of 64.0–0.125 µg/mL. RPMI 1640 medium with no fungal cells served as a blank control. The fungal suspensions containing DMSO and FCZ served as the negative control and the positive control, respectively. C. albicans, C. glabrata, C. parapsilosis, C. tropicalis, and C. auris cells were incubated at 30 °C for 24 h, C. neoformans and A. fumigatus cells were incubated at 30 °C for 48 h. The optical density (OD630) at 630 nm was measured by a spectrophotometer, which was used to calculate the MIC80 values of each compound.
GC-MS analysis of sterol composition
Overnight cultured C. albicans SC5314 fungal cells were collected and diluted to 5 × 106 cells/mL in YEPD medium. The fungal suspension was transferred to 50 ml conical flasks, then 8.0 µg/mL of compounds or fluconazole were added. The fungal cells were cultured by shaking (200 rpm) at 30 °C. After incubation for 8 h, the fungal suspension was centrifuged (3000 rpm, 1 min), harvested and washed twice with distilled water carefully. The saponifying agent (6 ml of 15% NaOH resolved in 90% ethanol) was added to the precipitate. After 1 h of saponification at 80 °C, sterols were extracted with hexane (3 × 5 ml). The combined extracts were evaporated in a water bath and washed once with water. The residue was dried in a 60 °C water bath to obtain the total sterols. Finally, the total sterol was analysed by GC-MS and the structures of each sterol content were identified and compared with the National Institute of Standards and Technology (NIST) database.
Haemolytic assays
Briefly, 100 µL of fresh rabbit red cell (RBCs) suspension in 96-well plates was mixed with 100 µL of a solution of compounds, FCZ or AmB to the final concentrations of 128–1 µg/mL. RBCs incubated with PBS (containing an equal volume of DMSO) were used as the negative control, and those incubated with distilled water were used as the positive control (100% haemolysis). After incubation for 1 h at 37 °C, the supernatants were centrifuged, collected, and the absorbance (Abs) was measured by using a spectrophotometer at 405 nm. The percentage of haemolysis was calculated using the following equation: haemolysis % = (AbsS − Abs0)/(Abs100 − Abs0) × 100%, where AbsS represented the average absorbance of the sample, Abs0 was the average absorbance of the negative control, and Abs100 mean the average absorbance of the positive control.
Computational methodology
The crystal structures of C. albicans CYP51 (PDB ID: 5TZ1) were downloaded from the protein database bank. The docking studies were carried out with SYBYL-X 2.0 software. Protein preparation using the standard Tripos molecular mechanics force field with a Gasteiger–Hückel charge, and other staged minimisation was set as default. Then, the ligands and water were removed, and hydrogens were added to the amino acid residues. The binding site was defined as whole residues within a 10 Å radius around the original ligand. Next, the active pocket was generated, and the compound was docked through the Surface Dock module. Finally, the best conformation was chosen to analyse the ligand-protein interaction. The image was further plotted by Pymol.
ADME/T prediction
Tested compounds were predicted using DS-ADMET and DS-TOPKAT modulesCitation19,Citation20. The operation process was performed as follows: the structures of tested compounds were prepared by Chemdraw, and then imported into the program. Two modules ADMET descriptors and Toxicity Prediction were selected. The prediction items, including aqueous solubility, plasma protein binding, intestinal absorption, blood-brain barrier penetration, Ames mutagenicity, FDA rodent carcinogenicity, Skin_Irritancy, Skin_sensitisation, Ocular_Irritancy and DTP (development toxicity potential) were set as research objects. Subsequently, running the program obtains the corresponding prediction results.
X-ray crystallography
Single crystals of amine 4 were obtained by slow evaporation of dichloromethane solution amine 4 at room temperature. The X-ray single crystal diffraction data for amine 4 were collected on Bruker APEX DUO diffractometers with Mο Kα radiation (λ = 1.34139 Å) at 173 ± 2 K in the ω-2θ scanning mode. The structures were solved by direct methods using the SHELXS-97A program and refined by full-matrix least-squares techniques (SHELXL-97) on F2. Anisotropic thermal parameters were assigned to all non-hydrogen atoms. The organic hydrogen atoms were generated geometrically CCDC-2259250 (amine 4) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Supplemental Material
Download PDF (5.4 MB)Disclosure statement
The authors declare no conflicts of interest.
