
Abstract
A range of 3H-1,2-benzoxaphosphepine 2-oxide aryl derivatives with various substitution patterns at positions 7, 8, or 9 of the scaffold was synthesised in five steps from the commercially available salicylaldehydes. All of the newly obtained compounds were studied for their inhibition potency against carbonic anhydrase (CA) isoforms I, II, IX, and XII. Delightfully, these compounds showed a striking selectivity for the cancer-associated CA IX and XII over the cytosolic CA I and II, whose inhibition may lead to side-effects. Overall, a structure–activity relationship (SAR) revealed that 7- and 8-substituted aryl derivatives were more effective inhibitors of CA IX and XII than 9-substituted derivatives. In addition, the fluorine-containing analogues emerged as the most potent CA IX/XII inhibitors in this series.
Graphical Abstract

Introduction
Cancer is a devastating group of diseases which is one of the major causes of death worldwide, accounting for nearly 10 million deaths in 2020Citation1. By approaching to the middle of the 21st century, the cancer incidence and mortality rates are expected to increase to 29.5 million and 16.4 million per year, respectivelyCitation2. The development of novel and improved therapies to combat cancer is therefore of a paramount priority. Carbonic anhydrase (CA, EC 4.2.1.1) isoforms CA IX and CA XII are presently serving as biomarkers and anticancer drug targetsCitation3. Both of these isozymes are highly overexpressed in various cancer types and may contribute to the growth of cancer, subsequent metastasis, as well as impaired therapeutic responseCitation4.
CA IX and CA XII are transmembrane zinc metalloenzymes that belong to the α-CA familyCitation5. Humans have 15 α-CA isoforms with different expression patterns, molecular features and kinetic propertiesCitation6. These enzymes are involved in many important physiological processes (e.g. respiration, homeostasis, and metabolism), as they catalyse the reversible hydration of CO2Citation6. Hence, the development of selective CA IX and XII inhibitors is highly desired to prevent possible side effects.
At present, none of the clinically used CA inhibitors displays selectivity for a specific isoformCitation7. Due to the high level of structural homology between the CA isoforms and sequence similarities within the active site, the design and development of isoform-specific CA inhibitors remain challengingCitation8. However, to our knowledge, to date, several molecules have been reported as potent and selective CA IX and XII inhibitors, including coumarinsCitation9–12, isocoumarinsCitation13, thiocoumarinsCitation9,Citation12, sulfocoumarinsCitation9,Citation14–17, and their congeners—homosulfocoumarinsCitation18 (3H-1,2-benzoxathiepine 2,2-dioxides).
In the course of our research, devoted to discovering novel chemotypes acting as selective CA IX/XII inhibitors that could be employed in cancer chemotherapy, we previously designed and synthesised a series of 3H-1,2-benzoxaphosphepine 2-oxides as bioisosteres of homosulfocoumarinsCitation19. These new compounds showed excellent selectivity and good inhibitory activity against both CA IX and XII. Our interest in phosphorus heterocycles stems from the fact that phosphorus functionalities can improve the pharmacokinetics profile, bioavailability and water solubility of drugsCitation20. Moreover, several groups reported the use of organophosphorus compounds as CA inhibitorsCitation21.
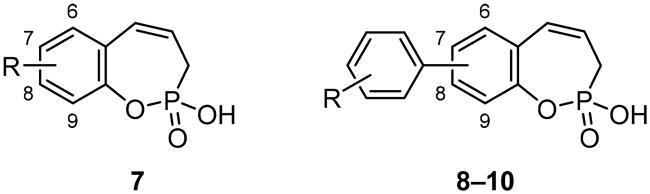
In continuation of our previous work on the development of benzoxaphosphepine 2-oxides as CA inhibitorsCitation19, the current study is aimed to investigate the chemical space around this novel chemotype. In this paper, we present the synthesis and biological evaluation of 7-, 8- and 9-aryl-substituted benzoxaphosphepine 2-oxides.
Materials and methods
Chemistry
The air- or moisture-sensitive reactions were performed under an argon atmosphere using dry glassware. All reagents, starting materials and solvents were purchased from commercial sources and used as received. TLC was performed on silica gel plates (60 F254) and visualised under UV light (254 and 365 nm). Reversed-phase chromatography was done on a Biotage Isolera One system using Biotage SNAP KP-C18-HS cartridges. Melting points were determined on an OptiMelt MPA100 apparatus. IR spectra were recorded on a Shimadzu FTIR IR Prestige-21 spectrophotometer. 1H, 13C and 31P NMR spectra were recorded on a Bruker Avance Neo 400 MHz spectrometer. The chemical shifts (δ) were reported in parts per million (ppm) relative to the residual solvent peak as an internal reference (DMSO-d6: 1H 2.50, 13C 39.52; CD3OD: 1H 3.31, 13C 49.00). 31P shifts were referenced externally to H3PO4. The coupling constants (J) were expressed in Hertz (Hz). HRMS was performed on a Q-TOF Micro mass spectrometer.
The synthesis and characterisation of 4-iodo-2-vinylphenol (2a), 2-bromo-6-vinylphenol (2c), ethyl allylphosphonochloridate (3), ethyl (4-iodo-2-vinylphenyl) allylphosphonate (4a), 2-bromo-6-vinylphenyl ethyl allylphosphonate (4c) together with corresponding 3H-1,2-benzoxaphosphepine 2-oxides 6a,c and 7a,c are reported by our group in the previous paperCitation19.
5-Bromo-2-vinylphenol (2b)
The titled compound 2b was obtained according to the general procedure previously reportedCitation19 using MePPh3Br (16.35 g, 45.77 mmol), tBuOK (5.25 g, 46.8 mmol), and 4-bromo-2-hydroxybenzaldehyde (4.00 g, 19.9 mmol) as a yellowish solid (3.21 g, 81%). The NMR spectra are consistent with the literatureCitation19. 1H NMR (400 MHz, DMSO-d6) δ = 5.24 (dd, 1H, J = 11.3, 1.6 Hz), 5.79 (dd, 1H, J = 17.6, 1.6 Hz), 6.87 (dd, 1H, J = 17.6, 11.3 Hz), 6.95 (dd, 1H, J = 8.3, 2.0 Hz), 7.00 (d, 1H, J = 2.0 Hz), 7.37 (d, 1H, J = 8.3 Hz), and 10.13 (s, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 114.6, 118.3, 120.8, 122.0, 123.5, 128.0, 130.8, and 155.7 ppm.
5-Bromo-2-vinylphenyl ethyl allylphosphonate (4b)
The titled compound 4b was obtained according to the general procedure previously reportedCitation19 using 5-bromo-2-vinylphenol (2b) (3.87 g, 19.4 mmol), ethyl allylphosphonochloridate (3) (3.46 ml, 23.3 mmol) and NEt3 (3.38 ml, 24.3 mmol) as a colourless oil (4.23 g, 66%). IR (thin film, cm−1): 1265 (P = O), 1218 (P = O), 1181 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 25.50 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.20 (t, 3H, J = 7.0 Hz), 2.94 (dt, 1H, J = 7.3, 1.2 Hz), 2.99 (dt, 1H, J = 7.3, 1.2 Hz), 4.02–4.18 (m, 2H), 5.20–5.33 (m, 2H), 5.42 (dd, 1H, J = 11.2, 1.0 Hz), 5.70–5.83 (m, 1H), 5.91 (dd, 1H, J = 16.6, 1.0 Hz), 6.84–6.93 (m, 1H), 7.38–7.43 (m, 1H), 7.48–7.50 (m, 1H), and 7.62–7.66 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.1 (d, JP,C = 5.6 Hz), 30.9 (d, JP,C = 137 Hz), 62.8 (d, JP,C = 6.8 Hz), 117.3, 120.5 (d, JP,C = 1.4 Hz), 120.6 (d, JP,C = 15.0 Hz), 123.7 (d, JP,C = 2.6 Hz), 127.3 (d, JP,C = 11.6 Hz), 128.0, 128.2 (d, JP,C = 5.0 Hz), 129.2, and 147.8 (d, JP,C = 9.1 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C13H17O3PBr: 331.0099, found 331.0114.
