
Abstract
Significant advancements have been made in the domain of targeted anticancer therapy for the management of malignancies in recent times. VEGFR-2 is characterised by its pivotal involvement in angiogenesis and subsequent mechanisms that promote tumour cells survival. Herein, novel N-arylmethyl-aniline/chalcone hybrids 5a–5n were designed and synthesised as potential anticancer and VEGFR-2 inhibitors. The anticancer activity was evaluated at the NCI-USA, resulting in the identification of 10 remarkably potent molecules 5a–5j that were further subjected to the five-dose assays. Thereafter, they were explored for their VEGFR-2 inhibitory activity where 5e and 5h emerged as the most potent inhibitors. 5e and 5h induced apoptosis with cell cycle arrest at the SubG0-G1 phase within HCT-116 cells. Moreover, their impact on some key apoptotic genes was assessed, suggesting caspase-dependent apoptosis. Furthermore, molecular docking and molecular dynamics simulations were conducted to explore the binding modes and stability of the protein–ligand complexes.
Introduction
Cancer is a dynamic and complex disease primarily characterised by the uncontrolled growth and spread of aberrant cellsCitation1. When a solid tumour grows to a volume of more than 1–2 mm3, the resources available in the surrounding tissue become insufficient to sustain further tumour growthCitation2. As a result, a microenvironment gradually develops within the tumour tissue, releasing abundant growth factors and cytokines that stimulate angiogenesis and lymphangiogenesis to meet the increased demands of tumour growth and metabolismCitation3. Pro- and anti-angiogenic factors control the vascular network within the tumour, assisting in the transportation of nutrition, and oxygenCitation4. The process of tumour angiogenesis, which is controlled by the balance between pro-angiogenic factors and anti-angiogenic factors created by the host cells, as well as the tumour cells, is directly responsible for the growth and metastasis of the tumourCitation5. Several growth factors have been shown to control angiogenesis, including vascular endothelial growth factor (VEGF), basic fibroblast growth factor (BFGF), and platelet-derived growth factors (PDGFs)Citation6.
The VEGF family mainly consists of VEGF-A, PlGF (placenta growth factor), VEGF-B, VEGF-C, VEGF-D, VEGF-E; each VEGF isoform playing distinct roles in vascular patterning and arterial development through the activation of receptor tyrosine kinases (RTKs), including VEGFR1, VEGFR2, and VEGFR3Citation7,Citation8. Particularly, the VEGF-A/VEGFR-2 signalling pathways appear to be crucial in mediating the cellular responses involved in angiogenesisCitation9. Several types of cancer including cervical cancerCitation10, colorectal cancerCitation11, non-small cell lung cancerCitation12, hepatocellular carcinomaCitation13, and renal carcinomaCitation14 have shown VEGFR-2 overexpression predictors with poor prognosis and survivalCitation15–17. Additionally, suppression of molecular pathways related to angiogenesis has been recognised as a useful strategy for the generation of novel anticancer agents. Therefore, suppression of the VEGF/VEGFR-2 signalling pathway has emerged as a promising era of drug development to reduce the rate of angiogenesis as a potential mechanism of anticancer activity.
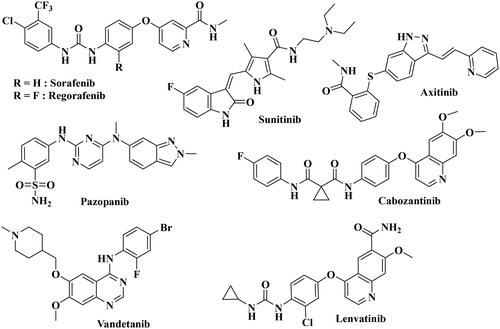
Several VEGFR-2 inhibitors have been developed in the last ten years (). FDA has licenced antibodies and small compounds as VEGF inhibitors. Bevacizumab (Avastin®)Citation18 binds to all isoforms of VEGF-A and is now approved for the treatment of non-small cell lung cancer, breast cancer, advanced colorectal cancer, metastatic renal cell carcinoma, as well as recurrent glioblastomaCitation19. Aflibercept (Zaltrap®) is a second example of an approved anti-VEGF drug, consisting of a soluble decoy receptor which binds VEGF, without activating the endogenous receptors and which has been indicated for metastatic colorectal cancer treatmentCitation20–22. Ramucirumab (Cyramza®) is an antibody directed against the VEGFR-2 extracellular domain, preventing the binding of VEGF ligands, and hence blocking receptor activation. It is now indicated to treat metastatic non-small cell lung cancer (NSCLC)Citation23,Citation24. Alongside these biologics, small molecules have also been developed as VEGFR inhibitors. Currently, there are eight FDA-approved anti-angiogenic RTK inhibitors, with the VEGF receptor being their main targetCitation25. These are Sorafenib, Sunitinib, Pazopanib, Regorafenib, Cabozantinib, Axitinib, Vandetanib, and Lenvatinib. All these standard VEGF inhibitors are able to induce cellular apoptosis and inhibit cellular growth with activation of key pro-apoptotic markersCitation26,Citation27. However, some adverse effects, such as cardiac and thyroid toxicities, were reported over the use of VEGF, suggesting that the urgent need for the development of new VEGF inhibitors with potent activity and minimal cytotoxicity.
Figure 1. Representative examples of FDA-approved VEGFR inhibitors.
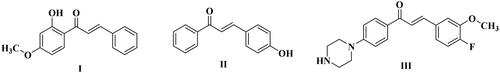
Among the great variety of pharmacophoric features used to design VEGFR-2 inhibitors, the chalcone motif bearing a variety of aromatic and/or heterocyclic ring has already proved to lead to potent anticancer compoundsCitation28–30 such as compounds I–III ().
Figure 2. Examples of reported chalcone VEGFR inhibitors.
In line with our previous works to develop new anticancer compoundsCitation31–36, we are reporting here the design and the synthesis of new N-arylmethyl-aniline/chalcone hybrids, incorporating the same essential pharmacophoric features of Sorafenib, a clinically used VEGF inhibitor ()Citation37. First, a heteroaromatic moiety interacts through hydrogen bonds with the hinge region of the ATP binding site. Second, an aromatic linker group can fill up an area between the DFG domain and the hinge region. Third, the pharmacophoric moiety can form hydrogen bonds and interact with the DFG domain of the kinase. Fourth, the hydrophobic group can interact with the allosteric hydrophobic pocket, adjacent to the ATP binding site.
Figure 3. New synthesised compound rational design.
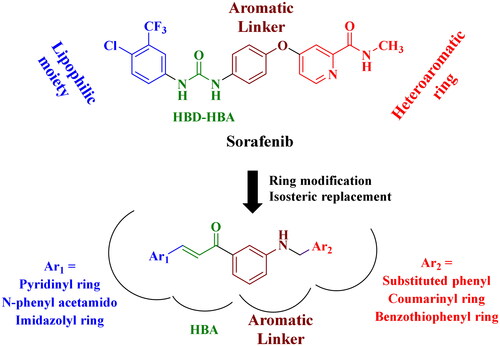
Our concept includes molecularly altering the VEGFR-2 inhibitor (Sorafenib) at four separate places in order to consider the four primary VEGFR-2 pharmacophoric properties and to utilise bioisosteric modulation techniques. The heterocyclic moiety in Sorafenib was changed to a variety of aryl/fused heteroaromatic rings with respect to the first site. Second, the urea linker was changed into an α, β-unsaturated carbonyl group of the chalcone moiety, while the core aromatic linker (phenoxy moiety) was substituted with an N-methylene aniline ring. Three different aromatic groups (Ar2), pyridine, N-phenyl acetamide, and imidazole, were chosen to evaluate how their different physicochemical properties might impact the activity of the newly developed compounds.
All targeted compounds were tested in vitro for anticancer efficacy against the 60-cell line panel of the National Cancer Institute (NCI, Bethesda, USA). The VEGFR-2 enzymatic inhibitory potency of the most active compounds was assessed for further exploration of their putative anticancer mechanism, followed by DNA cell cycle analysis and capacity to trigger apoptosis. The levels of key apoptotic proteins, such as Cytochrome-C (Cyt. C), Bax, Bcl-2, and caspase-3 were investigated for the most potent derivatives in the HCT-116 cancer cell line. Finally, using Sorafenib as a reference ligand, the synthesised compounds were subjected to molecular docking and dynamic investigations against VEGFR.
2. Results and discussion
2.1. Chemistry
The synthesis of the final compounds 5a–5n was started by Boc-protection of amino group of 3-aminoacetophenone 1 which was then reacted with the appropriate aromatic aldehyde to afford the chalcones 3i–iii. Boc-deprotection of chalcones 3i–iii followed by reductive amination with the appropriate aromatic aldehyde produced the final compounds 5a–5n with approximate yields ranging from 72% to 90% (Scheme 1).
Scheme 1. Synthesis of target derivatives 5a–5n
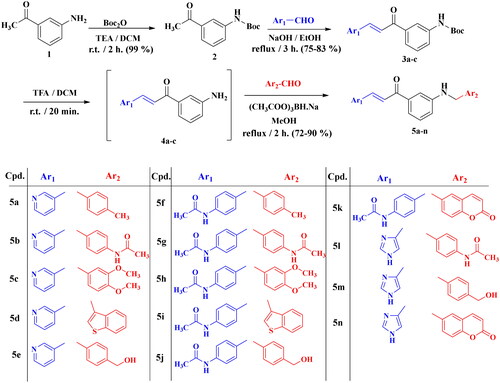
The proposed structures of all final compounds were confirmed with different spectral analyses, NMR spectroscopy (1H-NMR, 13C-NMR), LC/MS mass spectroscopy, and high-resolution mass spectrometry. All 1H-NMR spectra of compounds 5a–5n displayed a doublet peak at δ 4.26–4.59 ppm corresponding to the N-methylene group and a D2O exchangeable triplet peak at δ 6.44–6.67 ppm corresponding to anilino NH protons. In addition to the D2O exchangeable peaks corresponding to acetamido- NH protons for compounds 5b, 5f–5l. Compounds 5e, 5j, and 5 m showed a significant singlet peak corresponding to the O-methylene group at δ 4.46 ppm, and a D2O exchangeable broad singlet peak at δ 5.09 − 5.10 ppm corresponding to OH protons. Also, the imidazole containing derivatives 5 l–5n showed an extra D2O exchangeable peak at δ 12.43–12.54 ppm corresponding to imidazolo-NH protons.
In addition, singlet peaks corresponding to methyl, acetamido-methyl or methoxy groups appeared at the expected range for compounds bearing such groups. For instance, compound 5h displayed three singlet peaks integrated for three protons each at δ 3.73, 3.71, and 2.08 ppm corresponding to two methoxy groups and one acetamido-methyl group, respectively. 13C-NMR spectra for compounds 5a–5n showed peaks corresponding to chalcone carbonyl group carbons at δ 189.3 − 190.2 ppm. Acetamido-containing derivatives displayed another carbonyl group peaks at δ 168.0 − 168.7 ppm. In addition, compounds 5k and 5n bearing coumarin nucleus displayed an extra carbonyl carbon peak at δ 159.9 − 160.7 ppm, respectively. Also, peaks corresponding to methylene, methyl, acetamido-methyl or methoxy groups appeared at the expected range for compounds bearing such groups. For instance, compound 5h displayed four peaks in the aliphatic region at δ 55.5, 55.4, 46.3, and 24.1 ppm corresponding to two methoxy groups, methylene group, and one acetamido-methyl group, respectively. Finally, the ESI-high-resolution mass spectrometry for all final compounds 5a–5n confirmed the molecular weight as all measured values were within 0.0001 − 0.001 difference range from the calculated values.
