
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Two new neolignans, myrifralignans F–G (14 and 18), four new diarylnonanoid derivatives, myrifragranones A–D (21–24), and 18 known compounds were isolated and structurally elucidated from nutmeg (Myristica fragrans Houtt.) seeds. The absolute configurations of these secondary metabolites were determined using the electronic circular dichroism technique. The inhibitory potential of these isolated compounds on soluble epoxide hydrolase (sEH) was investigated for the first time. Among them, malabaricones B and C (19 and 20) and four new compounds 21–24 displayed inhibitory activities against sEH, with IC50 values ranging from 14.24 to 46.35 µM. Additionally, the binding mechanism, key binding interactions, stability, and dynamic behaviour of the active compounds with the sEH enzyme were analysed using in silico molecular docking and dynamics simulations. Our findings suggest that nutmeg could become a promising natural source for discovering and developing new sEH inhibitors.
Graphical abstract

Introduction
Epoxy fatty acids are a type of lipid produced by the cytochrome-mediated oxidation of polyunsaturated fatty acidsCitation1. They are primarily metabolised through epoxide ring hydrolysis, leading to the formation of vicinal diols, such as dihydroxy-eicosatrienoic acidCitation2. Soluble epoxide hydrolase (sEH), an enzyme that catalyses the hydrolysis of various substrates, including epoxyeicosatrienoic acids (EETs) and epoxides of docosahexaenoic acid, predominantly facilitates this important metabolic pathwayCitation3,Citation4.
Among the substrates that sEH converts, EETs have numerous beneficial physiological effects, such as vasodilatory, vasoprotective, and anti-inflammatory properties. They can dilate blood vessels and inhibit platelet aggregation, thereby lowering blood pressure and reducing thrombosis risk, respectivelyCitation5,Citation6. Furthermore, EETs possess anti-inflammatory properties, which can help to mitigate inflammation-associated tissue damageCitation7. Given the important biological activities associated with EETs, sEH inhibition represents a promising therapeutic strategy for various human diseases. sEH inhibitors have been used to effectively treat cardiovascular diseases, hypertension, inflammation, and painCitation8–10.
In Asian countries, natural products and herbal medicine have a longstanding history of being utilised for the treatment of various diseases because of their safety and low risk of side effectsCitation11. Nutmeg (Myristica fragrans Houtt.), a member of the Myristicaceae family, is an evergreen tropical tree, that is grown commercially in Asian countries, including China, Japan, Korea, and VietnamCitation12. Nutmegs have a unique, aromatic scent accompanied by a warm and slightly sweet flavourCitation13. They are approximately oval (2–3 cm long), complex, meaty, white, and have red-brown veins. They have been used as a natural spice in culinary preparation and play a primary role in treating rheumatism, cholera, psychosis, stomach cramps, nausea, and diarrhoeaCitation14. Reportedly, nutmegs have been associated with various pharmacological properties, including anti-inflammatory, anticancer, antioxidant, and hepatic detoxificationCitation12,Citation15. They also contain several phytochemicals, including neolignans, phenylpropanoids, alkaloids, diarylnonanoids, terpenoids, alkanes, and fatty acidsCitation12. Among them, neolignans and diarylnonanoids, which have various therapeutic characteristics, are regarded as the primary bioactive constituents in nutmeg.
As part of our ongoing research on medicinal plants in Korea, this study involved a phytochemical investigation on dried nutmeg. As a result, two new neolignans (14 and 18), four new diarylnonanoid derivatives (21–24), and 18 known compounds were isolated. The structure and configurations of these metabolites were elucidated through the application of advanced spectroscopic techniques, such as NMR, mass, and electronic circular dichroism (ECD) spectra. Additionally, in vitro assays were used to investigate the inhibitory effect and mode of enzyme inhibition of the isolated compounds against sEH for the first time. Notably, the diarylnonanoids, malabaricones B and C (19 and 20), and their derivatives, myrifragranones A–D (21–24), exhibited strong effects against sEH. Subsequently, the key binding interactions and conformational stability of the sEH- active compound complexes were elucidated by conducting in silico studies.
Materials and methods
Plants
Nutmeg samples were obtained from a traditional medicine market located in Daegu, Korea, in March 2017. Authentication of the samples was conducted by Professor Byung Sun Min, Daegu Catholic University. A herbarium voucher (21 A-MF) was stored at the Pharmacognosy Lab, College of Pharmacy, Kyungpook National University.
Extraction and isolation
Nutmegs (20.0 kg) were subjected to reflux extraction using MeOH (20 L × 3). The solution was concentrated under a vacuum. The MeOH extract (3.5 kg) was successfully suspended in water (4 L) and divided with n-hexane and EtOAc. Following the evaporation of the solvent, the EtOAc extract (397.0 g) was subjected to vacuum liquid chromatography, using CH2Cl2–MeOH (gradient 99:1–10:1, v/v) as mobile phase to yield fractions 1 A–1G. Fractions 1 C (120 g) and 1D (12 g) were combined and fractionated using CC with n-hexane–EtOAc (gradient 10:1–1:1, v/v) to yield seven fractions (2 A–2G). Compound 20 (3.6 g) was produced by recrystallizing fraction 2 A (9.0 g) from CH2Cl2 at room temperature (20–25 °C). Fraction 2D (22.0 g) was passed out silica gel column and eluted with n-hexane–EtOAc (3:1, v/v) to separate five fractions (2D1–2D5) and compounds 9 (289.1 mg) and 10 (17.5 mg). Fraction 2D2 (16.95 g) was separated via RP-18 gel CC using MeOH–H2O (3:1, v/v) to have compounds 1 (6.1 g), 3 (2.8 g), and 19 (179.8 mg). Fraction 2D4 (715.3 mg) was purified using preparative HPLC (Waters Alliance system containing a Waters 1525 pump, Waters 2998 PDA detector, and Phenomenex Gemini C18 column, 30 × 250 mm, 5 μm) using 65% ACN in H2O to obtain compounds 2 (67.2 mg), 4 (2.0 mg), and 5 (2.0 mg). Fraction 2 F (21.3 g) was separated via RP-18 gel CC using MeOH–H2O (1:1, v/v) as a mobile phase to yield nine fractions (2F1–2F9). Compounds 7 (4.7 mg) and 18 (15.6 mg) were purified from fraction 2F4 (823.8 mg) via RP-18 CC, MeOH–H2O (2:1, v/v). Compounds 11 (4.4 mg), 12 (12.7 mg), and 13 (2.0 mg) were purified from 2F8 (1.34 g) under the same condition as compounds 7 and 18. Compounds 6 (158.8 mg), 8 (258.3 mg), and 14 (2.0 mg) were obtained by subjecting 2F6 (2.6 g) to RP-18 CC and eluting it with acetone–H2O (1:1, v/v). Compounds 16 (4.0 mg) and 17 (79.3 mg) were isolated from fraction 2F7 (618.6 mg) via RP-18 CC, MeOH–H2O (2.5:1, v/v). Compounds 15 (2.0 mg), 21 (5.0 mg), 22 (22.0 mg), 23 (21.9 mg), and 24 (3.5 mg) were purified by subjecting fraction 2F9 (1.02 g) to RP-18 CC, MeOH–H2O (5:1, v/v), followed by Sephadex column using MeOH–H2O (2:1, v/v).
Myrifralignan F (14): colourless oil; [α] −16.0 (c 0.02, CHCl3); UV (CHCl3) λmax (logε) 273 (4.04) nm; ECD (CHCl3) mdeg (λmax) −2.88 (272 nm), +2.66 (236 nm); IR νmax 3314, 2941, 2832, 1448, 1116, 1024 cm−1; NMR data, ; HR-EI-MS m/z 420.1784 [M]+ (calcd for C22H28O8, 420.1784).
Table 1. NMR data (500 MHz, CDCl3) of compounds 14, 16, and 18.
