
Abstract
In this study, new benzothiazole–pyrimidine hybrids (5a–c, 6, 7a–f, and 8–15) were designed and synthesised. Two different functionalities on the pyrimidine moiety of lead compound 4 were subjected to a variety of chemical changes with the goal of creating various functionalities and cyclisation to further elucidate the target structures. The potency of the new molecules was tested against different tuberculosis (TB) strains. The results indicated that compounds 5c, 5b, 12, and 15 (MIC = 0.24–0.98 µg/mL) are highly active against the first-line drug-sensitive strain of Mycobacterium tuberculosis (ATCC 25177). Thereafter, the anti-tubercular activity was evaluated against the two drug-resistant TB strains; ATCC 35822 and RCMB 2674, where, many compounds exhibited good activity with MIC = 0.98–62.5 3 µg/mL and 3.9–62.5 µg/mL, respectively. Compounds 5c and 15 having the highest anti-tubercular efficiency towards sensitive strain, displayed the best activity for the resistant strains by showing the MIC = 0.98 and 1.95 µg/mL for MDR TB, and showing the MIC = 3.9 and 7.81 µg/mL for XDR TB, consecutively. Finally, molecular docking studies were performed for the two most active compounds 5c and 15 to explore their enzymatic inhibitory activities.
Introduction
Tuberculosis (TB) is a lethal transmissible disease and is one of the most contagious killer diseases worldwideCitation1,Citation2. Nearly, 10 million persons get the active stage of TB each year, as described by the World Health OrganisationCitation3. Isoniazid, ethambutol, rifampicin and streptomycin as first-line drugs have shown effective potency towards Mycobacterium tuberculosisCitation4,Citation5. Nevertheless, the TB strains unsusceptible to first line drugs, mostly the multidrug-resistant TB (MDR TB) is a continuous grave problem. The high incidence of a second threat from TB is observed specially in the immune-compromised patients and is known as extensively drug-resistant TB (XDR TB)Citation6,Citation7. Due to the silent form of Mycobacterium tuberculosis strains, various current frontline therapeutics have become ineffective and more people are prone to be infected with (MDR-TB) strains every yearCitation8. Hence, the development of new drugs able to combat this TB epidemic is crucial. As a result, there is a challenge to discover new effective second-line anti-tubercular drugs in the treatment of MDR and XDR TB.
Decaprenylphosphoryl-β-d-ribose-2′-epimerase (DprE1) is an enzyme that is included in the mycobacterial decaprenylphosphoryl-d-arabinose (DPA) biosynthesis. DprE1 is a necessary component for the growth and survival of the mycobacterial cell, so it is considered to be an effective drug targetCitation9–11. The thymidine monophosphate kinase of Mycobacterium tuberculosis (TMPKmt) is another potential target for TB. TMPK is related to the nucleoside monophosphate kinase (NMPK) family that is pivotal for growth in various organisms, involving mycobacteriaCitation12–14. TMPK enzyme is important in the metabolism of M. tuberculosis and has a crucial and unique role in the formation of DNA essential for M. tuberculosis. Hence, it was expected that the discovery of a new series of drugs having dual enzyme inhibitory activity against both DprE1 and TMPKmt may get over the problem of the acquired mycobacterial resistance and provide a synergistic activity.
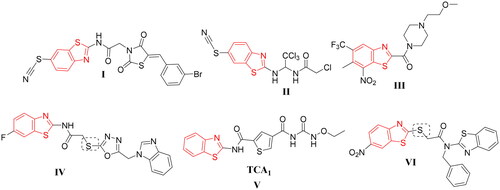
Benzothiazole-based small molecules have a prominent potency towards several microbesCitation15–18. In addition, various analogues with benzothiazole moiety such as compounds I–VI () were effective as remarkable anti-tubercular agentsCitation19–24. Furthermore, Wang et al. reported the benzothiazole derivative V as a selective inhibitor of DprE1. Also, the benzothiazole derivative VI was highly selective against DprE1 ().
Figure 1. Scaffolds of some reported benzothiazole with potent anti-mycobacterial activity (I–VI).
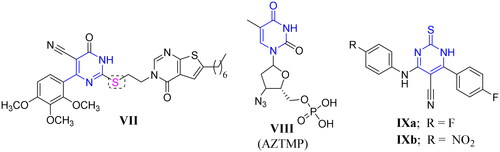
On the other hand, pyrimidines and their related structures were known as building blocks and privileged scaffolds for the discovery of new antibacterial, antiviral, and antifungal candidatesCitation25,Citation26. The related thiouracil molecules were well-reported as promising candidates with promising antimicrobial and antiviral activitiesCitation27,Citation28. Also, it is worth mentioning that alkaloids and alkaloid-like molecules are important scaffolds in therapeutic drugs for the treatment of TBCitation29. Moreover, several pyrimidine-based derivatives demonstrated significant anti-mycobacterial properties towards both M. tuberculosis H37Rv and INH resistant clinical strain as shown in compounds VIIa–b, VIII, and IXa–bCitation13,Citation30,Citation31 (). Interestingly, compound VIII was identified as a competitive inhibitor of TMPKmtCitation12,Citation13,Citation32. Also, compounds IXa–b emerged as good TMPKmt inhibitors that demonstrated an effective anti-mycobacterial activityCitation33 ().
Figure 2. Structures of some reported pyrimidine or thiouracil-based derivatives (VII, VIII, and IXa–b) as potent anti-mycobacterial agents.
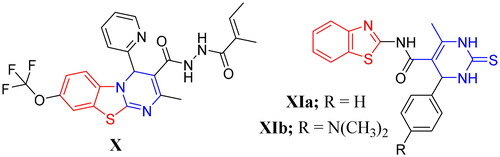
Furthermore, hybridisation or fusion of both benzothiazole and pyrimidine moieties into a single entity resulted in diverse series of small molecules endowed with many pharmacological activities against bacteria, fungi, cancer and inflammation in addition to the mycobacteriaCitation13,Citation33–41. For example, the 4H-pyrimido[2,1-b]benzothiazole X and the hybridised benzothiazolyl pyrimidine derivatives XIa–b showed a remarkable activity against TBCitation13,Citation14 (). Also, the hybrids XIa–b were highly selective against DprE1Citation13,Citation14.
Figure 3. Examples of some benzothiazole derivatives (X and XIa–b) incorporating pyrimidine moiety as potent anti-mycobacterial agents.
Upon consideration of the findings stated above and by using the tethering technique, new series of benzothiazole–pyrimidine conjugates were designed and synthesised. First, the design and synthesis of the lead compound 4 were performed through hybridising the benzothiazole ring with the pyrimidine moiety with thioacetamido linkage ().
Figure 4. Design of the lead compound 4 and the target hybrids.
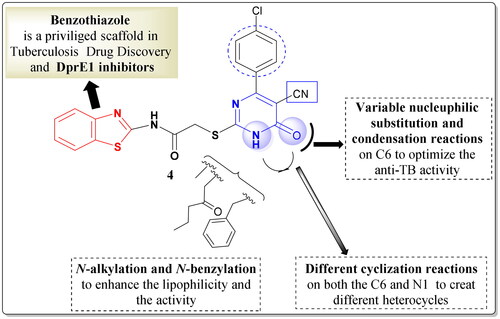
Thereafter, the effect of different substitutions on N-1 and C-6 positions of the pyrimidine motif was explored to help in studying the SAR of these new hybrids as potential anti-tubercular agents and as dual target enzyme inhibitors (). Hence, the N-1 position was substituted with different lipophilic moieties; alkyl, alkylaryl, or ester groups. Moreover, different nucleophilic substitution reactions on C-6 were done in addition to the creation of fused heterocyclic membered rings using both N-1 and C6 positions. The herein described 18 new hybridised benzothiazolyl pyrimidine derivatives (5, 6a–c, 7a–f, and 8–15) were then tested for their anti-TB efficiency towards the first line drugs-sensitive strain (ATCC 25177), in addition to the MDR (ATCC 35822), and XDR (RCMB 2674) M. tuberculosis strains. Subsequently, the dual enzyme inhibitory activity of the most active compounds was explored using the molecular docking studies depending on the ligand–protein interactions.
Results and discussion
Chemistry
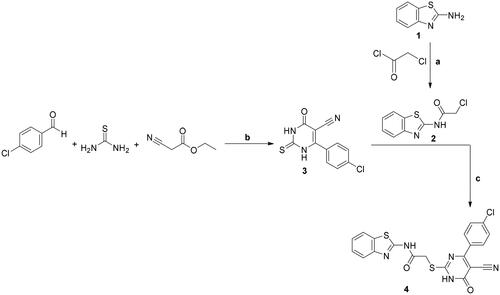
A new series of benzothiazole–pyrimidine hybrids 5, 6a–c, 7a–f, and 8–15 were described in the drawn Schemes 1–4. The elemental analyses with the spectral data confirmed the structures of the new target compounds. The lead compound 4 was synthesised through the reaction between the two key intermediates; the 2-chloroacetamide derivative 2 and the 6-(4-chlorophenyl)-tetrahydropyrimidine-5-carbonitrile derivative 3. The synthesis of compound 2 was performed through the chloroacetylation of the 2-aminobenzothiazole 1 with the chloroacetyl chloride using equimolar quantitiesCitation42. The 6-(4-chlorophenyl)-tetrahydropyrimidine-5-carbonitrile derivative 3 was prepared by using equimolar quantities of ethyl cyanoacetate, 4-chlorobenzaldehyde, and thioureaCitation43–45. Then, the pyrimidine derivative 3 was reacted with the chloroacetamide derivative 2 to give the target compound 4Citation46 (Scheme 1).
Scheme 1. Synthesis of compounds 2–4; conditions and reagents: (a) dry benzene, anhydrous K2CO3, reflux for 12 h; (b) absolute ethyl alcohol, anhydrous K2CO3, reflux for 10–12 h; (c) dry acetone, anhydrous K2CO3, reflux for 8–10 h.
The pyrimidine-bearing benzothiazole hybrid 4 was subjected to a number of reactions on the N-1 and/or the C6 reactive sites of the pyrimidine moiety. The N-alkylated target compounds 5a–c, were synthesised using alkylating agents such as CH3I, ClCH2COOC2H5, and C6H5CH2Cl, respectively, using an anhydrous K2CO3 in DMF. The 4-chloropyrimidine derivative 6 was produced through the reflux of hybrid 4 with excess POCl3 (Scheme 2).