2.2. Biological Evaluation
2.2.1. In vitro single-dose cellular antiproliferative assay
In vitro antiproliferative activity against NCI 60-cell lines for 14 newly synthesised compounds was tested for their in vitro anticancer activity by the National Cancer Institute (NCI, Maryland, USA), under the Developmental Therapeutic Program (DTP)Citation38–40. Single dose concentration of 10 µM of compounds was tested on a panel of 60 tumour cell lines. The antiproliferative activity was presented as growth inhibition percentage (GI%) (). While four compounds (5k–5n) showed low mean growth inhibition against the full panel with a GI% between 24.1 and 39.0%, the remaining 10 compounds (5a–5j) exhibited significant mean growth inhibition against the full panel with a GI% between 61.8 and 123.2%. Among the highly potent compounds (5a–5j), compounds 5e and 5h showed significant antiproliferative activities against most of the tested cancer cell lines. Compound 5e exhibited significant antiproliferative activity against 57 cancer cell lines with GI% ranging from 14.66 to 99.08%. It showed potent growth inhibitory effects with GI% from 16.44 to 79.27% against NSCLC cell lines, 14.66 to 98.45% against CNS Cancer cell lines, 17.22 to 91.93% against Melanoma cell lines, 32.6–97.21% against Ovarian Cancer cell lines, 15.35–94.43% against renal cancer cell lines, 55.05–62.6% against prostate cell lines and 33.46–98.12% against breast cancer cell lines (). On the other hand, compound 5h exhibited antiproliferative activity against 59 cancer cell lines with GI% ranging from 11.47 to 99.09%. It showed potent growth inhibitory effects with GI% from 78.26 to 98.31% against Leukaemia cell lines, 21.2–94.9% against NSCLC Cancer cell lines, 56.87–84.14% against CNS cell lines, 53.74–61.5% against Melanoma Cancer cell lines, 75.45–96.8% against ovarian cancer cell lines, 11.47–60.74% against renal cancer cell lines, 75.42–84.95% against prostate cancer cell lines and 89.39–99.09% against breast cancer cell lines. Regarding sensitivity to individual cell lines, it was noticed that all the newly synthesised compounds exhibited high antiproliferative activity against 13 different cell lines with a GI mean greater than 100% (), namely RPMI-8236 (Leukaemia), NCI-H522 (Lung), HCT-116, HCT-15, HT29, KM12, SW-620 (Colon), SF-539, U251 (CNS), LOX-IMVI (Melanoma), 786–0 (Renal), MCF7, and MDA-MB-438 (Breast).
Figure 4. Cell line sensitivity to compounds 5a–5n. The average of growth inhibition (GI) obtained with all compounds of the series was calculated for each cell line of the panel. Red bars correspond to an average of GI greater than 100%.
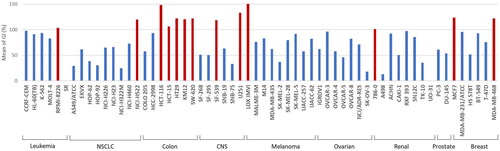
Table 1. Anticancer effects of compounds 5a–5n versus NCI-60 cancer cell lines presented as percent growth inhibition.
2.2.2. In vitro five-doses cellular anti-proliferation assay
Based on the results of full panel mean growth inhibition, ten of the final compounds (5a–5j) that met the predetermined threshold inhibition criteria in a minimum number of cell lines were selected for five-dose screening for further determination of half maximum growth inhibition concentration (GI50) (). Total growth inhibition (TGI) and half maximum lethal concentration (LC50) were also calculated, in addition to, the mean graph midpoints (MG-MID) for the different subpanels and for the full panel have been calculated for the GI50 parameter, providing an average activity parameter (all graphs are presented in the supplementary data). The results of sorafenib as a reference compound were obtained from NCI Data (NSC: 800934)Citation41 (). The results revealed that all compounds showed significant growth inhibitory activity against different cancer cell lines with full panel GI50 MG-MID ranging from 4.48 to 10.82 µM, compared to sorafenib (full panel GI50 MG-MID = 3.00 µM).
Table 2. In vitro anticancer activity presented as GI50 (μM) of selected active compounds (5a–5j) and Sorafenib (NSC: 800934) versus different cell lines.
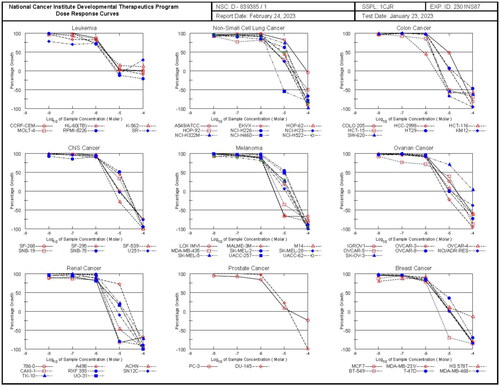
In general, compound 5h had comparatively homogenous anti-proliferative effect throughout the whole NCI panel and elicited low micromolar inhibitory dose values, with a full panel GI50 MG-MID of 4.48 µM and subpanel GI50 MG-MID ranging between 2.36 µM (Leukaemia), and 8.91 µM (NSCLC cancer) (; ). This compound showed significant growth inhibitory activity with single-digit micromolar GI50 value against 53 different cell lines among which 22 cell lines showed superior activity to sorafenib. The best potency of compound 5h was against HCT-116 (colon cancer) with a sub-micromolar GI50 value of 0.78 µM. Compounds 5b and 5d were next in potency order and showed similar activity pattern to compound 5h with a full panel GI50 MG-MID of 4.65 and 4.74 µM, respectively. Also, compounds 5b and 5d showed significant activity against HCT-116 with GI50 value of 1.80 and 1.82 µM, respectively (all graphs are presented in the supplementary data). Another potent compound is hybrid 5e potent compound against most of the examined cancer cell lines with a full panel GI50 MG-MID of 9.96 µM and subpanel GI50 MG-MID ranging between 3.29 µM (Leukaemia) and 13.62 µM (NSCLC cancer). The highest potency of 5e was shown against HCT-116 (colon cancer) with a GI50 of 1.88 µM suggesting that this cell line was particularly sensitive to this series of compounds ().
Figure 5. The dose response curves of NCI full panel for compound 5h (the graphs of the remaining compounds are presented in the supplementary data).
Concerning the safety of the new N-Arylmethyl-aniline/Chalcone hybrids, the cytotoxicity impact towards the normal fibroblast WI-38 cell line was evaluated for calculation of safety index. The results revealed that all the synthetic analogues exhibited safety index values much greater than 1 for all cancer cell lines compared to normal fibroblast WI-38. Anticancer compounds with higher safety index values (particularly when safety index > 1) are widely recognised to be more selective and safer for upcoming in-vitro and in-vivo researchCitation42–44. According to the SI data in , all the evaluated compounds have a comparatively safe impact when compared to all cancer cell lines.
On the other hand, regarding subpanel selectivity, the index obtained by dividing the full panel MG-MID (µM) of the compounds by their individual subpanel MG-MID (µM) is considered as a measure of compound selectivity. Ratios greater than 6 refers to high selectivity towards the corresponding cell line, ratios between 3 and 6 indicates moderate selectivity, while compounds not meeting either of these measures are considered non-selectiveCitation45. In this context, the selectivity index was calculated for compounds 5b, 5d, 5e, and 5h as represent to the examined series. All the tested compounds proved to be non-selective with broad spectrum anticancer activity against all cancer subpanels tested at GI50 level, except for compound 5e that showed moderate activity against leukaemia cancer cell lines. For compounds 5b, 5d, and 5h the selectivity ratios ranging from 0.62–1.90, 0.69–1.76, and 0.50–1.90, respectively, compared to sorafenib (0.79–1.45) (). These results suggest that this series of compounds shared sorafenib to be non-selective with broad spectrum anticancer activity towards different cancer cells.
Table 3. Median growth inhibitory concentrations (MG-MID-GI50, μM) and selectivity index of in vitro subpanel tumour cell lines for compounds 5b, 5d, 5e, and 5h and sorafenib (NSC: 800934).
2.2.3. Structure–activity relationship
In this series of compounds, two different modulations were studied: the aromatic group at the chalcone side (Ar1 moiety) which occupy the allosteric pocket of the kinase and a second aromatic group at the aniline side (Ar2 moiety), which should interact with the hinge region. Concerning Ar1, the results showed that substitution by adding an N-acetamido-phenyl ring (compounds 5f–5j) or a pyridine (5a–5e) dramatically increased the activity compared to the heteroaromatic imidazolyl ring (5l–5n), as confirmed by the NCI screening by promoting compounds (5a–5j) to 5-dose assay screening. As well known, the amide functionality is very interesting because it has the unusual capacity to generate important hydrogen bonding interactions, with the C = O and NH functioning as the hydrogen bond acceptor (HBA) and hydrogen bond donor (HBD), respectively. In this context, all compounds bearing acetamido group at the chalcone side showed excellent to outstanding anticancer activity, except for compounds 5k that bearing a coumarin ring at the other side. Moreover, adding highly basic and polar (amidic bioisoterism) imidazolyl group with both free N, shifted down the activities (as 5l–5n showed low to moderate activities). This could be explained by the hydrophilic character of this group, which is not tolerated by the allosteric pocket of VEGFR-2 ().
Figure 6. Structure–activity relationship for compounds 5a–5n.
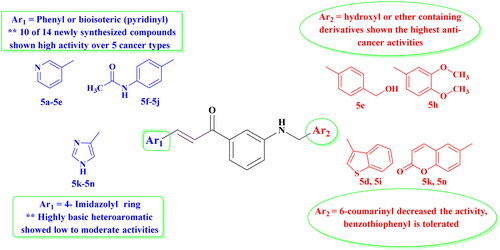
On the other hand, substitution at the aniline side with various functional aromatic groups (Ar2) showed significant activity, except for the coumarin containing compounds. In addition, by adding oxygen-bearing derivatives, another impact was seen, particularly with the two most active compounds (5e; Ar2= 4-hydroxymethylphenyl) and (5h; Ar2= 3,4-dimethoxyphenyl), probably due to the possibility to increase hydrogen bond interactions in the ATP pocket of the kinase. This point will be discussed in the docking part of the study.
2.2.3. VEGFR-2-TK inhibitory activity
VEGFR, as one of the tyrosine kinases that regulates angiogenesis is considered a potential target for the development of anti-cancer candidatesCitation46–48. Therefore, all compounds promoted to 5 doses NCI assay 5a–5j were further evaluated for their ability to inhibit VEGFR-2 using colorimetric assay of Human VEGFR-2 Enzyme-Linked Immunosorbent Assay (ELISA) using the clinically known VEGFR-2 inhibitor, Sorafenib, as a reference drug. Results were expressed as half-maximal inhibitory concentration (IC50) values and presented in . The IC50 of the tested compounds ranged from 0.31 μM to 2.0 μM and compounds 5e and 5h showed the highest potential of VEGFR-2 inhibitory effect among the tested compounds with an IC50 value of 0.37 μM and 0.31 μM, comparable to Sorafenib IC50 = 0.27 μM (). These results highly supported that VEGFR-2 could be a possible target for anti-tumour activity of this series of compounds.