(7R,8R)-1–(4′-hydroxy-3′-methoxyphenyl)-1-methoxy-2–(2″-methoxy-4″-(1‴-(E)-propenyl)-phenoxy) propane (16): colourless oil; [α] −24.4 (c 0.02, CHCl3), −27.6 (c 0.02, MeOH); UV (CHCl3) λmax (logε) 263 (4.24) nm; ECD (CHCl3) mdeg (λmax) −0.72 (237 nm); IR νmax 3680, 3324, 2943, 2831, 1453, 1114, 1032, 1023 cm−1; NMR data, ; HR-EI-MS m/z 358.1780 [M]+ (calcd for C21H26O5, 358.1780).
Myrifralignan G (18): colourless oil; [α] +14.9 (c 0.02, CHCl3); UV (CHCl3) λmax (logε) 235 (4.10), 280 (3.59) nm; ECD (CHCl3) mdeg (λmax) −0.35 (276 nm), −1.40 (240 nm), +3.45 (208 nm); IR νmax 3324, 2943, 2832, 1449, 1116, 1024 cm−1; NMR data, ; HR-EI-MS m/z 392.1835 [M]+ (calcd for C22H28O7, 392.1835).
Myrifragranone A (21): yellow oil; [α] +25.2 (c 0.02, CHCl3); UV (CHCl3) λmax (logε) 275 (4.02) nm; ECD (CHCl3) mdeg (λmax) +1.55 (273 nm), −0.65 (240 nm), +2.70 (220 nm), +5.40 (205 nm); IR νmax 3680, 3324, 2942, 2831, 1453, 1115, 1024 cm−1; NMR data, ; HR-FAB-MS m/z 707.3202 [M + Na]+ (calcd. for C41H48O9Na, 707.3196).
Table 2. NMR data (500 MHz, CD3OD) of compounds 21–24.
Myrifragranone B (22): yellow oil; [α] +42.9 (c 0.02, CHCl3); UV (CHCl3) λmax (logε) 239 (4.57), 260 (4.49) nm; ECD (CHCl3) mdeg (λmax) +1.15 (285 nm), −0.25 (260 nm), +7.70 (235 nm), +4.80 (215 nm); IR νmax 3680, 3324, 2943, 2831, 1453, 1116, 1024 cm−1; NMR data, ; HR-FAB-MS m/z 737.3309 [M + Na]+ (calcd for C42H50O10Na, 737.3302).
Myrifragranone C (23): yellow oil; [α] −22.3 (c 0.02, CHCl3); UV (CHCl3) λmax (logε) 239 (4.63), 261 (4.56) nm; ECD (CHCl3) mdeg (λmax) +0.85 (270 nm), −6.50 (240 nm), +1.25 (215 nm); IR νmax 3852, 3814, 3680, 3324, 2942, 2831, 1453, 1115, 1024 cm−1; NMR data, ; HR-FAB-MS m/z 737.3306 [M + Na]+ (calcd for C42H50O10Na, 737.3302).
Myrifragranone D (24): yellow oil; [α] +40.8 (c 0.02, CHCl3); UV (CHCl3) λmax (logε) 234 (4.60), 265 (4.65) nm; ECD (CHCl3) mdeg (λmax) −0.65 (275 nm), −0.33 (240 nm), +1.98 (207 nm); IR νmax 3680, 3324, 2942, 2831, 1453, 1115, 1024, 831 cm−1; NMR data, ; HR-FAB-MS m/z 491.2800 [M − H2O + H]+ (calcd for C31H39O5, 491.2797).
Snatzke’s method for identifying the configuration of propanyl-1,2-diol moiety in compound 18
The CD of compound 18 was evaluated in DMSO at a concentration of 0.7 mM. The solution of 18 in DMSO was combined with dimolybdenum tetraacetate [Mo2(OAc)4] at a ratio of 1:1 at 20–25 °C. The reaction achieved a stationary state approximately 30–40 min after mixing. Afterward, CD spectrum of the mixture was recorded. The Mo2(OAc)4-induced CD (ICD) spectrum, with its signature located in the diagnostic region at 305 nm, was used to determine the absolute configuration of the propanyl-1,2-diol moiety in compound 18 16.
sEH inhibition assay
sEH inhibition assay was conducted based on a previous study with minor modificationsCitation16, and described in Supplementary data. AUDA (10007927, Cayman Chemical, MI, USA), a sEH inhibitor, was selected as a positive control in this assayCitation17. The experiments were conducted three times independently, and the data were stated as the mean ± standard deviation, p values <0.05 was considered statistically significant.
Molecular docking simulation
AutoDock Vina 1.1.2 was used to accomplish the docking procedure. The human sEH enzyme structure was retrieved from the RCSB PDB database (ID: 3ANS) at a resolution of 1.98 ÅCitation18. The binding inhibitor and water molecules were eliminated from the enzyme structure via BIOVIA Discovery Studio Visualiser 21 (Dassault Systèmes, MA, USA). The 3D chemical structures of compounds 19–24 were constructed using Spartan’18 (Wavefunction Inc., CA, USA). The ligand energy is minimised by computing Gasteiger charges using AutoDockTools 1.5.6 before exporting in the ‘.pdbqt’ format for the docking process. Blind docking was utilised in this studyCitation19–21. The grid box dimensions were set to x = 96, y = 96, z = 124; the grid position was set to x = 27.058, y = 28.677, z = 108.904; an exhaustiveness value of 100 was used, along with a number of binding modes of 1000, to comprehensively explore the protein-ligand interaction. The docking diagrams were examined and analysed using Discovery Studio Visualiser 21Citation22.
Molecular dynamics
To simulate the dynamic behaviour of the protein-ligand complex, molecular dynamics (MD) simulations were conducted based on our previously published studyCitation23. The dynamics system was prepared with the CHARMM36 force field using the web-based CHARMM-GUI platform. The orthorhombic TIP3P solvation model was employed for water modelling. Finally, 50 ns of MD trajectory (time step of 0.02 fs) were carried out using the GROMACS 2022.1 program.
Results and discussion
Structural elucidation of compounds isolated from nutmeg
The EtOAc extract of nutmeg was subjected to repeated CC and preparative HPLC, yielding 24 compounds, including two new neolignans (14 and 18) and four new diarylnonanoid derivatives (21–24). By comparing spectroscopic results with those previously published literature, the known metabolites were identified as: myrislignan (1)Citation24, erythro-(7S,8R)-Δ8′-7-hydroxy-3,4,3′,5′-tetramethoxy-8-O-4′-neolignan (2)Citation25, (7S,8R)-rhaphidecursinol B (3)Citation15, erythro-(7S,8R)-3,4-methylenedioxy-7-hydroxy-1′-allyl-3′,5′-dimethoxy)-8-O-4′-neolignans (4)Citation26, erythreo-(7S,8R)-1–(4′-hydroxy-3′methoxyphenyl)-1-methoxy-2–(2″-methoxy-4″-(1‴-(E)-propenyl)-phenoxy) propane (5)Citation27, erythro-(7R,8S)-Δ8′-4,7-dihydroxy-3,3′,5′-trimethoxy-8-O-4′-neolignan (6)Citation28, erythro-(7R,8S)-Δ8′-4,7-dihydroxy-3,5,3′-trimethoxy-8-O-4′-neolignan (7)Citation29, maceneolignan F (8)Citation30, maceneolignan H (9)Citation30, erythro-(7R,8S)-Δ8′-7-acetoxy-3,4-methylenedioxy-3′,5′-dimethoxy-8-O-4′-neolignan (10)Citation26, myrifralignan C (11)Citation15, surinamensin (12)Citation29, myrislignanometin E (13)Citation31, myrifralignan E (15)Citation15, (+)-(4-hydroxy-3-methoxy-l′-allyl-3′,5′-dimethoxy)-8-O-4′-neolignan (17)Citation32, malabaricone B (19)Citation33, and malabaricone C (20)Citation33 (). In addition, the ECD technique was employed to determine the absolute configuration of new compounds and compound 16.
Figure 1. Structures of compounds from nutmeg 1–24.