Scheme 2. Synthesis of 5a–c and 6; conditions and reagents: (a) dry DMF, CH3I/C6H5CH2Cl/ClCH2COOC2H5, anhydrous K2CO3, reflux for 12 h; (b) POCl3, reflux for 3 h.
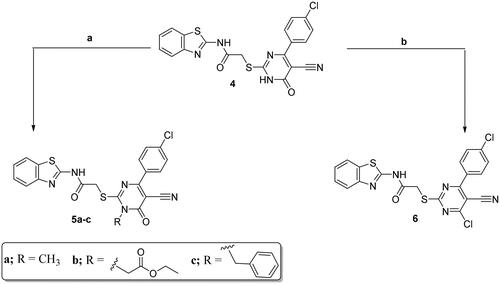
IR and NMR (1H and 13C) spectral data of molecules 5a–c and 6 were matched with the suggested structures. Noticeably, the proton spectra of compounds 5a–c displayed new singlets at δ 3.82, 5.34, and 5.64 ppm due to the N-CH3, N-CH2-COOEt, and N-CH2-C6H5 protons, respectively. In addition, 5b displayed triplet signal at δ 0.86 ppm and a quartet signal at δ 1.45 ppm related to CH3 and OCH2 protons, respectively. Also, the 1H NMR spectrum for 5c showed an increase in the aromatic protons integration.
Furthermore, a new series of benzothiazole derivatives was synthesised by the nucleophilic substitution reactions of the chloro derivative 6 with variable nucleophiles. The reaction between compound 6 and different amine derivatives (primary or secondary) in absolute ethyl alcohol using the triethylamine (TEA) yielded the compounds 7a–f. 1H NMR spectra of 7a–d showed the exchangeable singlets of NH proton of the anilino moiety around δ 9.72 to 12.64 ppm, whereas, the singlet signals for the OCH3 and COOH group protons appeared at δ 3.67 and 11.94 ppm for 7b and 7d, respectively. The proton spectrum of compound 7e displayed two triplet signals at δ = 1.80, 3.70 ppm related to the eight protons of the 2-methyl (CH2) in addition to the 2N-methyl (N-CH2) groups, respectively. The eight protons of CH2-N-CH2 and CH2-S-CH2 of compound 7f were confirmed through the 1H NMR spectrum by showing two broad signals at δ 2.68 and 4.14 ppm, respectively.
Further, reflux of 98% hydrazine hydrate with chloropyrimidine derivative 6 in absolute ethyl alcohol gave the hydrazine-pyrimidine hybrid 8. Confirming of the hybrid 8 was done through its IR spectrum through showing three stretching bands at 3376 and 3296 and 3187 cm–1 attributed to the stretching vibrations of NH and NH2, respectively. In addition, two singlets at δ = 5.04 and 12.47 ppm were observed by its proton spectrum, that were matched with the protons of NH2 and NH, respectively. The replacing of the Cl atom in 6 with mercapto functionality was performed upon using thiourea and reflux in boiling ethanol to give 9. The dissociation of the non-stable isothiouronium salt was considered to be the reason for the formation of compound 9. The IR spectrum of the C═S functionality displayed an absorption band at 1278 cm–1. Proton and carbon NMR spectra were also consistent with its structure.
Also, the chloro derivative 6 was used as a reactive key precursor for ring cyclisation giving imidazopyrimidine 11, quinazolinopyrimidine 12, and tetrazolopyrimidine 13 derivatives. First, non-cyclised glycine bearing derivative 10 is formed by the reaction of the chloropyrimidine derivative 6 with glycine in butanol. Thereafter, the heating of compound 10 with acetic anhydride led to the intramolecular cyclisation giving the corresponding imidazopyrimidine derivative 11.
For compound 10, its IR spectrum displayed the carboxylic C═O vibrating at 1715 cm–1. Also, 10 was confirmed with its proton spectrum that displayed D2O exchangeable singlets at δ 8.38 and 12.69 ppm related to the amino (NH) and hydroxyl (OH) protons of glycine motif. The 1H NMR spectrum confirmed the structure of 11 by displaying a signal that is singlet at δ 4.04 ppm due to protons of CH2 of the imidazole moiety. Also, the proton spectrum lacked any D2O exchangeable protons for the NH and OH protons of glycine motif.
Further, cyclic pyrimidoquinazoline derivative 12 was yielded through the fusion between the chloro derivative 6 and the anthranilic acid in the oil bath. The 1H NMR spectrum of 12 lacked any deuterium oxide exchangeable singlets due to NH and OH protons of anthranilic acid confirming their contribution in the cyclisation process giving the cyclised quinazoline derivative 12. Also, 1H NMR spectrum of 12 displayed increasing in the aromatic protons integration confirming its structure. Regarding the tetrazolo derivative 13, its synthesis was performed through the reflux of the 4-chloro pyrimidine 6 with NaN3 in glacial CH3COOH (Scheme 3).
Scheme 3. Synthesis of compounds 7a–f and 8–13; conditions and reagents: (a) absolute ethyl alcohol, primary or secondary amine, TEA, room temperature for 24 h, then the heating under reflux for 6–12 h; (b) hydrazine hydrate, abs. ethanol, reflux, 6 h; (c) thiourea, abs. ethanol, reflux, 6 h; (d) n-butanol, glycine, reflux for 3 h; (e) reflux for 2 h with acetic anhydride; (f) fusion with the anthranilic acid in the oil bath at 190 °C, for 2 h; (g) glacial acetic acid, sodium azide, reflux for 3 h.
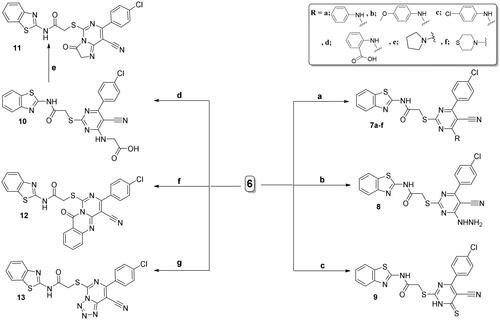
Compound 8 was utilised as another key precursor for further heterocyclisation affording pyrazolo ring fused to the pyrimidine nucleus (derivatives 14 and 15). Upon reacting acetylacetone with hydrazine derivative 8 in refluxing glacial acetic acid, 3,5-dimethyl-N-substituted pyrazole 14 was afforded. Moreover, the hydrazine derivative 8 was heated under reflux with ethylacetoacetate in sodium-ethoxide solution in absolute ethanol to afford the corresponding pyrazolone derivative 15 (Scheme 4).
Scheme 4. Synthesis of 14 and 15; conditions and reagents: (a) glacial acetic acid and acetyl acetone, reflux for 6 h. (b) Reflux with the ethyl acetoacetate in NaOC2H5, for 4 h.
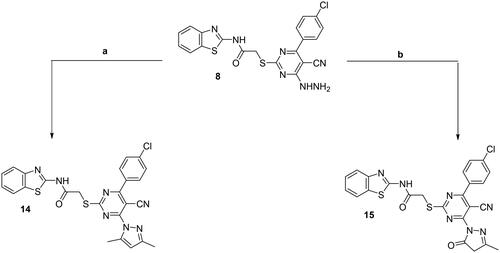
Compounds 14 and 15 were revealed by the IR and 1H NMR spectral data through the disappearance of both the forked band and the exchangeable singlet signal characterising the NH2 of the hydrazine functionality. For compound 14, 1H NMR spectrum showed two singlet signals at δ = 2.52 and 2.64 ppm matched with the two CH2 groups of the pyrazole moiety and showed a signal that was singlet at δ = 7.53 ppm related to the 4-H-pyrazole moiety. While for compound 15, the IR spectrum showed the carbonyl stretching band of pyrazolone ring vibrating at 1696 cm–1. Also, 1H NMR spectrum displayed two bands at δ 2.42 and 4.04 ppm due to methyl (CH3) and methylene (CH2) groups of pyrazolone ring, respectively.
Biological evaluations
Anti-tuberculosis effect
The anti-TB effects of 5a–c, 6, 7a–f, and 8–15 besides the lead compound 4 were tested for the sensitive strain of M. tuberculosis (ATCC 25177) by the assay of Microplate Alamar Blue and using INH as the reference. describes the results and the minimum inhibitory concentration (MIC) was used for their expression. SAR depended on the obtained results and on the comparison of the new compounds with the lead compound 4 in addition to the reference INH.
Table 1. MIC in µg/mL for 4, 5a–c, 6, 7a–f, and 8–15, as well as INH against the sensitive M. tuberculosis (ATCC 25177).
As shown in , the majority of the tested compounds displayed an effective anti-TB activity by having MIC in the range of 0.24–7.81 µg/mL. Compound 5c was the highest active derivative by having MIC of 0.24 µg/mL and showed an equal potency to INH and showed a potency with about 16-fold of the lead compound 4 (MIC = 3.9 µg/mL). In addition, compounds 5b, 12, and 15 revealed high potent activity by having MIC values of 0.98, 0.98, and 0.48 µg/mL, respectively. Whereas a moderate activity was observed for 4, 5a, 6, 7e, 7f, and 8, 9, and 14 by having MIC values in the range from 1.95 to 7.81 µg/mL. Regarding the other derivatives, a slight to fair anti-TB activity was observed by having MIC in the range from 15.63 to 125 µg/mL
The lead benzothiazolyl pyrimidine hybrid 4 showed a moderate effect with MIC of 3.9 µg/mL relative to INH. Introducing lipophilic moieties on N-1 of pyrimidine ring, shown by compounds 5a–c, led to more effective anti-tubercular activity with MIC in the range of 0.24–1.95 µg/mL in comparison to compound 4 having MIC of 3 µg/mL. Generally, the N-benzyl counterpart 5c demonstrated the highest activity by having MIC of 0.24 µg/mL not only among the N-substituted derivatives 5a–c, but also among all the synthesised molecules herein. The N-ethyl acetate bearing analogue 5b was the second active drug of the N-substituted derivatives (MIC = 0.98 µg/mL) compared to 4 and INH. Compound 5a bearing methyl group displayed the lowest activity in this series (MIC = 1.95 µg/mL) compared to 4 and INH. Clearly, it was assumed that the increasing of the lipophilic side chain on N-1 of pyrimidine ring increased the bioavailability and penetration and was mostly advantageous for the anti-tubercular activity. Hence, the activity of the N-substituted derivatives decreased in the order of N-CH2-C6H5 > N-CH2-CO-C2H5 > N-CH3.