Table 4. In vitro assessment of VEGFR inhibitory activity of derivatives 5a–5j as measured IC50 compared to Sorafenib as positive standard.
2.2.3. Apoptotic assay of compounds 5e and 5h
Since the newly synthesised analogues showed significant antiproliferative activity across different cancer cell lines, the two most potent analogues both for enzyme inhibition and antiproliferative activity, compounds 5e and 5h, were selected and tested for their potential apoptotic effect and interference with the cell cycle progression on HCT-116 (colon cancer), the most sensitive cancer cell line of the NCI panel. Doxorubicin was used as positive standard control in both apoptosis and cell cycle assays due to its known potency to suppress DNA topoisomerase and inducing several DNA damage mechanisms causing double-strand DNA breaks in different types of growing cancer cellsCitation49–55 and its synergistic anti-cancer effect with other compoundsCitation56–60. 0.1% DMSO was used as a negative control.
The results revealed that Doxorubicin, compounds 5e and 5h were all able to induce reduction in the percentage of viable cells with significant elevation in the late apoptotic cells percentage for compounds 5e and 5h, while Doxorubicin-induced cellular necrosis in comparison to the control (DMSO) (; ). This clearly indicates that cell death resulting from the antiproliferative action of the target derivatives 5e and 5h was attributable to apoptosis.
Figure 7. Contour plots measuring the percentage of viable (LL), early apoptotic (LR), late apoptotic (UL), and necrotic cells (UR) by AV/PI assay using flow cytometry. The assay was performed after the treatment of HCT-116 (colon cancer) for 24h with doxorubicin, 5e and 5h compared to 0.1% DMSO negative control.
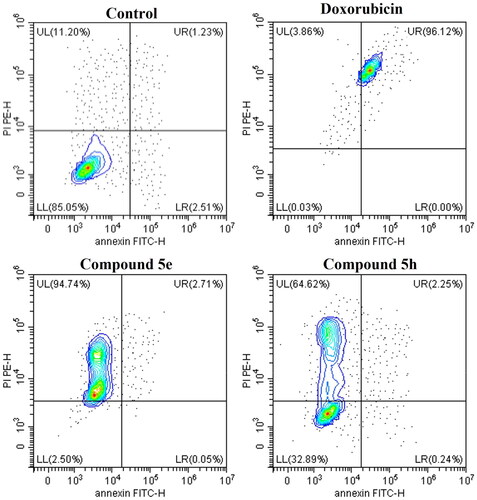
Table 5. Apoptosis assay measuring the percentage of viable, apoptotic, late apoptotic, and necrotic cells by AV/PI assay using flow cytometry. The assay was performed after the treatment of HCT-116 (colon cancer) for 24 h with doxorubicin (positive control), 5e and 5h compared to 0.1% DMSO negative control. Data represented as mean ± standard error of the mean (SEM), n = 3.
2.2.4. Cell cycle analysis
Continuous cell division is necessary for cancer cell growth, specification, and motility. This process is regulated through cell cycle phases that serve as important checkpoints for continuous proliferationCitation61,Citation62. Our results showed that treatment of HCT-116 cancer cells with compounds 5e and 5h resulted in interfering with the cell cycle distribution, causing cell growth arrest at SubG0-G1 (; ). Additionally, compounds 5e and 5h showed reduction in G0-G1, S and G2/M phases compared to the control and Doxorubicin. This indicates that compounds 5e and 5h arrested the cell cycle progression of HCT-116 cells mainly in the SubG0-G1 phase, which coincided with the previously observed induction of apoptosis.
Figure 8. Histograms for cell cycle analysis measuring the percentage of SubG0-G1, G0-G1, S-, and G2M phases by PI assay using flow cytometry. The assay was performed after the treatment of both HCT-116 (colon cancer) for 24h with doxorubicin, 5e and 5h compared to 0.1% DMSO negative control. Data represented as mean ± standard error of the mean (SEM), n = 3.
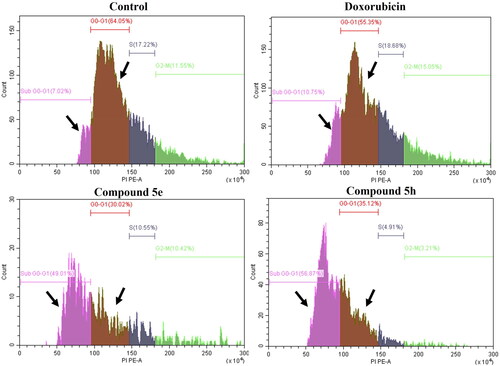
Table 6. Cell cycle analysis of HCT-116 (colon cancer) treated for 24 h with doxorubicin (positive control), 5e and 5h compared to 0.1% DMSO negative control showing the DNA content at different cycle phases.
2.2.5. Effect of compounds 5e and 5h on Bax, Bcl-2, caspase-3, Cyt-c, and TNF-α
The Bcl-2 family has been identified as essential proteins in controlling the mitochondrial pathway. Among them are pro-apoptotic proteins (e.g., Bax, Bid, Bim, and Bak) and anti-apoptotic proteins (e.g., Bcl-2, Bcl-XL, and Mcl-1). Bax specifically affects the mitochondria, leading to an elevation into mitochondrial permeability, which subsequently triggers the release of specific cellular components like cytochrome-c (Cyt-c), leading to caspase activation, including caspase-3, and finally to cell apoptosisCitation63,Citation64.
In order to further explore the mechanisms of the induced apoptosis by compounds 5e and 5h, the expression of some key regulatory genes of Cyt-c, Bcl-2, Bax, TNF- α, and caspase-3 were assessed in HCT-116 cells by RT-qPCR, after incubation with 5e and 5h, using Doxorubicin as the positive control. The results are indicated in . Compounds 5e and 5h greatly induced the expression of the pro-apoptotic gene Bax by 5- and 30 folds higher than Doxorubicin. Cyt-c was also overexpressed after treatment by compounds 5e and 5h (around 2.3-folds) but remains smaller than that obtained with Doxorubicin. An increase of caspase-3 expression was also observed after treatment by only compound 5e (2.28 folds compared to Doxorubicin). Furthermore, compounds 5e and 5h also increased the level of pro-inflammatory mediator TNF-α. Particularly compound 5e induced a high increase of the expression of this pro-inflammatory cytokine (372 folds), compared to compound 5h (2.2-folds) and Doxorubicin (2.33 folds). Additionally, the level of the anti-apoptotic protein Bcl-2 is virtually unchanged after treatment with either compound 5e or 5h, respectively compared to Doxorubicin. Consequently, a higher increase of the Bax/Bcl-2 ratio was observed for compounds 5e and 5h compared to Doxorubicin, supporting the possible apoptotic effect and anticancer activity of compounds 5e and 5h against the HCT-116 cell line.
Table 7. Gene expression fold change in HCT-116 (colon cancer) treated for 24h with doxorubicin (positive control), 5e and 5h compared to 0.1% DMSO negative control showing the expression fold change in n = 3 using 2-ΔΔct method.
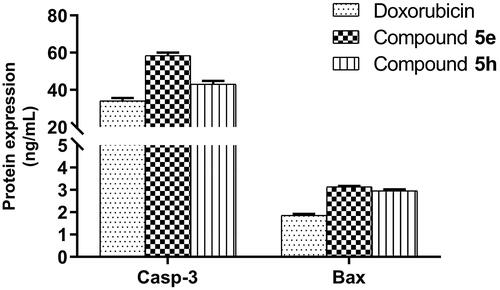
To investigate the effect of compounds 5e and 5h on final protein levels, caspase-3 and Bax proteins were measured using ELISA assay. As observed at the mRNA level for the Bax gene, an increase of the Bax protein level was also detected after treatment of HCT-116 cells by compounds 5e and 5h in the same proportions (3.14- and 2.96 folds respectively), and higher than with Doxorubicin (1.86-fold) (), suggesting that these compounds have Bax-induced apoptotic cell death. Regarding caspase-3, compound 5e showed parallel expression in both gene and protein (58.44-fold) levels. However, in interesting observation, compound 5h showed minimal gene expression of caspase-3 with much elevated level on protein (43.12-fold) in a way matched to Doxorubicin (34.15-fold). This difference in observation despite the optimisation of procedures might suggest some explanations, such as the gene expression of caspase-3 is not the major determinant of its protein level, and there might be other contributing regulatory factors controlling the level of caspase-3 and hence the observed potency of compound 5h in apoptosis. Also, compound 5h might influence other regulatory processes controlling mRNA translation, miRNA targeting species and post-translational modifications, like phosphorylation, acetylation, and glycosylation and hence, increasing the stability of mRNA and protein productsCitation65 and this is something we are working-on currently in the project.
Figure 9. Measurement of caspase-3 (Casp-3) and Bax protein levels after treatment of compounds 5e and 5h in HCT-116 cancer cells compared to 0.1% DMSO (negative control). The cells were treated with the IC50 concentrations of compounds for 24h and data were shown as mean ± SEM of three independent experiments (n = 3).
It was shown that apoptosis plays a critical determinant role in network vascularity and cancer microvascular remodellingCitation46,Citation66. The studies documented that capillary network formation is a dynamic process involving cell-cell matrix interactions and cells that fail to be incorporated within this network will show characteristic morphological features of cell death. Additionally, inhibition of angiogenesis will induce cellular hypoxia and production of reactive oxygen species that induce cellular apoptosisCitation46,Citation67. These observations may explain the antiproliferative effects of compounds 5e and 5h and correlate with VEGFR-inhibition and apoptosis.
2.3. Molecular modeling
2.3.1. Molecular docking
A docking simulation experiment was carried out utilising the most powerful derivatives, 5e and 5h, considering the strong anticancer activity shown by the synthesised derivatives above mentioned. The purpose of the docking was to provide important insights into the ways that compounds 5e and 5h bind to the VEGFR-2 enzyme. To carry out and visualise each docking stage, the molecular operating environment (MOE) 2019.02 was used. Two scenarios were proposed as the compounds contain a Michael system that may results in irreversible inhibition of the VEGFR-2 enzyme Accordingly, both the docking approaches, covalent and non-covalent docking were applied to elucidate the most potential mode of binding of the proposed compounds interestingly, the covalent docking approach have failed as the Michael system remained far away from the reactive thiol of the cys919, and no poses were retrieved. In the opposite hand, the non-covalent approach yielded successful binding poses with similar binding pattern with the reported VEGFR-2 inhibitors.
After that, the co-crystallized pose of Sorafenib was re-docked to the active sites of their corresponding targets in order to validate the docking investigation. For Sorafenib, the calculated RMSD values between the co-crystallized and the docked poses were found to be 1.01 Å, demonstrating the accuracy of the used docking techniques. The docking scores (S) for Sorafenib were −18.5 kcal/mol. The Sorafenib docking score was used as a standard for the docked compounds 5e and 5h ( and ).
