
Abstract
Series of 1,3-disubstituted ureas and diadamantyl disubstituted diureas with fluorinated and chlorinated adamantane residues were shown to inhibit human soluble epoxide hydrolase (sEH) with inhibition potency ranging from 40 pM to 9.2 nM. The measured IC50 values for some molecules were below the accuracy limit of the existing in vitro assays. Such an increase in activity was achieved by minimal structural modifications to the molecules of known inhibitors, including 4-[trans-4-(1-adamantylcarbamoylamino)cyclohexyl]oxybenzoic acid. For the chlorinated homologue of the latter the sharp jump in inhibitory activity can be (according to molecular dynamics data) the result of interactions – Cl-π interaction. Considering the extremely high inhibitory activity, acceptable solubility and partial blockage of metabolically sensitive centres in their structures, some compounds are of interest for further in vivo biotesting.
Introduction
A wide range of mammalian biochemical pathways are regulated by enzyme soluble epoxide hydrolase (sEH), which is involved in the metabolism of epoxyeicosatrienoic acids (EETs) to the corresponding diols.Citation1 Experimental manipulations of sEH are frequently achieved by its inhibition. It has been shown that such inhibition leads to improved microcirculation and promotes tissue repair after myocardial infarction and ischaemic stroke. It also reduces systemic arterial pressure, increases diuresis, and exerts anti-inflammatory, neurogenic and antidepressant-like effects.Citation2–9
Most of the potent sEH inhibitors published to date contain urea moiety, which mimics epoxide transition state in the active site of the enzyme.Citation10 Among the highly active inhibitors are 1,3-disubstituted ureas containing an adamantane residue, for example, 1-(1-acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea (APAU)Citation11 (4-[trans-4–(1-adamantylcarbamoylamino)cyclohexyl]oxybenzoic acid (t-AUCB, 1a)Citation12 and its homo- or chlorinated derivatives (1b, 1c respectively)Citation13 or diadamantyl disubstituted diureas of general formula 2.Citation13,Citation14 Some of them have inhibitory activity in vitro in the low nanomolar concentration range (IC50: 0.5–2 nМ).
Despite the high activity of adamantane-containing sEH inhibitors, their use as molecular tools for in vivo studies is limited, since they often have poor pharmacokinetic properties. The latter include low metabolic stability caused by the high lipophilicity of adamantane, which is prone to oxidation by metabolic enzymes.Citation15,Citation16 Different attempts were undertaken to solve the problem: incorporation of hydrophilic groups into the adamantane coreCitation13,Citation17 or replacement of the latter with more polar groups (see, e.g. ref. Citation18) etc. These attempts met with limited success, since adamantane fits the hydrophobic pocket in the binding site of sEH, and the insertion of polar groups often leads to a decrease in activity. Recently, however, it has been shown that the replacement of the adamantane residue in t-AUCB with more lipophilic fluorinated and chlorinated benzohomoadamantane leads to compounds 3а and 3b with good metabolic stability and water solubility.Citation19 Perhaps this is due to the partial blocking of metabolically sensitive centres by halogen atoms, which is widely used in drug designCitation20–22 Previously, we synthesised series of fluorinated and chlorinated analogs of t-AUCB derivative 1b and diureas 2,Citation13,Citation14 namely compounds 9, 10, 11a–i, 12a–g and 12i.Citation23,Citation24 Given the remarkable microsomal stability and inhibitory efficiency of compounds 3а,b we proposed to carry out the primary bioscreening of the series 9, 10, 11a–i, 12a–g and 12i. In this communication, we present results of biotesting, which showed an unexpectedly sharp increase in inhibitory activity of some compounds to picomolar concentration range. Possible explanations of these results by molecular modelling methods and the data on the water solubility of the tested compounds are also presented.
Results and discussion
Series of 1,3-disubstituted ureas (9, 10) and diadamantyl disubstituted diureas with fluorinated and chlorinated adamantane residues (11a–i, 12a–g and 12i) was obtained previously by a multi-step procedure with a key stage being the reaction of 1-fluoro-3-(isocyanatomethyl)adamantane and 1-chloro-3-(isocyanatomethyl)adamantane with trans-4-((4-aminocyclohexyl)oxy)benzoic acid or the corresponding diamines.Citation23,Citation24 Some physicochemical characteristics of the compounds are presented in and .