8-Bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6b)
The titled compound 6b was obtained according to the general procedure previously reportedCitation19 using 5-bromo-2-vinylphenyl ethyl allylphosphonate (4b) (2.00 g, 6.04 mmol) and ruthenium catalyst 5 (CAS: 250220–36-1) (286 mg, 0.30 mmol) as a greenish dense oil (1.51 g, 83%). IR (thin film, cm−1): 1270 (P = O), 1237 (P = O), 1202 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 39.41 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.28 (t, 3H, J = 7.0 Hz), 2.68–2.93 (m, 2H), 4.15–4.25 (m, 2H), 5.94–6.04 (m, 1H), 6.66–6.70 (m, 1H), 7.28–7.32 (m, 1H), 7.43–7.48 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 16.2 (d, JP,C = 5.7 Hz), 25.6 (d, JP,C = 125 Hz), 62.5 (d, JP,C = 6.8 Hz), 121.3, 123.3 (d, JP,C = 12.2 Hz), 124.4 (d, JP,C = 3.5 Hz), 127.1, 128.0, 128.7 (d, JP,C = 8.8 Hz), 132.3, and 147.9 (d, JP,C = 7.8 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C11H13O3PBr: 302.9786, found 302.9781.
8-Bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b)
The titled compound 7b was obtained according to the general procedure previously reportedCitation19 using 8-bromo-2-ethoxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (6b) (1.13 g, 3.73 mmol) and TMSBr (2.93 ml, 22.4 mmol) as a white solid (0.88 g, 86%). Mp: 182–184 °C. IR (KBr, cm−1): 2519 (O = P-OH), 2245 (O = P-OH), 1250 (P = O), and 1205 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.97 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.58–2.68 (m, 2H), 5.91–6.04 (m, 1H), 6.57–6.63 (m, 1H), 7.24–7.29 (m, 1H), 7.31–7.34 (m, 1H), 7.38–7.42 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 120.9, 124.4 (d, JP,C = 12.0 Hz), 124.6 (d, JP,C = 3.2 Hz), 127.5, 128.2 (d, JP,C = 8.4 Hz), 132.2, and 148.5 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M–H]–: m/z calcd for C9H7O3PBr: 272.9316, found 272.9320.
General procedure for the synthesis of 3H-1,2-benzoxaphosphepine 2-oxide aryl derivatives 8–10
The corresponding 3H-1,2-benzoxaphosphepine 2-oxide halogen derivative 7 (200 mg, 1.0 eq) was placed in a pressure tube and dissolved in 1,4-dioxane (5 ml) followed by the addition of degassed water (1 ml). The corresponding boronic acid (1.5 eq), K2CO3 (2.0 eq) and Pd(dppf)Cl2 (10 mol% for iodo derivative 7a; 20 mol% in case of bromo derivatives 7b and 7c) were added to the solution. The reaction mixture was purged with argon for 5 min, the tube was sealed and heated for 16 h at 80 °C. Upon cooling to rt, the reaction mixture was filtered through a pad of celite, which was washed with MeCN. The pH of the filtrate was adjusted to 2 by addition of TFA. After that, the filtrate was concentrated in vacuo. The crude product was purified by reversed-phase flash chromatography (MeCN/water = 10 to 95%) and recrystallised from EtOAc.
2-Hydroxy-7-phenyl-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8a)
By following the general procedure, 8a was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), phenylboronic acid (114 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (101 mg, 60%). Decomp. > 205 °C. IR (KBr, cm−1): 2611 (O = P-OH), 2161 (O = P-OH), 1619 (O = P-OH), 1215 (P = O), and 1196 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.77 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.62 (d, 1H, J = 6.6 Hz), 2.67 (d, 1H, J = 6.6 Hz), 5.92–6.05 (m, 1H), 6.69–6.76 (m, 1H), 7.15–7.22 (m, 1H), 7.32–7.40 (m, 1H), 7.42–7.50 (m, 2H), and 7.56–7.70 (m, 4H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 122.3 (d, JP,C = 3.2 Hz), 124.0 (d, JP,C = 12.2 Hz), 126.7, 127.5 (d, JP,C = 2.4 Hz), 128.3, 128.8, 129.0, 129.1 (d, JP,C = 8.6 Hz), 136.4, 139.2, and 147.6 (d, JP,C = 7.5 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C15H14O3P: 273.0681, found 273.0685.
2-Hydroxy-7–(4-methoxyphenyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8b)
By following the general procedure, 8b was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (4-methoxyphenyl)boronic acid (142 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (115 mg, 61%). Mp: 214–216 °C. IR (KBr, cm−1): 2522 (O = P-OH), 2207 (O = P-OH), 1265 (P = O), and 1219 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.14 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.61 (d, 1H, J = 6.1 Hz), 2.66 (d, 1H, J = 6.1 Hz), 3.79 (s, 3H), 5.91–6.04 (m, 1H), 6.67–6.74 (m, 1H), 6.98–7.05 (m, 2H), 7.13–7.19 (m, 1H), and 7.50–7.64 (m, 4H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.2 (d, JP,C = 125 Hz), 55.2, 114.4, 122.2 (d, JP,C = 3.2 Hz), 123.8 (d, JP,C = 12.2 Hz), 126.9, 127.7, 128.1, 128.2, 129.2 (d, JP,C = 8.5 Hz), 131.5, 136.1, 147.0 (d, JP,C = 7.5 Hz), and 158.9 ppm. HRMS (ESI) [M + H]+: m/z calcd for C16H16O4P: 303.0786, found 303.0798.
Ethyl 4–(2-hydroxy-2-oxido-3H-benzo[f][1,2]oxaphosphepin-7-yl)benzoate (8c)
By following the general procedure, 8c was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (4-(ethoxycarbonyl)phenyl)boronic acid (182 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (126 mg, 59%). Mp: 250–252 °C. IR (KBr, cm−1): 2512 (O = P-OH), 2187 (O = P-OH), 1710 (C = O), 1286 (P = O), and 1218 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.17 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.34 (t, 3H, J = 7.0 Hz), 2.61 (d, 1H, J = 6.0 Hz), 2.66 (d, 1H, J = 6.0 Hz), 5.93–6.06 (m, 1H), 6.67–6.76 (m, 1H), 7.17–7.25 (m, 1H), 7.63–7.74 (m, 2H), 7.77–7.86 (m, 2H), and 7.97–8.07 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 14.2, 27.4 (d, JP,C = 125 Hz), 60.8, 122.6 (d, JP,C = 3.2 Hz), 124.3 (d, JP,C = 12.2 Hz), 126.8, 127.7, 128.6, 128.7, 128.8 (d, JP,C = 8.4 Hz), 129.2, 129.8, 134.9, 143.6, 148.4 (d, JP,C = 7.6 Hz), and 165.5 ppm. HRMS (ESI) [M + H]+: m/z calcd for C18H18O5P: 345.0892, found 345.0901.