Figure 10. Docking interaction (2D) with the VEGFR-2 enzyme for (A) Sorafenib, (B) 5e, and (C) 5h.
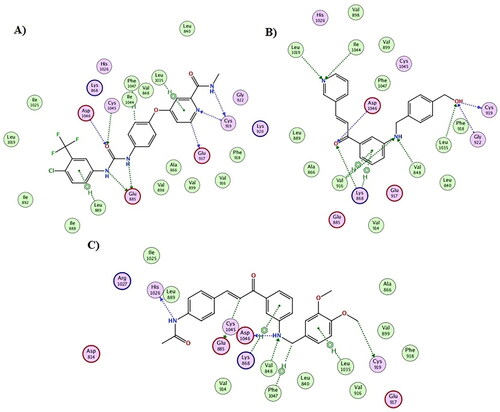
Figure 11. Docking interaction (3D form) for compounds (A) 5e and (B) 5h.
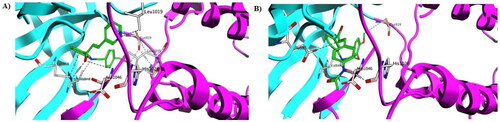
Sorafenib interacts with the VEGFR-2 enzyme by a variety of hydrophobic and hydrophilic (through hydrogen bonding) interactions, as shown in . Among the most common are those that interact with Cys1045, Asp1046, Glu885, and Cys919 via hydrogen bonds. The urea linker and terminal amide of sorafenib are responsible for its major hydrophilic interaction. The fact that compounds 5e and 5h were able to achieve docking scores of −16.9 and −17.7 kcal/mol, which are extremely similar and yet more affine to that of Sorafenib docking score (-18.5 kcal/mol), explains their powerful inhibitory actions. For instance, the terminal hydroxyl group for compound 5e formed three hydrogen bond interactions with Cys919, Gly922, and Leu1035, the nitrogen of the pyridine moiety formed two hydrogen bond interactions with Leu1019 and Ile1044, while the carbonyl group of the chalcone moiety interacted with Asp1046 and Lys868 via hydrogen bond interaction. Furthermore, the aniline NH group formed two hydrogen bonds with Val 848 and Val 916, as well as two hydrophobic interactions with Lys868 and Val916 ( and ). On the other hand, compound 5h showed similar binding mode and various interactions with the VEGFR-2 binding site as seen with Sorafenib and compound 5e. From and , the terminal methoxy group for compound 5h was engaged in a hydrogen bond interaction with Cys919; moreover, the aniline NH group was involved in two hydrogen bond interactions with Asp1046 and Val848. In addition, the olefinic carbon (HC = C*H-CO) and the terminal amidic nitrogen interacted with Glu885 and His1026, respectively. Besides, various hydrophobic interactions were also formed between compound 5h and VEGFR-2 residues such as Lys868, Leu1037, and Phe1047. Subsequently, both compounds 5e and 5h achieved adequate interactions with the amino acid residues of VEGFR-2, achieving a binding mechanism comparable to that shown for sorafenib, which explains their high enzymatic activity against VEGFR-2. Finally, molecular dynamic simulations were used to emphasise the docking results even more.
2.3.2. Molecular dynamics
2.3.2.1. RMSD and RMSF analysis
The results retrieved from the biology and docking studies presented our synthesised compounds, especially compound 5h, as promising anticancer agents. Therefore, further in silico insights were performed using molecular dynamic simulations (MDSs) and their applications using GROMACS 2.1.1 software. Well-established MDSs are privileged over others in silico studies in the precise estimation of the stability of a protein–ligand complex. Accordingly, this advantage was considered, and the binding pose of 5h with VEGFR-2 retrieved from the docking step were simulated for 100 ns. The Apo form of VEGFR-2, alongside the binding of each target to its corresponding co-crystallized ligand, was added to the simulation list to provide means of comparison and benchmarking.
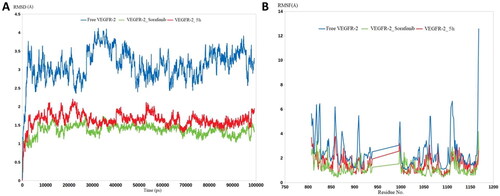
As shown in , the free VEGFR-2 enzyme demonstrated significant dynamicity, serving its intended function as primary oncogenic proteins. This was highlighted by RMSD calculations, in which the unbound proteins of VEGFR-2 achieved average RMSD values of 3.5 Å. Compound 5h demonstrated an excellent ability to restrict the dynamic nature of VEGFR-2, as evidenced by the lower RMSD values of 5h with VEGFR-2 (nearly 1.9 Å). The extent of the decrease in RMSD values for compound 5h was very close to sorafenib (1.4 Å). The RMSF values were similar to the RMSD values, in which the residues of the Apo proteins reached average fluctuations of 3.9 Å (). The binding of 5h and sorafenib to VEGFR-2 induced great stability for its residues, lowering their fluctuation to average RMSFs of 1.8 and 1.3 Å, respectively. In general, both the RMSD and RMSF calculations led to one conclusion: that compound 5h is capable of potently inhibiting VEGFR-2 owing to its ability to form strong and stable interactions with the active sites of its intended target.
Figure 12. Molecular dynamic simulation of two stable complexes of 5h-VEGFR-2 and Sorafenib-VEGFR-2; (A) RMSD analysis for the MD simulations of 5h–VEGFR-2 and sorafenib–VEGFR-2, in addition to the Apo form of VEGFR-2, (B) RMSF analysis for the MD simulations of 5h–VEGFR-2 and Sorafenib–VEGFR-2, in addition to the Apo form of VEGFR-2.
3. Conclusions
In summary, a series of fourteen N-arylmethyl-aniline/chalcone hybrids (5a–5n) was synthesised to target VEGFR-2 and impeded cancer cell growth. Evaluation against the NCI 60-cell line panel at 10 μM revealed ten highly effective compounds (5a–5j) with remarkable growth inhibition (mean GI% ranging from 61.8 to 123.2%) with minimal cytotoxic effect against normal fibroblast WI-38 cells (safety index greater than 1 for the whole 10 compounds). Further testing at five different concentrations identified compounds 5b, 5d, 5e, and 5h as the most potent, with full panel GI50 values range of 9.96 − 4.48 μM. HCT-116 colon cancer cells showed heightened sensitivity to compounds 5e and 5h, with GI50 values of 1.88 and 1.95 µM, respectively. Apoptotic and cell cycle analyses of compounds 5e and 5h on HCT-116 cells demonstrated significant reductions in viable cells and pronounced elevation in late apoptotic cells, arresting the cell cycle at the SubG0-G1 phase. Additionally, compounds 5e and 5h exhibited the most potent VEGFR-2 inhibitory activity with IC50 values of 0.37 and 0.31 μM, respectively. Mechanistic investigations for 5e and 5h revealed significant upregulation of apoptotic genes, particularly Bax, leading to an elevated Bax/Bcl-2 ratio (4.5 and 31.6, respectively) compared to the positive control Doxorubicin (2.0). Bax protein levels were upregulated after treatment with compounds 5e and 5h higher than Doxorubicin consistent with their gene expression data suggesting the Bax-induced apoptosis. Also, caspase-3 protein levels were upregulated with compounds 5e and 5h, suggesting caspase-dependent pathway with further investigations required to confirm the mechanistic effect. In terms of SAR, the incorporation of N-phenyl acetamide or a pyridinyl moiety within the lipophilic pocket of VEGFR-2 has been found to increase potency. Moreover, the introduction of a hydroxyl or methoxy groups into the hinge region also enhanced potency, as observed with compounds 5e and 5h, the most potent VEGFR-2 inhibitors of this series. Furthermore, molecular docking analysis of compounds 5e and 5h with VEGFR-2 revealed effective interactions with key residues Cys919 and Asp1046, with 5h additionally bound to another key residue, Glu885. Additionally, MDS involving compound 5h complexed with VEGFR-2 highlighted its enhanced stability (RMSD = 1.9 Å and RMSF = 1.8 Å) compared to free unbound VEGFR-2 (RMSD = 3.5 Å and RMSF = 3.9 Å), and comparable stability relative to the VEGFR-2-Sorafenib complex (RMSD = 1.4 Å and RMSF = 1.3 Å).
4. Experimental
4.1. Chemistry
Melting points (°C) of the novel compounds were uncorrected and were measured by using Electrothermal Stuart 5MP3. Following up the progress of the chemical reactions was performed using F254 - TLC Merck plates. The NMR data had been recorded using Bruker-Avance 500 NMR spectrometer (500 MHz for 1H-NMR and 101 MHz for 13C-NMR) and Bruker-Avance 600 NMR spectrometer (600 MHz for 1H-NMR and 126 MHz for 13C-NMR) in deuterated dimethylsulphoxide (DMSO–d6). The chemical shifts (δH) were reported relative to the solvent (DMSO-d6). Quattro microTM ESI triple quadrupole mass spectrometer (ESI+ electrospray ionisation mode) coupled to an Alliance HPLC system (Waters, Milford, USA) equipped with a Chromolith High Resolution RP-18e column (25 × 4.6 mm) was used for LC/MS analyses. The samples were previously separated using gradient solutions from 100% (H2O + 0.1% HCO2H) to 100% (ACN + 0.1% HCO2H) in 3 min at a flow rate of 3 ml/min and with UV detection at 214 nm. High resolution mass spectrometry (HRMS) analyses were measured at the Laboratoire de Mesures Physiques, University of Montpellier, France using a time-of-flight (TOF) spectrometer coupled to a positive electrospray ionisation (ESI+) source.
4.1.1. General method for synthesis of compounds 3a-3c
A mixture of 3-aminoacetophenone 1 (100 mmol, 13.5 g) and Boc2O (120 mmol, 26.16 g) in dichloromethane (DCM, 20 ml) was stirred in the presence of triethylamine (TEA, 200 mmol, 27.9 ml) at room temperature for 2 h. The solvent was evaporated, and the remaining was purified using silica gel column chromatography (DCM: Methanol/99:1) to afford the Boc-protected derivative 2. A mixture of compound 2 (10 mmol, 2.35 g) and appropriate aromatic aldehyde (11 mmol) in ethanolic NaOH solution was refluxed for 3 h. The reaction mixture was cooled, and the resulting precipitate compounds 3a–c were collected, washed with cold ethanol, and dried. The structures of compounds 3a–c were confirmed with different spectral analyses.
4.1.2. General method for synthesis of compounds 5a-5n
Compounds 3a–c (1 mmol) were Boc-deprotected by stirring in 1:1 mixture of DCM and trifluoracetic acid for 20 min to afford amines 4a–c. Without purification, compounds 4a–c were mixed with the appropriate aromatic aldehyde (1.1 mmol) and sodium triacetoxyborohydride (2 mmol, 0.424 mg) in methanol (30 ml) and were refluxed for 2 h. The solvent was evaporated, and the remaining was purified using silica gel column chromatography (DCM: Methanol/99:1) to afford the final compounds 5a–5n.