Table 1. Human sEH inhibition data and some physicochemical characteristics of t-AUCB analogs 9 and 10.
Table 2. Human sEH inhibition data and some physicochemical characteristics of compounds 11a–i, 12a–g, 12i.
Noteworthy is the irregularity in the change in the melting points of substances observed within each series. Disubstituted diureas containing fluorinated adamantane (11) generally have a much lower melting points than those with the chlorine substituent (12), while the melting points of the 1,3-disubstituted ureas 9 are 10 are reversed. The most obvious irregularity is a sharp jump in the melting point (confirmed in several independent measurements) for compound 11c, which turns out to be approximately twice as high as that for the other substances of series 11. These melting temperature aberrations obviously result from the influence of different structural factors (the length of the spacer between urea groups and the volume of the halogen substituent in adamantane) on a complex system of intra- and intermolecular interactions in the structures under consideration.
The water solubility of compounds measured in the turbidimetric assay is inversely related to their melting points in the series 11 and 12 (). The solubility of fluorine containing diureas 11 is twice as high (140–200 μM) as that of their chlorine containing analogues 12 (60–80 μM). Interestingly, the solubility of compounds 9 and 10 and t-AUCB is almost the same () and chlorine containing diurea 12e demonstrates equal solubility as its non-substituted analogue 1,1′-(hexane-1,6-diyl)bis(3-(adamantan-1-ylmethyl)urea) (DAMH, ). Basically, no correlation was observed between the experimentally measured values of water solubility and those calculated by ESol methodCitation25 integrated to Swiss ADME software,Citation26 as well as with the values of the logarithm of the octanol–water partition coefficient (logP) calculated in this software (see and ). As expected, in series 11 and 12, the calculated values systematically change depending on the number of methylene groups, while the measured values of water solubility within each series of diureas are practically independent on the length of the spacer between two urea groups. This trend is also linked to some features of intra- and intermolecular interactions of the considered structures in water.
The inhibitory potency of the compounds 9, 10, 11a–i, 12a–g and 12i against the human sEH was studied using a kinetic fluorescent assay,Citation27 trans-4-((4–(3-(4-(trifluoromethoxy)phenyl)ureido)cyclohexyl)oxy)benzoic acid (t-TUCB)Citation28 and DAMHCitation13 were used as positive controls. Both compounds were tested in our previous studiesCitation13,Citation28 and had IC50 values 0.4 nM and 1 nM respectively in the earlier version of the assay with a measurement limit of 0.4 nM. In the present study we used a test with increased measurement accuracy up to 0.04 nM and got the same IC50 values for DAMH () and for t-TUCB ().
The first interesting and unexpected result of the bioscreening was a sharp (by about an order of magnitude) increase in the inhibitory activity of chlorine containing compound 10 compared to its fluorinated (9) and unsubstituted analogues (1b). Thus, the activity of compound 10 is in the picomolar concentration range (). Theoretically, such a sharp increase in potency can be a consequence of both a decrease in the desolvation energy of the ligand with a bulky hydrophobic chlorine atom and the participation of the latter in additional interaction with the protein (hydrophobic, dipole–dipole, halogen, and, theoretically, even covalent bond etc.). To assume about the origin of the interactions we performed molecular dynamics simulations for the complex sEH − 10.
Proposed binding modes of 10 with the sEH (PDB ID: 5AM3) obtained by molecular dynamics and molecular docking are presented at the .
Figure 1. (A) Final view of the complex of 10 with sEH (PDB ID: 5AM3) predicted by molecular dynamics (in pink) and predicted by molecular docking (in light blue). Molecule of t-AUCB (1a) is shown in beige for comparison. The inhibitors are represented by the ball-and-stick model, selected amino acid residues are represented by coloured sticks, hydrogen atoms are omitted for clarity. Protein secondary structure is shown by ribbons. (B) A two-dimensional diagram of the interactions of compound 10 with sEH (Cl – π interaction is shown specially by a solid blue line).