The replacement of the carbonyl group with the chloro group in compound 6, increased the anti-tubercular effect (MIC = 1.95 µg/mL) compared to 4 by about twofold. Unexpectedly, the presence of an aniline moiety directly attached to the pyrimidine ring as in compounds 7a–d diminished the activity (MIC = 15.63–125 µg/mL) relative to compound 4 and the reference INH. Concerning the impact of substitution of the aniline moiety within hybrids 7b–d, the anti-tubercular activity decreased by 2–8 folds (MIC = 125, 125, and 31.25 µg/mL, respectively) compared to the unsubstituted analogue 7a (MIC = 15.63 µg/mL). It was noticed that the para substitution of the anilino motif with a methoxy (analogue 7b) or chlorine atom (analogue 7c) had the same impact (MIC = 125 µg/mL) and resulted in a diminished activity. However, the slightly better activity of 7d (MIC = 31.25 µg/mL) having the carboxylic acid functionality at the ortho site of the aniline ring, indicated that the ortho substitution could be more favourable than the para substitution. Overall, the steric factors of aniline derivatives 7a–d may be the reason for the visible decrease in the potency and hence the unsubstituted derivative 7a was more favourable than the substituted ones 7b–d. Conversely, the introduction of alicyclic amino groups (pyrrolidine and thiomorpholine) in compounds 7e and 7f manifested a better activity (MIC = 7.81 and 7.81 µg/mL, respectively) than anilino derivatives 7a–d (MIC = 125, 125, and 15.63 µg/mL, respectively). However, tethering the pyrimidine motif with pyrrolidine 7e and thiomorpholine 7f did not show an increase in the anti-tubercular activity compared to compound 4 and INH. Furthermore, the present study revealed that the presence of the hydrazino group in compound 8 or thione group in compound 9 at C-6 of the pyrimidine ring had not affected the anti-tubercular activity by having MIC of 3.9 µg/mL that was similar to 4.
It is worth to mention that neither the open aminoacetic acid derivative 10 nor its cyclised imidazolo derivative 11 showed an increase in the activity (MIC = 15.63 and 31.25 µg/mL, respectively) compared to the lead compound 4 and INH. Noticeably, the open aminoacetic acid derivative 10 has twice the activity of its cyclised imidazole derivative 11. Comparably, the fusion of the quinazolino moiety to the pyrimidine ring in 12 showed a remarkable increase in the activity (MIC = 0.98 µg/mL) by about fourfold compared to compound 4. Unlike the cyclised imidazolo derivative 11 and its open counterpart 10, the pyrimidoquinazoline derivative 12 (MIC = 0.98 µg/mL) showed more than 32-fold increase in the activity in comparison to its open anthranilic acid analogue 7d (MIC = 31.25 µg/mL). Fusion of pyrimidine ring with tetrazole moiety in compound 13 diminished the activity (MIC = 62.5 µg/mL) compared to compound 4 and INH. From the obtained results, it could be concluded that the fusion of pyrimidine ring with five membered heterocyclic rings as in compounds 11 and 13 is less favourable than that with higher ones as in compound 12. Both pyrazole 14 and pyrazolone 15 derivatives showed higher activity (MIC = 7.81 and 0.48 µg/mL, respectively) than their hydrazino counterpart 8, highlighting that the heterocyclisation of the hydrazine derivative 8 is favourable. Interestingly, the pyrazolone 15 was the second most active newly synthesised compound after 5c and showed half the activity (MIC = 0.48 µg/mL) of INH.
Finally, different factors influenced the activity of the newly synthesised molecules against different strains of TB described as N-substitution, nucleophilic substitution, or heterocyclisation at position 6 of the pyrimidine ring as well as ring cyclisation at N-1 and C-6. It was found that raising the lipophilic character for the pyrimidine moiety through N-1 alkyl substitution was more favourable for activity. Collectively, the N-benzyl derivative 5c showed the best activity that may be attributed to its high lipophilicity, whereas the pyrazolone derivative 15 was the second most active derivative. On the other hand, the anilino derivatives 7b and 7c were the least active ones followed by the tetrazole derivative 13.
Anti-tuberculosis effect against multidrug resistant M. tuberculosis (ATCC 35822)
In this work, the anti-tubercular activity for the most potent members in the previous assay (4, 5a–c, 6, 7e, 7f, 8, 9, 12, 14, and 15) was further assessed against MDR strain using MABA protocol. The tested MDR strain (ATCC 35822) demonstrates a resistance towards isoniazid, cycloserine, kanamycin, and rifampin.
Interpreting the obtained results for the target molecules demonstrated that most of the evaluated derivatives displayed good effect by having MIC values of 0.98–62.5 µg/mL (). Hybrids 5c and 15 with the most anti-TB efficiency for the sensitive M. tuberculosis (ATCC 27294), exhibited potent effect against the MDR strain (ATCC 35822) by having MIC values of 0.98 and 1.95 µg/mL, respectively. They also showed more than 128- and 64-fold rising in the potency in comparison with the lead hybrid 4 having MIC value equal to 125 µg/mL. Furthermore, hybrids 5a, 5b, and 12 exerted an excellent action by having MIC of 7.81 µg/mL, whereas derivatives 6 produced a good action by having MIC of 31.25 µg/mL. Additionally, compounds 7e, 8, and 9 showed a moderate action by having MIC of 62.5 µg/mL.
Table 2. MIC in µg/mL of 4, 5a–c, 6, 7e, 7f, 8, 9, 12, 14, and 15 against the MDR TB (ATCC 35822) and XDR TB (RCMB 2674) strains.
Anti-tuberculosis activity against extensively drug-resistant M. tuberculosis (RCMB 2674)
Furthermore, the anti-TB effect of 4, 5a–c, 6, 7e, 7f, 8, 9, 12, 14, and 15 against XDR strain is shown in . It was observed that the results of tested molecules showed a visible correlation for MDR and XDR strains. Many of the tested derivatives were observed to be of a good effect by having MIC values in the range of 3.9–62.5 µg/mL. Hybrids 5c and 15 having the most anti-TB efficiency against sensitive and MDR strains, showed also an excellent activity against XDR by having MIC values of 3.9 and 7.81 µg/mL, respectively, and by showing more than 16–32-fold enhancing in the efficiency in comparison with the lead hybrid 4. Moreover, compounds 5a, 5b, and 12 showed a moderate activity by having MIC of 31.25 µg/mL. In contrast, the derivative 6 produced a weak activity by having MIC of 62.5 µg/mL.
Molecular docking
In structural molecular biology and computer-assisted drug design, molecular docking is a critical tool. The goal of ligand–protein docking is to predict the most likely binding mode(s) of a ligand with a known three-dimensional structure of a protein. Successful docking methods effectively search high-dimensional spaces and employ a scoring function that correctly ranks candidate dockingsCitation47. In the present work, two of the most important biological targets in Mycobacterium tuberculosis were selected to perform the molecular docking study; decaprenylphosphoryl-d-ribose oxidase (DprE1) (pdb ID: 4KW5)Citation48 and TMPKmt (pdb ID: 1W2H)Citation49. Two synthesised derivatives were chosen to test their binding affinity with the two target enzymes (5c and 15). The two tested compounds showed good binding scores to both enzymes compared to the score of the co-crystallised ligands ().
Table 3. Binding energy results in kcal/mol for the tested hybrids versus the co-crystallised ligands.
For DprE1 binding site, 5c derivative showed good binding interactions through the main binding site amino acid residues. Several hydrogen bonding interactions were formed between the main functional groups in the tested 5c compound, nitrile N with Cys387, carbonyl oxygens with Cys387 and Pro316 and pyrimidone N with Gly117 with additional halogen bonding of Cl with His132 (). On the other hand, compound 15 showed also good binding interactions through hydrogen bonding of nitrile N with His132 and Tyr415, carbonyl oxygens with Gly117, pyrazolone with Lys418 and S with Tyr60 ().
Figure 5. 2D and 3D interactions of compound 5c with the binding position of DprE1 enzyme.
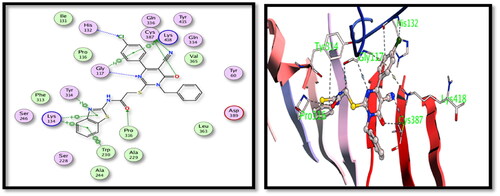
Figure 6. 2D and 3D interactions of compound 15 with the binding position of DprE1 enzyme.
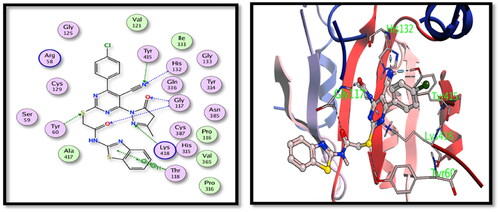
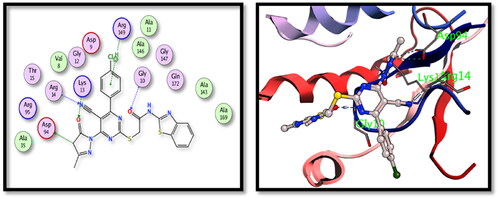
The two tested derivatives showed also a good binding affinity to TMPKmt enzyme. Both derivatives showed hydrogen bond interactions with the key amino acids Gly10 and Lys13. Compound 5c binds to Gly10 (nitrile N), Lys13 and Arg95 (carbonyl oxygens) () while compound 15 showed interactions with Arg14 (nitrile N), Gly10 and Lys13 (carbonyl oxygens) ().
Figure 7. The 2D and 3D interactions of compound 5c with the binding position of TMPKmt enzyme.
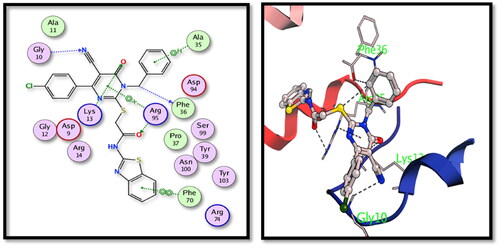
Figure 8. The 2D and 3D interactions for the compound 15 with the binding position of TMPKmt enzyme.