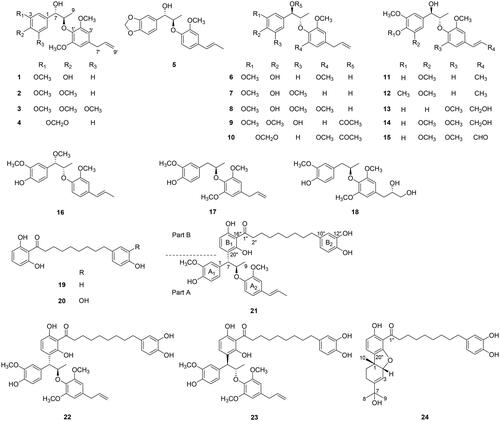
Myrifralignan F (14) was yielded as a colourless oil, [α] −16.0 (c 0.02, CHCl3). The HR-EI-MS spectrum of 14 displayed a molecular ion peak at m/z 420.1784 [M]+ (calcd. for C22H28O8, 420.1784) (Figure S1.10), indicating a molecular formula of C22H28O8. The 1D-NMR spectrum of compound 14 revealed signals of two tetrasubstituted benzene rings [δH 6.54 (2H, s, H-2, H-6) and 6.68 (2H, s, H-3′, H-5′)], two olefinic protons [δH 6.58 (1H, d, J = 15.8 Hz, H-7′) and 6.33 (1H, dt, J = 15.8, 5.7 Hz, H-8′)]; two oxygenated methine groups [δH 4.78 (1H, d, J = 4.8 Hz, H-7) and 4.35 (1H, dd, J = 5.6, 1.4 Hz, H-8)], a methylene group [δH 4.35 (2H, dd, J = 5.6, 1.4 Hz, H-9′)], four methoxy groups [δH 3.87 (6H, s, 3-, 5-OCH3) and 3.90 (6H, s, 2′-, 6′-OCH3)], and a methyl group [δH 1.12 (3H, d, J = 6.4 Hz, H-9)] (). By analysing the HMQC spectrum, these protons showed correlation with the corresponding carbons at δC 12.9 (C-9), 63.7 (C-9′), 56.5 (3-, 5-OCH3), 56.3 (2′-, 6′-OCH3), 73.8 (C-7), 82.8 (C-8), 102.9 (C-2, C-6), 103.7 (C-3′, C-5′), 131.1 (C-7′), and 128.6 (C-8′). Furthermore, eight quaternary carbons [δC 131.0 (C-1), 147.1 (C-3, C-5), 133.8 (C-4), 134.8 (C-1′), 153.9 (C-2′, C-6′), and 133.0 (C-4′)] were also observed in the carbon NMR of 14. The E-configuration of the C-7′–C-8′ double bond was established by observing a coupling constant (J = 15.8 Hz) between protons H-7′ and H-8′Citation22. The spectroscopic data of 14 were closely similar to those of myrislignanometin E (13)Citation31, except for the signal assigned to an additional methoxy group at C-5. This presence was demonstrated via the HMBC correlation between δH 3.87 (6H, s, 3-, 5-OCH3) and δC 147.1 (C-3, C-5) (). The relative erythyo-configuration of 14 was determined based on the small coupling constant between H-7 and H-8 (J = 2.8 Hz) as well as the chemical shifts of protons and carbons at C-7 and C-9Citation34. The absolute configuration of 14 was identified as 7 R,8S based on the optical rotation of [α]
−16.0 (c 0.02, CHCl3)Citation26, and a positive Cotton effect at 225–250 nm in the ECD spectrum (Figure S1.11)Citation15,Citation35.
Figure 2. Key HMBC (→) and COSY (bold) correlations of compounds 14, 16, 18, and 21–24.
Compound 16 was isolated as a colourless oil. The HR-EI-MS analysis indicated a molecular ion peak at m/z 358.1780 [M]+ (calcd. for C21H26O5, 358.1780) (Figure S2.10). The 1H-NMR spectrum of compound 16 displayed signals of two trisubstituted benzene rings [δH 6.91 (1H, d, J = 1.7 Hz, H-2), 6.88 (1H, d, J = 8.2 Hz, H-5), 6.88 (1H, d, J = 1.7 Hz, H-3′), 6.83 (2H, d, J = 8.2, 1.7 Hz, H-6, H-5′), and 6.90 (1H, d, J = 8.2 Hz, H-6′)], a double bond [δH 6.33 (1H, dd, J = 15.7, 1.6 Hz, H-7′) and 6.10 (1H, dq, J = 15.7, 6.6 Hz, H-8′)], two oxygenated methine groups [δH 4.28 (1H, d, J = 6.3 Hz, H-7), 4.45 (1H, quint, J = 6.4 Hz, H-8)/δC 86.7 (C-7), 79.2 (C-8)], three methoxy groups [δH 3.89 (3H, s, 3-OCH3), 3.28 (3H, s, 7-OCH3), 3.84 (3H, s, 2′-OCH3)/δC 56.1 (3-, 2′-OCH3), 57.3 (7-OCH3)], and a methyl group [δH 1.12 (3H, d, J = 6.4 Hz, H-9)]. The spectroscopic data of 16 were similar to those of the reported compound threo-1–(4′-hydroxy-3′-methoxyphenyl)-1-methoxy-2–(2″-methoxy-4″-(1‴-(E)-propenyl)-phenoxy) propaneCitation36 (). However, the absolute configuration of this compound has not yet been elucidated. The optical rotation of 16 ([α] −24.4 in CHCl3 and −27.6 in MeOH) was opposite to those of fargesiphenol A ([α]
+38.5 in MeOH), fargesiphenol B ([α]
+41.2 in MeOH), and fargesiphenol C ([α]
+42.5 in MeOH), suggesting 7 R,8R-configurationCitation37. The absolute configuration of 16 was verified through the presence of a negative Cotton effect observed in the 237–240 nm region instead of the positive effect in fargesiphenols A–C (Figure S2.11)Citation37. Thus, the structure of 16 was clearly established as (7 R,8R)-1–(4′-hydroxy-3′-methoxyphenyl)-1-methoxy-2–(2″-methoxy-4″-(1‴-(E)-propenyl)-phenoxy) propane.
Myrifralignan G (18) was yielded as a colourless oil, with [α] +14.9 (c 0.02, CHCl3). The molecular formula of 18 was identified as C22H28O7 by HR-EI-MS at m/z 392.1835 [M]+ (calcd. for C22H28O7, 392.1835) (Figure S3.10). The NMR spectra of compound 18 revealed the presence of two benzene rings [δH 6.75 (1H, d, J = 1.3 Hz, H-2), 6.79 (1H, d, J = 8.0 Hz, H-5), 6.68 (1H, dd, J = 8.0, 1.3 Hz, H-6), and 6.40 (2H, s, H-3′, H-5′)], two oxygenated methine groups [δH 3.08 (1H, dd, J = 13.6, 5.5 Hz, H-8), 3.93 (1H, dd, J = 6.5, 3.4 Hz, H-8′)/δC 80.0 (C-8), 73.2 (C-8′)], three methoxy groups [δH 3.34 (3H, s, 3-OCH3), 3.78 (6H, s, 2′-, 6′-OCH3)/δC 56.0 (3-OCH3), 56.2 (2′-, 6′-OCH3)], and a methyl group [δH 1.20 (3H, d, J = 6.1 Hz, H-9)]. These observed signals highly resembled those of (+)-(4-hydroxy-3-methoxy-l′-allyl-3′,5′-dimethoxy)-8-O-4′-neolignan (17), except that an allyl moiety was replaced by a propanyl-1,2-diol moietyCitation32, which was supported by the COSY correlations of H-7′/H-8′/H-9′ and HMBC cross-peaks of H-9′ with C-7′ and C-8′ (). In addition, the position of a propanyl-1,2-diol group at C-4′ was confirmed by the HMBC correlation of H-7′ with C-3′, C-4′, and C-5′ (). The negative Cotton effects of compound 18 at 240 and 276 nm in the ECD spectrum were similar to those of maceneolignan K and (−)-miliusfragranol B, revealing the absolute configuration of C-8 as R-formCitation38. The absolute configuration of C-8′ was assigned using the Snatzke’s methodCitation39. The positive effect at 305 nm in the ICD spectrum of compound 18 in Mo2(OAc)4 solution, provided evidence indicating the assignment of S configuration at C-8′ (Figure S3.11).