7–(4-Fluorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8d)
By following the general procedure, 8d was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (4-fluorophenyl)boronic acid (130 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (114 mg, 63%). Decomp. > 232 °C. IR (KBr, cm−1): 2514 (O = P-OH), 2177 (O = P-OH), 1648 (O = P-OH), 1214 (P = O), and 1200 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.88 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.59–2.70 (m, 2H), 5.91–6.06 (m, 1H), 6.67–6.75 (m, 1H), 7.15–7.22 (m, 1H), 7.24–7.33 (m, 2H), 7.55–7.63 (m, 2H), and 7.66–7.75 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 115.7 (d, JF,C = 21.0 Hz), 122.3 (d, JP,C = 3.2 Hz), 124.0 (d, JP,C = 12.2 Hz), 127.4, 128.3, 128.6 (d, JF,C = 8.2 Hz), 128.8, 129.1 (d, JP,C = 8.6 Hz), 135.4, 135.6 (d, JF,C = 2.4 Hz), 147.5 (d, JP,C = 7.4 Hz), and 162.0 (d, JF,C = 244 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C15H13O3PF: 291.0586, found 291.0586.
7–(4-Chlorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8e)
By following the general procedure, 8e was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (4-chlorophenyl)boronic acid (146 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (112 mg, 59%). Decomp. > 212 °C. IR (KBr, cm−1): 2089 (O = P-OH), 1637 (O = P-OH), 1201 (P = O), and 1117 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.74 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.62 (d, 1H, J = 6.0 Hz), 2.67 (d, 1H, J = 6.0 Hz), 5.92–6.06 (m, 1H), 6.67–6.75 (m, 1H), 7.16–7.23 (m, 1H), 7.47–7.54 (m, 2H), and 7.58–7.73 (m, 4H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 122.4 (d, JP,C = 3.2 Hz), 124.1 (d, JP,C = 12.2 Hz), 127.4, 128.4, 128.8, 128.9, 129.0 (d, JP,C = 8.6 Hz), 132.4, 135.0, 137.9, and 147.8 (d, JP,C = 7.5 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C15H13O3PCl: 307.0291, found 307.0290.
2-Hydroxy-7–(4-(trifluoromethyl)phenyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8f)
By following the general procedure, 8f was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (4-(trifluoromethyl)phenyl)boronic acid (177 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as an off-white solid (133 mg, 63%). Decomp. > 280 °C. IR (KBr, cm−1): 1669 (O = P-OH), 1222 (P = O), and 1197 (P = O). 31P NMR (162 MHz, CD3OD) δ = 40.27 ppm. 1H NMR (400 MHz, CD3OD) δ = 2.52 (d, 1H, J = 6.7 Hz), 2.57 (d, 1H, J = 6.7 Hz), 5.98–6.11 (m, 1H), 6.63–6.69 (m, 1H), 7.23–7.27 (m, 1H), 7.47–7.50 (m, 1H), 7.52–7.57 (m, 1H), 7.60–7.63 (m, 2H), and 7.83–7.87 (m, 2H) ppm. 13C NMR (101 MHz, CD3OD) δ = 29.2 (d, JP,C = 125 Hz), 121.7, 124.1 (d, JP,C = 3.0 Hz), 124.3 (q, JF,C = 3.6 Hz), 124.7 (q, JF,C = 3.5 Hz), 126.5 (d, JP,C = 12.2 Hz), 127.1, 128.4, 129.7 (d, JP,C = 8.3 Hz), 129.9, 130.7, 131.6, 132.2 (q, JF,C = 32.0 Hz), 136.3, 142.7, and 151.1 (d, JP,C = 7.5 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C16H13O3PF3: 341.0554, found 341.0566.
7–(3,5-Dichlorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8 g)
By following the general procedure, 8 g was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (3,5-dichlorophenyl)boronic acid (178 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (119 mg, 56%). Decomp. > 180 °C. IR (KBr, cm−1): 2508 (O = P-OH), 2255 (O = P-OH), 1230 (P = O), and 1206 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 34.87 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.61 (d, 1H, J = 5.8 Hz), 2.66 (d, 1H, J = 5.8 Hz), 5.92–6.05 (m, 1H), 6.66–6.73 (m, 1H), 7.15–7.21 (m, 1H), 7.55–7.60 (m, 1H), and 7.66–7.78 (m, 4H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.5 (d, JP,C = 125 Hz), 122.5 (d, JP,C = 3.2 Hz), 124.3 (d, JP,C = 12.2 Hz), 125.3, 126.8, 127.7, 128.6, 128.8 (d, JP,C = 8.5 Hz), 129.4, 133.2, 134.7, 142.6, and 148.5 (d, JP,C = 7.5 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C15H12O3PCl2: 340.9901, found 340.9906.
7–(3-Fluorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8h)
By following the general procedure, 8h was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (3-fluorophenyl)boronic acid (130 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (180 mg, 60%). Mp: 171–173 °C. IR (KBr, cm−1): 2579 (O = P-OH), 2308 (O = P-OH), and 1190 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.68 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.60–2.72 (m, 2H), 5.90–6.08 (m, 1H), 6.67–6.77 (m, 1H), 7.14–7.24 (m, 2H), 7.46–7.60 (m, 3H), and 7.62–7.70 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 113.4 (d, JF,C = 22.2 Hz), 114.2 (d, JF,C = 21.0 Hz), 122.4 (d, JP,C = 3.2 Hz), 122.7, 124.0 (d, JP,C = 12.2 Hz), 127.6, 128.4, 129.0, 129.1, 130.9 (d, JP,C = 8.6 Hz), 135.0, 141.6 (d, JF,C = 7.8 Hz), 148.0 (d, JP,C = 7.6 Hz), and 162.7 (d, JF,C = 243 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C15H13O3PF: 291.0586, found 291.0588.
2-Hydroxy-7-(o-tolyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8i)
By following the general procedure, 8i was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), o-tolylboronic acid (127 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (101 mg, 57%). Mp: 156–158 °C. IR (KBr, cm−1): 2585 (O = P-OH), 2303 (O = P-OH), 1623 (O = P-OH), and 1193 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.70 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.23 (s, 3H), 2.63 (d, 1H, J = 6.6 Hz), 2.68 (d, 1H, J = 6.6 Hz), 5.90–6.03 (m, 1H), 6.64–6.74 (m, 1H), and 7.14–7.32 (m, 7H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 20.2, 27.3 (d, JP,C = 125 Hz), 121.6 (d, JP,C = 3.2 Hz), 123.8 (d, JP,C = 12.2 Hz), 126.0, 127.5, 127.7, 129.1 (d, JP,C = 8.6 Hz), 129.6, 129.8, 130.4, 131.0, 134.8, 137.4, 140.2, and 147.0 (d, JP,C = 7.5 Hz) ppm. HRMS (ESI) [M + H]+: m/z calcd for C16H16O3P: 287.0837, found 287.0841.
Methyl 3–(2-hydroxy-2-oxido-3H-benzo[f][1,2]oxaphosphepin-7-yl)benzoate (8j)
By following the general procedure, 8j was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (3-(methoxycarbonyl)phenyl)boronic acid (168 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (117 mg, 57%). Mp: 145–147 °C. IR (KBr, cm−1): 2303 (O = P-OH), 1718 (C = O), 1252 (P = O), and 1220 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.62 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.62 (d, 1H, J = 6.6 Hz), 2.67 (d, 1H, J = 6.6 Hz), 3.89 (s, 3H), 5.93–6.06 (m, 1H), 6.71–6.76 (m, 1H), 7.19–7.24 (m, 1H), 7.59–7.68 (m, 3H), 7.93–7.97 (m, 2H), and 8.17–8.19 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.4 (d, JP,C = 125 Hz), 52.3, 122.5 (d, JP,C = 3.0 Hz), 124.2 (d, JP,C = 12.2 Hz), 127.1, 127.6, 128.1, 128.6, 128.9, 129.0, 129.5, 130.4, 131.5, 135.2, 139.7, 148.0 (d, JP,C = 7.6 Hz), and 166.1 ppm. HRMS (ESI) [M + H]+: m/z calcd for C17H16O5P: 331.0735, found 331.0734.