Conclusions
In the present research, variable benzothiazolyl pyrimidine hybrids 4, 5a–c, 6, 7a–f, and 8–15 were synthesised. The activity against the different TB strains namely sensitive M. tuberculosis (ATCC 25177), MDR strain of M. tuberculosis (ATCC 35822) and the XDR strain of M. tuberculosis (RCMB2674) were tested for the 18 newly synthesised derivatives in addition to the lead compound 4. Compounds 5c and 15 showed the best anti-TB effect in this work by having MIC = 0.24 and 0.48 µg/mL, showing minimal resistance against MDR strain by having MIC = 0.98 and 1.95 µg/mL and against XDR strain by having MIC = 3.9 and 7.81 µg/mL, respectively. The SAR analysis highlighted that enhancing the lipophilic nature regarding the pyrimidine ring through N-1 alkyl substitution was more favourable for activity, whereas introducing the amino moieties in compounds 7a–d through the nucleophilic substitution, produced a diminished anti-mycobacterial activity. Finally, the two most active compounds 5c and 15 showed good binding scores to DprE1 and TMPKmt enzymes compared to the score of the co-crystallised ligands.
Experimental work
Chemistry
General methods regarding chemistry
No additional purification was done for the used commercial solvents and chemical agents that were reagent-grade. The apparatus of IA 9100 MK digital melting point griffin was used to measure the total melting points and they were not corrected and the open capillary tubes were used for measuring. At the Micro Analytical Center, Science College, in Cairo, Egypt, the IR spectral data were done on Bruker Vector 22 FTIR Spectrometer. At Pharmacy College, at Beni-Suef University, in Egypt, the proton and carbon spectral data were made on a Bruker Avance III at 400 MHz and at 100 MHz for NMR (1H and 13C), respectively. At Mycology and Biotechnology Regional Center, at the University of Al Azhar, in Cairo, Egypt, inlet-part to single quadrupole mass analyzer was used to record the mass spectra. Compounds 2–4 were synthesised based on the reported proceduresCitation42–46. Compound 1 can be obtained commercially.
General method for synthesis of N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-1-substituted-6-oxo-1,6-dihydropyrimidin-2yl)thio) acetamide derivatives (5a–c)
A mixture of 5 mmol of 4 (2.26 g) and 73 mmol of anhydrous K2CO3 (1 g) in 20 mL of dry DMF was mixed with 20 mmol of the suitable alkyl halide. The mixture underwent 12 h reflux followed by cooling and pouring on cold H2O. The precipitate was collected through filtration, left to dry and then recrystallised using CHCl3/C2H5-OH mixture.
N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-1-methyl-6-oxo-1,6 dihydro pyrimidin-2-yl)thio)acetamide (5a)
Yield 73%; melting point: 167–170 °C. IR (ʋmax/cm–1): 3424 (stretching NH), 3197 (aromatic CH), 2921 (aliphatic CH), 2212 (C≡N), 1694 (C═O), 1579 (C═N), 1530 (C═C). 1H NMR (DMSO-d6) δ ppm: 3.85 (s, 3H, N-CH3), 4.13 (s, 2H, CH2S), 7.35 (t, 1H, C5-H, J = 7.2 Hz), 7.47 (d, 2H, C2′-H and C6′-H, J = 7.2 Hz), 7.52 (t, 1H, C6-H, J = 8.00 Hz), 7.64 (d, 1H, C4-H, J = 8.4 Hz), 7.78 (d, 2H, C3′-H and C5′-H, J = 8 Hz), 7.89 (d, 1 H, C7-H, J = 4 Hz). 13C NMR (DMSO-d6) δ ppm: 22.45, 32.82, 79.21, 113.74, 118.16, 122.15, 123.28, 123.58, 124.64, 126.08, 128.49, 130.47, 132.65, 135.22, 140.62, 152.75, 158.62, 159.56, and 172.87. MS m/z (%): 467 (M+, 20.97), 469 (M++2, 13.95). Anal. Calcd. for C21H14ClN5O2S2: C 53.90, H 3.02, N 14.9; found: C 53.91, H 3.06, N 14.91.
Ethyl-2-(2-((2-(benzo[d]thiazol-2-ylamino)-2-oxoethyl)thio)-4-(4-chlorophenyl)-5-cyano-6-oxopyrimidin-1(6H)-yl)acetate (5b)
Yield 68%; melting point: 178–180 °C. IR (ʋmax/cm–1): 3392 (stretching NH), 3061 (aromatic CH), 2922 (aliphatic CH), 2206 (C≡N), 1702, 1688 (C═O), 1533 (C═N), 1459 (C═C). 1H NMR (DMSO-d6) δ ppm: 1.17 (t, 3 H, CH3, ethyl, J = 8.4 Hz), 4.11 (q, 2 H, CH2, ethyl, J = 6.8 Hz), 4.31 (s, 2 H, CH2S), 5.20 (s, 2 H, N-CH2), 7.38 (t, 1 H, C5-H, J = 6.4 Hz), 7.51 (d, 2 H, C2′-H and C6′-H, J = 6.2 Hz), 7.67 (d, 2 H, C2′-H and C6′-H, J = 9.6 Hz), 7.80 (d, 1H, C4-H, J = 8.4 Hz), 7.84 (t, 1 H, C6-H, J = 7.4 Hz), 7.94 (d, 1 H, C7-H, J = 7.6 Hz). 13C NMR (DMSO-d6) δ ppm: 23.08, 34.16, 44.38, 63.05, 96.20, 112.70, 114.32, 118.58, 122.33, 126.61, 128.18, 130.92, 131.51, 133.76, 134.18, 140.51, 148.27, 159.04, 162.36, 168.27, 169.93, and 173.46. MS m/z (%): 539 (M+, 10.98), 540 (M++1, 2.66), 541 (M++2, 3.07). Anal. Calcd. for C24H18ClN5O4S2: C 53.38, H 3.36, N 12.97; found: C 53.36, H 3.40, N 13.02.
N-(benzo[d]thiazol-2-yl)-2-((1-benzyl-4-(4-chlorophenyl)-5-cyano-6-oxo-1,6-dihydro pyrimidin-2-yl)thio)acetamide (5c)
Yield 73%; melting point: 201–203 °C. IR (ʋmax/cm–1): 3363 (stretching NHs); 2977 (aromatic CH), 2917 (aliphatic CH), 2213 (C≡N), 1692 (C═O), 1590 (C═N), 1547 (C═C). 1H NMR (DMSO-d6) δ ppm: 4.26 (s, 2H, CH2S), 5.14 (s, 2H, CH2-phenyl), 7.32 (t, 1H, C5-H, J = 7.2 Hz), 7.43 (t, 3H, C3″-H, C5″-H and C6-H, J = 6.8 Hz), 7.46 (t, 1H, C4″-H, J = 5.6 Hz), 7.76 (d, 2H, C2″-H and C6″-H, J = 6.2 Hz), 7.79 (d, 2H, C2′-H, C6′-H, J = 8.2 Hz), 7.83 (d, 3H, C3′-H, C5′-H, and C4-H, J = 8.6 Hz), 7.96 (d, 1H, C7-H, J = 8.00 Hz), 12.44 (s, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.53, 56.50, 89.51, 114.90, 120.71, 121.83, 121.95, 126.91, 128.02, 129.07, 129.31, 130.21, 132.08, 132.31, 133.56, 135.07, 151.72, 157.30, 163.03, 166.81, 169.17, 169.63, and 173.83. MS m/z (%): 543 (M+, 34.58), 545 (M++2, 19.79). Anal. Calcd. for C27H18ClN5O2S2: C 59.61, H 3.33, N 12.87; found: C 59.63, H 3.35, N 12.80.
Synthesis of N-(benzo[d]thiazol-2-yl)-2-((4-chloro-6–(4-chlorophenyl)-5-cyanopyrimidin-2-yl)thio)acetamide (6)
A 10 mmol of compound 4 (4.51 g) was dissolved in 30 mL of phosphorus oxychloride and then underwent 3 h reflux. Then, leaving the reaction mixture to cool was done, and was followed by pouring onto cold water with continuous agitation. The formed precipitate was subjected to filtration, and recrystallisation using aqueous ethyl alcohol.
Yield 77%; melting point: 185–187 °C. IR (ʋmax/cm–1): 3435 (stretching NH), 3199 (aromatic CH), 2918 (aliphatic CH), 2217 (C≡N), 1696 (C═O), 1580 (C═N), 1536 (C═C). 1H NMR (DMSO-d6) δ ppm: 4.08 (s, 2H, CH2S), 7.26 (d, 2H, C2′-H and C6′-H, J = 4.0 Hz), 7.32 (t, 1H, C5-H, J = 7.0 Hz), 7.46 (t, 1H, C6-H, J = 7.8 Hz), 7.71 (d, 2H, C3′-H and C5′-H, J = 6.8 Hz), 7.70 (d, 1H, C4-H, J = 8.0 Hz), 7.97 (d, 1H, C7-H, J = 7.6 Hz), 12.38 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.11, 93.76, 116.15, 120.67, 121.79, 128.04, 128.70, 129.09, 132.01, 132.05, 133.62, 135.34, 146.89, 157.39, 161.34, 165.80, 167.09, and 167.55. MS m/z (%): 470 (M+, 60.60), 472 (M++2, 20.43). Anal. Calcd. for C20H11Cl2N5OS2: C 50.85, H 2.35, N 14.83; found: C 50.87, H 2.40, N 14.91.
General method for synthesis of N-(benzo[d]thiazol-2-yl)-2-(4-substituted-5-cyano-6-(phenylamino)pyrimidin-2-yl)thio)acetamide (7a–f)
Few drops of TEA were added to a mixture of 10 mmol of 6 (4.70 g) and 10 mmol of primary or secondary amino derivative in 30 mL absolute ethyl alcohol and then was allowed to stir without heating for 24 h. Further, the reaction mixture underwent 6–12 h reflux. The mixture was leaved to cool and was then concentrated using reduced pressure. The residue was then treated with a plenty of water and the separated precipitate was obtained by filtration. The collected solid was subjected to washing with water, drying, and recrystallisation from methyl alcohol.
