
Abstract
Oxytocin (OT) is a neurohypophyseal peptide hormone containing a disulphide-bridged pseudocyclic conformation. The biomedical use of OT peptides is limited amongst others by disadvantageous pharmacokinetic parameters. To increase the stability of OT by replacing the disulphide bridge with the stable and more rigid [1,2,3]triazol-1-yl moiety, we employed the Cu2+-catalysed side chain-to-side chain azide-alkyne 1,3-cycloaddition. Here we report the design, synthesis, conformational analysis, and in vitro pharmacological activity of a homologous series of Cα1-to-Cα6 side chain-to-side chain [1,2,3]triazol-1-yl-containing OT analogues differing in the length of the bridge, location, and orientation of the linking moiety. Exploiting this macrocyclisation approach, it was possible to generate a systematic series of compounds providing interesting insight into the structure-conformation-function relationship of OT. Most analogues were able to adopt similar conformation to endogenous OT in water, namely, a type I β-turn. This approach may in the future generate stabilised pharmacological peptide tools to advance understanding of OT physiology.
Graphical Abstract
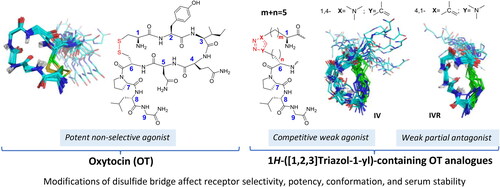
Introduction
Oxytocin (OT)Citation1 is a neurohypophyseal heterodetic cyclic nonapeptide centrally expressed in the hypothalamus and secreted by the posterior pituitary gland. It has important biological functions mediated by interaction with its cognate oxytocin receptor (OTR) – a member of the G protein-coupled receptors (GPCRs). In the central nervous system (CNS) OT regulates complex social behaviours such as social bonding, pair attachment, maternal care, stress and anxietyCitation2. In the periphery, OT is produced in several organs such as the reproductive systemCitation3,Citation4, GI tractCitation5, heartCitation6 and boneCitation7 and is involved in uterine smooth muscle contractility during parturition and milk ejection during lactationCitation8,Citation9, and has also analgesic/anti-inflammatory activityCitation10,Citation11.
Targeting peripheral OTR has been implemented in the clinic as drug for induction of labour, prevention of postpartum haemorrhage and lactation enhancerCitation12. Latest research suggests that OTR is an attractive target for drug development in many therapeutic areas for instance in cancerCitation13,Citation14. OT is not selective for the OTR; it also interacts with the related vasopressin (VP) receptors (VP receptors, i.e. V1a, V1b and V2R. The two endogenous neuropeptides OT and VP have a conserved Cys(1)-to-Cys(6) disulphide-bridge and differ only in 2 amino acid residues IleCitation3 and LeuCitation7 (in OT) and PheCitation3 and ArgCitation7 (in VP)Citation15. The four OT/VP receptors are widely distributed, highly conserved and are partially co-localised in central and peripheral neurons and in many peripheral organsCitation1. As a result, this non-selective and integrated system, which is comprised of OT/VP and their cognate receptors, activates a plethora of central and peripheral activities that are difficult to discern physiologically and target pharmacologicallyCitation16.
The development of OTR selective and functionally biased ligands and the correspondence of their cell-based pharmacology to in vivo animal pharmacology and eventually clinical trials are a major challenge due to species and tissue specific signalling properties of the OT/OTR system. Moreover, overcoming the limited stability in aqueous solution, the high susceptibility to metabolic degradation and the inherent chemical reactivity of the disulphide bond without compromising the putative bioactive conformation are formidable tasks. Evidently, the heterodetic cyclic structure of OT is indispensable and is essential for binding to OTR and functional activityCitation17. Oxytocin preferentially adopts a type I β-turn conformation in water, centred on Ile(3)-Gln(4) residues, while the predominant conformations presented both in the crystal structure and in DMSO solution are type II β-turns stabilised by a hydrogen bond between the C = O of Tyr(2) and the N-H of Asn(5) and a type III β-turn stabilised by a hydrogen bond between C = O of Cys(6) and N-H of Gly(9). In addition, another hydrogen bond between the C = O of Asn(5) side chain and the N-H of Leu(8) is observed in DMSOCitation18–22. Linearised analogues [Ala(1,6)]- and [Ser(1,6)]OT for instance are devoid of any contractile activity on rat mammary gland and isolated uterusCitation17. The putative deficiencies of the disulphide-containing peptide pharmaceuticals are their transitory nature resulting from poor in vivo stability due to the inherent reactivity of the disulphide function. Disulphide-containing peptides may be subjected to reduction by enzymes such as glutathione reductase and thioredoxin reductase, or attack by nucleophilic and basic agents leading to β-elimination, homolytic cleavage, sequestration to other thio-containing molecules and polymerisationCitation23.
Several approaches to mitigate the aforementioned drawbackCitation24–40 involved substitution of the disulphide containing bridge connecting Cα(1)-to-Cα(6) in OT by more stable tethers such as amidesCitation41, thioethersCitation42, diselenidesCitation39, ditelluridesCitation43,Citation44, and mono- or di-carbon-based bridgesCitation25,Citation26 and were sometimes accompanied by contracting or expanding the endogenous disulphide containing 20-membered ring. In general, these modifications led to a variable loss of biological activity but resulted in improvement of metabolic plasma stabilityCitation39.
The emergence of the disubstituted 1H-[1,2,3]triazol-1-yl moiety, generated by the copper-catalysed azide-alkyne cycloaddition (CuAAC) also known as the copper-catalysed Huisgen cycloaddition, is an attractive isosteric and bioequivalent surrogate of the amide bond and a mimetic of the disulphide functionCitation45–52. Hence we explored this technology as a replacement of the disulphide for side chain-to-side chain macrocyclisation of OTCitation53. Synthetically, the employment of α-amino acid building blocks presenting side chains modified by either ω-azido or ω-ethynyl functions, which are orthogonal to the commonly employed protecting groups used in peptide synthesis, and the selective and mild orthogonal CuAAC that is compatible with a wide range of peptide synthetic methodologies both in solution and solid state are very appealingCitation52–54. Moreover, the numerous, versatile and subtle structural permutations accessible in a single given pairwise bridgehead location of the side chain-to-side chain 1H-[1,2,3]triazol-1-yl-bridged modification are very attractive. These include the size of the bridge, the location within, and the orientation, either 1,4- or 4,1-1H-[1,2,3]triazol-1-yl moiety, in the bridge (). As such, it allows access to careful fine tuning of molecular flexibility, side chain-to-side chain, side chain-to-backbone and intermolecular interactions. In addition, the chemical and metabolic stability of the 1H-[1,2,3]triazol-1-yl moiety surpasses the inherent susceptibility of the disulphide bond and even the amide functionCitation51. Our leading examples of side chain-to-side chain (1H-[1,2,3]triazol-1-yl)-bridged cyclopeptidesCitation47,Citation53,Citation55–57 together with numerous reports from other laboratories attest to the great potential of this side chain-to-side chain bridging modality. For example, introduction of Cα(i)-to-Cα(i + 4) 1,4-(1H-[1,2,3]triazol-1-yl)-containing bridge into hPTHrP-derived sequence stabilised preferentially turn-helix structuresCitation53. Bridging positions 5 and 10 in Nα-Ac[Nle(4),D-Phe(7)]αMSH(4-10)NH2 by either 1,4- or 4,1-(1H-[1,2,3]triazol-1-yl])-containing bridges of the same size but different in the location of the triazolyl moiety demonstrated how subtle changes differentially affect the critical type I β-turn and the biological activity on 4 human melanocortin receptor subtypesCitation56. Application of a similar approach to octreotide, a potent agonist of the somatostatin receptor, yielded 1H-[1,2,3]triazol-1-yl containing analogues that reproduced closely only the β-turn conformation and were lacking the dynamic 310-helix – β-turn conformational equilibrium observed in the parent octreotideCitation57.
Figure 1. All permutations replacing the disulphide bridge in OT by the side chain-to-side chain 1H-[1,2,3]triazol-1-yl-bridged modification. (A) Partial schematic structure of the 1,4- and 4,1-(1H-[1,2,3]triazol-1-yl) containing permutations included in this study, where m + n = 2–5 and m = 1–4 and n = 1–4 and (B) OT schematic structure.
![Figure 1. All permutations replacing the disulphide bridge in OT by the side chain-to-side chain 1H-[1,2,3]triazol-1-yl-bridged modification. (A) Partial schematic structure of the 1,4- and 4,1-(1H-[1,2,3]triazol-1-yl) containing permutations included in this study, where m + n = 2–5 and m = 1–4 and n = 1–4 and (B) OT schematic structure.](/cms/asset/02ec7ee4-ce8d-4b5e-b350-8c92a738e705/ienz_a_2254019_f0001_c.jpg)
Here, we evaluated the replacement of the disulphide-containing bridge in OT by 1H-[1,2,3]triazol-1-yl-containing bridge to serve an unmet need for metabolically stable OT-derived drug candidates and chemical probes with desired pharmacological activities such as improved subtype selectivity. Based on our accumulated expertise in employing the CuAAC methodology introducing intramolecular side chain-to-side chain macrocyclisation we carried out a systematic replacement of the side chain-to-side chain disulphide bridge in OT by extensive series of homologous permutations (). We report the design, synthesis, conformational and in vitro biological characterisation of a series of Cα(1)-to- Cα(6)side chain-to-side chain disubstituted-1,4-/4,1-(1H-[1,2,3]triazol-1-yl)-bridged OT analogues presenting a systematic permutation of ring size, location and orientation of the triazolyl moiety. Our study offers insight on the impact of these structural modifications on the in vitro activity and solution conformation of the novel 1H-[1,2,3]triazol-1-yl)-bridged series of OT analogues.