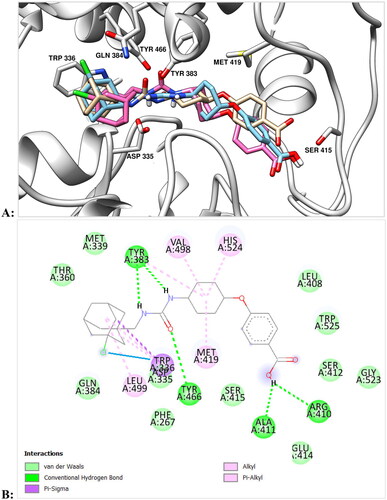
As can be seen from the , urea moiety of 10 is located within the active site of enzyme and forms hydrogen bonds with Tyr383, Tyr466, and Asp335 residues. Adamantane moiety is exposed within the hydrophobic pocket formed by Trp336, Met 339, Ile363, Phe381, Leu499 residues. According to the docking data, the position of the adamantane ring in compound 10 is almost identical to that for t-AUCB, with the chlorine atom also being in the mentioned hydrophobic pocket. Simulation of molecular dynamics reveals, however, some changes in the position of the ligand from that predicted on the basis of molecular docking, namely, a slight compaction of the molecule and a deviation of the position of adamantane in depth (). In this case, the position of the chlorine atom is close to orthogonal with respect to the benzene ring of the Trp336 residue (at the distance 3.5 Å). This location is very close to the “face-on” Cl – π interaction, which, according to calculations, can provide an additional gain in binding energy of 2.01 kcal/mol (see in details in Ref. Citation29) Note that the fluorine substituent can theoretically also form a similar interaction, but it is more sensitive to the electronic characteristics of the aromatic fragment and occurs at a shorter distance.Citation30
The presence of a methylene spacer between the adamantane and urea moieties gives molecule 10 some conformational flexibility compared to t-AUCB (1a) and seems to be important for the “face-on” Cl – π interaction (). This conclusion is indirectly confirmed by the fact that the IC50 value for the chlorine derivative of t-AUCB, which does not contain the indicated methylene bridge (1с), is 1.4 nM,Citation13 i.e. more than an order of magnitude higher than for compound 10. Thus, according to molecular modelling data, the high inhibitory activity observed for compound 10 is the result of not a covalent or halogen bonding, but interactions – Cl-π interaction. This makes it interesting to further examine the sEH–10 complex by X-ray crystallography.
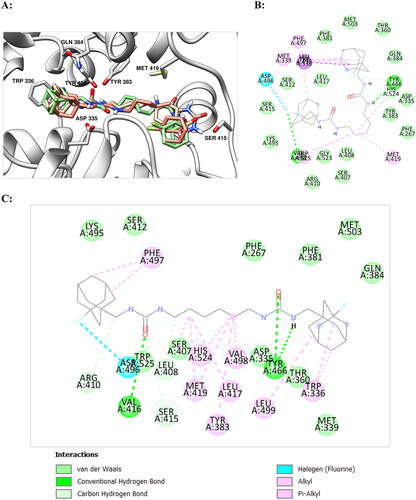
In the series 11 and 12 diureas with ethylene spacer between two urea groups (11a and 12a) possess the lowest activity among the studied compounds (). This data is in accordance with the results of our previous study for the series of structurally similar unsubstituted diureasCitation18 and is explained with the hydrophobic nature of the sEH active site. All other compounds of the series 11 and 12 were highly active, eleven diadamantyl disubstituted diureas with fluorinated and chlorinated adamantane residues demonstrated IC50 values as low as 0.04 nM. Chlorine containing compounds were more potent against sEH than their fluorine analogues, but the latter, starting with compound 11e with six methylene spacer, also show picomolar activity. According to molecular modelling data, the difference in activities by an order of magnitude for compounds 11d and 11e can be explained by the possibility of the latter to form markedly more essential interactions with residues of the active site of the enzyme (see ).
Figure 2. (А) Positions of compounds 11d (in green) and 11e (in salmon) with sEH predicted by molecular docking (in light blue). Molecule of t-AUCB (1a) is shown in beige for comparison. The inhibitors are represented by the ball-and-stick model, selected amino acid residues are represented by coloured sticks, hydrogen atoms are omitted for clarity. Protein secondary structure is shown by ribbons. (B,C) Two-dimensional diagrams of the interactions of compounds 11d (B) and 11e (C) with sEH. More essential interactions with residues of the active site for the second compound are clearly visible.