N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-6-(phenylamino)pyrimidin-2-yl)thio)acetamide (7a)
Yield 81%; melting point: 222–224 °C. IR (ʋmax/cm–1): 3423 (2 stretching NHs); 3161 (aromatic CH), 2964 (aliphatic CH), 2227 (C≡N), 1672 (C═O), 1604 (C═N), 1552 (C═C). 1H NMR (DMSO-d6) δ ppm: 4.22 (s, 2H, CH2S), 7.27 (d, 2H, C3′-H and C5′-H, J = 8.2 Hz), 7.42 (t 1H, C5-H, J = 7.6 Hz), 7.53 (t, 1H, C4″-H, J = 7.2 Hz) 7.63 (d, 2H, C2′-H and C6′-H, J = 9.4 Hz), 7.70 (d, 3H, C4-H, C2″-H and C6″-H, J = 8.6), 7.82 (t, 3H, C6-H, C3″-H and C5″-H, J = 8.4 Hz), 7.97 (d, 1H, C7-H, J = 6.8 Hz), 10.15 (s, 1H, NH, D2O exchangeable), 12.37 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.66, 85.09, 113.96, 116.25, 120.75, 122.07, 123.67, 125.77, 126.44, 128.53, 128.96, 130.98, 132.08, 135.03, 136.41, 149.28, 157.07, 159.53, 160.46, 167.14, 168.18, and 173.25. Mass spectrum m/z (%): 528 (M+, 5.72), 529 (M++1, 1.22), 530 (M++2, 2.28). Anal. Calcd. for C26H17ClN6OS2: C 59.03, H 3.24, N 15.89; found: C 59.02, H 3.20, N 15.94.
N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-6-((4-methoxyphenyl) amino)pyrimidin-2-yl)thio)acetamide (7b)
Yield 75%; melting point: 236–238 °C. IR (ʋmax/cm–1): 3424, 3321 (2 stretching NHs), 3040 (aromatic CH), 2919 (aliphatic CH), 2213 (C≡N), 1692 (C═O), 1607 (C═N), 1549 (C═C). 1H NMR (DMSO-d6) δ ppm: 3.67 (s, 3H, OCH3), 4.16 (s, 2H, CH2S), 6.81 (d, 2H, C3″-H and C5″-H, J = 8.0 Hz), 7.29 (d, 2H, C3′-H and C5′-H, J = 8.4 Hz), 7.38 (t, 1H, C5-H, J = 6.6 Hz), 7.41 (d, 2H, C2″-H, C6″-H J = 6.8 Hz), 7.49 (t, 1H, C6-H, J = 7.4 Hz), 7.66 (d, 2H, C2′-H and C6′-H, J = 8.4 Hz), 7.77 (d, 1H, C4-H, J = 8.4 Hz), 7.81 (d, 1H, C7-H, J = 7.6 Hz), 9.72 (s, 1H, NH, D2O exchangeable), 12.53 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.66, 55.47, 85.09, 113.96, 116.25, 120.75, 122.07, 123.67, 125.77, 126.44, 128.53, 128.96, 130.63, 130.98, 132.08, 135.03, 136.41, 149.28, 157.07, 160.46, 167.14, 168.18, and 173.25. MS m/z (%): 558 (M+, 21.57), 559 (M++1, 5.20), 560 (M++2, 8.93). Anal. Calcd. for C27H19N6O2S2: C 62.44, H 4.12, N 15.60; found: C 62.45, H 4.16, N 15.66.
N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-6-((4-chlorophenyl)amino)-5-cyano pyrimidin-2-yl)thio)acetamide (7c)
Yield 81%; melting point: 212–214 °C. IR (cm–1): 3435, 3256 (2 stretching NHs, stretching), 3172 (aromatic CH), 2913 (aliphatic CH), 2212 (C≡N), 1682 (C═O), 1644 (C═N), 1535 (C═C), 756 (C-Cl).1H NMR (DMSO-d6) δ ppm: 4.21 (s, 2H, CH2S), 7.29 (t 1H, C5-H, J = 10.4 Hz), 7.46 (t 1H, C6-H, J = 10.6 Hz), 7.55 (d, 4H, C3″-H and C5″, C3′-H and C5′-H, J = 7.2 Hz), 7.81 (d, 4H, C2″-H and C6″-H, C2′-H and C6′-H J = 9.2 Hz), 7.85 (d, 1H, C4-H, J = 7.6 Hz), 7.99 (d, 1H, C7-H, J = 6.4 Hz), 9.91 (s, 1H, NH, D2O exchangeable), 12.54 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.28, 85.97, 116.03, 121.08, 122.18, 124.06, 125.56, 126.64, 128.75, 128.98, 129.39, 131.01, 131.90, 134.81, 136.58, 136.84, 149.07, 158.27, 160.42, 167.34, 167.59, and 173.13. MS m/z (%): 562 (M+, 7.40), 563 (M++1, 2.84), 564 (M++2, 1.76). Anal. Calcd. for C26H16Cl2N6OS2: C 55.42, H 2.86, N 14.91; found: C 55.44, H 2.89, N 14.98.
2-((2-((2-(Benzo[d]thiazol-2-ylamino)-2-oxoethyl)thio)-6–(4-chlorophenyl)-5-cyano pyrimidin-4-yl)amino)benzoic acid (7d)
Yield 76%; melting point: 281–282 °C. IR (ʋmax/cm–1): 3415, 3224 (2 stretching NHs), 3160 (aromatic CH), 2955, 2836 (aliphatic CH), 2227 (C≡N), 1721, 1673 (2C═O), 1590 (C═N), 1569 (C═C). 1H NMR (DMSO-d6) δ ppm: 4.33 (s, 2H, CH2S), 7.18 (t, 1H, C4″-H, J = 7.2 Hz), 7.33 (t, 1H, C5-H, J = 7.6 Hz), 7.45 (t, 1H, C6-H, J = 8.2 Hz), 7.61 (t, 1H, C5″-H, J = 8.4 Hz), 7.82 (d, 3H, C4-H, C3′-H and C5′-H, J = 7.2 Hz), 7.89 (d, 3H, C3″-H, J = 8.4 Hz), 7.98 (d, 2H, C2′-H and C6′-H, J = 7.6 Hz), 8.02 (d, 1H, C7-H, J = 8.0 Hz), 8.63 (d, 1H, C6″-H, J = 8.4 Hz), 11.94 (s, 1H, COOH, D2O exchangeable), 12.64 (br, 2H, NH and NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.10, 93.79, 114.96, 116.15, 120.66, 121.76, 128.02, 128.69, 128.97, 129.09, 129.21, 132.00, 133.61, 146.87, 157.40, 157.47, 161.34, 165.77, 167.07, 167.55, 167.63, 168.48, 169.93, 172.55, and 173.88. MS m/z (%): 572 (M+, 94.03), 573 (M++1, 15.72), 574 (M++2, 34.99). Anal. Calcd. for C27H17ClN6O3S2: C 56.59, H 2.99, N 14.67; found: C 56.57, H 3.03, N 14.75.
N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-6-(pyrrolidin-1-yl) pyrimidin-2-yl)thio)acetamide (7e)
Yield 77%; melting point: 234–236 °C. IR (ʋmax/cm–1): 3391 (stretching NH), 3060 (aromatic CH), 2968, 2925 (aliphatic CH), 2208 (C≡N), 1682 (C═O), 1545 (C═N), 1463 (C═C). 1H NMR (DMSO-d6) δ ppm:1.84 (br, 4H, CH2-N-CH2), 3.72 (br, 4H, CH2CH2), 4.23 (s, 2H, CH2S), 7.32 (t, 1H, C5-H, J = 7.2 Hz), 7.48 (d, 3H, C4-H, 3′-H and C5′-H, J = 7.6 Hz), 7.75 (t, 1H, C6-H, J = 8 Hz), 7.88 (d, 2H, C2′-H and C6′-H, J = 5.2 Hz), 7.97 (d, 1H, C7-H, J = 7.6 Hz), 12.60 (br, 1H, NH, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 22.01, 35.66, 49.05, 83.63, 118.45, 121.20, 122.18, 124.12, 126.69, 128.76, 130.22, 131.53, 135.37, 136.34, 148.90, 158.41, 168.12, 168.91, 171.96, and 172.94. MS m/z (%): 506 (M+, 12.18), 507 (M++1, 1.65), 508 (M++2, 3.65). Anal. Calcd. for C24H19ClN6OS2: C 56.85, H 3.78, N 16.57; found: C 56.87, H 3.81, N 16.65.
N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-6-thiomorpholino pyrimidin-2-yl) thio)acetamide (7f)
The yield was of 84%; melting point: 187–189 °C. IR (ʋmax/cm–1): 3222 (stretching NH), 3155, 3050 (aromatic CH), 2925, 2860 (aliphatic CH), 2227 (C≡N), 1695 (C═O), 1610 (C═N), 1537 (C═C). 1H NMR (DMSO-d6) δ ppm: 2.68 (br, 4H, CH2-N-CH2 thiomorpholine), 4.14 (br, 4H, CH2-S-CH2, thiomorpholine), 4.27 (s, 2H, CH2S), 7.34 (br, 1H, C5-H), 7.47 (br, 1H, C6-H), 7.82 (br, 3H, C4-H, C3′-H and C5′-H), 7.99 (br, 3H, C7-H, C2′-H and C6′-H), 12.55 (br, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 38.25, 49.07, 61.87, 92.98, 112.69, 116.59, 123.46, 125.40, 128.81, 129.12, 132.05, 134.49, 134.54, 135.68, 166.66, 167.27, 167.30, 167.47, and 176.73. MS m/z (%): 538 (M+, 13.28), 539 (M++1, 1.18), 539 (M++2, 2.98). Anal. Calcd. for C24H19ClN6OS3: C 53.47, H 3.55, N 15.59; found: C 53.46, H 3.51, N 15.65.