Results and discussion
Synthesis strategy
We designed a series of 1,4-/4,1-disubstituted-(1H-[1,2,3]triazol-1-yl)-bridged i-to-i + 5 side chain-to-side chain OT homologues that vary in the size of the heterodetic ring-forming linker connecting Cα(1)-to-Cα(6), the location of the [1,2,3]triazole-1-yl moiety in the linker Cα(1)-(CH2)m-1,4-/4,1-disubstituted-(1H-[1,2,3]triazol-1-yl)-(CH2)n-Cα(6) where m + n = 5 (m and n = 1, 2, 3 and 4), m + n = 3 (m and n = 1 or 2) or m + n = 2 (m and n = 1) and its orientation in the linker (Schemes 1 and 2). The synthesis was carried out by microwave-assisted solid-phase peptide synthesis (MW-SPPS) following our Fmoc/tBu published methodology as previously reportedCitation52,Citation56. We used Nα-Fmoc-ω-azido- and Nα-Fmoc-ω-ynoic-α-amino acids as building blocks to generate two set of precursors that will differ in the order of incorporating these two complementing blocks. Placing the Nα-Fmoc-ω-azido- and Nα-Fmoc-ω-ynoic-α-amino acids in position 1 and 6, respectively, will generate the set of 1,4-disubstituted-(1H-[1,2,3]triazol-1-yl)-bridged analogues (Scheme 1, precursors I’–VII’). Placing the same complementing building blocks in the reversed order, namely positions 6 and 1, respectively, will generate the set of 4,1-disubstituted-(1H-[1,2,3]triazol-1-yl)-bridged analogues (Scheme 2, precursors IR’–VIIR’).
Scheme 1. Sequences of the linear precursors H-Xaa1-Tyr2-Ile3-Gln4-Asn5-Yaa6-Pro7-Leu8-Gly9-NH2 (I’–VII’) and of the 1,4-(1H-[1,2,3]triazol-1-yl) containing cyclopeptides (I–VII), {[H-Ala(&1)-Tyr2-Ile3-Gln4-Asn5-Ala(&2)-Pro7-Leu8-Gly9-NH2]} {[1-[&1(CH2)m]-1H-1,2,3-triazol-4-yl]-(CH2)n&1]} Citation53 with m + n = 5, 3 or 2 and m and n = 1–4, 1–2, or 1. All amino acids are of the L configuration.
![Scheme 1. Sequences of the linear precursors H-Xaa1-Tyr2-Ile3-Gln4-Asn5-Yaa6-Pro7-Leu8-Gly9-NH2 (I’–VII’) and of the 1,4-(1H-[1,2,3]triazol-1-yl) containing cyclopeptides (I–VII), {[H-Ala(&1)-Tyr2-Ile3-Gln4-Asn5-Ala(&2)-Pro7-Leu8-Gly9-NH2]} {[1-[&1(CH2)m]-1H-1,2,3-triazol-4-yl]-(CH2)n&1]} Citation53 with m + n = 5, 3 or 2 and m and n = 1–4, 1–2, or 1. All amino acids are of the L configuration.](/cms/asset/caa55c46-d7af-43f2-90f5-945966121a3f/ienz_a_2254019_sch0001_c.jpg)
Scheme 2. Sequences of the linear precursors H-Yaa1-Tyr2-Ile3-Gln4-Asn5-Xaa6-Pro7-Leu8-Gly9-NH2 (IR’–VIIR’) and of the 4,1-(1H-[1,2,3]triazolyl containing cyclopeptides (IR–VIIR), {[H-Ala(&1)-Tyr2-Ile3-Gln4-Asn5-Ala(&2)-Pro7-Leu8-Gly9-NH2]} {[1-[&1(CH2)m]-1H-1,2,3-triazol-4-yl]-(CH2)n&1]} Citation53 with m + n = 5, 3, or 2 and m and n = 1–4, 1–2, or 1. All amino acids are of the L configuration.
![Scheme 2. Sequences of the linear precursors H-Yaa1-Tyr2-Ile3-Gln4-Asn5-Xaa6-Pro7-Leu8-Gly9-NH2 (IR’–VIIR’) and of the 4,1-(1H-[1,2,3]triazolyl containing cyclopeptides (IR–VIIR), {[H-Ala(&1)-Tyr2-Ile3-Gln4-Asn5-Ala(&2)-Pro7-Leu8-Gly9-NH2]} {[1-[&1(CH2)m]-1H-1,2,3-triazol-4-yl]-(CH2)n&1]} Citation53 with m + n = 5, 3, or 2 and m and n = 1–4, 1–2, or 1. All amino acids are of the L configuration.](/cms/asset/6fc885a1-5cc7-4962-83e8-5d8753395f84/ienz_a_2254019_sch0002_c.jpg)
Deprotection and cleavage from resin generated the linear precursors I’–VII’ and IR’–VIIR’ that were then submitted to CuAAC-mediated solution phase intramolecular macrocyclisation procedure as previously reportedCitation58,Citation59. Following this procedure we converted successfully the linear precursors I’–VI’ and IR’–VIR’ to the respective cyclic OT homologous series I–VI and IR–VIR with yields ranging from 15 to 45% (Scheme 3). However, the same procedure when applied to the linear precursors VII’ and VIIR’ failed to generate the anticipated respective VII and VIIR. In this series the cyclic analogues VII and VIIR present the shortest bridge (–CH2-1,4-/4,1-1H-([1,2,3]triazol-1-yl)-CH2–), which could lead to a slow and inefficient solution phase intramolecular cyclisation. On the other hand, performing the intramolecular on-resin MW-assisted CuAAC-mediated macrocyclisation following the method reported by D’Ercole et al.Citation52, yielded the anticipated macrocyclic products VII and VIIR in 15% and 40%, respectively (Scheme 3). The critical on-resin "infinite" dilution of the resin-bound linear precursor enabled the effective CuAAC-mediated intramolecular cyclisation and yielded the anticipated products. The OT used in this study as a reference peptide in the biological assays was synthesised by Fmoc/tBu SPPS strategy. The purity of each peptide in the entire series of 1,4- and 4,1-(disubstituted (1H-[1,2,3]-triazol-1-yl)-containing OT analogues I–VII and IR–VIIR was established by RP-HPLC-MS analysis and was ≥95% (). The structural identity and integrity of OT and analogues was confirmed by LC − MS (see Figure S1 A-Q) as well as 1H-, 13C-, and 15N-NMR (see Tables S1–S15).
Scheme 3. General procedure CuAAC intramolecular macrocyclisation of either 1,4- or 4,1-disubstituted-(1H-[1,2,3]triazol-1-yl) containing OT analogues (A and B respectively) carried out in solution (I–VI and IR–VIR) (a)Citation47,Citation56 (R = NH2) or by on-resin microwave-assisted strategy (VII–VIIR) (b)Citation52 (X= tBu; Y = Trt; R = Tentagel® S RAM resin).
![Scheme 3. General procedure CuAAC intramolecular macrocyclisation of either 1,4- or 4,1-disubstituted-(1H-[1,2,3]triazol-1-yl) containing OT analogues (A and B respectively) carried out in solution (I–VI and IR–VIR) (a)Citation47,Citation56 (R = NH2) or by on-resin microwave-assisted strategy (VII–VIIR) (b)Citation52 (X= tBu; Y = Trt; R = Tentagel® S RAM resin).](/cms/asset/b8bd4d6b-327d-4dd3-8fe3-4ec3badef799/ienz_a_2254019_sch0003_c.jpg)
Table 1. Analytical characterisation data for peptide I–VII and IR–VIIR.
Pharmacodynamic properties of OT analogues
For pharmacological characterisation of the 1H-[1,2,3]triazol-1-yl-containing oxytocin analogues (I–VII and IR–VIIR) we analysed their affinity via radioligand binding assays in cell membranes stably expressing the human oxytocin receptor. Functional receptor Gq activation was measured utilising the homogeneous time resolved fluorescence (HTRF) inositol-1-phosphate (IP-1) protocol with human OTR in a stable HEK293 cell lineCitation60,Citation61.
All peptides were screened in a one-point radioligand displacement assay to identify lead compounds for in-depth pharmacological characterisation. Compounds II, IIR, III, IIIR, VI, VIR and VIIR (each at 10 µM) did not displace the radioligand from the receptor with >30% efficacy and were hence considered as “inactive.” On the other hand, compounds IV, IVR, V and VII displaced tritiated OT from the OTR with an efficacy of >50% (). The most promising compounds from the pre-screen, i.e. IV and IVR (as our lead pair), as well as I, VI, VR, VII and VIIR (for comparison) were measured in competitive radioligand binding experiments to determine their affinity (). Compound IV and IVR displaced tritiated OT with a Ki of ∼2.80 µM and 0.68 µM, respectively (, ). The two lead analogues were further analysed in second messenger functional assays for potency and efficacy to activate the OTR. OTR-dependent activation of the Gq-pathway was measured by detecting the accumulation of IP-1, a downstream metabolite of D-myo-inositol 1,4,5-trisphosphate (IP-3). Compound IV activated the OTR with an EC50 of 0.65 μM and Emax of 32.6% suggesting partial agonism, whereas very low level of receptor activation by IVR was observed for concentrations up to 10 µM (), suggesting that it may be an inhibitor. To investigate the mechanism of antagonism (competitive vs. non-competitive), we performed Schild regression analysis of IVR on the oxytocin receptor. Here, we measured concentration-dependent activation of the receptor by its endogenous ligand OT alone or in presence of several concentrations of IVR (100 nM, 300 nM, 1 µM and 3 µM). As expected for a competitive antagonist, we observed a dextral shift of the oxytocin potency without affecting the efficacy (, ) resulting in a linear regression slope of 1.0 ± 0.12 and a pA2 value of 7.1 (∼79 nM functional inhibitory affinity) ().