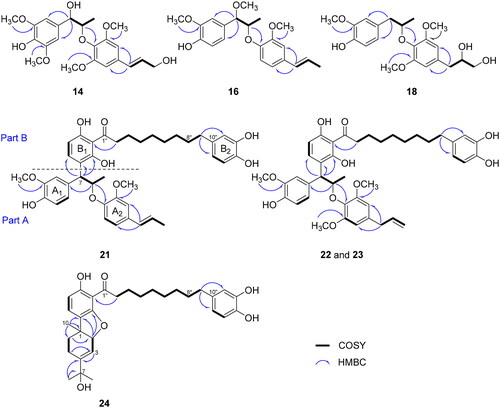
Myrifragranone A (21) was isolated as a yellow oil. Based on the HR-FAB-MS spectrum, the molecular formula was demonstrated as C41H48O9, with a positive ion at m/z 707.3202 [M + Na]+ (calcd. for C41H48O9Na, 707.3196) (Figure S4.10). According to the 1D NMR data, compound 21 comprised two phenylpropyl groups (part A) and a diphenylnonanoid moiety (part B), which was validated by 2D NMR spectra (). In part A, the NMR spectrum showed signals of two trisubstituted benzene rings [δH 6.97 (1H, d, J = 1.8 Hz, H-2), 6.68 (1H, d, J = 8.2 Hz, H-5), 6.77 (1H, dd, J = 8.2, 1.8 Hz, H-6), 6.91 (1H, d, J = 1.8 Hz, H-2′), 6.82 (1H, dd, J = 8.3, 1.8 Hz, H-5′), and 6.87 (1H, d, J = 8.3 Hz, H-6′)], a double bond [δH 6.32 (1H, dd, J = 15.7, 1.6 Hz, H-7′), 6.12 (1H, dd, J = 15.7, 6.6 Hz, H-8′)/δC 132.0 (C-7′), 124.4 (C-8′)], an oxygenated proton [δH 5.14 (1H, dt, J = 7.5, 6.0 Hz, H-8)], two methoxy groups [δH 3.77 (3H, s, 3-OCH3), 3.72 (3H, s, 2′-OCH3)/δC 56.3 (3-OCH3), 56.7 (2′-OCH3)], and two methyl groups [δH 1.22 (3H, d, J = 6.0 Hz, H-9) and 1.85 (3H, dd, J = 6.6, 1.5 Hz, H-9′)] (). The linkage of a propyl moiety between the A1 and A2 rings and a trans double bond with C-4′ of A2 ring was established by HMBC cross-peaks of H-8 with C-1 and C-1′, and H-7′ with C-3′ and C-5′, respectively ().
In part B, two ortho protons [δH 6.28 (1H, d, J = 8.5 Hz, H-18″) and 7.57 (1H, d, J = 8.5 Hz, H-19″)] exhibited an HMBC correlation with C-16″ (δC 111.0), C-17″ (δC 160.2), and C-21″ (δC 162.6), thereby establishing a tetrasubstituted benzene ring (B1). Another trisubstituted benzene ring (B2) was assigned by the HMBC cross-peaks of H-11″ with C-13″ (δC 144.0) and C-15″ (δC 120.6) and of H-14″ with C-10″ (δC 135.8) and C-13″ (δC 144.0). Additionally, the NMR spectrum showed the existence of a -(CH2)8- group [δH 1.33 (8H, m, H-4″–H-7″), 1.56 (2H, d, J = 6.7 Hz, H-8″), 1.67 (2H, m, H-3″), and 2.45 (2H, m, H-9″)], which was further supported by the COSY analysis (). The association between two aromatic rings via -(CH2)8- and carbonyl groups was established by analysing the HMBC spectrum. Finally, part B was accommodated to part A through the connection between C-7 and C-20″, as confirmed by the HMBC cross-peaks of H-7 with C-19″, C-20″, and C-21″ (). The relative configuration of 21 was determined as threo by NOESY correlation between H-7 and H-9 and the large coupling constant (J = 7.5 Hz) of H-7 and H-8 (Figure S8 and ). The absolute configuration of 21 was identified by integrating measured and calculated ECD. Gaussian 16 software was employed at the IEFPCM/B3LYP/6-311G+(d,p) level in MeOHCitation40. The ECD spectrum of compound 21 displayed positive Cotton effects at 220 and 273 nm and a negative effect at 240 nm, which were in accordance with the calculated ECD spectrum for the 7 R,8R configuration. By contrast, the 7S,8S configuration showed the opposite results (Figure S4.11).
Myrifragranone B (22) was obtained as a yellow oil, and its HR-FAB-MS spectrum revealed a positive ion at m/z 737.3309 [M + Na]+ (calcd for C42H50O10Na, 737.3302) (Figure S5.10), in agreement with the formula C42H50O10. The NMR data of 22 showed highly similarities to those of myrifragranone A (21), except for the addition of a methoxy group [δH 3.67 (3H, s, 6′-OCH3)/δC 56.3 (6′-OCH3)] and presence of an allyl group [δH 3.25 (1H, d, J = 6.6 Hz, H-7′), 5.92 (1H, ddt, J = 15.7, 10.0, 6.6 Hz, H-8′), 5.04 (2H, dd, J = 6.6, 1.5 Hz, H-9′)/δC 41.3 (C-7′), 138.7 (C-8′), 115.9 (C-9′)] in compound 22 instead of a propenyl group in compound 21 (). This evidence was confirmed by the COSY correlation of H-7′/H-8′/H-9′ and HMBC cross-peaks of H-7′ with C-3′, C-5′, and C-9′ (). The position of the additional methoxy group at C-6′ was determined by the correlation signal of 6′-OCH3 (δH 3.67) and C-6′ (δC 154.4) in HMBC spectrum. The threo-configuration of 22 was deduced based on the large coupling constants of (J = 7.7 Hz) H-7 and H-8 (). The ECD spectrum of 22 exhibited positive Cotton effects at 235 and 285 nm and a negative Cotton effect at 260 nm, similar to the calculated ECD spectrum of 7 R,8R (Figure S5.11). This similarity led to the conclusion that the absolute configuration of 22 is 7 R,8R.
Myrifragranone C (23) was obtained as a yellow oil. It has the same molecular formula (C42H50O10) as myrifragranone B (22) with a positive ion peak at m/z 737.3306 [M + Na]+ (calcd for C42H50O10Na, 737.3302) observed in the HR-FAB-MS spectrum (Figure S6.10). shows that the NMR data of 23 were closely similar to those of 22, except for the difference in the chemical shift of H-7 and H-8. The coupling constant of H-7 and H-8 (J = 8.2 Hz) indicated the threo configuration of 23. Moreover, the opposite optical rotation value of 23 ([α] −22.3, c 0.02 in CHCl3) instead of 22 ([α]
+42.9, c 0.02 in CHCl3) suggests that 22 and 23 are stereoisomers. By analysing the measured and calculated ECD spectra of 23, it was determined that the absolute configuration of 23 was 7S,8S (Figure S5.11).