2-Hydroxy-7–(3-nitrophenyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8k)
By following the general procedure, 8k was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (3-nitrophenyl)boronic acid (156 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a yellow solid (120 mg, 61%). Mp: 231–233 °C. IR (KBr, cm−1): 2580 (O = P-OH), 1646 (O = P-OH), and 1214 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.39 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.63 (d, 1H, J = 6.6 Hz), 2.68 (d, 1H, J = 6.6 Hz), 5.94–6.07 (m, 1H), 6.71–6.77 (m, 1H), 7.21–7.26 (m, 1H), 7.72–7.78 (m, 3H), 8.12–8.23 (m, 2H), and 8.43–8.46 (m, 1H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.3 (d, JP,C = 125 Hz), 121.0, 122.1, 122.6 (d, JP,C = 3.2 Hz), 124.3 (d, JP,C = 12.2 Hz), 127.8, 128.6, 128.8 (d, JP,C = 8.5 Hz), 129.3, 130.5, 133.2, 133.9, 140.7, and 148.3, 148.4 ppm. HRMS (ESI) [M + H]+: m/z calcd for C15H13NO5P: 318.0531, found 318.0537.
7–(4-(tert-Butyl)phenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (8 l)
By following the general procedure, 8 l was prepared from 2-hydroxy-7-iodo-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7a) (200 mg, 0.62 mmol), (4-(tert-butyl)phenyl)boronic acid (166 mg, 0.93 mmol), K2CO3 (172 mg, 1.24 mmol), and Pd(dppf)Cl2 (45 mg, 0.062 mmol) as a white solid (110 mg, 54%). Decomp. > 210 °C. IR (KBr, cm−1): 2162 (O = P-OH), 1654 (O = P-OH), 1220 (P = O), and 1195 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 31.16 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.31 (s, 9H), 2.41–2.47 (m, 1H), 5.83–5.98 (m, 1H), 6.53–6.63 (m, 1H), 7.01–7.11 (m, 1H), 7.41–7.51 (m, 4H), and 7.52–7.60 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 28.7 (d, JP,C = 125 Hz), 31.1, 34.2, 122.5 (d, JP,C = 2.8 Hz), 125.3 (d, JP,C = 11.4 Hz), 125.6, 126.2, 126.7, 128.3, 128.4 (d, JP,C = 8.4 Hz), 128.7, 135.3, 136.6, 148.6 (d, JP,C = 7.4 Hz), and 149.6 ppm. HRMS (ESI) [M + H]+: m/z calcd for C19H22O3P: 329.1307, found 329.1307.
2-Hydroxy-8-phenyl-3H-benzo[f][1,2]oxaphosphepine 2-oxide (9a)
By following the general procedure, 9a was prepared from 8-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b) (200 mg, 0.73 mmol), phenylboronic acid (133 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (121 mg, 61%). Mp: 155–157 °C. IR (KBr, cm−1): 2582 (O = P-OH), 2162 (O = P-OH), 1255 (P = O), and 1184 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 34.14 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.60 (d, 1H, J = 6.4 Hz), 2.65 (d, 1H, J = 6.4 Hz), 5.87–6.01 (m, 1H), 6.60–6.67 (m, 1H), 7.34–7.41 (m, 3H), 7.44–7.52 (m, 3H), and 7.67–7.72 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.7 (d, JP,C = 125 Hz), 119.6 (d, JP,C = 2.8 Hz), 122.4, 124.0 (d, JP,C = 11.6 Hz), 126.6, 127.1, 128.0, 128.6 (d, JP,C = 8.0 Hz), 129.0, 131.3, 138.7, 141.0, and 148.7 (d, JP,C = 7.1 Hz) ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H12O3P: 271.0524, found 271.0536.
2-Hydroxy-8–(4-methoxyphenyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (9b)
By following the general procedure, 9b was prepared from 8-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b) (200 mg, 0.73 mmol), (4-methoxyphenyl)boronic acid (166 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (147 mg, 67%). Mp: 205–207 °C. IR (KBr, cm−1): 2556 (O = P-OH), 2305 (O = P-OH), 1248 (P = O), and 1183 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 34.96 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.63 (d, 1H, J = 6.4 Hz), 2.68 (d, 1H, J = 6.4 Hz), 3.80 (s, 3H), 5.85–6.00 (m, 1H), 6.61–6.67 (m, 1H), 7.00–7.06 (m, 2H), 7.32–7.36 (m, 2H), 7.45–7.49 (m, 1H), and 7.63–7.69 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.4 (d, JP,C = 125 Hz), 55.2, 114.5, 118.9 (d, JP,C = 3.2 Hz), 122.1, 123.3 (d, JP,C = 11.8 Hz), 126.2, 127.8, 128.8 (d, JP,C = 8.2 Hz), 130.9, 131.3, 140.8, 148.4 (d, JP,C = 7.2 Hz), and 159.3 ppm. HRMS (ESI) [M–H]–: m/z calcd for C16H14O4P: 301.0630, found 301.0641.
Ethyl 4–(2-hydroxy-2-oxido-3H-benzo[f][1,2]oxaphosphepin-8-yl)benzoate (9c)
By following the general procedure, 9c was prepared from 8-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b) (200 mg, 0.73 mmol), (4-(ethoxycarbonyl)phenyl)boronic acid (212 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (145 mg, 58%). Mp: 189–191 °C. IR (KBr, cm−1): 2577 (O = P-OH), 2287 (O = P-OH), 1708 (C = O), 1283 (P = O), and 1192 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.08 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.34 (t, 3H, J = 7.1 Hz), 2.65 (d, 1H, J = 6.2 Hz), 2.70 (d, 1H, J = 6.2 Hz), 4.34 (q, 2H, J = 7.1 Hz), 5.92–6.06 (m, 1H), 6.64–6.72 (m, 1H), 7.39–7.51 (m, 2H), 7.57–7.65 (m, 1H), 7.85–7.91 (m, 2H), and 8.01–8.06 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 14.2, 27.4 (d, JP,C = 125 Hz), 60.8, 119.9 (d, JP,C = 3.2 Hz), 122.9, 124.2 (d, JP,C = 12.0 Hz), 126.9, 127.9, 128.6 (d, JP,C = 8.2 Hz), 129.1, 129.9, 131.5, 139.6, 143.0, 148.5 (d, JP,C = 7.2 Hz), and 165.5 ppm. HRMS (ESI) [M–H]–: m/z calcd for C18H16O5P: 343.0735, found 343.0750.
8–(4-Chlorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (9d)
By following the general procedure, 9d was prepared from 8-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b) (200 mg, 0.73 mmol), (4-chlorophenyl)boronic acid (171 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (134 mg, 60%). Mp: 203–205 °C. IR (KBr, cm−1): 2583 (O = P-OH), 2292 (O = P-OH), 1218 (P = O), and 1187 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.06 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.64 (d, 1H, J = 6.4 Hz), 2.69 (d, 1H, J = 6.4 Hz), 5.90–6.03 (m, 1H), 6.63–6.70 (m, 1H), 7.36–7.42 (m, 2H), 7.50–7.55 (m, 3H), and 7.72–7.77 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.4 (d, JP,C = 125 Hz), 119.6 (d, JP,C = 3.2 Hz), 122.6, 123.9 (d, JP,C = 12.0 Hz), 127.3, 128.4, 128.7 (d, JP,C = 8.4 Hz), 129.0, 131.4, 132.9, 137.4, 139.7, and 148.4 (d, JP,C = 7.2 Hz) ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H11O3PCl: 305.0134, found 305.0143.