Figure 2. Pharmacology of oxytocin analogues. (A) One-point radioligand displacement experiments. Percentage specific displacement of [3H]-OT from the OTR (n = 2) by compounds I–VII and IR–VIIR (10 μM) normalised to control (10 μM OT). Specific displacement is displayed as a heatmap in greyscale ranging from white (0% specific displacement) to black (100% specific displacement); radioligand bound, i.e. [3H]-OT at 0% displacement corresponds to an average of 3775 fmoles/mg protein, respectively. (B) Concentration-dependent displacement of [3H]-OT from the OTR by compounds I (n = 2), IV (n = 2), IVR (n = 3), V (n = 4), VR (n = 2), VII (n = 4) and VR (n = 2). Eleven semi-logarithmic spaced concentrations were measured in the range from 1 nM to 100 μM. Specific binding was calculated by subtracting the non-specific binding (determined with 10 μM OT) from the total binding. The data were normalised to 100% specific binding of [3H]-OT, which refers to an average of 1000–1500 fmoles/mg protein for oxytocin receptor and fitted by nonlinear regression (slope = 1). (C) Ligand induced activation of the Gq-pathway. Concentration-dependent formation of inositol-1-phosphate (IP-1) by compound IV (n = 3) and IVR (n = 3). The ligand induced formation of IP-1 was measured in HEK293 cells (104 cells/well) stable expressing the oxytocin receptor after stimulation for 1h at 37 °C with ligands. Data are mean ± SD of three independent experiments with three technical replicates. (D) Accumulation of IP-1 by stimulation of OTR with OT (30 pM–1 µM) in the absence or presence of 100 nM, 300 nM, 1 µM or 3 µM of IVR. Data are normalised to percentage of maximal receptor activation, measured at the highest OT concentration, and are shown as mean ± SEM (n = 4). (E) Schild regression analysis of IVR at the OTR: A = EC50 of OT alone; A′ = EC50 of OT in presence of IVR; B = logarithm of IVR concentration; Schild slope = 1.0 ± 0.2 (SEM), R2 = 0.96.
![Figure 2. Pharmacology of oxytocin analogues. (A) One-point radioligand displacement experiments. Percentage specific displacement of [3H]-OT from the OTR (n = 2) by compounds I–VII and IR–VIIR (10 μM) normalised to control (10 μM OT). Specific displacement is displayed as a heatmap in greyscale ranging from white (0% specific displacement) to black (100% specific displacement); radioligand bound, i.e. [3H]-OT at 0% displacement corresponds to an average of 3775 fmoles/mg protein, respectively. (B) Concentration-dependent displacement of [3H]-OT from the OTR by compounds I (n = 2), IV (n = 2), IVR (n = 3), V (n = 4), VR (n = 2), VII (n = 4) and VR (n = 2). Eleven semi-logarithmic spaced concentrations were measured in the range from 1 nM to 100 μM. Specific binding was calculated by subtracting the non-specific binding (determined with 10 μM OT) from the total binding. The data were normalised to 100% specific binding of [3H]-OT, which refers to an average of 1000–1500 fmoles/mg protein for oxytocin receptor and fitted by nonlinear regression (slope = 1). (C) Ligand induced activation of the Gq-pathway. Concentration-dependent formation of inositol-1-phosphate (IP-1) by compound IV (n = 3) and IVR (n = 3). The ligand induced formation of IP-1 was measured in HEK293 cells (104 cells/well) stable expressing the oxytocin receptor after stimulation for 1h at 37 °C with ligands. Data are mean ± SD of three independent experiments with three technical replicates. (D) Accumulation of IP-1 by stimulation of OTR with OT (30 pM–1 µM) in the absence or presence of 100 nM, 300 nM, 1 µM or 3 µM of IVR. Data are normalised to percentage of maximal receptor activation, measured at the highest OT concentration, and are shown as mean ± SEM (n = 4). (E) Schild regression analysis of IVR at the OTR: A = EC50 of OT alone; A′ = EC50 of OT in presence of IVR; B = logarithm of IVR concentration; Schild slope = 1.0 ± 0.2 (SEM), R2 = 0.96.](/cms/asset/5f9b4317-8a00-47b8-b998-8dcd51417de2/ienz_a_2254019_f0002_b.jpg)
Table 2. Pharmacological properties of clicked OT analogues at OTR.
Table 3. Antagonist properties of oxytocin analogues IVR at OTR.
Stability of triazolyl constrained analogues IV and IVR in serum of pregnant woman
To confirm the increased enzymatic stability of the triazolyl constrained analogues of OT, we tested the serum stability of the partial agonist and competitive antagonist IV and IVR, respectively. We selected serum of woman at 40th week of pregnancy, containing high concentration of oxytocinase, a specific aminopeptidase reported to be the main player of OT plasma degradationCitation62–65. Peptides IV, IVR, and OT (as control) were incubated at 37 °C in phosphate buffer at pH 7.2 and in the selected serum. Their stability was evaluated at different points (1, 3, 5, 20, 24, and 48 h) by RP-HPLC-MS ().
Figure 3. Serum stability. Stability of OT native, IV and IVR analogues in woman serum at 40th week of pregnancy (n = 2).
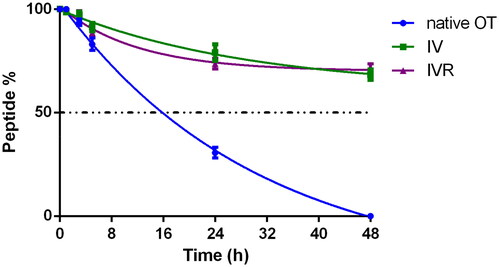
In phosphate buffer the native OT exhibited a half-time (t½) of 24 h and after 72 h no traces of OT were observed in RP-HPLC. In woman serum at 40th week of pregnancy, native OT showed a shorter half-life of 16 h and after 48 h it was not detectable by analytical RP-HPLC. On the other hand, both IV and IVR analogues exhibited a stronger stability as compared to native OT. After incubation at 37 °C in the selected serum, the half-life time of analogues IV and IVR was greater than 48 h demonstrating greater stability than the native OT (t1/2 = 16 h). Only after 10 days of incubation in serum both [1,2,3]triazol-1-yl-containing analogues were not detectable by analytical RP-HPLC (not shown). As previously reported by Muttenthaler et al.Citation39, no traces of linear reduced analogues were observed.
Conformational analysis in aqueous solution
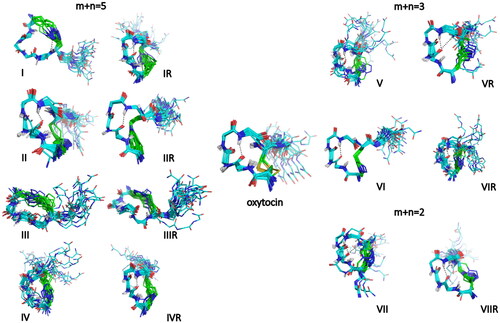
The conformational space of OT and of the 14 analogues was analysed by CD and NMR spectroscopy in water. NMR structures were generated by molecular dynamics calculation under distance and dihedral angle restraints derived from 1H-1H rotating-frame Overhauser effects (ROEs) and 3JHNHα coupling constants, respectively. presents the conformational ensembles obtained for all peptides of this study. A superimposition of each analogue conformer displaying the lowest root-mean-square deviation with OT structure is also provided in Supplementary Information (Figure S5 and Table S18), to show the degree of structural similarity (or difference) to OT. RMS deviations are indicated in Table S18.
Figure 4. Superimposition of the 20 lowest energy structures of OT and analogues in water. Backbone atoms are shown with the following colours: carbon in cyan, nitrogen in blue, oxygen in red, amide hydrogen in white. The side-chain atoms of the bridging residues (1,6) are also displayed (carbon in green, sulphur in yellow). When present, hydrogen bonds are highlighted in black. As in OT, analogues IR, II and IIR, IVR, V and VR, VI and VIR, and VII and VIIR adopt the type I β-turn conformation centred on Ile(3)-Gln(4) (the H-bond between Tyr(2) CO and Asn(5) HN groups is highlighted in black). Peptide I exhibits a H-bond between residue 6 amide proton and one nitrogen of the triazole ring. Peptides III, IIIR and VIR present an inverse γ-turn on Asn(5). Peptide IV has a flexible skeleton.