4.1.2.1. (E)-1–(3-((4-methylbenzyl)amino)phenyl)-3-(pyridin-3-yl)prop-2-en-1-one (5a)
Orange powder (yield 78% − 0.78 mmol, 255 mg); m.p. 114–116 °C; IR (υmax./cm−1) 3303 (N-H), 1656 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.61 (dd, J = 1.53, 4.73 Hz, 1H), 8.29 (td, J = 1.89, 7.97 Hz, 1H), 7.89 (d, J = 15.87 Hz, 1H), 7.68 (d, J = 15.72 Hz, 1H), 7.49 (dd, J = 4.73, 7.93 Hz, 1H), 7.36 (d, J = 7.78 Hz, 1H), 7.27 (s, 1H), 7.22 − 7.26 (m, 3H), 7.13 (d, J = 7.78 Hz, 2H), 6.87 (dd, J = 2.06, 7.86 Hz, 1H), 6.52 (t, J = 6.03 Hz, 1H, exchangeable with D2O), 4.29 (d, J = 5.95 Hz, 2H), 2.26 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.4, 150.8, 150.1, 149.0, 139.8, 138.0, 136.6, 135.7, 134.9, 130.5, 129.1, 128.8, 127.1, 124.3, 123.8, 117.3, 116.5, 111.3, 46.1, 20.6. MS (ESI) m/z: 329 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 329.1648, Found, 329.1651.
4.1.2.2. (E)-N-(4-(((3–(3-(pyridin-3-yl)acryloyl)phenyl)amino)methyl)phenyl)acetamide (5b)
Orange powder (yield 85% − 0.85 mmol, 315 mg); m.p. 194–196 °C; IR (υmax./cm−1) 3289 and 3196 (N-H), 1670 and 1603 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 9.88 (s, 1H, exchangeable with D2O), 8.98 (s, 1H), 8.61 (dd, J = 1.60, 4.81 Hz, 1H), 8.29 (td, J = 1.79, 8.01 Hz, 1H), 7.90 (d, J = 15.72 Hz, 1H), 7.69 (d, J = 15.72 Hz, 1H), 7.52 (d, J = 8.54 Hz, 2H), 7.48 (dd, J = 4.81, 8.01 Hz, 1H), 7.37 (d, J = 7.93 Hz, 1H), 7.29 (d, J = 8.54 Hz, 2H), 7.22 − 7.26 (m, 2H), 6.87 (dd, J = 2.14, 7.93 Hz, 1H), 6.49 (t, J = 5.87 Hz, 1H, exchangeable with D2O), 4.28 (d, J = 5.80 Hz, 2H), 2.01 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.3, 168.0, 150.8, 150.1, 149.0, 139.8, 138.0, 138.0, 134.9, 134.1, 130.5, 129.1, 127.5, 124.3, 123.8, 119.0, 117.3, 116.5, 111.3, 46.0, 23.8. MS (ESI) m/z: 372 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 372.1707, Found, 372.1711.
4.1.2.3. (E)-1–(3-((3,4-dimethoxybenzyl)amino)phenyl)-3-(pyridin-3-yl)prop-2-en-1-one (5c)
Orange powder (yield 77% − 0.77 mmol, 288 mg); m.p. 126–128 °C; IR (υmax./cm−1) 3300 (N-H), 1666 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 8.98 (s, 1H), 8.61 (d, J = 4.43 Hz, 1H), 8.29 (d, J = 7.78 Hz, 1H), 7.91 (d, J = 15.72 Hz, 1H), 7.69 (d, J = 15.72 Hz, 1H), 7.48 (dd, J = 4.35, 7.55 Hz, 1H), 7.37 (d, J = 7.63 Hz, 1H), 7.28 (s, 1H), 7.25 (t, J = 7.86 Hz, 1H), 7.01 (s, 1H), 6.86 − 6.93 (m, 3H), 6.45 (t, J = 5.65 Hz, 1H, exchangeable with D2O), 4.26 (d, J = 5.65 Hz, 2H), 3.73 (s, 3H), 3.71 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.4, 150.9, 150.2, 149.1, 148.7, 147.7, 139.9, 138.0, 134.9, 132.1, 130.5, 129.1, 124.3, 123.9, 119.3, 117.4, 116.5, 111.8, 111.4, 55.5, 55.4, 46.2. MS (ESI) m/z: 375 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 375.1703, Found, 375.1704.
4.1.2.4. (E)-1–(3-((benzo[b]thiophen-3-ylmethyl)amino)phenyl)-3-(pyridin-3-yl)prop-2-en-1-one (5d)
Yellow powder (yield 87% − 0.87 mmol, 323 mg); m.p. 132–134 °C; IR (υmax./cm−1) 3282 (N-H), 1671 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 8.99 (d, J = 2.44 Hz, 1H), 8.61 (dd, J = 1.53, 4.73 Hz, 1H), 8.29 (td, J = 1.83, 8.09 Hz, 1H), 7.97 − 8.00 (m, 2H), 7.93 (d, J = 15.87 Hz, 1H), 7.70 (d, J = 15.72 Hz, 1H), 7.63 (s, 1H), 7.48 (dd, J = 4.73, 7.93 Hz, 1H), 7.37 − 7.44 (m, 3H), 7.36 (t, J = 1.83 Hz, 1H), 7.28 (t, J = 7.86 Hz, 1H), 6.98 (dd, J = 2.06, 7.86 Hz, 1H), 6.56 (t, J = 5.72 Hz, 1H, exchangeable with D2O), 4.59 (d, J = 5.49 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 189.4, 150.9, 150.3, 149.0, 140.1, 140.0, 138.1, 138.0, 135.0, 134.1, 130.5, 129.3, 124.5, 124.3, 124.1, 123.9, 123.8, 122.9, 122.2, 117.3, 116.7, 111.3, 41.2. MS (ESI) m/z: 371 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 371.1213, Found, 371.1210.
4.1.2.5. (E)-1–(3-((4-(hydroxymethyl)benzyl)amino)phenyl)-3-(pyridin-3-yl)prop-2-en-1-one (5e)
Yellow powder (yield 83% − 0.83 mmol, 285 mg); m.p. 131–133 °C; IR (υmax./cm−1) 3304 (N-H), 1661 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.61 (d, J = 4.43 Hz, 1H), 8.28 (d, J = 7.93 Hz, 1H), 7.89 (d, J = 15.72 Hz, 1H), 7.68 (d, J = 15.72 Hz, 1H), 7.45 − 7.52 (m, 1H), 7.31 − 7.38 (m, 3H), 7.21 − 7.29 (m, 4H), 6.88 (d, J = 8.09 Hz, 1H), 6.53 (t, J = 5.42 Hz, 1H, exchangeable with D2O), 5.09 (br. s., 1H, exchangeable with D2O), 4.46 (s, 2H), 4.33 (d, J = 5.80 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 189.4, 150.8, 150.1, 149.0, 141.0, 139.8, 138.1, 138.0, 134.9, 130.5, 129.1, 126.9, 126.5, 124.3, 123.8, 117.3, 116.5, 111.3, 62.7, 46.2. MS (ESI) m/z: 345 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 345.1598, Found, 345.1599.
4.1.2.6. (E)-N-(4–(3-(3-((4-methylbenzyl)amino)phenyl)-3-oxoprop-1-en-1-yl)phenyl)acetamide (5f)
Orange powder (yield 83% − 0.83 mmol, 319 mg); m.p. 89–91 °C; IR (υmax./cm−1) 3314 and 3269 (N-H), 1670 and 1628 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 10.17 (s, 1H, exchangeable with D2O), 7.74 − 7.77 (d, J = 8.70 Hz, 2H), 7.65 − 7.69 (d, J = 8.70 Hz, 2H), 7.61 (d, J = 2.14 Hz, 2H), 7.25 − 7.31 (m, 3H), 7.19 − 7.24 (m, 2H), 7.13 (d, J = 7.78 Hz, 2H), 6.84 (dd, J = 2.06, 7.86 Hz, 1H), 6.50 (t, J = 5.95 Hz, 1H, exchangeable with D2O), 4.29 (d, J = 5.80 Hz, 2H), 2.26 (s, 3H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.5, 168.7, 149.0, 143.2, 141.5, 138.6, 136.8, 135.7, 129.6, 129.3, 129.1, 128.9, 127.2, 120.6, 118.8, 117.0, 116.2, 111.3, 46.1, 24.1, 20.7. MS (ESI) m/z: 385 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 385.1911, Found, 385.1906.
4.1.2.7. (E)-N-(4–(3-(3-((4-acetamidobenzyl)amino)phenyl)-3-oxoprop-1-en-1-yl)phenyl) acetamide (5 g)
Orange powder (yield 85% − 0.85 mmol, 363 mg); m.p. 109–111 °C; IR (υmax./cm−1) 3290 and 3184 (N-H), 1660 and 1601 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 10.17 (s, 1H, exchangeable with D2O), 9.89 (s, 1H, exchangeable with D2O), 7.6 (d, J = 8.70 Hz, 2H), 7.66 (d, J = 8.54 Hz, 2H), 7.62 (d, J = 5.34 Hz, 2H), 7.52 (d, J = 8.39 Hz, 2H), 7.28 − 7.32 (m, 3H), 7.20 − 7.24 (m, 2H), 6.84 (dd, J = 2.06, 8.01 Hz, 1H), 6.48 (t, J = 5.87 Hz, 1H, exchangeable with D2O), 4.27 (d, J = 5.80 Hz, 2H), 2.07 (s, 3H), 2.02 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.5, 168.5, 168.0, 148.9, 143.1, 141.4, 138.5, 137.9, 134.2, 129.5, 129.3, 129.0, 127.5, 120.6, 119.0, 118.8, 116.8, 116.2, 111.4, 46.0, 24.0, 23.8. MS (ESI) m/z: 428 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 428.1969, Found, 428.1959.
4.1.2.8. (E)-N-(4–(3-(3-((3,4-dimethoxybenzyl)amino)phenyl)-3-oxoprop-1-en-1-yl)phenyl) acetamide (5h)
Orange powder (yield 90% − 0.9 mmol, 387 mg); m.p. 94–96 °C; IR (υmax./cm−1) 3292 and 3271 (N-H), 1662 and 1642 (C = O); 1H NMR (600 MHz, DMSO-d6) δ 10.17 (s, 1H, exchangeable with D2O), 7.76 (d, J = 8.80 Hz, 2H), 7.68 (d, J = 8.51 Hz, 2H), 7.63 (d, J = 2.35 Hz, 2H), 7.31 (d, J = 7.63 Hz, 1H), 7.26 (s, 1H), 7.23 (t, J = 7.78 Hz, 1H), 7.01 − 7.03 (m, 1H), 6.86 − 6.92 (m, 3H), 6.44 (t, J = 5.72 Hz, 1H, exchangeable with D2O), 4.26 (d, J = 5.28 Hz, 2H), 3.73 (s, 3H), 3.71 (s, 3H), 2.08 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 189.5, 168.6, 149.0, 148.8, 147.7, 143.2, 141.5, 138.5, 132.1, 129.6, 129.3, 129.1, 120.5, 119.3, 118.9, 117.0, 116.2, 111.7, 111.4, 111.3, 55.5, 55.4, 46.3, 24.1. MS (ESI) m/z: 431 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 431.1965, Found, 431.1965.