8–(3,5-Dichlorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (9e)
By following the general procedure, 9e was prepared from 8-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b) (200 mg, 0.73 mmol), (3,5-dichlorophenyl)boronic acid (208 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (141 mg, 57%). Mp: 136–138 °C. IR (KBr, cm−1): 2533 (O = P-OH), 2262 (O = P-OH), 1221 (P = O), and 1196 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 33.47 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.59 (d, 1H, J = 6.4 Hz), 2.64 (d, 1H, J = 6.4 Hz), 5.90–6.03 (m, 1H), 6.60–6.66 (m, 1H), 7.35–7.39 (m, 1H), 7.46–7.49 (m, 1H), 7.54–7.61 (m, 2H), and 7.74–7.78 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.8 (d, JP,C = 125 Hz), 120.2, 122.6, 124.7, 125.3, 127.2, 128.3, 128.4, 131.4, 134.7, 137.8, 142.2, and 148.8 ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H10O3PCl2: 338.9745, found 338.9760.
2-Hydroxy-8–(2-nitrophenyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (9f)
By following the general procedure, 9f was prepared from 8-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7b) (200 mg, 0.73 mmol), (2-nitrophenyl)boronic acid (182 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a yellowish solid (159 mg, 69%). Mp: 218–220 °C. IR (KBr, cm−1): 2604 (O = P-OH), 2240 (O = P-OH), 1185 (P = O), and 1126 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 34.84 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.65 (d, 1H, J = 6.4 Hz), 2.70 (d, 1H, J = 6.4 Hz), 5.92–6.05 (m, 1H), 6.64–6.70 (m, 1H), 7.07–7.10 (m, 1H), 7.16 (dd, 1H, J = 8.0, 1.6 Hz), 7.38 (d, 1H, J = 8.0 Hz), 7.57–7.61 (m, 1H), 7.62–7.68 (m, 1H), 7.75–7.81 (m, 1H), and 8.01 (dd, 1H, J = 8.0, 1.0 Hz) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.4 (d, JP,C = 125 Hz), 120.9 (d, JP,C = 3.3 Hz), 123.9, 124.2 (d, JP,C = 11.6 Hz), 124.3, 127.7, 128.6 (d, JP,C = 8.6 Hz), 129.3, 131.3, 131.8, 133.1, 133.7, 137.8, 148.0 (d, JP,C = 7.2 Hz), and 148.7 ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H11NO5P: 316.0375, found 316.0384.
2-Hydroxy-9-phenyl-3H-benzo[f][1,2]oxaphosphepine 2-oxide (10a)
By following the general procedure, 10a was prepared from 9-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7c) (200 mg, 0.73 mmol), phenylboronic acid (133 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (166 mg, 84%). Mp: 184–186 °C. IR (KBr, cm−1): 2592 (O = P-OH), 2261 (O = P-OH), 1255 (P = O), and 1204 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 33.69 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.67 (dd, 1H, J = 6.7, 1.0 Hz), 2.72 (dd, 1H, J = 6.7, 1.0 Hz), 5.86–5.99 (m, 1H), 6.62–6.68 (m, 1H), 7.23–7.39 (m, 4H), 7.40–7.46 (m, 2H), and 7.60–7.64 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.8 (d, JP,C = 125 Hz), 123.3 (d, JP,C = 11.6 Hz), 124.4, 127.2, 128.0, 128.5, 129.3 (d, JP,C = 8.6 Hz), 129.6, 130.5 (d, JP,C = 4.6 Hz), 134.3 (d, JP,C = 3.4 Hz), 137.4, and 144.8 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H12O3P: 271.0524, found 271.0527.
2-Hydroxy-9–(4-methoxyphenyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (10b)
By following the general procedure, 10b was prepared from 9-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7c) (200 mg, 0.73 mmol), (4-methoxyphenyl)boronic acid (166 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (196 mg, 89%). Mp: 199–201 °C. IR (KBr, cm−1): 2573 (O = P-OH), 2257 (O = P-OH), 1248 (P = O), and 1205 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 34.24 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.66 (d, 1H, J = 6.4 Hz), 2.71 (d, 1H, J = 6.4 Hz), 3.80 (s, 3H), 5.85–6.00 (m, 1H), 6.61–6.67 (m, 1H), 6.95–7.02 (m, 2H), 7.20–7.34 (m, 3H), and 7.54–7.60 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.6 (d, JP,C = 125 Hz), 55.1, 113.5, 123.3 (d, JP,C = 11.3 Hz), 124.4, 128.4, 129.4 (d, JP,C = 7.8 Hz), 129.7, 130.0, 130.4, 130.8, 134.0, 144.8 (d, JP,C = 7.4 Hz), and 158.6 ppm. HRMS (ESI) [M–H]–: m/z calcd for C16H14O4P: 301.0630, found 301.0636.
Ethyl 4–(2-hydroxy-2-oxido-3H-benzo[f][1,2]oxaphosphepin-9-yl)benzoate (10c)
By following the general procedure, 10c was prepared from 9-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7c) (200 mg, 0.73 mmol), (4-(ethoxycarbonyl)phenyl)boronic acid (212 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (208 mg, 83%). Mp: 194–196 °C. IR (KBr, cm−1): 2566 (O = P-OH), 2272 (O = P-OH), 1710 (C = O), 1285 (P = O), and 1190 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 34.39 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 1.34 (t, 3H, J = 7.0 Hz), 2.66 (d, 1H, J = 6.1 Hz), 2.71 (d, 1H, J = 6.1 Hz), 4.35 (q, 2H, J = 7.0 Hz), 5.88–6.02 (m, 1H), 6.64–6.70 (m, 1H), 7.25–7.42 (m, 3H), 7.72–7.78 (m, 2H), and 7.98–8.03 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 14.2, 27.6 (d, JP,C = 125 Hz), 60.8, 123.6 (d, JP,C = 11.6 Hz), 124.6, 128.6, 128.8, 129.1 (d, JP,C = 8.4 Hz), 130.0, 130.3, 131.2, 133.2 (d, JP,C = 3.4 Hz), 142.2, 144.8 (d, JP,C = 7.4 Hz), and 165.7 ppm. HRMS (ESI) [M–H]–: m/z calcd for C18H16O5P: 343.0735, found 343.0740.
9–(4-Chlorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (10d)
By following the general procedure, 10d was prepared from 9-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7c) (200 mg, 0.73 mmol), (4-chlorophenyl)boronic acid (171 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (161 mg, 72%). Mp: 217–219 °C. IR (KBr, cm−1): 2568 (O = P-OH), 2261 (O = P-OH), 1256 (P = O), 1205 (P = O), and 1190 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 37.90 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.61 (d, 1H, J = 6.2 Hz), 2.66 (d, J = 6.2 Hz), 5.84–5.97 (m, 1H), 6.58–6.65 (m, 1H), 7.21–7.27 (m, 1H), 7.28–7.35 (m, 2H), 7.44–7.50 (m, 2H), and 7.62–7.68 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 28.0 (d, JP,C = 125 Hz), 123.9 (d, JP,C = 10.4 Hz), 124.3, 127.5, 128.0, 128.7, 129.0 (d, JP,C = 8.2 Hz), 130.2, 130.8, 131.5, 133.0 (d, JP,C = 2.4 Hz), 136.0, and 145.1 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H11O3PCl: 305.0134, found 305.0143.