First of all, OT conformation was studied at 5 °C in water as a reference to validate our molecular mechanics calculation protocol. As in the previously published NMR PDB solution structure of OT (2MGO), we confirmed that oxytocin preferentially adopts a type I β-turn conformation in water centred on Ile(3)-Gln(4) residues. This is supported by the observation of characteristic ROEs (Ile(3) HN/Gln(4) HN, Gln(4) HN/Asn(5) HN, and Tyr(2) Hδ/Gln(4) HN ROEs (Table S16). A typical hydrogen bond between the carbonyl group of Tyr(2) and the HN group of Asn(5) is observed in the NMR structures (). The analysis of amide proton temperature coefficients indicates that HN protons in Asn(5) and Cys(6) are more sequestered from solvent than in other residues (ΔδHN/ΔT of −5.5 and −5.1 ppb/°C, respectively, in comparison with an average value of −7.5 ppb/°C) (Table S17). This is in agreement with the orientation of both HN groups towards the interior of the peptide macrocycle in the NMR structures. The CD spectrum of OT shows a negative band around 195 nm and a positive band around 225 nm, which reflects the presence of folded conformations (Figure S2). However it differs from the signature observed for short model peptides adopting a canonical type I β-turn structureCitation66,Citation67, indicating that the type I β-turn contribution of residues 2–5 may be masked by contributions of other residues of OT to the CD spectrum.
The NMR analysis of 1H-[1,2,3]triazol-1-yl-containing OT analogues reveals some similarities with OT in Hα and Cα chemical shifts (Figure S4), together with the presence of sequential HN/HN ROEs and some medium-range ROEs (Table S16) indicative of the presence of folded conformations. Interestingly, all peptides show a deshielding of Tyr(2) Hα, a shielding of Ile(3) and Gln(4) Hα protons, and a deshielding of Ile(3) and Gln(4) Cα carbons (Tables S15–S16 and Figure S4). However, differences can be observed in the set of ROE correlations and the amide proton temperature coefficients, reflecting different turn propensities and stabilities. As in OT peptide, a type I β-turn conformation centred on Ile(3)-Gln(4) is observed in the NMR structures of 10 out of the 14 analogues studied ( and Tables S1–S17). Some analogues also populate inverse γ-turn conformations on Asn(5) (III, IIIR, VIR) or a type I β-turn shifted on Gln(4)-Asn(5) (VII). The analogue IR has a 310 helical folding with a double turn conformation, in agreement with the helical CD signature, which departs from that of other analogues (Figures S2 and S3). In all structures, the exocyclic C-terminal Pro(7)-Leu(8)-Gly(9)-NH2 extension is highly flexible, as supported by near random coil values of the Hα and Cα chemical shifts (Figure S4) and very negative temperature coefficients of amide protons (Table S17). OT analogues IV and IVR have the most interesting pharmacological properties. In both analogues the disulphide bridge is replaced by a longer 1H-[1,2,3]triazol-1-yl-containing bridge, where m = 1, n = 4 and m + n = 5, as compared with the –CH2-S-S-CH2- in OT and the location of the 1H-[1,2,3]triazol-1-yl moiety in the bridge is shifted by only one methylene from the N-proximal bridgehead. The only difference between IV and IVR is the 1H-[1,2,3]triazol-1-yl orientation 1,4- vs 4,1-, respectively. They share a common CD signature with oxytocin, with a similar positive contribution around 230 nm indicative of folded conformations (Figure S2). Nevertheless, a different picture emerges from the NMR analysis. While peptide IVR shows all diagnostic ROEs of a type I β-turn centred on Ile(3)-Gln(4), similar to OT, peptide IV on the other hand lacks characteristic Ile(3) HN/Gln(4) HN and Tyr(2) Hδ/Gln(4) HN ROE correlations (Table S16), thus appearing to be more flexible, as compared to IVR.
Comparison with the active conformation of oxytocin in the OTR binding site
Recent publication of the structure of the oxytocin receptor (OTR)-OT-G-protein complex (PDB: 7QVM) allows the comparison of the receptor-bound conformation of OT to the NMR structure in aqueous solution. Even if a β-turn conformation is observed in both structures, it is important to note that the turn position is shifted from Ile(3)-Gln(4) in water to Tyr(2)-Ile(3) in the stabilised mutant [D153Y]OTR-OT bound conformation. In it, Ile(3) is buried in a hydrophobic pocket formed by side-chain residues of transmembrane helices (TMs) II–VI and VII and the Tyr(2) penetrates deeply into a pocket lined up by TMs II, III, VI and VIICitation68. In addition, the cryo-electron microscopy (cryo-EM) structure of wild-type OTR in complex with OT and G protein (PDB: 7RYC) also reveals that Tyr(2) of OT sits deep within the receptor core alongside TM VII and TM II, and Ile(3) occupies a hydrophobic pocket formed on TM VCitation69. Activation of the OTR leads to a perturbation of TM VII that results in partial unfolding and formation of a kink in proximity to the extracellular receptor sideCitation68. This indicates that the predominant conformation of OT in water may not be the bioactive conformation and that binding to OTR implies conformational selection of otherwise a weakly populated conformer in solution and/or an induced fit conformational transition.
Therefore, the divergent pharmacological properties of peptides IV and IVR cannot be directly inferred from the differences in the conformational space in solution. To get insight on the steric impact of the 1H-([1,2,3]triazol-1-yl)-containing cyclization on the putative bound conformation, we decided to model peptides IV and IVR conformations to fit to the receptor-bound OT conformation. We also applied the same superimposition protocol on peptide IIR that is inactive () and in which the triazolyl ring is shifted by –(CH2)3– from the N-proximal bridgehead that corresponds to Cys(1) in OT. The superimposition of peptide IIR adopting the receptor-bound conformation of OT indicates that the 1H-[1,2,3]triazol-1-yl ring would be centred at the Cα atom of Cys(6) in OT, but it will impose a steric clash between Pro(7) in OT and W(188) located in the extracellular loop 2 (ECL2) connecting TMs IV-V (). Conversely, in the active analogues IV and IVR, the triazole moiety like the disulphide bridge is shifted only by one –CH2- from the N-proximal bridgehead and occupies the position of Sγ atom in Cys(1) of OT, and like OT it is facing the apolar OTR residues M(315) and L(316) in TM VII. Comparing the orientations of the exocyclic C-terminal tripeptide extension in the inactive IIR with the orientation of the same tripeptides in the active analogues IV and IVR reveals the latter are closer aligned with that of OT and unlike the one in IIR does not experience the steric hindrance imposed by the proximity to W(188). Apparently, the exocyclic C-terminal amidated extension of OT is facing ECL2 and ECL3 of the cognate receptor and is critical for its activation. Nevertheless, lack of perfect overlap of the C-terminal extension of OT with those of the weakly active analogues IV and IVR may account for their weak affinity to the OTR.
Figure 5. Superimposition of peptides IIR (yellow), IV (orange), IVR (pink) onto OT (cyan) in the OTR receptor (PDB 7QVM). Residues of OTR in close contact with OT ligand (distance < 5 Å) are coloured in green. Only the side chains of W188, M315 and L316 are shown. Analogues II, IV, IVR were modelled by constraining dihedral angles to adopt the bioactive conformation of OT. The 1H-[1,2,3]triazol-1-yl ring in peptides II, IV, IVR is indicated by an arrow.
![Figure 5. Superimposition of peptides IIR (yellow), IV (orange), IVR (pink) onto OT (cyan) in the OTR receptor (PDB 7QVM). Residues of OTR in close contact with OT ligand (distance < 5 Å) are coloured in green. Only the side chains of W188, M315 and L316 are shown. Analogues II, IV, IVR were modelled by constraining dihedral angles to adopt the bioactive conformation of OT. The 1H-[1,2,3]triazol-1-yl ring in peptides II, IV, IVR is indicated by an arrow.](/cms/asset/3c27708b-6fb9-48c6-b0e8-493c28022cfe/ienz_a_2254019_f0005_c.jpg)
The stronger binding affinity and the negligible efficacy of analogue IVR relative to analogue IV ( and ) raised the interest to characterise the antagonistic properties of analogue IVR. Indeed, analogue IVR did behave as a competitive weak antagonist able to shift dose dependently the binding curve to the right ( and ) and generated a Schild regression plot with a slop of 1 that is characteristic for a competitive antagonist (). Examination of the superimposition of analogues IV and IVR onto the OT bound conformation when bound to the active conformation of OTRCitation68 () does not provide any insight that can explain the antagonistic properties of IVR. Since the main conformational characteristics of analogue IVR (but not analogue IV) are similar to those of OT (Tables S16 and S17) and formally, the only structural difference between analogues IV and IVR is the reversed orientation of the 1H-[1,2,3]triazol-1-yl moiety in the bridge (1,4- and 4,1-, respectively), it is plausible to attribute the weak antagonism in the latter to this structural discrepancy.