4.1.2.9. (E)-N-(4–(3-(3-((benzo[b]thiophen-3-ylmethyl)amino)phenyl)-3-oxoprop-1-en-1-yl) phenyl) acetamide (5i)
Orange powder (yield 78% − 0.78 mmol, 332 mg); m.p. 99–101 °C; IR (υmax./cm−1) 3302 and 3275 (N-H), 1672 and 1642 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 10.17 (s, 1H, exchangeable with D2O), 7.99 (d, J = 7.78 Hz, 2H), 7.75 (d, J = 8.70 Hz, 2H), 7.60 − 7.69 (m, 5H), 7.37 − 7.45 (m, 2H), 7.30 − 7.35 (m, 2H), 7.25 (t, J = 7.78 Hz, 1H), 6.92 − 6.99 (dd, J = 2.21, 8.16 Hz, 1H), 6.54 (t, J = 5.65 Hz, 1H, exchangeable with D2O), 4.59 (d, J = 5.65 Hz, 2H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.4, 168.7, 149.0, 143.2, 141.5, 140.1, 138.6, 138.0, 134.1, 129.6, 129.3, 129.2, 124.5, 124.1, 123.8, 122.9, 122.2, 120.5, 118.8, 116.9, 116.4, 111.4, 41.2, 24.1. MS (ESI) m/z: 427 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 427.1475, Found, 427.1468.
4.1.2.10. (E)-N-(4–(3-(3-((4-(hydroxymethyl)benzyl)amino)phenyl)-3-oxoprop-1-en-1-yl) phenyl) acetamide (5j)
Yellow powder (yield 75% − 0.75 mmol, 300 mg); m.p. 139–141 °C; IR (υmax./cm−1) 3300 (N-H), 1661 and 1643 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 10.16 (s, 1H, exchangeable with D2O), 7.76 (d, J = 8.54 Hz, 2H), 7.66 (d, J = 8.54 Hz, 2H), 7.62 (d, J = 3.05 Hz, 2H), 7.31 − 7.35 (m, 2H), 7.25 − 7.31 (m, 3H), 7.20 − 7.24 (m, 2H), 6.84 (dd, J = 2.21, 8.16 Hz, 1H), 6.52 (t, J = 5.95 Hz, 1H, exchangeable with D2O), 5.10 (br. s., 1H, exchangeable with D2O), 4.46 (br. s., 2H), 4.32 (d, J = 5.80 Hz, 2H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.5, 168.6, 148.9, 143.1, 141.5, 141.0, 138.5, 138.2, 129.6, 129.3, 129.1, 126.9, 126.5, 120.6, 118.8, 116.9, 116.2, 111.3, 62.7, 46.2, 24.1. MS (ESI) m/z: 401 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 401.1860, Found, 401.1850.
4.1.2.11. (E)-N-(4–(3-oxo-3–(3-(((2-oxo-2H-chromen-6-yl)methyl)amino)phenyl)prop-1-en-1-yl)phenyl)acetamide (5k)
Yellow powder (yield 83% − 0.83 mmol, 364 mg); m.p. 153–155 °C; IR (υmax./cm−1) 3316 and 3274 (N-H), 1673 and 1629 (C = O); 1H NMR (500 MHz, DMSO-d6) δ 10.16 (s, 1H, exchangeable with D2O), 8.06 (d, J = 9.61 Hz, 1H), 7.75 (d, J = 8.54 Hz, 2H), 7.71 (s, 1H), 7.59 − 7.68 (m, 5H), 7.38 (d, J = 8.54 Hz, 1H), 7.33 (d, J = 7.48 Hz, 1H), 7.21 − 7.26 (m, 2H), 6.86 (dd, J = 2.21, 8.01 Hz, 1H), 6.62 (t, J = 5.95 Hz, 1H, exchangeable with D2O), 6.47 (d, J = 9.46 Hz, 1H), 4.41 (d, J = 5.80 Hz, 2H), 2.07 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 189.4, 168.5, 159.9, 152.4, 148.7, 144.1, 143.1, 141.4, 138.6, 136.3, 130.9, 129.5, 129.2, 129.1, 126.5, 120.6, 118.8, 118.5, 116.8, 116.5, 116.3, 116.2, 111.4, 45.6, 24.0. MS (ESI) m/z: 439 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 439.1652, Found, 439.1640.
4.1.2.12. (E)-N-(4-(((3–(3-(1H-imidazol-4-yl)acryloyl)phenyl)amino)methyl)phenyl)acetamide (5 l)
Yellow powder (yield 72% − 0.72 mmol, 260 mg); m.p. 94–96 °C; IR (υmax./cm−1) 3292 and 3185 (N-H), 1661 and 1602 (C = O); 1H NMR (600 MHz, DMSO-d6) δ 12.43 (br. s., 1H, exchangeable with D2O), 9.87 (s, 1H, exchangeable with D2O), 7.81 (br. s., 1H), 7.48 − 7.62 (m, 5H), 7.28 (d, J = 8.51 Hz, 2H), 7.15 − 7.23 (m, 3H), 6.81 (d, J = 7.04 Hz, 1H), 6.49 (t, J = 5.87 Hz, 1H, exchangeable with D2O), 4.26 (d, J = 5.87 Hz, 2H), 2.01 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 189.9, 168.6, 149.5, 139.2, 138.5, 138.0, 134.7, 130.2, 129.6, 128.0, 122.1, 119.5, 119.2, 118.8, 116.8, 116.2, 112.0, 46.5, 24.4. MS (ESI) m/z: 361 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 361.1659, Found, 361.1653.
4.1.2.13 (E)-1–(3-((4-(hydroxymethyl)benzyl)amino)phenyl)-3-(1H-imidazol-4-yl)prop-2-en-1-one (5 m)
Yellow powder (yield 85% − 0.85 mmol, 283 mg); m.p. 104–106 °C; IR (υmax./cm−1) 3358 and 3316 (N-H), 1656 (C = O); 1H NMR (600 MHz, DMSO-d6) δ 12.43 (br. s., 1H, exchangeable with D2O), 7.83 (br. s., 1H), 7.50 − 7.62 (m, 3H), 7.31 − 7.34 (d, J = 7.92 Hz, 2H), 7.25 − 7.28 (d, J = 8.22 Hz, 2H), 7.12 − 7.24 (m, 4H), 6.81 (d, J = 7.92 Hz, 1H), 6.55 (t, J = 6.02 Hz, 1H, exchangeable with D2O), 5.10 (br. s., 1H, exchangeable with D2O), 4.46 (s, 2H), 4.31 (d, J = 5.87 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 190.2, 149.3, 141.2, 139.1, 138.7, 138.0, 137.0, 129.7, 127.4, 127.1, 122.0, 119.0, 117.2, 116.5, 113.3, 111.7, 63.1, 46.4. MS (ESI) m/z: 334 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 334.1550, Found, 334.1543.
4.1.2.14. (E)-6-(((3–(3-(1H-imidazol-4-yl)acryloyl)phenyl)amino)methyl)-2H-chromen-2-one (5n)
Yellow powder (yield 83% − 0.78 mmol, 308 mg); m.p. 104–106 °C; IR (υmax./cm−1) 3299 (N-H), 1674 and 1642 (C = O); 1H NMR (600 MHz, DMSO-d6) δ 12.54 (br. s., 1H, exchangeable with D2O),8.06 (d, J = 9.39 Hz, 1H), 7.82 (s, 1H), 7.71 (d, J = 2.05 Hz, 1H), 7.58 − 7.64 (m, 2H), 7.57 (s, 1H), 7.50 − 7.55 (m, 1H), 7.38 (d, J = 8.51 Hz, 1H), 7.17 − 7.25 (m, 3H), 6.82 (dd, J = 2.35, 7.92 Hz, 1H), 6.67 (t, J = 6.02 Hz, 1H, exchangeable with D2O), 6.47 (d, J = 9.68 Hz, 1H), 4.40 (d, J = 5.87 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 190.1, 160.7, 152.8, 149.0, 144.8, 139.2, 136.9, 132.6, 131.4, 131.3, 129.8, 129.7, 127.0, 119.0, 118.9, 117.2, 116.8, 116.7, 116.7, 113.3, 111.8, 45.9. MS (ESI) m/z: 372 [M + H]+; HRMS (ESI) m/z [M + H]+: Calcd. 372.1343, Found, 372.1335.
4.2. Biological assays
4.2.1. Preparation of compounds stock solutions
DMSO (Sigma-Aldrich, St. Louis, MO, USA) was used as solvent for either negative control or for preparation of 1 mM stocks of the synthetic analogues and stored at −20 °C.
4.2.2. Assay for anticancer activity
The newly synthesised analogues were sent to the National Cancer Institute, Germantown, MD, USA for testing their potential anti-cancer activity after 48 h using sulforhodamine B assayCitation68. Parameters for cellular growth inhibition including 50% growth inhibitory concentration (GI50) were determined in 60 different cancer cell lines represent nine human cancers: breast, central nervous system, colon, kidney, leukaemia, lung, melanoma, ovary, and prostateCitation69 deposited in National Cancer Institute, USA as presented in and . The one-dose results resemble the mean graphs from the 5-dose experiment in appearance and presented as a mean graph of the treated cells’ growth percentage. The one-dose assay’s reports figure percent growth of treated cells in comparison to both the time zero cell count and the no-drug control. This makes it possible to identify growth inhibition (values between 0 and 100) and lethality (values less than 0). The cancer screening panel’s human tumour cell lines are cultured in RPMI 1640 media supplemented with 2 mM L-glutamine and 5% foetal bovine serum. Depending on the doubling time of each cell line, 100 μL of cells are plated at plating densities ranging from 5,000 to 40,000 cells/well for a standard screening experiment in 96-well microtiter plates. Before adding the compounds, the microtiter plates are incubated for 24 h at 37° C, 5% CO2, 95% air, and 100% relative humidity following cell inoculationCitation40. 10 µM was used as initial testing concentration followed by single dose analysis. Compounds which exhibit significant growth inhibition in the one-dose screen are evaluated against the 60-cell panel at five concentration levels at five dose concentrations: 0.01 μM–100 µM using the same previously mentioned protocol. For cytotoxicity assessment, WI-38 normal human fibroblast cells were used for the calculation of safety index which is the ratio of IC50 compound (WI-38)/IC50 compound (cancer cell line)Citation70. Increasing SI value above 1 indicates a more effective and safer drug as an anticancer compared to normal tissuesCitation71. Synthetic analogues that showed promising anti-proliferation activity with low cytotoxicity at low concentrations were considered the most effective compounds.
4.2.3. Cell culture
The HCT-116 cancer cell line was obtained from the tissue culture unit’s cell culture bank at the Holding company for the production of vaccines, sera, and drugs (VACSERA), Giza, Egypt, and grown, maintained, observed for growth and further assays in the Centre of Scientific Excellence "Helwan Structural Biology Research, (HSBR)" according to the previous protocolCitation72,Citation73. Briefly, HCT-116 cells were cultured in McCoys5a culturing media supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in a humidified atmosphere containing 5% CO2 and kept for observation until 90% confluency and being ready for splitting using 0.25% Trypsin.