Synthesis of 2-((5-cyano-4-hydrazinyl-6-phenylpyrimidin-2-yl) thio)-N-(6-methylbenzo[d]thiazol-2-yl)acetamide (8)
Equimolar quantities (10 mmol) of hydrazine hydrate (98%) (0.5 g) and 6 (4.70 g) were dissolved in 30 mL of absolute ethanol. After 6 h reflux, the separated material was then cooled by filtration and watery washing. The obtained solid was air dried and underwent recrystallisation using acetic acid and methyl alcohol. Yield 71%; melting point: 235–237 °C. IR (ʋmax/cm–1): 3460 (2 stretching NH), 3291, 3206 (NH2), 3056 (aromatic CH), 2919 (aliphatic CH), 2215 (C≡N), 1703 (C═O), 1585, (NH2 bend, C═N), 1461 (C═C). 1H NMR (DMSO-d6) δ ppm: 4.29 (s, 2H, CH2S), 5.04 (s, 2H, NH2, D2O exchangeable), 7.27 (d, 2H, C3′-H and C5′-H, J = 8.4 Hz), 7.41 (t, 1H, C5-H, J = 7.6 Hz), 7.51 (t, 1H, C6-H, J = 7.2 Hz), 7.66 (d, 3H, C4-H, C2′-H and C6′-H, J = 8.4 Hz), 7.75 (d, 1H, 4-H, J = 7.6 Hz), 7.80 (d, 1H, C7-H, J = 7.6 Hz), 12.47 (s, 2H, NH and NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.20, 100.40, 120.65, 121.76, 128.01, 129.11, 129.43, 131.02, 132.04, 133.61, 136.70, 146.95, 148.27, 155.75, 157.56, 161.73, 166.98, and 168.26. MS m/z (%): 467 (M+, 23.37), 469 (M++2, 9.62), Anal. Calcd. for C20H14ClN7OS2: C 51.33, H 3.02, N 20.95; found: C 51.35, H 3.06, N 21.01.
Synthesis of N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-6-thioxo-1,6-dihydropyrimidin-2-yl)thio)acetamide (9)
The reflux of equimolar quantities (10 mmol) of 6 (4.70 g) as well as thiourea (0.76 g) was done in 20 mL absolute ethyl alcohol for 6 h. The deposit was yielded by filtration off. The collected deposit was then leaved to dry and recrystallised using dioxane. Yield 76%; melting point: 280–282 °C. IR (ʋmax/cm–1): 3441, 3203 (2 stretching NHs), 3033 (aromatic CH), 2920 (aliphatic CH), 2218 (C≡N), 1692 (C═O), 1540 (C═N), 1472 (C═C), 1278 (C═S). 1H NMR (DMSO-d6) δ ppm: 4.38 (s, 2H, CH2S), 7.27 (d, 2H, C3′-H and C5′-H, J = 7.6 Hz), 7.36 (t, 1H, C5-H, J = 7.8 Hz), 7.54 (t, 1H, C6-H, J = 7.4 Hz), 7.66 (d, 2H, C2′-H and C6′-H, J = 8.4 Hz), 7.83 (d, 1H, C4-H, J = 8.0 Hz), 7.88 (d, 1H, C7-H, J = 8.0 Hz), 12.59 (s, 2H, 2NH, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.06, 93.80, 116.13, 120.71, 121.81, 128.06, 128.71, 129.01, 129.08, 129.22, 132.05, 133.65, 146.96, 161.38, 165.80, 167.06, 167.61, 173.88. MS m/z (%): 468 (M+, 11.33), 469 (M++1, 6.02), 470 (M++2, 1.43). Anal. Calcd. for C20H12ClN5OS3: C 51.11, H 2.57, N 14.90; found: C 51.13, H 2.61, N 14.87.
Synthesis of 2-((2-((2-(benzo[d]thiazol-2-ylamino)-2-oxoethyl)thio)-6-(4-chlorophenyl)-5-cyanopyrimidin-4-yl)amino)acetic acid (10)
The reflux of equimolar amounts 10 mmol of both compound 6 (4.70 g) in addition to glycine (0.75 g) was performed in 30 mL n-butyl alcohol for 3 h. The formed solid was separated by filtration and was air dried. Yield 80%; melting point: 270–272 °C. IR (ʋmax/cm−1): 3431, 3127 (2 stretching NH), 3015 (aromatic CH), 2833 (aliphatic CH), 2223 (C≡N), 1715, 1669 (2C═O), 1598 (C═N), 1557 (C═C).1H NMR (DMSO-d6) δ ppm: 4.12 (s, 2H, CH2-NH), 4.24 (s, 2H, CH2S), 7.38 (br, 1H, C5-H), 7.48 (br, 1H, C6-H), 7.82 (br, 3H, C4-H, C3′-H and 5′-H), 7.99 (br, 3H, C7-H, C2′-H and C6′-H), 8.38 (br, 1H, NH-CH2, D2O exchangeable), 12.69 (br, 2H, NHCO and COOH, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.39, 49.06, 84.87, 118.30, 120.80, 121.66, 128.00, 128.89, 129.68, 131.08, 131.98, 133.52, 136.38, 146.97, 157.49, 167.11, 166.82, 167.68, 170.67, and 172.00. MS m/z (%): 510 (M+, 27.28), 512 (M++2, 18.43). Anal. Calcd. for C22H15ClN6O3S2: C 51.71, H 2.96 N, 16.45; found: C 51.73, H 3.01, N 16.53.
Synthesis of N-(benzo[d]thiazol-2-yl)-2-((7-(4-chlorophenyl)-8-cyano-3-oxo-2,3-dihydroimidazo[1,2-c]pyrimidin-5-yl)thio)acetamide (11)
Compound 10 (5.10 g, 10 mmol) and 5 mL of acetic anhydride were heated under reflux for 2 h. After cooling, the precipitate was separated with filtration off and was then subjected to airily drying. Finally, compound 11 was formed through recrystallisation from acetic acid. Yield 68%; melting point: 267–269 °C. IR (ʋmax/cm−1): 3294 (stretching NH), 3053 (aromatic CH), 2968, 2916 (aliphatic CH), 2211 (C≡N), 1693 (2C═O), 1605 (C═N), 1563 (C═C).1H NMR (DMSO-d6) δ ppm: 4.04 (s, 2H, CH2, imidazole), 4.39 (s, 2H, CH2S), 7.29 (d, 2H, C3′-H and C5′-H, J = 8.0 Hz), 7.38 (t, 1H, C5-H, J = 7.6 Hz), 7.54 (t, 1H, C6-H, J = 7.2 Hz), 7.67 (d, 2H, C2′-H and C6′-H, J = 8.0 Hz), 7.83 (d, 1H, C4-H, J = 8.4 Hz), 7.89 (d, 1H, C7-H, J = 8.0 Hz), 12.66 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.53, 56.50, 89.51, 114.90, 120.71, 121.83, 121.95, 126.91, 128.02, 129.07, 129.31, 130.21, 132.31, 133.56, 135.07, 151.72, 157.45, 167.01, 169.17, 169.63, and 173.83. MS m/z (%): 492 (M+, 62.79), 493 (M++1, 13.85), 494 (M++2, 18.47). Anal. Calcd. for C22H13ClN6O2S2: C 53.60, H 2.66, N 17.05; found: C 53.62, H 2.62, N 17.12.
Synthesis of N-(benzo[d]thiazol-2-yl)-2-((3-(4-chlorophenyl)-4-cyano-10-oxo-10H-pyrimido[6,1-b]quinazolin-1-yl)thio)acetamide (12)
Compound 12 was formed by the fusion of 10 mmol of both the anthranilic acid (1.37 g) and the compound 6 (4.70 g) in a bath of oil at 190 °C for 2 h. After cooling, the mixture was poured on ice and the solid was separated by filtration. The separated solid was subjected to airily drying and recrystallisation using benzene. Yield 69%; melting point: 190–192 °C. IR (ʋmax/cm−1): 3200 (stretching NH), 3065 (aromatic CH), 2971, 2916 (aliphatic CH), 2207 (C≡N), 1689 (2C═O), 1603 (C═N), 1540 (C═C). 1H NMR (DMSO-d6) δ ppm: 4.30 (s, 2H, CH2S), 7.32 (t, 1H, C5-H, J = 7.4 Hz), 7.38 (t, 1H, C7″-H, pyrimido quinazoline, J = 8.4 Hz), 7.43 (d, 1H, C9″-H (pyrimido quinazoline), J = 8.4 Hz), 7.49 (t, 2H, C6-H and C8″-H, J = 7.6 Hz), 7.64 (d, 1H, C6″-H (pyrimido quinazoline), J = 7.6 Hz), 7.71 (d, 2H, C3′-H and C5′-H, J = 7.6 Hz), 7.81 (d, 2H, C2′-H and C6′-H, J = 8.4 Hz), 7.84 (d, 1H, C4-H, J = 8.4 Hz) 7.98 (d, 1H, C7-H, J = 7.6 Hz), 12.69 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 35.10, 93.79, 120.66, 121.76, 128.02, 128.69, 128.97, 129.09, 129.21, 132.00, 133.61, 146.87, 157.40, 157.47, 161.34, 165.77, 167.07, 167.29, 167.55, 167.63, 168.48, 169.93, 172.55, and 173.88. MS m/z (%): 554 (M+, 32.18), 556 (M++2, 12.19). Anal. Calcd. for C27H15ClN6O2S2: C 58.43, H 2.72, N 15.14; found: C 58.45, H 2.77, N 15.20.
Synthesis of N-(benzo[d]thiazol-2-yl)-2-((7-(4-chlorophenyl)-8-cyanotetrazolo[1,5-c]pyrimidin-5-yl)thio)acetamide (13)
A 50 mmol of NaN3 (3.25 g) was added to a solution of 10 mmol of 6 (4.70 g) in 30 mL glacial CH3COOH and was then allowed for 3 h reflux. After cooling and filtration, the separated solid was dried and the acetic acid was used for recrystallisation to yield 13. Yield 70%; melting point: 259–261 °C. IR (ʋmax/cm−1): 3296 (stretching NH), 3070 (aromatic CH), 2918 (aliphatic CH), 2211 (C≡N), 1694 (C═O), 1603 (C═N), 1536 (C═C). 1H NMR (DMSO-d6) δ ppm: 4.38 (s, 2H, CH2S), 7.31 (t, 1H, C5-H, J = 7.4 Hz), 7.44 (t, 1H, C6-H, J = 8.4 Hz), 7.77 (d, 2H, C3′-H and C5′-H, J = 8.0 Hz), 7.88 (d, 3H, C4-H, C2′-H and C6′-H, J = 8.4 Hz), 7.97 (d, 1H, C7-H, J = 7.6 Hz), 12.62 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 36.02, 93.81, 111.87, 120.73, 121.83, 128.07, 128.85, 129.09, 129.49, 129.82, 132.03, 143.54, 146.79, 151.88, 157.88, 167.11, 170.97, and 172.63. MS m/z (%): 478 (M+, 31.04), 480 (M++2, 12.24). Anal. Calcd. for C20H11ClN8OS2: C 50.16, H 2.31, N 23.40; found: C 50.14, H 3.35, N 23.48.