Myrifragranone D (24) was obtained as a colourless oil. The molecular formula of 24 was determined as C31H40O6 based on the HR-FAB-MS spectrum at m/z 491.2800 [M–H2O + H]+ (calcd for C31H39O5, 491.2797) (Figure S7.10). The 1H- and 13C-NMR data of 24 revealed the presence of a diphenylnonanoid group (part B), similar to those of myrifragranones A–C (21–23). However, compound 24 had a monoterpene moiety (part A) instead of two phenylpropyl groups in compounds 21–23. This evidence was confirmed by the signals of a vinyl proton [δH 5.99 (1H, d, J = 4.0 Hz, H-3)], an oxygenated methine [δH 4.78 (1H, d, J = 4.0 Hz, H-2)], three methyl groups [δH 1.34 (3H, s, H-8), 1.36 (3H, s, H-9), 1.28 (3H, s, H-10)/δC 28.7 (C-8), 28.8 (C-9), 25.0 (C-10)], and two methylene groups [δH 2.04 (2H, m, H-5), 1.74 (2H, t, J = 4.0 Hz, H-6)/δC 21.9 (C-5), 32.8 (C-6)] in the 1H-NMR spectrum, along with the signals of 10 carbons, including three quaternary carbons [δC 41.3 (C-1), 151.1 (C-4), 73.5 (C-7)] in the 13C-NMR spectrum (). The correlation signals of H-2/H-3 and H-5/H-6 in the COSY spectrum and HMBC cross-peaks of H-2 with C-4, C-6, C-20′, and C-21′; H3-8 with C-4 and C-7; and H-10 with C-1, C-6, and C-20′ enabled us to confirm the position of the monoterpene moiety (). The NOESY spectrum of 24 revealed the correlation between H-2 and H-10, suggesting the cis-configuration of these protons (Figure S8). The absolute configuration of 24 was determined using the TD-DFT method at IEFPCM/B3LYP/6-311G+(d,p) level for (1S,2S)-24 and (1 R,2R)-24. The measured ECD of 24 showed two negative Cotton effects at 240 and 275 nm, which were consistent with those of (1 R,2R)-24 (Figure S7.11). This indicates that the absolute configuration of 24 was 1 R,2R.
Inhibitory effect of isolated compounds on sEH
The sEH inhibition of compounds from nutmegs (1 − 24) was investigated at a concentration of 100 µM. The results are summarised in , which shows that neolignans 1–18 exhibited inhibitory effects on sEH with inhibition rates lower than 65% at 100 µM. Among them, compounds 6 and 17 exhibited the strongest inhibitory effects (IC50 = 71.73 ± 0.49 and 67.74 ± 3.07 µM, respectively). However, they exhibited much weaker activity compared with the diarylnonanoid derivative group.
Table 3. sEH inhibitory activity, type of inhibitions, and inhibition constants of all isolated compounds (1–24).
Malabaricone B (19) demonstrated strong sEH inhibitory effects with 93.47% inhibition and an IC50 value of 18.58 ± 2.42 µM. Malabaricone C (20), which has an additional hydroxy group linked with C-12″, displayed weaker inhibitory effects compared to compound 19, with an IC50 value of 29.95 ± 2.01 µM. New compounds 21–24 also showed interesting sEH inhibition results. Compounds 21 and 24 displayed 67.59% and 76.65% sEH inhibition, with IC50 values of 46.35 ± 2.72 and 39.58 ± 3.74 µM, respectively. Compounds 22 and 23, which have functional groups in part A similar to those of compounds 6 and 17, two neolignans with potent sEH inhibitory activities, exhibited a significant increase in their inhibitory activity against sEH, with 85.59% and 90.32% inhibition and IC50 values of 23.56 ± 2.94 and 14.24 ± 0.91 µM, respectively. Moreover, the higher sEH inhibition of compound 23 compared with compound 22 emphasises the crucial role of the absolute configuration of 7S,8S in the chemical structure of diarylnonanoid derivatives in influencing their sEH inhibitory activity. This finding underscores the significance of the absolute configuration of these compounds in developing more potent sEH inhibitors.
sEH enzyme kinetics
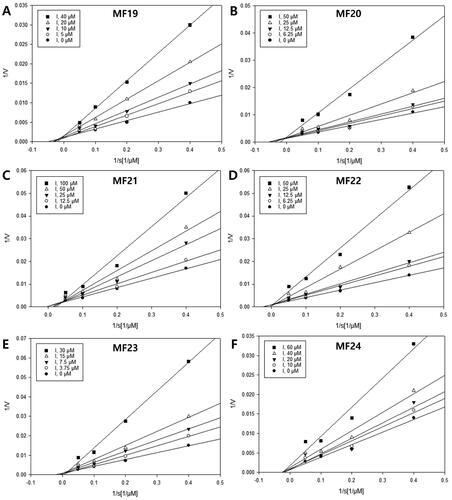
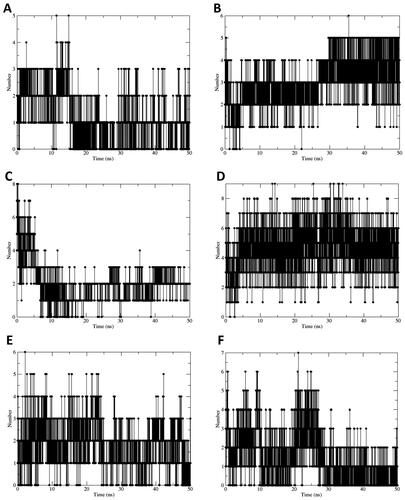
Enzyme kinetics studies were carried out to identify the inhibition mode and inhibition constant (represented by the Ki value) between active compounds (19–24) and sEH enzyme, using the Lineweaver–Burk and Dixon graphical methodsCitation41. shows that the Lineweaver–Burk plots of compounds 19–23 displayed a family of straight lines intersecting with the y- or 1/V axis. This evidence indicates that these compounds have a competitive inhibition mode. Meanwhile, the Lineweaver–Burk plots of compound 24 showed an intersection of straight lines with the x-axis, indicating that compound 24 functions as a non-competitive sEH inhibitor. The result suggests that the inhibition mode of compound 24 may be influenced by the presence of a monoterpene moiety.
Figure 3. Enzyme kinetics analysis. (A–F) Lineweaver–Burk plots of compounds 19–24, respectively.
The Ki values of active compounds were determined based on the Dixon plotsCitation42. The Ki values of two diarylnonanoids 19 and 20 were 20.9 and 11.1 µM, respectively (Figure S9). The Ki values of active compounds 21–23 binding with sEH were 23.3, 12.8, and 9.7 µM, respectively. On the other hand, compound 24, acting as a non-competitive sEH inhibitor, exhibited an inhibition constant with the sEH enzyme similar to the sEH-substrate complex, with a Ki value of 23.1 µM.
Molecular docking
Virtual screening techniques utilising molecular docking and MD methods have recently been used to identify potential sEH inhibitors by predicting the binding interaction and energy between sEH proteins and ligands. In this study, we conducted docking simulations using AutoDock Vina 1.1.2. The structure of human sEH (entry ID: 3ANS) was retrieved from the RCSB PDB databaseCitation18. The human sEH enzyme is composed of two folded domains that work independently: the N-terminus with phosphatase activity, and the C-terminusCitation43,Citation44. The latter is divided into the core domain (Ser235–Met369 and Met469–Asp545) and cap domain (Ser370–Asn468) and has hydrolase activity. These domains are connected by the NC loop. The active site of human sEH is situated within an L-shaped pocket buried inside the protein core. The pocket is entirely hydrophobic, and the branches are joined by a narrow bottleneck where the catalytic triad (Asp335, Asp496, and His524) and two stabilising residues (Tyr383 and Tyr466) are detectedCitation43,Citation45.
Diarylnonanoids 19 and 20, identified as competitive inhibitors, exhibited binding affinities to the active site of the sEH enzyme with values of −8.5 and −8.9 kcal/mol, respectively (, S10, and S11). The benzene ring and -(CH2)8- group of both compounds interacted hydrophobically with amino acid residues Leu408, Met419, Val498, and Trp525. Moreover, they formed van der Walls interaction with residue Asp496 of the catalytic triad. The long chain and benzene ring of compound 19 showed alkyl interactions with two essential residues, His524 and Tyr383, of the sEH active site, respectively. Whereas these two residues formed with compound 20 through hydrogen bonding interactions (Table S1).
Figure 4. (A–F) Docking interaction diagrams of sEH inhibition by compounds 19–24, respectively. (Green lines: hydrogen bonding interactions, pink lines: alkyl and π–alkyl interactions, magenta lines: π–π T-shaped interactions, and light green: van der Walls interactions with the corresponding residues of sEH).