Conclusions
Our systematic replacement of the disulphide-containing bridge in OT with 1H-[1,2,3]triazol-1-yl containing bridges generated a heterodetic series of 2 sets of 7 analogues. The first set is comprised of analogues presenting the 1,4-1H-[1,2,3]triazol-1-yl (I–VII) while the second set is comprised of analogues presenting the reversed orientation 4,1-1H-[1,2,3]triazol-1-yl (IR–VIIR). Each set duplicates the same permutations in terms of size of the bridge and the location of the of 1H-[1,2,3]triazol-1-yl moiety within the bridge. The aim of the present study is to understand how the replacement of the native –CH2-S-S-CH2– bridge by –(CH2)m-1,4-/4,1-1H-([1,2,3]triazol-1-yl)-(CH2)n– will affect binding affinity, receptor selectivity, potency of signalling, conformation and metabolic stability. Interestingly, the design of this study that included an extensive and systematic scope of structural permutations provides an appreciation to how minor structural modifications have such major impacts on potency, conformation, and serum stability. Specifically, the most interesting analogues emerging from our study are IV and IVR, a weak partial agonist and a competitive weak antagonist, respectively. In both analogues the disulphide bridge is replaced by a longer 1H-[1,2,3]triazol-1-yl-containing bridge, where m = 1, n = 4 and m + n = 5, as compared with the –CH2-S-S-CH2- in OT and the location of the 1H-[1,2,3]triazol-1-yl moiety in the bridge is shifted by only one methylene from the N-proximal bridgehead. The only structural difference between analogues IV and IVR is the reversed orientation of the 1H-[1,2,3]triazol-1-yl moiety in the bridge (1,4- and 4,1-, respectively). The conformational ensembles obtained for all peptides of this study by a detailed NMR analysis in solution reveal some similarities with OT in Hα and Cα chemical shifts together with the presence of sequential HN/HN ROEs and some medium-range ROEs indicative of the presence of folded conformations. Differences were observed in the set of ROE correlations and the amide proton temperature coefficients, reflecting different turn propensities and stabilities. In particular while peptide IVR shows all diagnostic ROEs of a type I β-turn centred on Ile(3)-Gln(4), similar to OT, peptide IV lacks characteristic Ile(3) HN/Gln(4) HN and Tyr(2) Hδ/Gln(4) HN ROE correlations. However, the divergent pharmacological properties of peptides IV and IVR cannot be directly inferred from the differences in the conformational space in solution, as previously demonstrated by the structure of the receptor (OTR)-OT-G-protein complex (PDB: 7QVM) and the cryo-EM structure (PDB: 7RYC). Modelling of peptides IV and IVR conformations to fit to the receptor-bound OT conformation, compared to inactive peptide IIR, demonstrated that in the active analogues the triazole moiety like the S-S bridge is shifted only by one –CH2- from the N-proximal bridgehead and occupies the position of Sγ atom in Cys(1) of OT, and like OT it is facing the apolar OTR residues M(315) and L(316) in TM VII. Moreover, orientations of the exocyclic C-terminal tripeptide extension (common to both active and inactive analogues) in the active analogues reveal that are closer aligned with that of OT and unlike the one in IIR do not experience the steric hindrance imposed by the proximity to W(188). Nevertheless, lack of perfect overlap of the C-terminal extension of OT with those of IV and IVR may account for their weak affinity to the OTR. Anyway, competitive weak antagonistic activity of IVR compared to IV can be explained with its main conformational characteristics similar to OT.
In conclusion it is plausible to attribute the weak antagonism in IVR to this structural discrepancy. However, it is relevant to point out that both analogues exhibit significant increase in metabolic stability relative to OT, when incubated in serum taken from pregnant women at week 40. Therefore, we are very encouraged by the insight gained in this study and are planning to launch a second round that will add local conformational constraints to the 1H-[1,2,3]triazol-1-yl-containg bridges reported herein. Better understanding of the structural features contributed by the 1H-[1,2,3]triazol-1-yl moiety relative to those of the disulphide function will allow to improve the design of cyclic peptides endowed with tailored biological and pharmacological profiles.
Experimental section
Materials
Fmoc-l-Pra-OH, (2S)-2-(Fmoc-amino)-5-hexynoic acid (Fmoc-l-Hex(5-ynoic)-OH), Fmoc-l-β-azido-Ala-OH, Fmoc-l-4-azido-homoalanine (Fmoc-l-hAla(γ-N3)-OH), Fmoc-l-Nva(δ-N3)-OH and Fmoc-l-Nle(δ-N3)-OH were purchased by MERCK KGaA (Darmstadt Germany); (2S-2-Fmoc-amino)-7-octynoic acid (Fmoc-l-Oct(7-ynoic)-OH) was purchased by Acrotein ChemBioInc (Hoover, USA); (2S)-2-(Fmoc-amino)-6-hexynoic acid (Fmoc-l-Hept(6-ynoic)-OH) was purchased by Ambeed (Arlington, USA). Fmoc-protected amino acids and Rink-amide Wang resin, N,N′-Diisopropylcarbodiimide (DIC), oxyma (Ethyl cyanohydroxyiminoacetate), 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were purchased from Iris Biotech GmbH (Marktredwitz, Germany); Tentagel® S RAM resin was purchased from Rapp Polymere (Tuebingen, Germany); Peptide-synthesis grade N,N-dimethylformamide (DMF), acetonitrile (ACN), trifluoroacetic acid (TFA) were purchased from Carlo Erba (Milan, Italy). Piperidine, diisopropyl ether (iPr2O) and triisopropyl silane (TIS) were purchased from Sigma-Aldrich (Milan, Italy).
Sample collection
One serum sample from a 40th week pregnant woman was used as control, and obtained within the study protocol 2020_0119314 approved by the AVEN ethics committee in Reggio Emilia (Italy) on 15th October 2020. The study was performed according to the Declaration of Helsinki, and written informed consent was obtained as appropriate.
Synthesis of all linear precursors H-Xxx1-Tyr2-Ile3-Gln4-Asn5-Yyy6-Pro7-Leu8-Gly9-NH2 (I’–VII’ and IR’–VIIR’)
The OT linear precursor peptides I’–VI’ and IR’–VIR’ were performed on Rink-amide Wang resin (0.64 mmol/g, 250 mg) and in case of linear peptides VII' and VIIR’ on Tentagel® S RAM (0.23 mmol/g, 435 mg) by microwave-assisted solid-phase peptide synthesis (MW-SPPS) following the Fmoc/tBu strategy, using the Liberty BlueTM automated microwave peptide synthesiser (CEM Corporation, Matthews, NC, U.S.A.). Fmoc-deprotections were performed with a solution of 20% piperidine in DMF (2 M). Peptide assembly was performed by repeating the MW-SPPS standard coupling cycle for each amino acid, using Fmoc-protected amino acids (2.5 equiv, 0.4 M in DMF), oxyma pure (2.5 equiv, 1 M in DMF) and DIC (2.5 equiv, 3 M in DMF). The coupling of unnatural amino acids Nα-Fmoc-Xxx(ω-N3)-OH was performed using the protocol described by D’Ercole et al.Citation52 Both deprotection and coupling reactions were performed in a Teflon vessel with microwave energy and nitrogen bubbling. Cleavage of OT linear peptides (I’–VI’ and IR’–VIR’) from the resin and simultaneous deprotection of the amino-acid side chains were carried out with a mixture of TFA/TIS/H2O (95:2.5:2.5, v/v/v, 1 mL/100 mg of resin-bound peptide) for 3 h with vigorous shaking at room temperature. Resin was filtered and washed with TFA. The filtrate was concentrated under N2, addition of cold iPr2O resulted in a precipitate that was separated by centrifugation, dissolved in H2O, and lyophilised. Lyophilised crude linear peptides (I’–VI’ and IR’–VIR’) were purified by Biotage® Isolera™ Systems instrument (Uppsala, Sweden) on SNAP Ultra C18 (25 g) column at 20 mL/min using a gradient 0–60% of B. The used solvent systems: A (0.1% TFA in H2O) and B (0.1% TFA in ACN).
Synthesis of {[H-Xxx(&1)-Tyr-Ile-Gln-Asn-Yyy(&2)-Pro-Leu-Gly-NH2] [1-[&1(CH2)m]-1H-1,2,3-triazol-4-yl]-(CH2)n&2]} with m + n = 5 or 3 and m or n = 1–4 (I–VI) and {[H-Xxx(&1)-Tyr-Ile-Gln-Asn-Yyy(&2)-Pro-Leu-Gly-NH2] [1-[&1(CH2)m]-1H-1,2,3-triazol-4-yl]-(CH2)n&2]} with m + n = 5 or 3 and m or n = 1–4 (IR–VIR)
The nomenclature used herein was formulated by Spengler et al and was referenced and used by us previouslyCitation52. Purified linear peptide precursors I’–VI’ and IR’–VIR’ (>95% purity) were subjected to CuAAC cycloaddition using the same conditions to those reported previously by usCitation56. The intermolecular macrocyclisation was monitored by RP-HPLC ESI-MS. Complete conversion of all linear precursors into the desired (1H-[1,2,3]triazol-4-yl)-containing cyclopeptides I–VI (Scheme 1) and (1H-[1,2,3]triazol-1-yl)-containing cyclopeptides IR–VIR (Scheme 2) was achieved after 18 h. The crude (1H-[1,2,3]triazolyl-containing cyclopeptides I–VI and IR–VIR were purified by Biotage® Isolera™ Systems instrument (Uppsala, Sweden) on SNAP Ultra C18 (25 g) column at 20 mL/min using a gradient 0–60% of B. The used solvent systems: A (0.1% TFA in H2O) and B (0.1% TFA in ACN). The copper salts were removed by elution with H2O before the initiation of the gradient. Characterisation of the OT cyclopeptides was performed by analytical HPLC using a Waters ACQUITY HPLC coupled to a single quadrupole ESI-MS (Waters® ZQ Detector, Waters Milford, MA, U.S.A.) supplied with a BEH C18 (1.7 μm 2.1 × 50 mm) column at 35 °C, at 0.6 mL/min with solvent system A (0.1% TFA in H2O) and B (0.1% TFA in ACN). Gradient elution was performed with a flow of 0.6 mL/min and started at 10% B, with a linear increasing to 90% B in 5 min. Data were acquired and processed using MassLynx software (Waters, Milford, MA, U.S.A.). The analytical data are reported as detail in , Figure S1 B-G and S1 I-N.