Synthesis of N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-6-(3,5-dimethyl-1H-pyrazol-1-yl)pyrimidin-2-yl)thio)acetamide (14)
The reflux of a solution of equimolar amounts (10 mmol) of both compound 8 (4.67 g) and acetylacetone (1.0 g, 1.02 mL) was done in glacial CH3COOH for 6 h. After cooling and filtration, the aqueous ethanol was used for recrystallisation of compound 14. Yield 76%; melting point: 223–225 °C. IR (ʋmax/cm−1): 3367 (stretching NH), 3197 (aromatic CH), 2919 (aliphatic CH), 2210 (C≡N), 1697 (C═O), 1556 (C═N), 1465 (C═C). 1H NMR (DMSO-d6) δ ppm: 2.52 (s, 3H, CH3, pyrazole), 2.63 (s, 3H, CH3, pyrazole), 4.42 (s, 2H, CH2S), 7.32 (t, 1H, C5-H, J = 7.2 Hz), 7.47 (t, 1H, C6-H, J = 7.6 Hz), 7.53 (s, 1H, CH, pyrazole), 7.55 (d, 2H, C3′-H and C5′-H, J = 8.2 Hz), 7.79 (d, 1H, C4-H, J = 8.2 Hz), 7.96 (d, 1H, C7-H, J = 6.0 Hz), 8.51 (d, 2H, C2′-H and C6′-H, J = 8.4 Hz), 12.56 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 21.45, 26.86, 35.33, 86.57, 116.13, 120.77, 122.61, 128.92, 129.23, 132.05, 133.17, 133.51, 135.98, 142.36, 147.15, 157.30, 160.45, 167.47, 168.69, and 173.19. MS m/z (%): 531 (M+, 23.35), 532 (M++1, 16.81), 533 (M++2, 10.47). Anal. Calcd. for C25H18ClN7OS2: C 56.44, H 3.41, N 18.43; found: C 56.45, H 3.45, N 18.38.
Synthesis of N-(benzo[d]thiazol-2-yl)-2-((4-(4-chlorophenyl)-5-cyano-6-(3-methyl-5-oxo-4,5-dihydro-1H-pyrazol-1-yl)pyrimidin-2-yl)thio) acetamide (15)
A 10 mmol of ethylacetoacetate (1.30 g, 1.27 mL) was added to a 10 mmol of compound 8 (4.67 g) and in a solution of sodium ethoxide that was formed by mixing 10 mmol metallic Na (0.23 g) with 30 mL absolute ethyl alcohol. After 4 h reflux, the mixture was leaved to cool and was added to ice water. The formed precipitate upon neutralisation with acetic acid was separated with filtration off. The mixture of chloroform and methyl alcohol was used for recrystallisation of compound 15. Yield 64%; melting point: 235–236 °C. IR (ʋmax/cm−1): 3198 (stretching NH), 3048 (aromatic CH), 2954, 2916 (aliphatic CH), 2226 (C≡N), 1696 (C═O), 1607 (C═N), 1581 (C═C). 1H NMR (DMSO-d6) δ ppm: 2.41 (s, 3H, CH3, pyrazolone), 4.04 (s, 2H, CH2, pyrazolone), 4.39 (s, 2H, CH2S), 7.27 (d, 2H, C3′-H and C5′-H, J = 8.4 Hz), 7.38 (t, 1H, C5-H, J = 6.8 Hz), 7.56 (t, 1H, C6-H, J = 7.0 Hz), 7.67 (d, 2H, C2′-H and C6′-H, J = 8.2 Hz), 7.84 (d, 1H, C4-H, J = 6.4 Hz), 7.89 (d, 1H, C7-H, J = 7.6 Hz), 12.56 (s, 1H, NHCO, D2O exchangeable). 13C NMR (DMSO-d6) δ ppm: 21.46, 25.95, 35.53, 89.51, 114.90, 120.71, 121.83, 121.95, 126.91, 128.02, 129.07, 129.31, 130.21, 132.31, 133.56, 135.07, 151.72, 167.01, 169.17, 169.63, 173.83. MS m/z (%): 533 (M+, 24.70), 535 (M++2, 10.15). Anal. Calcd. for C24H16ClN7O2S2: C 53.98, H 3.02, N 18.36; found: C 54.00, H 3.07, N 18.43.
Biological evaluation
Cultures of mycobacteria
The first line drug sensitive M. tuberculosis ATCC 25177/H37Ra and the first line drug resistant M. tuberculosis ATCC 35822 were obtained from ATCC (Manassas, VA). The first and second line drug resistant M. tuberculosis RCMB 2674, as well as the MDR clinically isolated strains of M. tuberculosis were obtained from the RCMB. Dubos medium treated with 50 µmol of NaNO3 was used as a medium for the growth of all used mycobacteria strains. The cultures were grown to exponential phase optical absorbance, where the absorbance was measured at 595 nm in an aerobic environment at 37 °C. The sonication of TB strains was allowed for 2 min in a watery bath sonicator (Ultrasonic, Freeport, IL), that as the mycobacteria prefer to grow in aggregated clusters.
Minimum inhibitory concentration assay
The MABA assay was used for the determination of MICs of all tested hybrids 4, 5a–c, 6, 7a–f, and 8–15 against three strains of M. tuberculosisCitation50,Citation51. INH was used as reference control. The final testing concentrations used for stock solutions of tested compounds were in the range of 1000–0.003 µg/mL. M. tuberculosis was allowed to grow to late exponential phase in Broth of Difco Middlebrook 7H9 enriched with 0.2% vol/vol of glycerol, 0.05% Tween 80, and 10% vol/vol of albumin, dextrose, catalase (7 H9–ADC–TG). Preparation of twofold serial dilutions of tested hybrids was done in 100 µL of 7H9–ADC–TG in 96-well clear-bottom micro plates. The ending evaluating volume of 200 µL of M. tuberculosis was yielded by addition of 100 µL having 2 × 105 CFU. Then, the incubation of plates was done at 37 °C for 16–24 h. On the seventh day of the incubation, all wells were supplemented with 20 µL of alamar blue and 12.5 µL of 20% Tween 80. After incubation at 37 °C, the reading of fluorescence was taken at values of 530 and 590 nm. The lowest concentration producing a decrease in the 100% fluorescence related to the average of controls of the replicate bacterium only was used for the definition of the MICCitation33.
Molecular docking
In the present work, molecular operating environment (MOE, 2019.0102) softwareCitation52 was operated for doing the molecular docking, energy minimised structures were gained by applying MMFF94x force field until the reaching of RMSD gradient of 0.1 kcal·mol–1 Å–1. The X-ray crystallographic structures of DprE1 and TMPKtm enzymes were downloaded from protein data bank (PDB) with the following ID: 4KW5 and 1W2H. Each protein file’s co-crystallised ligand was used to identify the essential binding features to the enzymes using triangle matcher as a placement method and London dG as a scoring algorithmCitation52.
Supplemental Material
Download PDF (1.8 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Sandhu GK. Tuberculosis: current situation, challenges and overview of its control programs in India. J Glob Infect Dis. 2011;3(2):143–150.
- Glaziou P, Floyd K, Raviglione MC. Global epidemiology of tuberculosis. Semin Respir Crit Care Med. 2018;39(3):271–285.
- Chakaya J, Khan M, Ntoumi F, Aklillu E, Fatima R, Mwaba P, Kapata N, Mfinanga S, Hasnain SE, Katoto PDMC, et al. Global Tuberculosis Report 2020 – reflections on the global TB burden, treatment and prevention efforts. Int J Infect Dis. 2021;113(Suppl. 1):S7–S12.
- Feuerriegel S, Oberhauser B, George AG, Dafae F, Richter E, Rüsch-Gerdes S, Niemann S. Sequence analysis for detection of first-line drug resistance in Mycobacterium tuberculosis strains from a high-incidence setting. BMC Microbiol. 2012;12(1):90.
- Montoro E, Lemus D, Echemendia M, Martin A, Portaels F, Palomino JC. Comparative evaluation of the nitrate reduction assay, the MTT test, and the resazurin microtitre assay for drug susceptibility testing of clinical isolates of Mycobacterium tuberculosis. J Antimicrob Chemother. 2005;55(4):500–505.
- Coninx R, Mathieu C, Debacker M, Mirzoev F, Ismaelov A, de Haller R, Meddings DR. First-line tuberculosis therapy and drug-resistant Mycobacterium tuberculosis in prisons. Lancet. 1999;353(9157):969–973.
- Ferguson LA, Rhoads J. Multidrug-resistant and extensively drug-resistant tuberculosis: the new face of an old disease. J Am Acad Nurse Pract. 2009;21(11):603–609.
- Tuyiringire N, Tusubira D, Munyampundu JP, Tolo CU, Muvunyi CM, Ogwang PE. Application of metabolomics to drug discovery and understanding the mechanisms of action of medicinal plants with anti-tuberculosis activity. Clin Transl Med. 2018;7:1–12.
- Ribeiro ALdJL, Degiacomi G, Ewann F, Buroni S, Incandela ML, Chiarelli LR, Mori G, Kim J, Contreras-Dominguez M, Park Y-S, et al. Analogous mechanisms of resistance to benzothiazinones and dinitrobenzamides in Mycobacterium smegmatis. PLOS One. 2011;6(11):e26675.
- Trefzer C, Škovierová H, Buroni S, Bobovská A, Nenci S, Molteni E, Pojer F, Pasca MR, Makarov V, Cole ST, et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-d-ribofuranose 2′-oxidase DprE1. J Am Chem Soc. 2012;134(2):912–915.
- Makarov V, Lechartier B, Zhang M, Neres J, van der Sar AM, Raadsen SA, Hartkoorn RC, Ryabova OB, Vocat A, Decosterd LA, et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol Med. 2014;6(3):372–383.
- De La Sierra IL, Munier-Lehmann H, Gilles AM, Bârzu O, Delarue M. X-ray structure of TMP kinase from Mycobacterium tuberculosis complexed with TMP at 1.95 Å resolution. J Mol Biol. 2001;311(1):87–100.
- Vanheusden V, Munier-Lehmann H, Pochet S, Herdewijn P, Van Calenbergh S. Synthesis and evaluation of thymidine-5′-O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg Med Chem Lett. 2002;12(19):2695–2698.
- Munier-Lehmann H, Chenal-Francisque V, Ionescu M, Chrisova P, Foulon J, Carniel E, Bârzu O. Relationship between bacterial virulence and nucleotide metabolism: a mutation in the adenylate kinase gene renders Yersinia pestis avirulent. Biochem J. 2003;373(Pt 2):515–522.