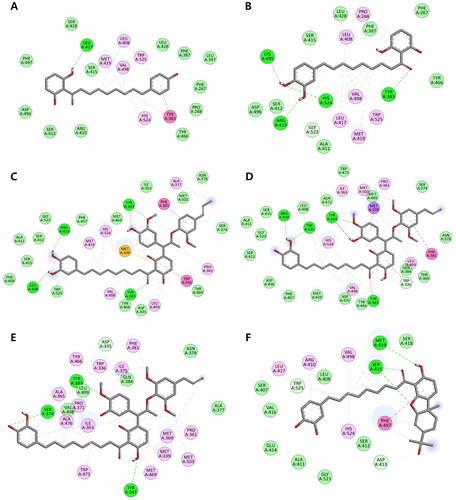
The docking simulations also showed that compounds 21–23 had binding affinity values of −10.5, −10.1, and −8.1 kcal/mol, respectively, and interacted with the sEH active site. The methoxy and carbonyl groups of compound 21 formed hydrogen bonds with Tyr343, Leu408, Arg410, and stabilising residue Tyr383. Its long chain and aromatic rings established alkyl interactions with Pro361, Ala377, Met419, Val498, and Leu499. In addition, compound 21 exhibited van der Waals and hydrophobic interactions with Asp335 in the catalytic triad and Tyr383, a stabilising residue, respectively. Compounds 22 and 23 demonstrated the most potent sEH inhibition as they can interact with two stabilising residues, Tyr383 and Tyr466, through hydrogen bonding and alkyl interactions, respectively. Moreover, compound 22 could interact with Asp496 and His524 (two residues of the catalytic triad) via van der Walls and alkyl interactions. Compound 23 exhibited hydrogen bonding interactions with the critical residue Asp335 and formed hydrophobic interactions with many critical residues in the C-terminus and active site, including Trp336, Met339, Pro361, Ile363, Ala365, Met369, Pro371, Ile375, Phe381, Ala476, Trp473, Tyr466, Met469, and Met503.
According to kinetic studies, compound 24 has been identified as a non-competitive sEH inhibitor, suggesting that it can bind to the allosteric site of the target enzyme. The allosteric sites of the human sEH enzyme are located outside the active site and situated in two regions around the two allosteric amino acid residues Cys423 and Cys522Citation46. The docking results show that compound 24 interacts with the allosteric site of sEH with a binding affinity value of −9.8 kcal/mol. Two aromatic rings of the compound display hydrogen bonding interactions with Met419 and Trp525, and alkyl interactions with Arg410 and Leu417 (). The long chain of this compound is connected with Val498 and a catalytic triad residue His524 via alkyl interactions. The monoterpene moiety of compound 24 exhibits hydrogen bonds and a π–π T-shaped interaction with residues Ser415 and Phe497, respectively. However, the moderate sEH inhibitory activity of this compound may be partially explained by its lack of interaction with the critical allosteric residue Cys522.
MD Simulations
MD simulations were carried out to explore the stability and dynamic behaviour of the protein-ligand complex simulation systemsCitation47. Various parameters, including potential energy, root mean square deviation (RMSD), root mean square fluctuation (RMSF), number of hydrogen bonds, centre of mass (CoM) distance, and superposition of active compounds (19–24), were examined throughout the 50 ns trajectory.
The potential interaction energy profiles between the active compounds and the sEH enzyme are depicted in . The average energy values for compounds 19–24 were determined to be −515570, −515345, −513837, −513912, −513471, and −515273 kJ/mol, respectively. These energy values remained stable throughout the 50 ns MD simulation. The barebone RMSD values of the complexes between the test compounds and sEH exhibited a rapid increase from 0.1 nm to 0.23 nm during the initial 7 ns of the equilibration phase in the MD process (). Subsequently, the RMSD values gradually increased from 0.23 nm to 0.25 nm between 8 ns and 24 ns. The RMSD values remained approximately 0.24 nm for the complexes of compounds 21–24 and 0.30 nm for the complex of compound 20 until the completion of the MD procedures. Whereas the RMSD value of sEH-compound 19 complex notably increased from 0.25 nm to 0.3 nm at 24 ns, followed by a gradual increase from 0.3 nm to 0.35 nm between 25 ns and 40 ns, and remained around 0.37 nm until the end of the simulation. Throughout the 50 ns MD simulation, the complexes formed by compounds 21–24 with the sEH enzyme exhibited a maximum RMSD value of 0.26 nm. This evidence indicates that these complexes maintained stability during the simulation period. Additionally, the linkage between the neolignan and monoterpene moieties in the chemical structure of compounds 21–24 contributed to the increased stability in their complexes with sEH compared with compounds 19 and 20.
Figure 5. Molecular dynamics simulation of compounds 19–24 with sEH protein. (A) Potential energy, (B) RMSD of protein backbone (compounds 19: black, 20: red, 21: green, 22: blue, 23: yellow, and 24: maroon), (C–H) RMSF of residues in the complexes between sEH and compounds 19–24, respectively.
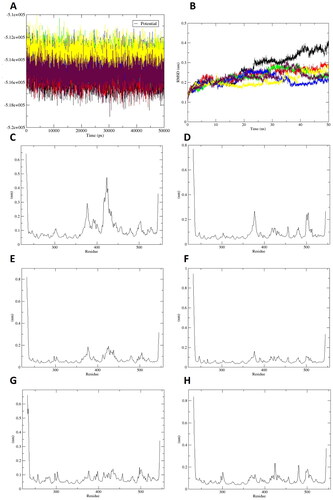
Greater flexibility during the MD simulation is demonstrated by higher RMSF valuesCitation48,Citation49. shows that the RMSF value of the sEH-compound 19 complex was approximately 0.3 nm at residue Tyr383 and 0.45 nm at residues Leu408–Met419. In the case of the sEH-compound 20 complex (), the RMSF value was 0.24 nm at residue Tyr383 and 0.23 nm at residues Phe497–Val498. The stabilising residue Tyr383 exhibited high RMSF value in compounds 19 and 20. Conversely, the complexes between sEH and compounds 21–24 showed RMSF values lower than 0.2 nm, indicating that these active compounds formed stable complexes with sEH in either the active or allosteric sites throughout the MD simulation ().
To gain a comprehensive understanding of the stability of the docked complexes, the hydrogen bonds formed within a distance of 0.35 nm between the sEH enzyme and the test compounds were calculated throughout the 50 ns MD simulations (). The results showed that compounds 19 and 20 formed 1–3 and 1–4 hydrogen bonds with the active site of sEH, respectively. Compounds 21 and 23 bound to the active site of sEH with 1–6 hydrogen bonds. In addition, compound 22 exhibited strong binding to the active site of sEH with 2–8 hydrogen bonds. Whereas compound 24 formed 1–6 hydrogen bonds with the allosteric binding site of the sEH enzyme. The CoM distance was analysed, and the results are depicted in Figure S12. The average CoM distances between the target enzyme and compounds 19–23 ranged from 1.0 nm to 1.5 nm, indicating relatively tight binding. Compound 24 exhibited a less tight binding to sEH than compounds 19–23, with an average CoM distance of 1.8 nm. The superposition of compounds 19–24 with the binding site of sEH during the 50 ns MD procedure is presented in Figure S13. Compared with the molecular docking results, the MD simulation results for all test compounds showed a consistent binding mode, indicating the high reliability of the MD simulation conducted in this study.
Figure 6. (A–F) Number of hydrogen bonds formed during molecular dynamics simulation by the complexes of sEH with compounds 19–24, respectively.
Physicochemical and pharmacokinetic properties
Physicochemical and pharmacokinetic properties of active compounds 19–24 were investigated using SwissADME web-based applicationCitation50. The assessment of drug-likeness properties often relies on rules, with Lipinski′s Rule of Five (Pfizer) being the most well-knownCitation51,Citation52. This rule suggests that a compound could be a good candidate for a drug if it does not violate more than two of the following criteria: the molecular weight (MWT) should not exceed 500, the Moriguchi Log P (MLogP) should be smaller than 4.15, hydrogen-bond donors (HBD) should be ≤ 5, and hydrogen-bond acceptors (HBA) should be ≤ 10Citation51–54. According to the predicted results presented in , drug-like properties revealed that all active compounds passed the Lipinski filter with no more than two violations.