4.2.4. Flow cytometry analysis
Apoptosis and cell cycle were performed according to the standard protocolsCitation74–77. Briefly, HCT-116 cancer cell line was incubated at a density of 1 × 106 cells per 25 cm2 flask for 24 h. the IC50 of the Doxorubicin (positive control) or synthetic compounds 5e and 5h were employed in this investigation. To find cells with a sub-G1 DNA content, cells were concentrated at 1500 × g and suspended in 50 g/ml propidium iodide (PI) staining solution and 20 g/ml RNaseACitation78. The Cytoflex flow cytometer (Beckman Coulter, USA) was used for the measurement of cell flow. This will be followed by flow cytometric analysis using a Beckman Coulter CytExpert software (version 2.4.0.28). The gating strategy used includes characterising heterogeneous large population by plotting population against 2D- plot forward scattered beam area (FSC-A) against forward side scattered beam height (SSC-H) to eliminate doublets followed by plotting the resulted collective population by plotting 2D contour plot to show the core cell population spread of Annexin-FITC-H against PI PE-H to spread cell populations in four quadrants known as Q1(LL), Q2 (LR), Q3 (UL), and Q4 (UR)Citation79,Citation80. For cell cycle analysis, treated HCT-116 cells were trypsinized and washed with PBS and fixed with ethanol (70%) for approximately an hour followed by washing with PBS. The cells were finally resuspended in solution of propidium iodide (PI) (50 µg/ml) and RNase1 (250 µg/ml) with incubation for a period of 30 min at room temperature and final investigation Cytoflex flow cytometer (Beckman Coulter, USA).
4.2.5. VEGFR-2 inhibition assay
All newly synthetic derivatives were investigated in vitro for their inhibitory activities against VEGFR-2 as a potential molecular target. The VEGFR-2 Kinase Assay Kit (BPS-Bioscience, #78857, USA)Citation81,Citation82 was used for testing the VEGFR tyrosine kinase activityCitation83,Citation84. Sorafenib was used as a positive control. Briefly, all synthesised compounds were evaluated for their ability to inhibit VEGFR-2 using 96-well plate seeded with a particular VEGFR-2 antibody, 100 µL of the standard solution (for preparation of standard curve) or the tested compounds were added, and all reactions was then incubated at room temperature for 2.5 h. Following a wash, 25 µL of the ADP-Glo™ reagent was added to each well, which was then incubated for an additional 45 min at room temperature before being washed. 50 µl of Kinase Detection reagent solution was then added, and the mixture was incubated at room temperature for 45 min. Immediately reading was performed in Luminoskan™ Ascent Microplate Luminometer (Thermofisher, USA, Cat. No. 2805621). Half-maximal inhibitory concentrations (IC50) were calculated from standard and samples as concentration–inhibition response curve. The developed signal was detected and quantified by a BioTek microplate reader for determination of IC50 of the tested compounds.
4.2.6. Gene expression analysis using RT-qPCR
Assessment for the gene expression analysis of key regulatory genes were performed using real-time quantitative PCR (RT-qPCR) as previously documentedCitation72,Citation73. Briefly, HCT-116 cancer cells were treated with IC50 of compounds 5e and 5h for 24 h and cells were harvested, and total RNA was extracted using Favor-PrepTM Blood/Cultured cell total RNA purification mini kit (Favorgen Biotech Corp., Ping-Tung, Taiwan). The purified RNA was then reverse transcribed into the first-strand cDNA using Revert Aid First-Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, USA). Key regulated genes (Caspase-3, Cyt-c, TNF-α, Bax, and Bcl-2) were assessed by gene expression analysis (primers sequence in ) using HERAPLUS SYBR® Green qPCR Kit (Willowfort, Nottingham, UK). Differential gene expression was performed using 2−ΔΔCT method using β-actin as reference geneCitation85.
Table 8. Primer’s sequence of apoptotic genes used for gene expression analysis.
4.2.7. Determination of caspase-3 and Bax levels using ELISA assay
The assays for caspase-3 (Abcam, ab39401)Citation86 and Bax activity (Abcam, ab199080)Citation87 are based on cleavage of the chromogenic substrate. Control and treated cells with IC50 of compounds 5e and 5h were harvested from 25cm2 flasks and lysed in HCT-116 cell lysis buffer (50 nmol/l, Tris-HCL, pH 7.5, 0.03% NP40, 1.0 mmol/l DTT) for 10 min. Lysates were then centrifuged at 12,000 g for 12 min and protein concentration of the supernatant (cytosolic extract) was estimated using the Biorad assay. Equal amounts of protein extracts were loaded onto a 96-well microplate and incubated with reaction buffer, DTT, DMSO. The assay is based on spectrophotometric detection of the chromophore p-nitroaniline (p-NA) after cleavage from the labelled substrate DEVD-pNA. The p-NA light emission was quantified colorimetry using BioTek microtiter plate reader (Biotek 800 TS) according to each kit requirement. Briefly, 50 µL of Reaction Mix (2X Reaction Buffer and DTT) was added to each sample, sample background control and the background wells that are pre-coated with primary antibodies specific to caspase-3 and Bax proteins. 5 μL of the 4 mM DEVD-pNA (200 μM final concentration) was added to each sample well, and the background well and incubate for 120 min and colour was determined colorimetry.
4.3. Molecular docking studies
4.3.1. Molecular docking
All the docking studies in the current work were carried out by implementing the docktite wizard of the Molecular Operating Environment (MOE 2019.02) packageCitation88,Citation89. The structural coordinates of the target enzyme were downloaded from the protein data bank: PDB IDs 4ASD for VEGFR-2 in complex with sorafenib. The two selected proposed ligands were sketched using the MOE builder and then energy minimisation and a conformational search were conducted using the default parameters of MOE software. The energy-minimized compounds 5e and 5h were saved to a single database file with the *mdb extension ready for docking conduction. By choosing the pocket around the binding domain of the co-crystallized ligand, the binding site where the docking is conducted was determined. To elucidate the most potential mode of binding for the proposed compounds, both covalent and non-covalent approach were applied the non-covalent approach gave the best results, suggesting a reversible binding mode Pose-retrieval docking step for the X-ray coordinates of the co-crystallized Sorafenib within its respective binding site was conducted to verify the applied docking shoed the best procedureCitation90. After that, compounds 5e and 5h were docked into the VEGFR-2 binding site with the validated protocol. Finally, docking results were further analysed through 2D interaction diagrams generated for the predicted binding mode between compounds 5e and 5h with VEGFR-2 enzyme.
4.3.2. Molecular dynamics
The two complexes 5h–VEGFR-2 and sorafenib–VEGFR-2, in addition to the Apo form of VEGFR-2, were subjected to molecular dynamic simulation (MDS) for 100,000 ps. The same MDS methodology as previously published by our group was applied in the current studyCitation90,Citation91. The receptor and ligand topologies were generated by PDB2gmx (embedded in GROMACS) and GlycoBioChem PRODRG2 Server respectively, both under GROMOS96 force fieldCitation92. After rejoining ligands and receptor topologies to generate three systems, the typical molecular dynamics scheme of GROMACS was applied for all the systems. This includes, solvation, neutralisation, energy minimisation under GROMOS96 43a1 force field and two stages of equilibration (NVT and NPT)Citation90,Citation91,Citation93. Finally, unrestricted production stage of 100 ns was applied for the four systems with the Particle Mesh Ewald (PME) method implemented to compute the long-range electrostatic values using 12 Å cut-off and 12 Å Fourier spacing. Indicative parameters, such as RMSD and RMSF, were calculated to evaluate the stability of all the complexes.
Author contributions
Hesham Haffez (biological experiments, project administration, data curation, formal analysis, writing – original draft, review and editing), Nosaiba A. Elsayed (project administration, chemistry experiments, software and writing – original draft), Marwa F. Ahmed (conceptualization and review and editing), Samar S. Fatahala (data curation, formal analysis, writing – original draft), Eman F. Khaleel (funding acquisition, resources and review and editing), Rehab Mustafa Badi (funding acquisition, resources and review and editing), Eslam B. Elkaeed (funding acquisition, resources and review and editing), Mahmoud A. El Hassab (molecular dynamics, software and writing – original draft, review and editing), Sherif F. Hammad (conceptualization and review and editing), Wagdy M. Eldehna (conceptualization, project administration, molecular dynamics, software and review and editing), Nicolas Masurier (conceptualization and writing – original draft, review and editing), and Radwan El-Haggar (chemistry experiments, project administration, data curation, formal analysis, supervision, funding acquisition, writing – original draft, review and editing).
Supplemental Material
Download PDF (8.4 MB)Acknowledgement
The authors are thankful to the Deanship of Scientific Research at King Khalid University for funding this work through Large Group Research Project under grant number (RGP2/321/44). The authors extend their appreciation to Scientific Research Fund - Vice president′s office for Graduate Studies and Research - Helwan University, and to Prof. Sameh Soror for giving them the opportunity to use the facilities/equipment available in HSBR laboratory. Also, authors would like to acknowledge the National Cancer Institute (NCI), developmental therapeutics program (DTP) of the United States for performing the anticancer screening.
Disclosure statement
The authors report no conflicts of interest.
References
- Diori K, Hamarat S. Reviewing cancer’s biology: an eclectic approach. J Egypt Natl Canc Inst. 2021;33:1–17.
- Gasparini G, Longo R, Toi M, Ferrara N. Angiogenic inhibitors: a new therapeutic strategy in oncology. Nat Clin Pract Oncol. 2005;2(11):562–577.
- Saman H, Raza SS, Uddin S, Rasul K. Inducing angiogenesis, a key step in cancer vascularization, and treatment approaches. Cancers (Basel). 2020;12(5):1172.
- Liang P, Ballou B, Lv X, Si W, Bruchez MP, Huang W, Dong X. Monotherapy and combination therapy using anti‐angiogenic nanoagents to fight cancer. Adv Mater. 2021;33(15):2005155.
- Haibe Y, Kreidieh M, El Hajj H, Khalifeh I, Mukherji D, Temraz S, Shamseddine A. Resistance mechanisms to anti-angiogenic therapies in cancer. Front Oncol. 2020;10:221.
- Qian Y, Han Q, Chen W, Song J, Zhao X, Ouyang Y, Yuan W, Fan C. Platelet-rich plasma derived growth factors contribute to stem cell differentiation in musculoskeletal regeneration. Front Chem. 2017;5:89.
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti-and pro-angiogenic therapies. Genes Cancer. 2011;2(12):1097–1105.
- Takahashi H, Shibuya M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin Sci (Lond)). 2005;109(3):227–241.
- Pauli SA, Tang H, Wang J, Bohlen P, Posser R, Hartman T, Sauer MV, Kitajewski J, Zimmermann R. The vascular endothelial growth factor (vegf)/vegf receptor 2 pathway is critical for blood vessel survival in corpora lutea of pregnancy in the rodent. Endocrinol. 2005;146(3):1301–1311.
- Dang Y-Z, Zhang Y, Li J-P, Hu J, Li W-W, Li P, Wei L-C, Shi M. High vegfr1/2 expression levels are predictors of poor survival in patients with cervical cancer. Medicine (Baltimore)). 2017;96(1):e5772.
- Dakowicz D, Zajkowska M, Mroczko B. Relationship between vegf family members, their receptors and cell death in the neoplastic transformation of colorectal cancer. Int J Mol Sci. 2022;23(6):3375.