It should be emphasised, that it is hard to define the exact correlation of inhibitory activity with the length of methylene spacer between the urea groups in the series of chlorine derivatives 12, because the measured IC50 values for some molecules were so low, that surpassed the accuracy limit of the assay. For example, for compound 12d IC50 was 0.002 nM (2 pM), which was then reduced to the value of the measurement limit. Considering the extremely high inhibitory activity and acceptable solubility of the obtained substances it is of interest to further study their in vivo activity. The presence of partially blocked metabolically sensitive centres in their structures, combined with data on good metabolic stability for compounds 3a,b,Citation19 suggests that at least the metabolic stability of the t-AUCB derivative, compound 10, may be better than that of the parent molecule.
Conclusions
Series of 1,3-disubstituted ureas and diadamantyl disubstituted diureas with fluorinated and chlorinated adamantane residues were shown to inhibit human soluble epoxide hydrolase with inhibition potency ranging from 40 pM to 9.2 nM. The measured IC50 values for some molecules were below the accuracy limit of the existing in vitro assays. Such an increase in activity was achieved by minimal structural modifications to the molecules of known inhibitors, and for the chlorinated homologue of t-AUCB, compound 10, it can be (according to molecular dynamics data) the result of interactions – Cl-π interaction. In general, some compounds of the presented series replenish the number of extremely potent inhibitors of human soluble epoxide hydrolase.
Experimental
The compounds 9, 10, 11a–i, 12a–g and 12i were obtained previously, following the method described in Refs. Citation23,Citation24.
Logarithm of the octanol–water partition coefficient (Log P, mean value) was estimated using Swiss ADME software,Citation26 aqueous solubility (Saq) was calculated by ESol methodCitation25 using Swiss ADME software.Citation26
Solubilities were measured in sodium phosphate buffer (pH 7.4, 0.1 M) containing 1% of DMSO by turbidimetric assay.
Recombinant human and recombinant rat sEH were produced in insect High Five cells using recombinant baculovirus expression vectors, and purified by affinity chromatography as reported previously.Citation31,Citation32 Each enzyme appeared as a single band (0.3 μg loading) with an estimated purity of more than 95% by Coomassie Brilliant Blue staining following SDS-PAGE separation. The final recombinant sEH preparations had no esterase or glutathione S-transferase activity which interferes with the CMNPC assay as described below. Human sEH was obtained by expressing cDNA in the baculovirus system in the cell line Spodoptera frugiperda 21. The recombinant protein is an analogue of soluble epoxide hydrolase isolated from human liver in molecular weight, hydrolytic activity, inhibition and immunoreactivity.
Determination of inhibitory potency (IC50) by fluorescent assay (CMNPC assay)Citation27
The enzyme (ca. 1 nM human sEH) was incubated at 30 °C with inhibitors ([I]final = 0.4–100,000 nM) for 5 min in 100 mM sodium phosphate buffer (200 μL, pH 7.4) containing 0.1 mg mL−1 of BSA and 1% of DMSO. The substrate (cyano(2-methoxynaphthalen-6-yl)methyl trans-(3-phenyloxyran-2-yl)methylcarbonate, CMNPC) was then added ([S]final = 5 μM). The activity was assessed by measuring the appearance of the fluorescent 6-methoxynaphthaldehyde product (λem = 330 nm, λex = 465 nm) at 30 °C during a 10 min incubation (Spectramax M2; Molecular Device, Inc., Sunnyvale, CA). The IC50 values, which are the concentrations of inhibitors that reduce activity by 50%, were calculated from at least five different concentrations, each in triplicate, with at least 2 on either side of 50% activity mark.