Table 4. Physicochemical and pharmacokinetic properties of compounds 19–24.
The blood-brain barrier (BBB) acts as a protective barrier, blocking neurotoxic effects from accessing the extracellular fluid of the central nervous systemCitation55. The results revealed that none of the active compounds are BBB permeable. Except for compound 24, all active compounds do not act as substrates for P-glycoprotein. This implies that the effectiveness of these compounds is unlikely to be influenced by P-glycoprotein over-expressionCitation54. Furthermore, compounds 19 and 20 were predicted to exhibit high absorption in the gastrointestinal (GI) tract, whereas compounds 21–24 were predicted to show low absorption in the GI tract.
Conclusions
A phytochemical investigation of nutmegs discovered two new neolignans (14 and 18), four new diarylnonanoid derivatives (21–24), and 18 known compounds. The ECD method identified that compound 16 had an absolute configuration. Moreover, the sEH inhibition of these metabolites was assessed using AUDA as a positive control (IC50 = 33.68 nM). The results showed that known compounds (19 and 20) and four new compounds (21–24) exhibited strong inhibitory effects on sEH, with IC50 values ranging from 14.24 to 46.35 µM. Enzyme kinetics and docking studies revealed that compounds 19–23 acted as competitive inhibitors and exhibited binding to the sEH active site, whereas compound 24 exhibited a non-competitive mode of inhibition and interacted with the allosteric site of the target enzyme. Compounds 19 and 20 exhibited strong inhibitory effects with IC50 values of 18.58 and 29.95 µM, respectively. However, these compounds have a less tight binding to the sEH enzyme as indicated by the high RMSD (0.3–0.35 nm) and RMSF values for the stabilising residue Tyr383. By contrast, the complexes of sEH with new compounds 21–24 showed tight binding to the active or allosteric site of the target enzyme, with an RMSD value of approximately 0.24 nm and RMSF value lower than 0.2 nm at the binding site. This suggests that the presence of neolignan and monoterpene moieties in the structure of these metabolites may play a significant role in increasing the stability of their complexes with sEH compared with compounds 19 and 20. Our findings provide a preliminary understanding of the sEH inhibition of active compounds 19–24 as potential treatment from natural sources. Based on the findings of this study, further investigations could be conducted to develop and discover new sEH inhibitors.
Author contributions
Vu Thi Oanh: Investigation, Writing- Original draft. Nguyen Viet Phong: Investigation, Writing- Original draft. Byung Sun Min: Resources. Seo Young Yang: Methodology, Software, Formal analysis, Writing-Review and Editing. Jeong Ah Kim: Conceptualisation, Writing-Review and Editing, Supervision.
Supplemental Material
Download PDF (8.9 MB)Acknowledgements
The authors are thankful to the Korea Basic Science Institute – Daegu centre for the MS and CD measurement services.
Disclosure statement
The authors report there are no competing interests to declare.
Additional information
Funding
References
- Atone J, Wagner K, Hashimoto K, Hammock BD. Cytochrome P450 derived epoxidized fatty acids as a therapeutic tool against neuroinflammatory diseases. Prostaglandins Other Lipid Mediat. 2020;147:106385.
- Lukin A, Kramer J, Hartmann M, Weizel L, Hernandez-Olmos V, Falahati K, Burghardt I, Kalinchenkova N, Bagnyukova D, Zhurilo N, et al. Discovery of polar spirocyclic orally bioavailable urea inhibitors of soluble epoxide hydrolase. Bioorg Chem. 2018;80:655–667.
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43(1):55–90.
- Fretland AJ, Omiecinski CJ. Epoxide hydrolases: biochemistry and molecular biology. Chem Biol Interact. 2000;129(1–2):41–59.
- Huang H, Weng J, Wang M-H. EETs/sEH in diabetes and obesity-induced cardiovascular diseases. Prostaglandins Other Lipid Mediat. 2016;125:80–89.
- Briggs WH, Xiao H, Parkin KL, Shen C, Goldman IL. Differential inhibition of human platelet aggregation by selected Allium thiosulfinates. J Agric Food Chem. 2000;48(11):5731–5735.
- Halade GV, Lee DH. Inflammation and resolution signaling in cardiac repair and heart failure. eBioMedicine. 2022;79:103992.
- Gur Maz T, Koc B, Jordan PM, İbiş K, Çalışkan B, Werz O, Banoglu E. Benzoxazolone-5-urea derivatives as human soluble epoxide hydrolase (sEH) inhibitors. ACS Omega. 2023;8(2):2445–2454.
- Imig JD, Carpenter MA, Shaw S. The soluble epoxide hydrolase inhibitor AR9281 decreases blood pressure, ameliorates renal injury and improves vascular function in hypertension. Pharmaceuticals). 2009;2(3):217–227.
- Kim JH, Park JS, Lee YJ, Choi S, Kim YH, Yang SY. Inhibition of soluble epoxide hydrolase by phytochemical constituents of the root bark of Ulmus davidiana var. japonica. J Enzyme Inhib Med Chem. 2021;36(1):1049–1055.
- Phong NV, Zhao Y, Min BS, Yang SY, Kim JA. Inhibitory activity of bioactive phloroglucinols from the rhizomes of Dryopteris crassirhizoma on Escherichia coli β-glucuronidase: kinetic analysis and molecular docking studies. Metabolites. 2022;12(10):938.
- Ha MT, Vu NK, Tran TH, Kim JA, Woo MH, Min BS. Phytochemical and pharmacological properties of Myristica fragrans Houtt.: an updated review. Arch Pharm Res. 2020;43(11):1067–1092.
- Lee C-J, Huang C-W, Chen L-G, Wang C-C. (+)-Erythro-Δ8′-7S,8R-dihydroxy-3,3′,5′-trimethoxy-8-O-4′-neolignan, an anti-acne component in degreasing Myristica fragrans Houtt. Molecules. 2020;25(19):4563.
- Barceloux DG. Nutmeg (Myristica fragrans Houtt.). Dis Mon. 2009;55(6):373–379.
- Cao G-Y, Xu W, Yang X-W, Gonzalez FJ, Li F. New neolignans from the seeds of Myristica fragrans that inhibit nitric oxide production. Food Chem. 2015;173(:231–237.
- Wang CY, Lee S, Jang H-J, Su XD, Wang H-S, Kim YH, Yang SY. Inhibition potential of phenolic constituents from the aerial parts of Tetrastigma hemsleyanum against soluble epoxide hydrolase and nitric oxide synthase. J Enzyme Inhib Med Chem. 2019;34(1):753–760.
- Jiang J-x, Guan Y, Shen H-j, Jia Y-l, Shen J, Zhang L-h, Liu Q, Zhu Y-l, Xie Q-m. Inhibition of soluble epoxide hydrolase attenuates airway remodeling in a chronic asthma model. Eur J Pharmacol. 2020;868:172874.
- Tanaka D, Tsuda Y, Shiyama T, Nishimura T, Chiyo N, Tominaga Y, Sawada N, Mimoto T, Kusunose N. A practical use of ligand efficiency indices out of the fragment-based approach: ligand efficiency-guided lead identification of soluble epoxide hydrolase inhibitors. J Med Chem. 2011;54(3):851–857.
- Eawsakul K, Ongtanasup T, Ngamdokmai N, Bunluepuech K. Alpha-glucosidase inhibitory activities of astilbin contained in Bauhinia strychnifolia Craib. stems: an investigation by in silico and in vitro studies. BMC Complement Med Ther. 2023;23(1):25.
- Ongtanasup T, Prommee N, Jampa O, Limcharoen T, Wanmasae S, Nissapatorn V, Paul AK, Pereira ML, Wilairatana P, Nasongkla N, et al. The cholesterol-modulating effect of the new herbal medicinal recipe from yellow vine (Coscinium fenestratum (Goetgh.)), ginger (Zingiber officinale Roscoe.), and safflower (Carthamus tinctorius L.) on suppressing PCSK9 expression to upregulate LDLR expression in HepG2 cells. Plants. 2022;11(14):1835.