9–(3,5-Dichlorophenyl)-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (10e)
By following the general procedure, 10e was prepared from 9-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7c) (200 mg, 0.73 mmol), (3,5-dichlorophenyl)boronic acid (208 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a white solid (164 mg, 66%). Mp: 210–212 °C. IR (KBr, cm−1): 2542 (O = P-OH), 2228 (O = P-OH), 1259 (P = O), and 1192 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 36.19 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.64 (d, 1H, J = 6.4 Hz), 2.70 (d, 1H, J = 6.4 Hz), 5.90–6.03 (m, 1H), 6.64–6.70 (m, 1H), 7.25–7.31 (m, 1H), 7.34–7.43 (m, 2H), 7.60–7.63 (m, 1H), and 7.66–7.68 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.5 (d, JP,C = 125 Hz), 123.9 (d, JP,C = 12.0 Hz), 124.6, 126.9, 128.4, 129.0 (d, JP,C = 8.6 Hz), 130.4, 131.3, 131.5, 133.7, 140.8, and 144.7 (d, JP,C = 7.4 Hz) ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H10O3PCl2: 338.9745, found 338.9751.
2-Hydroxy-9–(2-nitrophenyl)-3H-benzo[f][1,2]oxaphosphepine 2-oxide (10f)
By following the general procedure, 10f was prepared from 9-bromo-2-hydroxy-3H-benzo[f][1,2]oxaphosphepine 2-oxide (7c) (200 mg, 0.73 mmol), (2-nitrophenyl)boronic acid (182 mg, 1.09 mmol), K2CO3 (201 mg, 1.45 mmol), and Pd(dppf)Cl2 (106 mg, 0.15 mmol) as a yellowish solid (161 mg, 70%). Mp: 226–228 °C. IR (KBr, cm−1): 2359 (O = P-OH), 1229 (P = O), and 1192 (P = O). 31P NMR (162 MHz, DMSO-d6) δ = 35.15 ppm. 1H NMR (400 MHz, DMSO-d6) δ = 2.66 (d, 1H, J = 6.4 Hz), 2.72 (d, 1H, J = 6.4 Hz), 5.90–6.03 (m, 1H), 6.65–6.72 (m, 1H), 7.29–7.35 (m, 1H), 7.38–7.45 (m, 2H), 7.86–7.91 (m, 2H), and 8.25–8.31 (m, 2H) ppm. 13C NMR (101 MHz, DMSO-d6) δ = 27.5 (d, JP,C = 125 Hz), 123.2, 123.9 (d, JP,C = 12.0 Hz), 124.7, 128.8, 129.0 (d, JP,C = 8.6 Hz), 130.4, 131.0, 131.7, 132.3 (d, JP,C = 3.4 Hz), 144.4, 144.8 (d, JP,C = 7.4 Hz), and 146.6 ppm. HRMS (ESI) [M–H]–: m/z calcd for C15H11NO5P: 316.0375, found 316.0385.
Carbonic anhydrase inhibition assay
The CA-catalysed CO2 hydration activity was assayed by using an applied photophysics stopped-flow apparatus as reported in previous papers from our groupCitation22. All CA isoforms were recombinant proteins, obtained as reported earlierCitation23–25.
Results and discussion
Chemistry
Our group recently developed a strategy for the synthesis of simple derivatives of 3H-1,2-benzoxaphosphepine 2-oxide from the commercially available salicylaldehydesCitation19. This methodology employs a ring-closing metathesis (RCM) reaction as a key step to construct benzo-fused oxaphosphepine ring. Following this synthetic routeCitation19, we have prepared iodo- and bromo-substituted analogues 7a–c (Scheme 1). First, halosalicylaldehydes 1a–c were converted to styrenes 2a–c. Subsequent phosphorylation and RCM gave the cyclised compounds 6a–c. Lastly, deprotection was achieved using TMSBr and the target compounds 7a–c were successfully obtained.
Scheme 1. Reagents and conditions: (1) MePPh3Br, tBuOK, THF, rt, 18 h; (2) NEt3, CH2Cl2, 0 °C to rt, 18 h; (3) 5 (CAS: 250220–36-1), PhMe, 70 °C, 4 h; (iv) TMSBr, CH2Cl2, rt, 24 h.
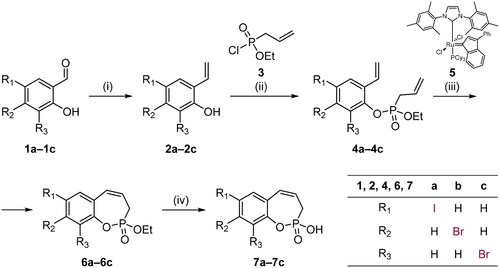
Scheme 2. Reagents and conditions: (1) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O (5:1), 80 °C, 16 h, 54–63%.
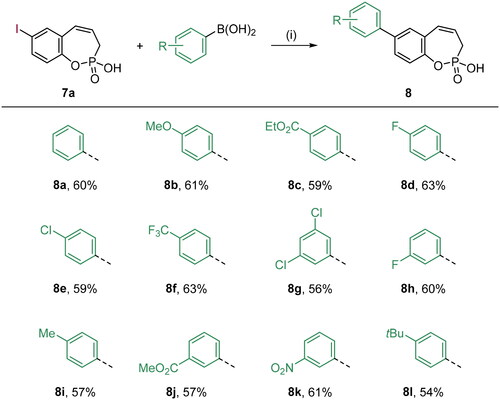
Scheme 3. Reagents and conditions: (1) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O (5:1), 80 °C, 16 h, 57–69%.
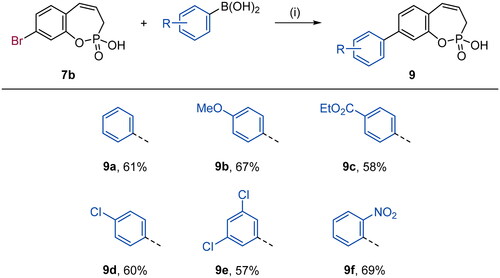
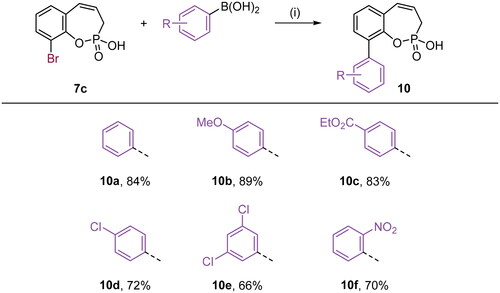
With the halo derivatives 7a–c in hand, we next proceeded to the Suzuki–Miyaura cross-coupling reaction, employing commercial arylboronic acids. As a result, 7-, 8-, and 9-aryl-substituted benzoxaphosphepine 2-oxides 8–10 were furnished in good to excellent yields (Schemes 2–4). Noteworthy, the electronic nature and substitution patterns of arylboronic acids did not significantly affect isolated yields. However, the coupling reactions occurring at position 9 of benzoxaphosphepine 2-oxide core displayed higher efficacy in yields, compared to positions 7 and 8.
Scheme 4. Reagents and conditions: (1) Pd(dppf)Cl2, K2CO3, 1,4-dioxane/H2O (5:1), 80 °C, 16 h, 66–89%.
Carbonic anhydrase inhibition
The newly synthesised compounds 7–10 were investigated for their CA inhibition activity against four pharmacologically relevant human CA isoforms—the ubiquitous cytosolic CA I and II as well as the cancer-associated CA IX and XII. In this study, CA I and CA II are considered off-target isoforms, that were tested in order to explore the selectivity of inhibitors towards the CA IX and CA XII isoforms. The clinically utilised acetazolamide (AAZ) was used as a reference drug. The following structure-activity relationship (SAR) can be deduced from the inhibition data reported in :
Table 1. Inhibition data of compounds 7–10 and the standard inhibitor acetazolamide (AAZ) against human CA isoforms I, II, IX and XII by the stopped-flow CO2 hydrase assay.
 .
.
Equally, as previously reported simple derivatives of benzoxaphosphepine 2-oxideCitation19, all aryl derivatives 8–10 reported here, as well as 7b, showed no inhibitory activity towards the off-target CA isoforms I and II (KI > 100 µM). In the context of cancer treatment with CA inhibitors, this is a desirable feature to prevent possible side effects, since CA I and CA II isoforms are found in many tissues throughout the bodyCitation6. The standard drug AAZ has a very good affinity for CA I and CA II.