Molecular modelling
Molecular dynamics simulations
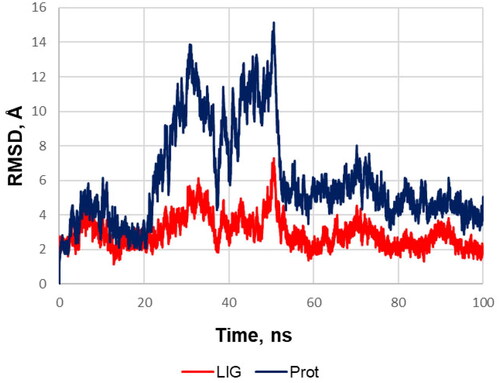
A three-dimensional model of the soluble epoxide hydrolase complex with t-AUCB (1a) (PDB ID: 5AM3)Citation33 was obtained from the Protein Data Bank (it was chosen due to a higher resolution compared to similar model of sEH – t-AUCB complex: PDB ID: 3WKECitation30) The molecule of t-AUCB and substances used for protein X-ray diffraction analysis were removed from the model. The starting structure of the sEH complex with compound 12 was obtained by means of molecular docking (see below). Molecular dynamics simulation was performed in the CHARMM36/CGenFF 4.4 force fieldCitation34,Citation35 using the GROMACS 2020.3 program.Citation36 The initial model of the system was built using the Ligand Reader & Modeller and Solution Builder modules of the CHARMM-GUI web service.Citation37 The protein molecule was built into a rectangular periodic box filled with water in the TIP3P model; the distance from the protein to the box boundary was at least 10 Å (approximate total size of the box was 120 × 120 × 120 Å3). Randomly selected water molecules were replaced by potassium and chlorine ions to ensure the electrical neutrality of the system and attain the total KCl concentration of 0.15 M. An initial molecular mechanics minimisation (up to 5000 steps) was performed on the central processor, and then the preliminary molecular dynamics equilibration was performed at 300 K temperature for 125 ps at constant volume using a v-rescale thermostat on an NVIDIA GeForce GTX 1080 GPU. Next, a working simulation of molecular dynamics was performed on a GPU at a constant pressure of 1 atm and a temperature of 300 K using a v-rescale thermostat and a Parrinello–Raman barostat (the movements of hydrogens were limited using the LINCS algorithm). The cpptraj softwareCitation38 in the AmberTools 18 packageCitation39 and UCSF Chimaera 1.15Citation40 were used to analyse and visualise the results. Mass-weighted root mean square deviations (RMSD) of the heavy protein atoms during the molecular dynamics simulation of the compound 12 and protein are presented at .
Figure 3. RMSD of the heavy protein atoms during the molecular dynamics simulation of the compound 12 (red) and protein (blue). The plot and the visual analysis of the trajectory indicates that during the simulation of molecular dynamics one monomeric subunit of sEH sharply deviates at the twentieth second and then returns to its original position; while the ligand shifts slightly to a more favourable position in the binding site and from about the fiftieth second the system stability is retained over the course of the production simulation.
Molecular docking
Molecular docking of the compounds was carried out in a 3D model of the sEH – t-AUCB (1a) complex (PDB: 5AM3) (substances used for protein X-ray diffraction analysis and t-AUCB molecule were preliminarily removed from the model). The protein atoms were charged according to the standard Kollman method using the AutoDock Tools 1.5.6 program. The 2D structures of the ligands were transformed into 3D ones, and the geometry was optimised by molecular mechanics in the Amber ff14SB force field using the Gasteiger charge model in the USCF Chimera 1.15 program.Citation40 The docking procedure was carried out using the AutoDock Vina 1.1.2 programCitation41 (grid box size: 12.75 Å × 15.0 Å × 21.75 Å, coordinates of the centre: x = 16.111 Å, y = 9.722 Å, z = 13.582 Å, exhaustiveness: = 20, energy range: = 4). The ligand – protein complexes with the best values of the scoring functions were selected. The structures of the complexes were visualised using the UCSF Chimaera 1.15 program.Citation40
2D interaction plots were obtained using Discovery studio visualiser (BIOVIA, San Diego, CA, USA).Citation42
Authors contributions
Conceptualisation, B.G., V.B.; writing—original draft preparation, B.G., O.Z.; investigation, V.B., D.D., V.D., C.M., B.H.; writing—review and editing, B.G., O.Z., G.B., N.Z.; project administration, G.B., O.Z.; software, N.Z.; resources, G.B.; visualisation, D.D, V.D., N.Z.; All authors have read and agreed to the published version of the manuscript.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Gautheron J, Jéru I. The multifaceted role of epoxide hydrolases in human health and disease. Int J Mol Sci. 2020;22(1):13.
- Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53(1):37–58.
- Oni-Orisan A, Alsaleh N, Lee CR, Seubert JM. Epoxyeicosatrienoic acids and cardioprotection: the road to translation. J Mol Cell Cardiol. 2014;74:199–208.
- Tacconelli S, Patrignani P. Inside epoxyeicosatrienoic acids and cardiovascular disease. Front Pharmacol. 2014;5:239.
- Huang H, Al-Shabrawey M, Wang MH. Cyclooxygenase- and cytochrome P450-derived eicosanoids in stroke. Prostaglandins Other Lipid Mediat. 2016;122:45–53.
- Anandan S-K, Webb HK, Chen D, Wang Y-X, Aavula BR, Cases S, Cheng Y, Do ZN, Mehra U, Tran V, et al. 1-(1-Acetyl-piperidin-4-yl)-3-adamantan-1-yl-urea (AR9281) as a potent, selective, and orally available soluble epoxide hydrolase inhibitor with efficacy in rodent models of hypertension and dysglycemia. Bioorg Med Chem Lett. 2011;21(3):983–988.
- Hammock BD, McReynolds CB, Wagner K, Buckpitt A, Cortes-Puch I, Croston G, Lee KSS, Yang J, Schmidt WK, Hwang SH. Movement to the clinic of soluble epoxide hydrolase inhibitor EC5026 as an analgesic for neuropathic pain and for use as a nonaddictive opioid alternative. J Med Chem. 2021;64(4):1856–1872.
- Borsini A. The role of soluble epoxide hydrolase and its inhibitors in depression. Brain Behav Immun Health. 2021;16 (16):100325.
- Shan J, Hashimoto K. Soluble epoxide hydrolase as a therapeutic target for neuropsychiatric disorders. Int J Mol Sci. 2022;23(9):4951.
- Nazari M, Rezaee E, Tabatabai SA. A comprehensive review of soluble epoxide hydrolase inhibitors evaluating their structure-activity relationship. Mini Rev Med Chem. 2023;23(1):99–117.
- Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, Patel DV, Anandan SK, Gless R, Webb HK. Pharmacokinetics and pharmacodynamics of AR9281, an inhibitor of soluble epoxide hydrolase, in single- and multiple-dose studies in healthy human subjects. J Clin Pharmacol. 2012;52(3):319–328.
- Hwang SH, Tsai H-J, Liu J-Y, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50(16):3825–3840.
- Burmistrov V, Morisseau C, Harris TR, Butov G, Hammock BD. Effects of adamantane alterations on soluble epoxide hydrolase inhibition potency, physical properties and metabolic stability. Bioorg Chem. 2018;76:510–527.
- Burmistrov VV, Butov GM, Karlov DS, Palyulin VA, Zefirov NS, Morisseau C, Hammock BD. Synthesis and properties of diadamantyl-containing symmetric diureas as target-oriented inhibitors of human soluble epoxide hydrolase. Russ J Bioorg Chem. 2016;42(4):404–414.
- Wanka L, Iqbal K, Schreiner PR. The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem Rev. 2013;113(5):3516–3604.
- Liu J-Y, Tsai H-J, Morisseau C, Lango J, Hwang SH, Watanabe T, Kim I-H, Hammock BD. In vitro and in vivo metabolism of N-adamantyl substituted urea-based soluble epoxide hydrolase inhibitors. Biochem Pharmacol. 2015;98(4):718–731.
- Du F, Sun W, Morisseau C, Hammock BD, Bao X, Liu Q, Wang C, Zhang T, Yang H, Zhou J, et al. Discovery of memantyl urea derivatives as potent soluble epoxide hydrolase inhibitors against lipopolysaccharide-induced sepsis. Eur J Med Chem. 2021;223:113678.
- Burmistrov V, Morisseau C, Karlov D, Pitushkin D, Vernigora A, Rasskazova E, Butov GM, Hammock BD. Bioisosteric substitution of adamantane with bicyclic lipophilic groups improves water solubility of human soluble epoxide hydrolase inhibitors. Bioorg Med Chem Lett. 2020;30(18):127430.