- Gjorgjieva M, Tomašič T, Kikelj D, Mašič LP. Benzothiazole-based compounds in antibacterial drug discovery. Curr Med Chem. 2018;25(38):5218–5236.
- Rouf A, Tanyeli C. Bioactive thiazole and benzothiazole derivatives. Eur J Med Chem. 2015;97:911–927.
- Gill RK, Rawal RK, Bariwal J. Recent advances in the chemistry and biology of benzothiazoles. Arch Pharm. 2015;348(3):155–178.
- Dar AA, Shadab M, Khan S, Ali N, Khan AT. One-pot synthesis and evaluation of antileishmanial activities of functionalized S-alkyl/aryl benzothiazole-2-carbothioate scaffold. J Org Chem. 2016;81(8):3149–3160.
- Shaikh FM, Patel NB, Sanna G, Busonera B, La Colla P, Rajani DP. Synthesis of some new 2-amino-6-thiocyanato benzothiazole derivatives bearing 2,4-thiazolidinediones and screening of their in vitro antimicrobial, antitubercular and antiviral activities. Med Chem Res. 2015;24(8):3129–3142.
- Mikulášek S, Odlerová Ž, Sutoris V, Perjessy A, Solčániová E. Benzothiazole compounds. XIII. Synthesis and antitubercular activity of some N-[1-(6-R-2-benzothiazolylamino)-2,2,2-trichloroethyl] formamide,-acetamide,-chloroacetamide, and-benzamide derivatives. Chem Pap. 1978;32:691–700.
- Landge S, Mullick AB, Nagalapur K, Neres J, Subbulakshmi V, Murugan K, Ghosh A, Sadler C, Fellows MD, Humnabadkar V, et al. Discovery of benzothiazoles as antimycobacterial agents: synthesis, structure–activity relationships and binding studies with Mycobacterium tuberculosis decaprenylphosphoryl-β-d-ribose 2′-oxidase. Bioorg Med Chem. 2015;23(24):7694–7710.
- Patel RV, Patel PK, Kumari P, Rajani DP, Chikhalia KH. Synthesis of benzimidazolyl-1,3,4-oxadiazol-2ylthio-N-phenyl (benzothiazolyl) acetamides as antibacterial, antifungal and antituberculosis agents. Eur J Med Chem. 2012;53:41–51.
- Chikhale RV, Bhole RP, Khedekar PB, Bhusari KP. Synthesis and pharmacological investigation of 3-(substituted 1-phenylethanone)-4-(substituted phenyl)-1,2,3,4-tetrahydropyrimidine-5-carboxylates. Eur J Med Chem. 2009;44(9):3645–3653.
- Gawad J, Bonde C. Design, synthesis and biological evaluation of some 2-(6-nitrobenzo [d] thiazol-2-ylthio)-N-benzyl-N-(6-nitrobenzo [d] thiazol-2-yl) acetamide derivatives as selective DprE1 inhibitors. Synth Commun. 2019;49(20):2696–2708.
- Mohamed MS, Awad SM, Sayed AI. Synthesis of certain pyrimidine derivatives as antimicrobial agents and anti-inflammatory agents. Molecules. 2010;15(3):1882–1890.
- Patil SB. Biological and medicinal significance of pyrimidines: a review. Int J Pharm Sci Res. 2018;9:44–52.
- Kamalakannan P, Venkappayya D. Synthesis and characterization of cobalt and nickel chelates of 5-dimethylaminomethyl-2-thiouracil and their evaluation as antimicrobial and anticancer agents. J Inorg Biochem. 2002;90(1–2):22–37.
- Ding Y, Girardet JL, Smith KL, Larson G, Prigaro B, Wu JZ, Yao N. Parallel synthesis of 5-cyano-6-aryl-2-thiouracil derivatives as inhibitors for hepatitis C viral NS5B-dependent RNA polymerase. Bioorg Chem. 2006;34(1):26–38.
- Kittakoop P, Mahidol C, Ruchirawat S. Alkaloids as important scaffolds in therapeutic drugs for the treatments of cancer, tuberculosis, and smoking cessation. Curr Top Med Chem. 2014;14(2):239–252.
- Mahran AM, Hassan NA, Osman DA, Ragab SS, Hassan AA. Synthesis and biological evaluation of novel pyrimidines derived from 6-aryl-5-cyano-2-thiouracil. Z Naturforsch C J Biosci. 2016;71(5–6):133–140.
- Pisal MM, Nawale LU, Patil MD, Bhansali SG, Gajbhiye JM, Sarkar D, Chavan SP, Borate HB. Hybrids of thienopyrimidinones and thiouracils as anti-tubercular agents: SAR and docking studies. Eur J Med Chem. 2017;127:459–469.
- Chitre TS, Bothara KG, Patil SM, Asgaonkar KD, Nagappa S, Kathiravan MK. Design, synthesis, docking and anti-mycobacterial activity of some novel thiouracil derivatives as thymidine monophoshate kinase (TMPKmt) inhibitors. Int J Res Pharm Biomed Sci. 2011;2:616–623.
- Mdluli K, Spigelman M. Novel targets for tuberculosis drug discovery. Curr Opin Pharmacol. 2006;6(5):459–467.
- Husseiny EM. Synthesis, cytotoxicity of some pyrazoles and pyrazolo [1,5-a] pyrimidines bearing benzothiazole moiety and investigation of their mechanism of action. Bioorg Chem. 2020;102:104053.
- Bondock S, Fadaly W, Metwally MA. Enaminonitrile in heterocyclic synthesis: synthesis and antimicrobial evaluation of some new pyrazole, isoxazole and pyrimidine derivatives incorporating a benzothiazole moiety. Eur J Med Chem. 2009;44(12):4813–4818.
- Keri RS, Keri RS, Patil MR, Patil SA, Budagumpi S. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur J Med Chem. 2015;89:207–251.
- Doma A, Kulkarni R, Garlapati A, Radha P. Synthesis and anti-inflammatory activity of novel pyrimidino benzothiazole amine derivatives. Pharmacophore. 2014;5:331–342.
- Azam MA, Dharanya L, Mehta CC, Sachdeva S. Synthesis and biological evaluation of some novel pyrazolopyrimidines incorporating a benzothiazole ring system. Acta Pharm. 2013;63(1):19–30.
- Diao PC, Lin WY, Jian XE, Li YH, You WW, Zhao PL. Discovery of novel pyrimidine-based benzothiazole derivatives as potent cyclin-dependent kinase 2 inhibitors with anticancer activity. Eur J Med Chem. 2019;179:196–207.
- Chikhale R, Menghani S, Babu R, Bansode R, Bhargavi G, Karodia N, Rajasekharan MV, Paradkar A, Khedekar P. Development of selective DprE1 inhibitors: design, synthesis, crystal structure and antitubercular activity of benzothiazolylpyrimidine-5-carboxamides. Eur J Med Chem. 2015;96:30–46.
- Bhoi MN, Borad MA, Jethava DJ, Acharya PT, Pithawala EA, Patel CN, Pandya HA, Patel HD. Synthesis, biological evaluation and computational study of novel isoniazid containing 4H-pyrimido [2,1-b] benzothiazoles derivatives. Eur J Med Chem. 2019;177:12–31.
- Patel RV, Kumari P, Rajani DP, Chikhalia KH. Synthesis of coumarin-based 1,3,4-oxadiazol-2ylthio-N-phenyl/benzothiazolyl acetamides as antimicrobial and antituberculosis agents. Med Chem Res. 2013;22(1):195–210.
- Ramesh B, Bhalgat CM. Novel dihydropyrimidines and its pyrazole derivatives: synthesis and pharmacological screening. Eur J Med Chem. 2011;46(5):1882–1891.
- Ram VJ. Chemotherapeutic agents, XVIII: synthesis of π-deficient pyrimidines and fused pyrimidines as leishmanicidal agents. Arch Pharm. 1990;323(11):895–899.
- Hassan GS, Awdallah FM, El-Saadi MT, Elgendi AO, Hemeda LR. Synthesis of some benzothiazole derivatives evaluated as antimicrobials and antibiofilms. J Chem Pharm. 2015;7:1125–1143.
- Hassan NW, Saudi MN, Abdel-Ghany YS, Ismail A, Elzahhar PA, Sriram D, Nassra R, Abdel-Aziz MM, El-Hawash SA. Novel pyrazine based anti-tubercular agents: design, synthesis, biological evaluation and in silico studies. Bioorg Chem. 2020;96:103610.
- Morris GM, Lim-Wilby M. Molecular docking. Methods Mol Biol. 2008;443:365–382.
- Wang F, Sambandan D, Halder R, Wang J, Batt SM, Weinrick B, Ahmad I, Yang P, Zhang Y, Kim J, et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc Natl Acad Sci USA. 2013;110(27):E2510–E2517.
- Fioravanti E, Adam V, Munier-Lehmann H, Bourgeois D. The crystal structure of Mycobacterium tuberculosis thymidylate kinase in complex with 3′-azidodeoxythymidine monophosphate suggests a mechanism for competitive inhibition. Biochemistry. 2005;44(1):130–137.
- Eldehna WM, El Hassab MA, Abdelshafi NA, Al-Zahraa Sayed F, Fares M, Al-Rashood ST, Elsayed ZM, Abdel-Aziz MM, Elkaeed EB, Elsabahy M, et al. Development of potent nanosized isatin–isonicotinohydrazide hybrid for management of Mycobacterium tuberculosis. Int J Pharm. 2022;612:121369.
- (a) Elsayed ZM, Eldehna WM, Abdel-Aziz MM, El Hassab MA, Elkaeed EB, Al-Warhi T, Abdel-Aziz HA, Abou-Seri SM, Mohammed ER. Development of novel isatin–nicotinohydrazide hybrids with potent activity against susceptible/resistant Mycobacterium tuberculosis and bronchitis causing-bacteria. J Enzyme Inhib Med Chem. 2021;36(1):384–392. (b) Al-Warhi T, Elimam DM, Elsayed ZM, Abdel-Aziz MM, Maklad RM, Al-Karmalawy AA, Afarinkia K, Abourehab MA, Abdel-Aziz HA, Eldehna WM. Development of novel isatin thiazolyl-pyrazoline hybrids as promising antimicrobials in MDR pathogens. RSC Adv. 2022;12(48):31466–31477.
- Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8(18):1555–1572.