Synthesis of {[H-Ala(&1)-Tyr-Ile-Gln-Asn-Ala(&2)-Pro-Leu-Gly-NH2] [1-[&1CH2]-1H-1,2,3-triazol-4-yl]-CH2&2]} with (VII) and {[H-Ala(&1)-Tyr-Ile-Gln-Asn-Ala(&2)-Pro-Leu-Gly-NH2] [1-[&1CH2]-1H-1,2,3-triazol-1-yl]-CH2&2]} with (VIIR)
On-resin orthogonally protected peptide VII’ and on-resin orthogonally protected peptide VIIR’ (0.1 mmol) were subjected to microwave-assisted CuAAC cycloaddition as reported by D’Ercole et al.Citation52 The monitoring of the intermolecular macrocyclisations was performed by a minicleavage. Each peptide was cleaved following the general cleavage protocol, purified and analysed as described for linear peptide precursors I’–VI’ and IR’–VIR’. The analytical data are reported as detail in , Figures S1-H and S1-O.
NMR conformational analysis
Lyophilised peptides were dissolved at a concentration of 2 to 3 mM in 550 µL of H2O/D2O (90:10 v/v) containing 50 mM deuterated sodium succinate, pH 5. Sodium 4,4-dimethyl-4-silapentane-1-sulfonate-d6 (DSS) was added at a final concentration of 0.11 mM for chemical shift calibration. NMR experiments were recorded on Bruker Avance III 500 MHz spectrometer equipped with a TCI 1H/13C/15N cryoprobe with Z-axis gradient. NMR spectra were processed with Topspin 3.2 software (Bruker) and analysed with NMRFAM-SPARKY programCitation70. 1H, 13C and 15N resonances were assigned using 1D 1H WATERGATE, 2D 1H-1H TOCSY (DIPSI-2 isotropic scheme of 72 ms duration), 2D 1H-1H ROESY (300 ms mixing time), 2D 1H-13C HSQC, 2D 1H-13C HSQC-TOCSY and 2D 1H-15N HSQC spectra recorded at 5 to 25 °C. 1H chemical shift was referenced against DSS. 1H signal and 13C and 15N chemical shifts were referenced indirectly. The chemical shift deviations were calculated as the differences between observed chemical shifts and random coil values reported in waterCitation70,Citation71. The temperature gradients of the amide proton chemical shifts were derived from 1D 1H WATERGATE experiments recorded from 5 to 25 °C. 3JHN-Hα were measured on 1D 1H WATERGATE experiments.
NMR assignment and conformational analysis
1H, 13C and 15N resonances of selected peptides were assigned using scalar coupling and dipolar coupling correlation experiments. The turn propensities of the different peptides were investigated by analysing 3JHN-Hα coupling constants, characteristic sequential and medium-range ROEs, and the temperature dependence of amide protons. Structures of major forms were calculated on the basis of ROE-based distance restraints and 3J coupling-based dihedral angle restraints.
NMR structure calculation
Inter-proton distance restraints were derived from ROESY cross-peak volumes integrated using NMRFAM-SPARKYCitation70. Upper bounds for proton pairs were calculated using the most intense inter-residual HN-Hα correlation as a reference (distance set to 2.2 Å). ROE cross-peak volumes involving chemically equivalent protons were divided by the multiplicity of protons, and distance restraints were calculated using r−6 average. An additional tolerance of 10% was applied to upper bounds for backbone protons and of 20% for side chain protons. The lower bounds were set to the sum of van der Waals radii of protons. Φ angle restraints were derived from 3JHN-Hα coupling constants using a Karplus relationshipCitation72. The force constants for distance and dihedral angle restraints were set to 20 kcal mol−1Å−2 and 50 kcal mol−1.rad−2, respectively. Structures were calculated using Amber 14 programCitation73. Noncanonical amino acid residues were parametrised using general Amber force field (gaff) atom types, and partial charges were computed via the AM1-BCC methodCitation74 implemented within Antechamber program. Peptides were built using the ff14SB force fieldCitation75. A set of 100 structures was calculated by simulated annealing using GBSA implicit solvation model and NMR-derived experimental restraints, as previously describedCitation76. The best 20 structures exhibiting the lowest potential energy and minimal restraint violations (< 0.2 Å and < 5° for distances and dihedral angles restraints, respectively) were selected to represent the final NMR ensemble.
Circular dichroism (CD)
CD spectra were recorded on JASCO J-815. The CD measurements were carried out in H2O. The spectra were registered with the following parameters: 0.2 nm resolution, 1.0 nm bandwidth, 20 mdeg sensitivity, 0.25 s response, 100 nm/min scanning speed, 5 scans, and 0.02 or 0.01 cm cuvette path length. The CD spectra of solvents were recorded and subtracted from the raw data. The spectra were corrected by a baseline that was measured with the identical solvent in the same cell. The CD intensity is given as mean residue molar ellipticity (θ) [deg·cm2·dmol−1].
Stability assay
Stock solutions (1 mg/mL) of native OT, IV and IVR triazolyl constrained analogues were prepared in H2O and stored at 4 °C until use. Three peptide solutions (0.1 mg/mL) were prepared by dilution of the respective stock solutions in phosphate buffer at pH 7.2. Each peptide (50 µL) was added to the 40th-week pregnant serum (300 µL) and incubated at 37 °C. A reference peptide was used as internal standard. At selected times (1, 3, 5, 20, 24, 48 h), an aliquot (30 μL) of each peptide was taken and quenched with a solution of H2O/CH3CN (1:1) with 0.1% TFA (70 μL). The vial containing the solution was cooled at 0 °C for 5 min. After centrifugation (11 000 rpm, 10 min), the supernatants were analysed (2 × 10 μL, injection) by RP-HPLC-MS at 215 nm. Data analysis (n = 2) was performed using Prism Version 6 a nonlinear fit one-phase decay model.
Membrane preparation
HEK293 cells stably expressing the OTRCitation61 were seeded in 10-cm dishes. At about 80% confluency the cell monolayer was rinsed with ice-cold PBS and mechanically detached with a cell scraper, resuspended in 5 mL ice-cold PBS and harvested by centrifugation at 300 × g and 4 °C. The pellet was resuspended in 1 L ice-cold hypotonic buffer (50 mM TRIS, 5 mM MgCl2, 0.1% BSA) supplemented with complete protease inhibitor cocktail. Cells were subsequently disrupted by four 10 s ultrasonication cycles using a benchtop ultrasonic cleaner filled with ice-cold water. Membranes were pelleted by centrifugation at 38 000 × g and 4 °C for 30 min, and resuspended in ice-cold hypotonic buffer, aliquoted and stored at −80 °C. The protein concentration was determined with the BCA assay.
Radioligand binding experiments
For radioligand binding experiments ligands were incubated with HEK293 cell membranes stably expressing oxytocin GFP-tagged receptors for 1 h at 37 °C. Unbound ligand was removed by rapid filtration over glass fibre filters using a cell harvester (Skatron). Non-specific binding was determined in the presence of 10 μM of unlabelled oxytocin. Data were expressed as % specific displacement of [3H]-OT was normalised between non-specific binding and total binding. One-point displacement experiments were measured in technical duplicates and heatmaps were created in Python 3 using the seaborn moduleCitation77. For concentration response experiments samples were measured in technical duplicates and fitted with a three parameter model (hill slope = 1) in GraphPad Prism.
Second messenger formation
The formation of second messanger IP-3 was measured with an IP1 Tb kit (Cisbio, Codolet, France) according to the manufacturer’s protocol with minor changes, as published previouslyCitation60. HEK293 cells stable expressing the GFP-tagged oxytocin receptor were seeded in white 348-well plates (Greiner bio-one) at a cell density of 104 cells per well. After 48 h the cell culture medium was removed and 5 μL of stimulation buffer were added followed by incubation for 30 min. Then the peptide solutions at indicated concentrations prepared in stimulation buffer were added, and the plate was incubated for 1 h at 37 °C and measured as outlined below. Antagonism of IVR at the oxytocin receptor was characterised by Schild regression analysis as previously describedCitation60,Citation78. Briefly, concentration-response curves of oxytocin were measured in the presence and absence of IVR. The cells were pre-treated with IVR (100 nM, 300 nM, 1 µM and 3 µM) for 30 min at 37 °C followed by co-incubation of oxytocin for an additional 30 min at 37 °C. The reaction was terminated by the addition of 5 μL of IP-1-d2 and 5 μL of Ab-cryptate. After 1 h of incubation at 37 °C fluorescence was measured on a Flexstation 3 (Molecular Devices, San Jose, CA, USA) and quantified using the 665/620 nm ratio. Samples were measured in technical triplicates. The data was normalised between the response of the positive control (10 μM oxytocin) and the negative control (stimulation buffer) and fitted by nonlinear regression using GraphPad Prism. The logarithm of the concentration-ratio (A′/A-1) was plotted against the logarithm of the respective concentration of inhibitor IVR (B) to generate the pA2 value (functional inhibitory affinity).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. FN, LM and AS synthesised the building blocks and linear peptides and performed the CuAAC cyclisation; AS, ML, OL and RL recorded the NMR and CD spectra; MES collected the selected serum; ML and OL interpreted the NMR data and performed conformational analysis; BR, NT and CWG carried out and analysed the pharmacological assays, AMP, CWG, MC and PR developed the project, designed the experiments and analysed all data. All authors participated in interpretation of their experimental data.