The cancer-associated CA IX isoform was inhibited by all of the tested compounds; however, 9-aryl-substituted derivatives 10 were weak inhibitors, with KI values ranging from 16.5 to 55.3 µM. In general, 7- and 8-aryl-substituted compounds 8 and 9 were much more effective inhibitors with good or moderate activity. The 7-aryl derivative with the 3-fluoro substituent 8h emerged as the most potent CA IX inhibitor with KI = 0.63 µM. The inclusion of –CO2R, –NO2 or –Cl substituent typically resulted in decreased inhibitory activity against CA IX.
Similarly, another cancer-associated isoform CA XII was inhibited by 7-aryl and 8-aryl derivatives 8 and 9, whereas 9-aryl-substituted derivatives 10 displayed weak or no inhibitory activity (KI: 25.5–65.3 µM for 10a,b,d–f and KI > 100 µM for 10c that bears the 4-CO2Et substituent). Only the 9-bromo derivative 7c had a moderate inhibition potency against both CA IX and XII. The 7-aryl derivative with the 4-fluoro substituent 8d was the most effective inhibitor against CA XII with KI = 0.25 µM. Other fluorine-containing compounds also exhibited good activity against CA XII (KI = 0.56 µM for 8h; KI = 0.59 µM for 8f). In contrast to CA IX inhibition profile, 7- and 8-aryl derivatives with the nitro group showed good inhibition of CA XII (KI = 0.64 µM for 8k; KI = 0.67 µM for 9f).
Collectedly, benzoxaphosphepine 2-oxide derivatives that are substituted with aryl groups in positions 7 or 8 displayed superior inhibition efficiency of CA IX and XII as compared to the 9-aryl-substituted derivatives. In comparison with the standard drug AAZ, which is a highly effective inhibitor of all the four CA isoforms considered in this study, the analogues 7–10 were less effective as CA IX and XII inhibitors. However, benzoxaphosphepine 2-oxide derivatives showed desirable selectivity as none of them inhibited the off-target CA I and CA II. The most potent inhibitors of both cancer-associated CA IX and CA XII were fluorine-containing compounds 8d and 8h.
It is worthwhile to underline here that a 6-h incubation of the enzyme and each compound 7–10 solutions is essential. When the incubation period of 15 min was used for assaying the inhibition, as generally done for the other types of CA inhibitors, only a weak inhibition was observed (data not shown). Furthermore, it appears to be the case with coumarins and related bioisosteresCitation9–18. These compounds were shown to act as prodrug inhibitors, being hydrolysed within the CA active site to the corresponding acids; subsequently, the obtained hydrolysis products bind within the enzyme active site cavityCitation10,Citation12,Citation14a. By considering aforementioned points, we assume that benzoxaphosphepine 2-oxides are likely to undergo the CA-mediated hydrolysis of oxaphosphepine ring with formation of phosphonic acid derivatives that act as CA inhibitors.
Conclusions
3H-1,2-Benzoxaphosphepine 2-oxides represent a novel chemotype acting as isoform-selective CA inhibitors. In this paper, we have expanded the chemical space around the benzoxaphosphepine scaffold by synthesising aryl derivatives. The latter were evaluated for their inhibitory activity against CA I, II, IX and XII. Most of the compounds tested manifested promising potency in inhibiting the cancer-associated CA isoforms IX and XII. Furthermore, none of the target compounds showed significant inhibition of the cytosolic CA I and II. The SAR studies indicated that 7- and 8-substituted aryl derivatives of 3H-1,2-benzoxaphosphepine 2-oxide were considerately more active CA IX/XII inhibitors than the 9-aryl derivatives. The introduction of aryl groups at the 9th position of the scaffold resulted in decreased potency. Among all the tested compounds, derivatives 8d,h with the fluoro-substituted aryl groups demonstrated the highest inhibitory activity against CA IX/XII, with KI values in the sub-micromolar range. Taking into account the efficiency and significant selectivity of these novel molecules, further development and evaluation will be pursued.
Supplemental Material
Download PDF (728.5 KB)Disclosure statement
No potential competing interest was reported by all authors except CTS. CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Long J, Ji Z, Yuan P, Long T, Liu K, Li J, Cheng L. Nut consumption and risk of cancer: a meta-analysis of prospective studies. Cancer Epidemiol Biomarkers Prev. 2020;29(3):565–573.
- (a) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 2011;10(10):767–777. (b) Supuran CT, Alterio V, Di Fiore A, D' Ambrosio K, Carta F, Monti SM, De Simone G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: three for the price of one. Med Res Rev. 2018;38(6):1799–1836. (c) Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019;38(1-2):65–77. (d) Singh S, Lomelino CL, Mboge MY, Frost SC, McKenna R. Cancer drug development of carbonic anhydrase inhibitors beyond the active site. Molecules. 2018;23:1045. (e) Waheed A, Sly WS, Doisy EA. Carbonic anhydrase XII functions in health and disease. Gene. 2017;623:33–40.
- (a) Robertson N, Potter C, Harris AL. Role of carbonic anhydrase IX in human tumor cell growth, survival, and invasion. Cancer Res. 2004;64(17):6160–6165. (b) Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, Brahimi-Horn MC, Pouysségur J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009;69(1):358–368. (c) Angeli A, Carta F, Nocentini A, Winum J-Y, Zalubovskis R, Akdemir A, Onnis V, Eldehna WM, Capasso C, Simone G. Carbonic anhydrase inhibitors targeting metabolism and tumor microenvironment. Metabolites. 2020;10:412.
- (a) Hewett-Emmett D, Tashian RE. Functional diversity, conservation, and convergence in the evolution of the alpha-, beta-, and gamma-carbonic anhydrase gene families. Mol Phylogenet Evol. 1996;5(1):50–77. (b) Hassan MI, Shajee B, Waheed A, Ahmad F, Sly WS. Structure, function and applications of carbonic anhydrase isozymes. Bioorg Med Chem. 2013;21(6):1570–1582.
- (a) Supuran CT. Structure and function of carbonic anhydrases. Biochem J. 2016;473(14):2023–2032. (b) Supuran CT. Carbonic anhydrases and metabolism. Metabolites. 2018;21:25.
- Kumar S, Rulhania S, Jaswal S, Monga V. Recent advances in the medicinal chemistry of carbonic anhydrase inhibitors. Eur J Med Chem. 2021;209:112923.
- (a)Alterio V, Di Fiore A, D'Ambrosio K, Supuran CT, De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem Rev. 2012;112(8):4421–4468. (b) Aggarwal M, Kondeti B, McKenna R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg Med Chem. 2013;21(6):1526–1533.
- Žalubovskis R. In a search for selective inhibitors of carbonic anhydrases: coumarin and its bioisosteres: synthesis and derivatization. Chem Heterocycl Comp. 2015;51(7):607–612.
- (a) Maresca A, Temperini C, Vu H, Pham NB, Poulsen S-A, Scozzafava A, Quinn RJ, Supuran CT. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc. 2009;131(8):3057–3062. (b) Supuran CT. Coumarin carbonic anhydrase inhibitors from natural sources. J Enzyme Inhib Med Chem. 2020;35(1):1462–1470.
- (a) Bonneau A, Maresca A, Winum J-Y, Supuran CT. Metronidazole-coumarin conjugates and 3-cyano-7-hydroxy-coumarin act as isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2013;28(2):397–401. (b) Sharma A, Tiwari M, Supuran CT. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J Enzyme Inhib Med Chem. 2014;29(2):292–296.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem. 2010;53(1):335–344.