- Codony S, Calvó-Tusell C, Valverde E, Osuna S, Morisseau C, Loza MI, Brea J, Pérez C, Rodríguez-Franco MI, Pizarro-Delgado J, et al. From the design to the in vivo evaluation of benzohomoadamantane-derived soluble epoxide hydrolase inhibitors for the treatment of acute pancreatitis. J Med Chem. 2021;64(9):5429–5446.
- Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. Applications of fluorine in medicinal chemistry. J Med Chem. 2015;58(21):8315–8359.
- Meanwell NA. Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design. J Med Chem. 2018;61(14):5822–5880.
- Zhou Y, Wang J, Gu Z, Wang S, Zhu W, Aceña JL, Soloshonok VA, Izawa K, Liu H. Next generation of fluorine-containing pharmaceuticals, compounds currently in phase II–III clinical trials of major pharmaceutical companies: new structural trends and therapeutic areas. Chem Rev. 2016;116(2):422–518.
- Danilov DV, Burmistrov VV, Kuznetsov YP, D’yachenko VS, Rasskazova EV, Butov GM. Synthesis and properties of N,N′-disubstituted ureas and their isosteric analogs containing polycyclic fragments: VIII. Synthesis and properties of N-[(3-chloroadamantan-1-yl)methyl]-N′-(fluoro, chlorophenyl)ureas and symmetrical bis-ureas. Russ J Org Chem. 2021; 57(3):327–337.
- Danilov DV, D’yachenko VS, Burmistrov VV, Butov GM, Novakov IA. Synthesis and properties of 1-[(3-fluoroadamantan-1-yl)methyl]-3-R-ureas and 1,1′-(alkan-1,n-diyl)bis{3-[(3-fluoroadamantan-1-yl)methyl]ureas} as promising soluble epoxide hydrolase inhibitors. Russ Chem Bull. 2022;71(1):107–113.
- Delaney JS. ESOL: estimating aqueous solubility directly from molecular structure. J Chem Inf Comput Sci. 2004;44(3):1000–1005.
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Jones PD, Wolf NM, Morisseau C, Whetstone P, Hock B, Hammock BD. Fluorescent substrates for soluble epoxide hydrolase and application to inhibition studies. Anal Biochem. 2005;343(1):66–75.
- Ulu A, Appt S, Morisseau C, Hwang SH, Jones PD, Rose TE, Dong H, Lango J, Yang J, Tsai HJ, et al. Pharmacokinetics and in vivo potency of soluble epoxide hydrolase inhibitors in cynomolgus monkeys. Br J Pharmacol. 2012;165(5):1401–1412.
- Imai YN, Inoue Y, Nakanishi I, Kitaura K. Cl-pi interactions in protein-ligand complexes. Protein Sci. 2008;17(7):1129–1137.
- Li P, Maier JM, Vik EC, Yehl CJ, Dial BE, Rickher AE, Smith MD, Pellechia PJ, Shimizu KD. Stabilizing fluorine-π interactions. Angew Chem Int Ed Engl. 2017;56(25):7209–7212.
- Beetham JK, Tian TG, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305(1):197–201.
- Wixtrom RN, Silva MH, Hammock BD. Affinity purification of cytosolic epoxide hydrolase using derivatized epoxy-activated Sepharose gels. Anal Biochem. 1988;169(1):71–80.
- Öster L, Tapani S, Xue Y, Käck H. Successful generation of structural information for fragment-based drug discovery. Drug Discov Today. 2015;20(9):1104–1111.
- Huang J, MacKerell AD. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34(25):2135–2145.
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31(4):671–690.
- Kim S, Lee J, Jo S, Brooks CL, Lee HS, Im W. CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J Comput Chem. 2017;38(21):1879–1886.
- Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29(11):1859–1865.
- Roe DR, Cheatham TE. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput. 2013;9(7):3084–3095.
- Salomon-Ferrer R, Case DA, Walker RC. An overview of the AMBER biomolecular simulation package. WIREs Comput Mol Sci. 2013;3(2):198–210.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461.
- Dassault Systèmes BIOVIA. Discovery studio modeling environment, release 2023. San Diego, CA: Dassault Systèmes, 2023.