Additional information
Funding
References
- Maddy AJ, Sanchez N, Shukla BS, Maderal AD. Dermatological manifestations of fungal infection in patients with febrile neutropaenia: a review of the literature. Mycoses. 2019;62(9):826–834.
- Kim J, Sudbery P. Candida albicans, a major human fungal pathogen. J Microbiol. 2011;49(2):171–177.
- Ngo HX, Garneau-Tsodikova S, Green KD. A complex game of hide and seek: the search for new antifungals. Medchemcomm. 2016;7(7):1285–1306.
- Yong L, Min C, Thomas H, Rong-Ya Y, Wan-Qing L. Epidemiology of opportunistic invasive fungal infections in China: review of literature. Chinese Medical Journal. 2013; 12602:361–368.
- Hoenigl M, Seidel D, Sprute R, Cunha C, Oliverio M, Goldman GH, Ibrahim AS, Carvalho A. COVID-19-associated fungal infections. Nat Microbiol. 2022;7(8):1127–1140.
- Xie F, Ni T, Zhao J, Pang L, Li R, Cai Z, Ding Z, Wang T, Yu S, Jin Y, et al. Design, synthesis, and in vitro evaluation of novel antifungal triazoles. Bioorg Med Chem Lett. 2017;27(10):2171–2173.
- Ni T, Pang L, Cai Z, Xie F, Ding Z, Hao Y, Li R, Yu S, Chai X, Wang T, et al. Design, synthesis, and in vitro antifungal evaluation of novel triazole derivatives bearing alkynyl side chains. Journal of Saudi Chemical Society. 2019;23(5):576–585.
- Wu J, Ni T, Chai X, Wang T, Wang H, Chen J, Jin Y, Zhang D, Yu S, Jiang Y. Molecular docking, design, synthesis and antifungal activity study of novel triazole derivatives. Eur J Med Chem. 2018; 143:1840–1846.
- Ding Z, Ni T, Xie F, Hao Y, Yu S, Chai X, Jin Y, Wang T, Jiang Y, Zhang D. Design, synthesis, and structure-activity relationship studies of novel triazole agents with strong antifungal activity against Aspergillus fumigatus. Bioorg Med Chem Lett. 2020;30(4):126951.
- CLSI. Reference method for broth dilution antifungal susceptibility testing of yeasts, M27-A3. 3rd ed. Wayne (PA): Clinical and Laboratory Standards Institute; 2009.
- CLSI. Performance standards for antifungal susceptibility testing of yeast. 1st ed. Wayne (PA): Clinical and Laboratory Standards Institute; 2017. p. M60.
- Stewart AG, Paterson DL. How urgent is the need for new antifungals? Expert Opin Pharmacother. 2021;22(14):1857–1870.
- Du H, Bing J, Hu T, Ennis CL, Nobile CJ, Huang G. Candida auris: epidemiology, biology, antifungal resistance, and virulence. PLOS Pathog. 2020;16(10):e1008921.
- Cortegiani A, Misseri G, Fasciana T, Giammanco A, Giarratano A, Chowdhary A. Epidemiology, clinical characteristics, resistance, and treatment of infections by Candida auris. J Intensive Care. 2018; 6:69.
- Borrull A, López-Martínez G, Poblet M, Cordero-Otero R, Rozès N. A simple method for the separation and quantification of neutral lipid species using GC-MS. Eur J Lipid Sci Technol. 2015;117(3):274–280.
- Xu H, Mou YH, Guo MB, Zhang R, Yan ZZ, An R, Wang X, Su X, Hou Z, Guo C. Discovery of novel selenium-containing azole derivatives as antifungal agents by exploiting the hydrophobic cleft of CYP51. Eur J Med Chem. 2022; 243:114707.
- Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, Lee PW, Tang Y. admetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model. 2012;52(11):3099–3105.
- Guan L, Yang H, Cai Y, Sun L, Di P, Li W, Liu G, Tang Y. ADMET-score - a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm. 2019;10(1):148–157.
- An Y, Dong Y, Liu M, Han J, Zhao L, Sun B. Novel naphthylamide derivatives as dual-target antifungal inhibitors: design, synthesis and biological evaluation. Eur J Med Chem. 2021; 210:112991.
- An Y, Liu W, Xie H, Fan H, Han J, Sun B. Construction and activity evaluation of novel benzodioxane derivatives as dual-target antifungal inhibitors. Eur J Med Chem. 2022; 227:113950.