Supplemental Material
Download PDF (1.5 MB)Acknowledgements
AS is a participant of BioTechNan project - Interdisciplinary Environmental Doctoral Studies KNOW in the field of Biotechnology and Nanotechnology, cofinanced by the European Union. The PhD of AS was performed in the context of a Cotutorate between the PhD Schools in Chemical Sciences of the University of Florence (XXXV Ciclo) and of the Wroclaw University of Science and Technology. A.M.P. thanks Ms. Thi Hong Ha Nguyen for her technical support in peptide synthesis and purification.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81(2):629–683.
- Lippert TH, Mueck AO, Seeger H, Pfaff A. Effects of oxytocin outside pregnancy. Horm Res. 2003;60(6):262–271.
- Chibbar R, Miller FD, Mitchell BF. Synthesis of oxytocin in amnion, chorion, and decidua may influence the timing of human parturition. J Clin Invest. 1993;91(1):185–192.
- Nicholson HD, Swann RW, Burford GD, Wathes DC, Porter DG, Pickering BT. Identification of oxytocin and vasopressin in the testis and in adrenal tissue. Regul Pept. 1984;8(2):141–146.
- Ohlsson B, Truedsson M, Djerf P, Sundler F. Oxytocin is expressed throughout the human gastrointestinal tract. Regul Pept. 2006;135(1-2):7–11.
- Gutkowska J, Jankowski M, Lambert C, Mukaddam-Daher S, Zingg HH, McCann SM. Oxytocin releases atrial natriuretic peptide by combining with oxytocin receptors in the heart. Proc Natl Acad Sci U S A. 1997;94(21):11704–11709.
- Colaianni G, Sun L, Zaidi M, Zallone A. Oxytocin and bone. Am J Physiol Regul Integr Comp Physiol. 2014;307(8):R970–R977.
- Blanks AM, Thornton S. The role of oxytocin in parturition. BJOG Int J Obstet. 2003;110(Suppl20):46–51.
- Monks DT, Palanisamy A. Oxytocin: at birth and beyond. A systematic review of the long-term effects of peripartum oxytocin. Anaesthesia. 2021;76(11):1526–1537.
- Rash JA, Aguirre-Camacho A, Campbell TS. Oxytocin and pain: a systematic review and synthesis of findings. Clin J Pain. 2014;30(5):453–462.
- Kim SH, Bennett PR, Terzidou V. Advances in the role of oxytocin receptors in human parturition. Mol Cell Endocrinol. 2017;449:56–63.
- Erickson EN, Myatt L, Danoff JS, Krol KM, Connelly JJ. Oxytocin receptor DNA methylation is associated with exogenous oxytocin needs during parturition and postpartum hemorrhage. Commun Med. 2023;3(1):11.
- Kodama Y, Tanaka I, Sato T, Hori K, Gen S, Morise M, Matsubara D, Sato M, Sekido Y, Hashimoto N. Oxytocin receptor is a promising therapeutic target of malignant mesothelioma. Cancer Sci. 2021;112(9):3520–3532.
- Liu H, Gruber CW, Alewood PF, Möller A, Muttenthaler M. The oxytocin receptor signalling system and breast cancer: a critical review. Oncogene. 2020;39(37):5917–5932.
- Liwo A, Tempczyk A, Oldziej S, Shenderovich MD, Hruby VJ, Talluri S, Ciarkowski J, Kasprzykowski F, Lankiewicz L, Grzonka Z. Exploration of the conformational space of oxytocin and arginine-vasopressin using the electrostatically driven Monte Carlo and molecular dynamics methods. Biopolymers. 1998;38(2):157–175.
- Carter CS, Kenkel WM, MacLean EL, Wilson SR, Perkeybile AM, Yee JR, Ferris CF, Nazarloo HP, Porges SW, Davis JM, et al. Is oxytocin “nature’s medicine”? Pharmacol Rev. 2020;72(4):829–861.
- Polácek I, Krejcí I, Nesvadba H, Rudinger J. Action of (1,6-di-alanine)-oxytocin and (1,6-di-serine)-oxytocin on the rat uterus and mammary gland in vitro. Eur J Pharmacol. 1970;9(2):239–245.
- Brewster AI, Hruby VJ, Spatola AF, Bovey FA. Carbon-13 nuclear magnetic resonance spectroscopy of oxytocin, related oligopeptides, and selected analogs. Biochemistry. 1973;12(8):1643–1649.
- Gruber CW, Koehbach J, Muttenthaler M. Exploring bioactive peptides from natural sources for oxytocin and vasopressin drug discovery. Future Med Chem. 2012;4(14):1791–1798.
- Kato T, Endo S, Fujiwara T, Nagayama K. Oxytocin solution structure changes upon protonation of the n-terminus in dimethyl sulfoxide. J Biomol NMR. 1993;3(6):653–673.
- Brewster AI, Hruby VJ. 300-mhz nuclear magnetic resonance study of oxytocin aqueous solution: conformational implications. Proc Natl Acad Sci U S A. 1973;70(12):3806–3809.
- Wood SP, Tickle IJ, Treharne AM, Pitts JE, Mascarenhas Y, Li JY, Husain J, Cooper S, Blundell TL, Hruby VJ. Crystal structure analysis of deamino-oxytocin: conformational flexibility and receptor binding. Science. 1986;232(4750):633–636.
- Trivedi MV, Laurence JS, Siahaan TJ. The role of thiols and disulfides on protein stability. Curr Protein Pept Sci. 2009;10(6):614–625.
- MacRaild CA, Illesinghe J, van Lierop BJ, Townsend AL, Chebib M, Livett BG, Robinson AJ, Norton RS. Structure and activity of (2,8)-dicarba-(3,12)-cystino alpha-imi, an alpha-conotoxin containing a nonreducible cystine analogue. J Med Chem. 2009;52(3):755–762.
- Yamanaka T, Hase S, Sakakibara S, Schwartz IL, Dubois BM, Walter R. Crystalline deamino-dicarba-oxytocin. Preparation and some pharmacological properties. Mol Pharmacol. 1970;6(5):474–480.
- Stymiest JL, Mitchell BF, Wong S, Vederas JC. Synthesis of oxytocin analogues with replacement of sulfur by carbon gives potent antagonists with increased stability. J Org Chem. 2005;70(20):7799–7809.
- Bondebjerg J, Grunnet M, Jespersen T, Meldal M. Solid-phase synthesis and biological activity of a thioether analogue of conotoxin g1. Chembiochem. 2003;4(2-3):186–194.
- Osapay G, Prokai L, Kim HS, Medzihradszky KF, Coy DH, Liapakis G, Reisine T, Melacini G, Zhu Q, Wang SH, et al. Lanthionine-somatostatin analogs: synthesis, characterization, biological activity, and enzymatic stability studies. J Med Chem. 1997;40(14):2241–2251.
- Schramme AR, Pinto CR, Davis J, Whisnant CS, Whitacre MD. Pharmacokinetics of carbetocin, a long-acting oxytocin analogue, following intravenous administration in horses. Equine Vet J. 2008;40(7):658–661.
- Sweeney G, Holbrook AM, Levine M, Yip M, Alfredsson K, Cappi S, Ohlin M, Schulz P, Wassenaar W. Pharmacokinetics of carbetocin, a long-acting oxytocin analog, in nonpregnant women. Curr Ther Res. 1990;47(3):528–540.
- Muttenthaler M, Nevin ST, Grishin AA, Ngo ST, Choy PT, Daly NL, Hu SH, Armishaw CJ, Wang CI, Lewis RJ, et al. Solving the alpha-conotoxin folding problem: efficient selenium-directed on-resin generation of more potent and stable nicotinic acetylcholine receptor antagonists. J Am Chem Soc. 2010;132(10):3514–3522.
- Walewska A, Zhang MM, Skalicky JJ, Yoshikami D, Olivera BM, Bulaj G. Integrated oxidative folding of cysteine/selenocysteine containing peptides: improving chemical synthesis of conotoxins. Angew Chem Int Ed Engl. 2009;48(12):2221–2224.
- Gowd KH, Yarotskyy V, Elmslie KS, Skalicky JJ, Olivera M, Bulaj G. Site-specific effects of diselenide bridges on the oxidative folding of a cystine knot peptide, omega-selenoconotoxin GVIA. Biochemistry. 2010;49(12):2741–2752.
- Metanis N, Keinan E, Dawson PE. Synthetic seleno-glutaredoxin 3 analogues are highly reducing oxidoreductases with enhanced catalytic efficiency. J Am Chem Soc. 2006;128(51):16684–16691.
- Armishaw CJ, Daly NL, Nevin ST, Adams DJ, Craik DJ, Alewood PF. Alpha-selenoconotoxins, a new class of potent alpha7 neuronal nicotinic receptor antagonists. J Biol Chem. 2006;281(20):14136–14143.
- Fiori S, Pegoraro S, Rudolph-Böhner S, Cramer J, Moroder L. Synthesis and conformational analysis of apamin analogues with natural and non-natural cystine/selenocystine connectivities. Biopolymers. 2000;53(7):550–564.
- Pegoraro S, Fiori S, Rudolph-Böhner S, Watanabe TX, Moroder L. Isomorphous replacement of cystine with selenocystine in endothelin: oxidative refolding, biological and conformational properties of [Sec3, Sec11, Nle7]-endothelin-1. J Mol Biol. 1998;284(3):779–792.