- Nasongkla N, Tuchinda P, Munyoo B, Eawsakul K. Preparation and characterization of MUC-30-loaded polymeric micelles against MCF-7 cell lines using molecular docking methods and in vitro study. Evid Based Complement Alternat Med. 2021;2021:5597681–5597689.
- Phong NV, Anh DTN, Chae HY, Yang SY, Kwon MJ, Min BS, Kim JA. Anti-inflammatory activity and cytotoxicity against ovarian cancer cell lines by amide alkaloids and piperic esters isolated from Piper longum fruits: In vitro assessments and molecular docking simulation. Bioorg Chem. 2022;128:106072.
- Khoa NM, Phong NV, Yang SY, Min BS, Kim JA. Spectroscopic analysis, kinetic mechanism, computational docking, and molecular dynamics of active metabolites from the aerial parts of Astragalus membranaceus Bunge as tyrosinase inhibitors. Bioorg Chem. 2023;134:106464.
- Li F, Yang X-W. Biotransformation of myrislignan by rat liver microsomes in vitro. Phytochemistry. 2008;69(3):765–771.
- Duan L, Tao H-W, Hao X, Gu Q-Q, Zhu W-M. Cytotoxic and antioxidative phenolic compounds from the traditional Chinese medicinal plant, Myristica fragrans. Planta Med. 2009;75(11):1241–1245.
- Zacchino SA, Badano H. Enantioselective synthesis and absolute configuration assignment of erythro(3,4,5-trimethoxy-7-hydroxy-1′-allyl-2′, 6′-dimethoxy)-8.0.4′-neolignan, isolated from mace (Myristica fragrans). J Nat Prod. 1988;51(6):1261–1265.
- Rye CE, Barker D. Asymmetric synthesis and anti-protozoal activity of the 8,4′-oxyneolignans virolin, surinamensin and analogues. Eur J Med Chem. 2013;60:240–248.
- Hattori M, Hada S, Shu Y-Z, Kakiuchi N, Namba T. New acyclic bis-phenylpropanoids from the aril of Myristica fragrans. Chem Pharm Bull. 1987;35(2):668–674.
- Francis SK, James B, Varughese S, Nair MS. Phytochemical investigation on Myristica fragrans stem bark. Nat Prod Res. 2019;33(8):1204–1208.
- Morikawa T, Hachiman I, Matsuo K, Nishida E, Ninomiya K, Hayakawa T, Yoshie O, Muraoka O, Nakayama T. Neolignans from the arils of Myristica fragrans as potent antagonists of CC chemokine receptor 3. J Nat Prod. 2016;79(8):2005–2013.
- Olajide OA, Ajayi FF, Ekhelar AI, Awe SO, Makinde JM, Alada ARA. Biological effects of Myristica fragrans (nutmeg) extract. Phytother Res. 1999;13(4):344–345.
- Kasahara H, Miyazawa M, Kameoka H. Biotransformation of an acyclic neolignan in rats. Phytochemistry. 1995;38(2):343–346.
- Kundu K, Nayak SK. Total syntheses of malabaricones B and C via a cross-metathesis strategy. J Nat Prod. 2017;80(6):1776–1782.
- Besombes S, Robert D, Utille J-P, Taravel FR, Mazeau K. Molecular modeling of syringyl and p-hydroxyphenyl β-O-4 dimers. Comparative study of the computed and experimental conformational properties of lignin β-O-4 model compounds. J Agric Food Chem. 2003;51(1):34–42.
- Arnoldi A, Merlini L. Asymmetric synthesis of 3-methyl-2-phenyl-1,4-benzodioxanes. Absolute configuration of the neolignans eusiderin and eusiderin C and D. J Chem Soc, Perkin Trans 1. 1985;17:2555–2557.
- Hada S, Hattori M, Tezuka Y, Kikuchi T, Namba T. New neolignans and lignans from the aril of Myristica fragrans. Phytochemistry. 1988;27(2):563–568.
- Gao X, Shen Y, Yang L, Shu L, Li G, Hu Q-F. 8-O-4′-neolignans from flower buds of Magnolia fargesii and their biological activities. J Braz Chem Soc. 2012;23(7):1274–1279.
- Morikawa T, Hachiman I, Ninomiya K, Hata H, Sugawara K, Muraoka O, Matsuda H. Degranulation inhibitors from the arils of Myristica fragrans in antigen-stimulated rat basophilic leukemia cells. J Nat Med. 2018;72(2):464–473.
- Di Bari L, Pescitelli G, Pratelli C, Pini D, Salvadori P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke’s method revisited. J Org Chem. 2001;66(14):4819–4825.
- Huong PTM, Phong NV, Huong NT, Trang DT, Thao DT, Cuong NX, Nam NH, Van Thanh N. Aplydactylonins A-C, three new sesquiterpenes from the Vietnamese sea hare Aplysia dactylomela and their cytotoxicity. J Nat Med. 2022;76(1):210–219.
- Whiteley CG. Enzyme kinetics: partial and complete non-competitive inhibition. Biochem Educ. 1999;27(1):15–18.
- Dixon M. The determination of enzyme inhibitor constants. Biochem J. 1953;55(1):170–171.
- Bzówka M, Mitusińska K, Hopko K, Góra A. Computational insights into the known inhibitors of human soluble epoxide hydrolase. Drug Discov Today. 2021;26(8):1914–1921.
- Zhao W-Y, Yan J-J, Zhang M, Wang C, Feng L, Lv X, Huo X-K, Sun C-P, Chen L-X, Ma X-C. Natural soluble epoxide hydrolase inhibitors from Inula britanica and their potential interactions with soluble epoxide hydrolase: insight from inhibition kinetics and molecular dynamics. Chem Biol Interact. 2021;345:109571.
- Gomez GA, Morisseau C, Hammock BD, Christianson DW. Structure of human epoxide hydrolase reveals mechanistic inferences on bifunctional catalysis in epoxide and phosphate ester hydrolysis. Biochemistry. 2004;43(16):4716–4723.
- Qiu Q, Abis G, Mattingly-Peck F, Lynham S, Fraternali F, Conte MR. Allosteric regulation of the soluble epoxide hydrolase by nitro fatty acids: a combined experimental and computational approach. J Mol Biol. 2022;434(17):167600.
- Jo AR, Kim JH, Yan X-T, Yang SY, Kim YH. Soluble epoxide hydrolase inhibitory components from Rheum undulatum and in silico approach. J Enzyme Inhib Med Chem. 2016;31(sup2):70–78.
- Phong NV, Yang SY, Min BS, Kim JA. Insights into the inhibitory activity and mechanism of action of flavonoids from the stems and branches of Acer mono Maxim. against α-glucosidase via kinetic analysis, molecular docking, and molecular dynamics simulations. J Mol Struct. 2023;1282:135188.
- Ashiru MA, Ogunyemi SO, Temionu OR, Ajibare AC, Cicero-Mfon NC, Ihekuna OA, Jagun MO, Abdulmumin L, Adisa QK, Asibor YE, et al. Identification of EGFR inhibitors as potential agents for cancer therapy: pharmacophore-based modeling, molecular docking, and molecular dynamics investigations. J Mol Model. 2023;29(5):128.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25.
- Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4(2):90–98.
- Ongtanasup T, Wanmasae S, Srisang S, Manaspon C, Net-Anong S, Eawsakul K. In silico investigation of ACE2 and the main protease of SARS-CoV-2 with phytochemicals from Myristica fragrans (Houtt.) for the discovery of a novel COVID-19 drug. Saudi J Biol Sci. 2022;29(9):103389.
- Ongtanasup T, Mazumder A, Dwivedi A, Eawsakul K. Homology modeling, molecular docking, molecular dynamic simulation, and drug-likeness of the modified alpha-mangostin against the β-tubulin protein of Acanthamoeba keratitis. Molecules. 2022;27(19):6338.
- Meng F, Xi Y, Huang J, Ayers PW. A curated diverse molecular database of blood-brain barrier permeability with chemical descriptors. Sci Data. 2021;8(1):289.