- Onyılmaz M, Koca M, Bonardi A, Degirmenci M, Supuran CT. Isocoumarins: a new class of selective carbonic anhydrase IX and XII inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):743–748.
- (a) Tars K, Vullo D, Kazaks A, Leitans J, Lends A, Grandane A, Zalubovskis R, Scozzafava A, Supuran CT. Sulfocoumarins (1,2-benzoxathiine-2,2-dioxides): a class of potent and isoform-selective inhibitors of tumor-associated carbonic anhydrases. J Med Chem. 2013;56(1):293–300. (b) Krasavin M, Žalubovskis R, Grandāne A, Domračeva I, Zhmurov P, Supuran CT. Sulfocoumarins as dual inhibitors of human carbonic anhydrase isoforms IX/XII and of human thioredoxin reductase. J Enzyme Inhib Med Chem. 2020;35(1):506–510.
- (a) Tanc M, Carta F, Bozdag M, Scozzafava A, Supuran CT. 7-Substituted-sulfocoumarins are isoform-selective, potent carbonic anhydrase II inhibitors. Bioorg Med Chem. 2013;21(15):4502–4510. (b) Nocentini A, Ceruso M, Carta F, Supuran CT. 7-Aryl-triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrase IX and XII. J Enzyme Inhib Med Chem. 2016;31(6):1226–1233.
- (a) Grandane A, Tanc M, Zalubovskis R, Supuran CT. Synthesis of 6-tetrazolyl-substituted sulfocoumarins acting as highly potent and selective inhibitors of the tumor-associated carbonic anhydrase isoforms IX and XII. Bioorg Med Chem. 2014;22(5):1522–1528. (b) Grandane A, Tanc M, Zalubovskis R, Supuran CT. 6-Triazolyl-substituted sulfocoumarins are potent, selective inhibitors of the tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem Lett. 2014;24(5):1256–1260. (c) Grandane A, Tanc M, Žalubovskis R, Supuran CT. Synthesis of 6-aryl-substituted sulfocoumarins and investigation of their carbonic anhydrase inhibitory action. Bioorg Med Chem. 2015;23(7):1430–1436. (d) Grandane A, Tanc M, Di Cesare Mannelli L, Carta F, Ghelardini C, Žalubovskis R, Supuran CT. 6-Substituted sulfocoumarins are selective carbonic anhdydrase IX and XII inhibitors with significant cytotoxicity against colorectal cancer cells. J Med Chem. 2015;58(9):3975–3983. (e) Grandāne A, Nocentini A, Domračeva I, Žalubovskis R, Supuran CT. Development of oxathiino[6,5-b]pyridine 2,2-dioxide derivatives as selective inhibitors of tumor-related carbonic anhydrases IX and XII. Eur J Med Chem. 2020;200:112300.
- Podolski-Renić A, Dinić J, Stanković T, Jovanović M, Ramović A, Pustenko A, Žalubovskis R, Pešić M. Sulfocoumarins, specific carbonic anhydrase IX and XII inhibitors, interact with cancer multidrug resistant phenotype through pH regulation and reverse P-glycoprotein mediated resistance. Eur J Pharm Sci. 2019;138:105012.
- (a) Pustenko A, Stepanovs D, Žalubovskis R, Vullo D, Kazaks A, Leitans J, Tars K, Supuran CT. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2017;32(1):767–775. (b) Pustenko A, Nocentini A, Balašova A, Alafeefy A, Krasavin M, Žalubovskis R, Supuran CT. Aryl derivatives of 3H-1,2-benzoxathiepine 2,2-dioxide as carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):245–254. (c) Pustenko A, Nocentini A, Balašova A, Krasavin M, Žalubovskis R, Supuran CT. 7-Acylamino-3H-1,2-benzoxathiepine 2,2-dioxides as new isoform-selective carbonic anhydrase IX and XII inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):650–656.
- Pustenko A, Balašova A, Nocentini A, Supuran CT, Žalubovskis R. 3H-1,2-Benzoxaphosphepine 2-oxides as selective inhibitors of carbonic anhydrase IX and XII. J Enzyme Inhib Med Chem. 2023;38(1):216–224.
- (a) Rodriguez JB, Gallo-Rodriguez C. The role of the phosphorus atom in drug design. ChemMedChem. 2019;14:190–216. (b)Yu H, Yang H, Shi E, Tang W. Development and clinical application of phosphorus-containing drugs. Med Drug Discov. 2020;8:100063. (c) Ceradini D, Shubin K. New methods for the synthesis of phosphono-δ-lactones (microreview). Chem Heterocycl Comp. 2021;57(12):1167–1169. (d) Balašova A, Žalubovskis R. Synthetic methods toward phosphacoumarins (microreview). Chem Heterocycl Comp. 2022;58(6-7):310–312.
- (a) Bonnac L, Innocenti A, Winum J-Y, Casini A, Montero J-L, Scozzafava A, Barragan V, Supuran CT. Carbonic anhydrase inhibitors: aliphatic N-phosphorylated sulfamates – a novel zinc-anchoring group leading to nanomolar inhibitors. J Enzyme Inhib Med Chem. 2004;19(3):275–278. (b) Winum J-W, Innocenti A, Gagnard V, Montero J-L, Scozzafava A, Vullo D, Supuran CT. Carbonic anhydrase inhibitors. Interaction of isozymes I, II, IV, V, and IX with organic phosphates and phosphonates. Bioorg Med Chem Lett. 2005;15(6):1683–1686. (c) Nocentini A, Alterio V, Bua S, Micheli L, Esposito D, Buonanno M, Bartolucci G, Osman SM, ALOthman ZA, Cirilli R, et al. Phenyl(thio)phosphon(amid)ate benzenesulfonamides as potent and selective inhibitors of human carbonic anhydrases II and VII counteract allodynia in a mouse model of oxaliplatin-induced neuropathy. J Med Chem. 2020;63(10):5185–5200.,. (d) Gülçin İ, Trofimov B, Kaya R, Taslimi P, Sobenina L, Schmidt E, Petrova O, Malysheva S, Gusarova N, Farzaliyev V, et al. Synthesis of nitrogen, phosphorus, selenium and sulfur-containing heterocyclic compounds: determination of their carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibition properties. Bioorg Chem. 2020;103:104171.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- (a) Supuran CT, Ilies MA, Scozzafava A. Carbonic anhydrase inhibitors. Part 29. Interaction of isozymes I, II and IV with benzolamide-like derivatives. Bioorg Med Chem. 1998;33:739–752. (b) Sentürk M, Gülçin I, Daştan A, Küfrevioğlu OI, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem. 2009;17(8):3207–3211.
- Leitans J, Kazaks A, Balode A, Ivanova J, Zalubovskis R, Supuran CT, Tars K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem. 2015;58(22):9004–9009.
- (a) Aspatwar A, Parvathaneni NK, Barker H, Anduran E, Supuran CT, Dubois L, Lambin P, Parkkila S, Winum J-W. Design, synthesis, in vitro inhibition and toxicological evaluation of human carbonic anhydrases I, II and IX inhibitors in 5-nitroimidazole series. J Enzyme Inhib Med Chem. 2020;35(1):109–117. (b) Pustenko A, Nocentini A, Gratteri P, Bonardi A, Vozny I, Žalubovskis R, Supuran CT. The antibiotic furagin and its derivatives are isoform-selective human carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):1011–1020. (c) Ivanova J, Carta F, Vullo D, Leitans J, Kazaks A, Tars K, Žalubovskis R, Supuran CT. N-substituted and ring opened saccharin derivatives selectively inhibit transmembrane, tumor-associated carbonic anhydrases IX and XII. Bioorg Med Chem. 2017;25(13):3583–3589.