- Muttenthaler M, Alewood PF. Selenopeptide chemistry. J Pept Sci. 2008;14(12):1223–1239.
- Muttenthaler M, Andersson A, de Araujo AD, Dekan Z, Lewis RJ, Alewood PF. Modulating oxytocin activity and plasma stability by disulfide bond engineering. J Med Chem. 2010;53(24):8585–8596.
- Dekan Z, Kremsmayr T, Keov P, Godin M, Teakle N, Dürrauer L, Xiang H, Gharib D, Bergmayr C, Hellinger R, et al. Nature-inspired dimerization as a strategy to modulate neuropeptide pharmacology exemplified with vasopressin and oxytocin. Chem Sci. 2021;12(11):4057–4062.
- Smith CW, Walter R, Moore S, Makofske RC, Meienhofer J. Replacement of the disulfide bond in oxytocin by an amide group. Synthesis and some biological properties of (cyclo-(1-l-aspartic acid,6-l-alpha,beta-diaminopropionic acid))oxytocin. J Med Chem. 1978;21(1):117–120.
- Hunter DJ, Schulz P, Wassenaar W. Effect of carbetocin, a long-acting oxytocin analog on the postpartum uterus. Clin Pharmacol Ther. 1992;52(1):60–67.
- de Araujo AD, Mobli M, Castro J, Harrington AM, Vetter I, Dekan Z, Muttenthaler M, Wan J, Lewis RJ, King GF, et al. Selenoether oxytocin analogues have analgesic properties in a mouse model of chronic abdominal pain. Nat Commun. 2014;5(1):3165.
- Moroder L. Isosteric replacement of sulfur with other chalcogens in peptides and proteins. J Pept Sci. 2005;11(4):187–214.
- Roice M, Johannsen I, Meldal M. High capacity poly(ethylene glycol) based amino polymers for peptide and organic synthesis. QSAR Comb Sci. 2004;23(8):662–673.
- Rečnik LM, Kandioller W, Mindt TL. 1,4-disubstituted 1,2,3-triazoles as amide bond surrogates for the stabilisation of linear peptides with biological activity. Molecules. 2020;25(16):3576.
- Testa C, Papini AM, Chorev M, Rovero P. Copper-catalyzed azide-alkyne cycloaddition (CuAAC)-mediated macrocyclization of peptides: impact on conformation and biological activity. Curr Top Med Chem. 2018;18(7):591–610.
- Jiang X, Hao X, Jing L, Wu G, Kang D, Liu X, Zhan P. Recent applications of click chemistry in drug discovery. Expert Opin Drug Discov. 2019;14(8):779–789.
- Horne WS, Stout CD, Ghadiri MR. A heterocyclic peptide nanotube. J Am Chem Soc. 2003;125(31):9372–9376.
- Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67(9):3057–3064.
- Holland-Nell K, Meldal M. Maintaining biological activity by using triazoles as disulfide bond mimetics. Angew Chem Int Ed Engl. 2011;50(22):5204–5206.
- D’Ercole A, Sabatino G, Pacini L, Impresari E, Capecchi I, Papini AM, Rovero P. On-resin microwave-assisted copper-catalyzed azide-alkyne cycloaddition of h1-relaxin b single chain ‘stapled’ analogues. Pept Sci . 2020;112(4):1–10.
- Cantel S, Le Chevalier Isaad A, Scrima M, Levy JJ, DiMarchi RD, Rovero P, Halperin JA, D'Ursi AM, Papini AM, Chorev M. Synthesis and conformational analysis of a cyclic peptide obtained via i to i + 4 intramolecular side-chain to side-chain azide-alkyne 1,3-dipolar cycloaddition. J Org Chem. 2008;73(15):5663–5674.
- Castro V, Rodríguez H, Albericio F. CuAAC: an efficient click chemistry reaction on solid phase. ACS Comb Sci. 2016;18(1):1–14.
- Scrima M, Grimaldi M, Di Marino S, Testa C, Rovero P, Papini AM, Chorev M, D'Ursi AM. Solvent independent conformational propensities of [1,2,3]triazolyl-bridged parathyroid hormone-related peptide-derived cyclo-nonapeptide analogues. Biopolymers. 2012;98(6):535–545.
- Testa C, Scrima M, Grimaldi M, D'Ursi AM, Dirain ML, Lubin-Germain N, Singh A, Haskell-Luevano C, Chorev M, Rovero P, et al. 1,4-disubstituted-[1,2,3]triazolyl-containing analogues of MT-II: design, synthesis, conformational analysis, and biological activity. J Med Chem. 2014;57(22):9424–9434.
- Testa C, D'Addona D, Scrima M, Tedeschi AM, D'Ursi AM, Bernhard C, Denat F, Bello C, Rovero P, Chorev M, et al. Design, synthesis, and conformational studies of [dota]-octreotide analogs containing [1,2,3]triazolyl as a disulfide mimetic. Pept Sci . 2018;110(5):e24071.
- Le Chevalier Isaad A, Barbetti F, Rovero P, D'Ursi AM, Chelli M, Chorev M, Papini AM. Nα-Fmoc-protected ω-azido- and ω-alkynyl-l-amino acids as building blocks for the synthesis of “clickable” peptides. Eur J Org Chem. 2008;2008(31):5308–5314.
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed. 2002;41(14):2596–2599.
- Di Giglio MG, Muttenthaler M, Harpsøe K, Liutkeviciute Z, Keov P, Eder T, Rattei T, Arrowsmith S, Wray S, Marek A, et al. Development of a human vasopressin V1a-receptor antagonist from an evolutionary-related insect neuropeptide. Sci Rep. 2017;7(1):41002.
- Koehbach J, O'Brien M, Muttenthaler M, Miazzo M, Akcan M, Elliott AG, Daly NL, Harvey PJ, Arrowsmith S, Gunasekera S, et al. Oxytocic plant cyclotides as templates for peptide G protein-coupled receptor ligand design. Proc Natl Acad Sci U S A. 2013;110(52):21183–21188.
- Rogi T, Tsujimoto M, Nakazato H, Mizutani S, Tomoda Y. Human placental leucine aminopeptidase/oxytocinase. A new member of type II membrane-spanning zinc metallopeptidase family. J Biol Chem. 1996;271(1):56–61.
- Yamahara N, Nomura S, Suzuki T, Itakura A, Ito M, Okamoto T, Tsujimoto M, Nakazato H, Mizutani S. Placental leucine aminopeptidase/oxytocinase in maternal serum and placenta during normal pregnancy. Life Sci. 2000;66(15):1401–1410.
- Kobayashi H, Nomura S, Mitsui T, Ito T, Kuno N, Ohno Y, Kadomatsu K, Muramatsu T, Nagasaka T, Mizutani S. Tissue distribution of placental leucine aminopeptidase/oxytocinase during mouse pregnancy. J Histochem Cytochem. 2004;52(1):113–121.
- Mohan S, McCloskey AG, McKillop AM, Flatt PR, Irwin N, Moffett RC. Development and characterisation of novel, enzymatically stable oxytocin analogues with beneficial antidiabetic effects in high fat fed mice. Biochim Biophys Acta Gen Subj. 2021;1865(3):129811.
- Perczel A, Kollat E, Hollosi M, Fasman GD. Synthesis and conformational analysis of N-glycopeptides. II. CD, molecular dynamics and NMR spectroscopic studies on linear N-glycopeptides. Biopolymers. 1993;33(4):665–685.
- Perczel A, Hollosi M. Turns. In: Fasman GD, editor. Circular dichroism and the conformational analysis of biomolecules. New York (NY): Plenum Press; 1996. p. 285–380.
- Waltenspühl Y, Ehrenmann J, Vacca S, Thom C, Medalia O, Plückthun A. Structural basis for the activation and ligand recognition of the human oxytocin receptor. Nat Commun. 2022;13(1):4153.
- Meyerowitz JG, Robertson MJ, Barros-Álvarez X, Panova O, Nwokonko RM, Gao Y, Skiniotis G. The oxytocin signaling complex reveals a molecular switch for cation dependence. Nat Struct Mol Biol. 2022;29(3):274–281.
- Lee W, Tonelli M, Markley JL. Nmrfam-sparky: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015;31(8):1325–1327.
- Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5(1):67–81.
- Vuister GW, Bax A. Quantitative j correlation: a new approach for measuring homonuclear three-bond J(HNHα) coupling constants in 15N -enriched proteins. J Am Chem Soc. 1993;115(17):7772–7777.
- Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, Darden TA, Duke RE, Gohlke H, et al. AMBER 14. San Francisco (CA): University of California. 2014.
- Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23(16):1623–1641.
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14sb: improving the accuracy of protein side chain and backbone parameters from ff99sb. J Chem Theory Comput. 2015;11(8):3696–3713.
- Byrne C, Belnou M, Baulieu E-E, Lequin O, Jacquot Y. Electronic circular dichroism and nuclear magnetic resonance studies of peptides derived from the fkbp52-interacting β-turn of the hERα ligand-binding domain. Pept Sci. 2019;111(4):1–14.
- Waskom ML. Seaborn: statistical data visualization. J Open Source Softw. 2021;6(60):3021.
- Duerrauer L, Muratspahić E, Gattringer J, Keov P, Mendel HC, Pfleger KDG, Muttenthaler M, Gruber CW. I8-arachnotocin-an arthropod-derived G protein-biased ligand of the human vasopressin V2 receptor. Sci Rep. 2019;9(1):19295.