- Ding M, Liu L, Hu C, Liu Y, Qiao Y, Jiang X. Expression of vegfr2 and nrp-1 in non-small cell lung cancer and their clinical significance. Chin J Cancer Res. 2014;26(6):669.
- Choi SB, Han HJ, Kim WB, Song TJ, Choi S. Vegf overexpression predicts poor survival in hepatocellular carcinoma. Open Med (Wars)). 2017;12(1):430–439.
- Song SH, Jeong IG, You D, Hong JH, Hong B, Song C, Jung WY, Cho YM, Ahn H, Kim C, et al. Vegf/vegfr2 and pdgf-b/pdgfr-β expression in non-metastatic renal cell carcinoma: A retrospective study in 1,091 consecutive patients. Int J Clin Exp Pathol. 2014;7(11):7681.
- Otrock ZK, Makarem JA, Shamseddine A. Vascular endothelial growth factor family of ligands and receptors. Blood Cells Mol Dis. 2007;38(3):258–268.
- Abdullaziz MA, Abdel-Mohsen HT, El Kerdawy AM, Ragab FA, Ali MM, Abu-Bakr SM, Girgis AS, El Diwani H. Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as vegfr-2 inhibitors. Eur J Med Chem. 2017;136:315–329.
- Kassab AE, Gedawy EM, Hamed MI, Doghish AS, Hassan R. Design, synthesis, anticancer evaluation, and molecular modelling studies of novel tolmetin derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. J Enzyme Inhib Med Chem. 2021;36(1):922–939.
- Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, Chinot O. Bevacizumab (avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 2020;86:102017.
- Lee C, Chen R, Sun G, Liu X, Lin X, He C, Xing L, Liu L, Jensen LD, Kumar A, et al. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. 2023;8(1):305.
- Ciombor KK, Berlin J. Aflibercept—a decoy vegf receptor. Curr Oncol Rep. 2014;16(2):368.
- De Groot AC. Dermatological drugs, topical agents and cosmetics. In: Side effects of drugs annual. Amsterdam, Netherlands: Elsevier; 1992.
- Stanel SC, Sjöberg J, Salmonson T, Foggi P, Caleno M, Melchiorri D, Gravanis I, Tzogani K, Pignatti F. European medicines agency approval summary: Zaltrap for the treatment of patients with oxaliplatin-resistant metastatic colorectal cancer. ESMO Open. 2017;2(2):e000190.
- Javle M, Smyth EC, Chau I. Ramucirumab: successfully targeting angiogenesis in gastric cancer. Clin Cancer Res. 2014;20(23):5875–5881.
- Cheng Y, Zhang T, Xu Q. Therapeutic advances in non‐small cell lung cancer: Focus on clinical development of targeted therapy and immunotherapy. MedComm (2020)). 2021;2(4):692–729.
- Ghalehbandi S, Yuzugulen J, Pranjol MZI, Pourgholami M. The role of vegf in cancer-induced angiogenesis and research progress of drugs targeting vegf. Eur J Pharmacol. 2023; 949:175586.
- Yang F, Jove V, Xin H, Hedvat M, Van Meter TE, Yu H. Sunitinib induces apoptosis and growth arrest of medulloblastoma tumor cells by inhibiting stat3 and akt signaling pathways. Mol Cancer Res. 2010;8(1):35–45.
- Grosse J, Warnke E, Wehland M, Pietsch J, Pohl F, Wise P, Magnusson NE, Eilles C, Grimm D. Mechanisms of apoptosis in irradiated and sunitinib-treated follicular thyroid cancer cells. Apoptosis. 2014;19(3):480–490.
- Varinska L, van Wijhe M, Belleri M, Mitola S, Perjesi P, Presta M, Koolwijk P, Ivanova L, Mojzis J. Anti-angiogenic activity of the flavonoid precursor 4-hydroxychalcone. Eur J Pharmacol. 2012;691(1-3):125–133.
- Lee YS, Lim SS, Shin KH, Kim YS, Ohuchi K, Jung SH, Bulletin P. Anti-angiogenic and anti-tumor activities of 2′-hydroxy-4′-methoxychalcone. Biol Pharm Bull. 2006;29(5):1028–1031.
- Ahmed MF, Santali EY, El-Haggar R. Novel piperazine–chalcone hybrids and related pyrazoline analogues targeting vegfr-2 kinase; design, synthesis, molecular docking studies, and anticancer evaluation. J Enzyme Inhib Med Chem. 2021;36(1):307–318.
- Eissa IH, El-Haggar R, Dahab MA, Ahmed MF, Mahdy HA, Alsantali RI, Elwan A, Masurier N, Fatahala S. Design, synthesis, molecular modeling and biological evaluation of novel benzoxazole-benzamide conjugates via a 2-thioacetamido linker as potential anti-proliferative agents, vegfr-2 inhibitors and apoptotic inducers. J Enzyme Inhib Med Chem. 2022;37(1):1587–1599.
- Ahmed MF, El‐Haggar R, Almalki AH, Abdullah O, El Hassab MA, Masurier N, Hammad S. Novel hydrazone‐isatin derivatives as potential egfr inhibitors: synthesis and in vitro pharmacological profiling. Arch. Pharm. 2023;356(9):2300244.
- Baccon-Sollier PL, Malki Y, Maye M, Ali LM, Lichon L, Cuq P, Vincent L-A, Masurier N. Imidazopyridine-fused [1, 3] diazepinones: Modulations of positions 2 to 4 and their impacts on the anti-melanoma activity. J Enzyme Inhib Med Chem. 2020;35(1):935–949.
- Bellet V, Lichon L, Arama DP, Gallud A, Lisowski V, Maillard LT, Garcia M, Martinez J, Masurier N. Imidazopyridine-fused [1, 3]-diazepinones part 2: structure-activity relationships and antiproliferative activity against melanoma cells. Eur J Med Chem. 2017;125:1225–1234.
- Peyressatre M, Arama DP, Laure A, González-Vera JA, Pellerano M, Masurier N, Lisowski V, Morris M. Identification of quinazolinone analogs targeting cdk5 kinase activity and glioblastoma cell proliferation. Front Chem. 2020;8:691.
- Gallud A, Vaillant O, Maillard LT, Arama DP, Dubois J, Maynadier M, Lisowski V, Garcia M, Martinez J, Masurier N. Imidazopyridine-fused [1, 3]-diazepinones: synthesis and antiproliferative activity. Eur J Med Chem. 2014;75:382–390.
- Modi SJ, Kulkarni V. Vascular endothelial growth factor receptor (vegfr-2)/kdr inhibitors: Medicinal chemistry perspective. Med Drug Discov. 2019;2:100009.
- Boyd MR, Paull K. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev Res. 1995;34(2):91–109.
- Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd M. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48(3):589–601.
- Shoemaker R. The nci60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6(10):813–823.
- USA: Developmental Therapeutic Program [2021; https://dtp.cancer.gov/.
- Koch A, Tamez P, Pezzuto J, Soejarto D. Evaluation of plants used for antimalarial treatment by the maasai of kenya. J Ethnopharmacol. 2005;101(1-3):95–99.
- Badisa R, Ayuk-Takem L, Ikediobi C, Walker E. Selective anticancer activity of pure licamichauxiioic-b acid in cultured cell lines. Pharm Biol. 2006;44(2):141–145.
- Wardihan Rusdi M, Alam G, Lukman , Manggau MA. Selective cytotoxicity evaluation in anticancer drug screening of boehmeria virgata (forst) guill leaves to several human cell lines: Hela, widr, t47d and vero. Dhaka Univ J Pharm Sci. 2014;12(2):87–90.
- Acton EM, Narayanan VL, Risbood PA, Shoemaker RH, Vistica DT, Boyd M. Anticancer specificity of some ellipticinium salts against human brain tumors in vitro. J Med Chem. 1994;37(14):2185–2189.
- Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P. Vegf, a prosurvival factor, acts in concert with tgf-β1 to induce endothelial cell apoptosis. Proc Natl Acad Sci U S A. 2006;103(46):17260–17265.
- Shibuya M. Vascular endothelial growth factor receptor‐2: its unique signaling and specific ligand, vegf‐e. Cancer Sci. 2003;94(9):751–756.
- Rizvi SU, Siddiqui HL, Nisar M, Khan N, Khan IJ. Discovery and molecular docking of quinolyl-thienyl chalcones as anti-angiogenic agents targeting vegfr-2 tyrosine kinase. Bioorg Med Chem Lett. 2012;22(2):942–944.
- Synowiec E, Hoser G, Bialkowska-Warzecha J, Pawlowska E, Skorski T, Blasiak J. Doxorubicin differentially induces apoptosis, expression of mitochondrial apoptosis-related genes, and mitochondrial potential in bcr-abl1-expressing cells sensitive and resistant to imatinib. Biomed Res Int. 2015;2015:673512–673519.
- Pilco-Ferreto N, Calaf G. Influence of doxorubicin on apoptosis and oxidative stress in breast cancer cell lines. Int J Oncol. 2016;49(2):753–762.
- Zinonos I, Labrinidis A, Liapis V, Hay S, Panagopoulos V, Denichilo M, Ponomarev V, Ingman W, Atkins GJ, Findlay D. Doxorubicin overcomes resistance to drozitumab by antagonizing inhibitor of apoptosis proteins (iaps). Anticancer Res. 2014;34(12):7007–7020.
- Tacar O, Sriamornsak P, Dass C. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157–170.
- Kwan YP, Saito T, Ibrahim D, Al-Hassan FMS, Ein Oon C, Chen Y, Jothy SL, Kanwar JR, Sasidharan S. Evaluation of the cytotoxicity, cell-cycle arrest, and apoptotic induction by euphorbia hirta in mcf-7 breast cancer cells. Pharm Biol. 2016;54(7):1223–1236.
- Kim HS, Lee YS, Kim DK. Doxorubicin exerts cytotoxic effects through cell cycle arrest and fas-mediated cell death. Pharmacology. 2009;84(5):300–309.
- Kciuk M, Gielecińska A, Mujwar S, Kołat D, Kałuzińska-Kołat Ż, Celik I, Kontek R. Doxorubicin—an agent with multiple mechanisms of anticancer activity. Cells. 2023;12(4):659.
- Fan LL, Sun GP, Wei W, Wang ZG, Ge L, Fu WZ, Wang H. Melatonin and doxorubicin synergistically induce cell apoptosis in human hepatoma cell lines. World J Gastroenterol. 2010;16(12):1473–1481.
- Mehraj U, Mir IA, Hussain MU, Alkhanani M, Wani NA, Mir MA. Adapalene and doxorubicin synergistically promote apoptosis of tnbc cells by hyperactivation of the erk1/2 pathway through ros induction. Front Oncol. 2022;12:938052.
- Amalina ND, Salsabila IA, Zulfin UM, Jenie RI, Meiyanto E. In vitro synergistic effect of hesperidin and doxorubicin downregulates epithelial-mesenchymal transition in highly metastatic breast cancer cells. J Egypt Natl Canc Inst. 2023;35(1):6.
- Miladiyah I, Yuanita E, Nuryadi S, Jumina J, Haryana SM, Mustofa M. Synergistic effect of 1, 3, 6-trihydroxy-4, 5, 7-trichloroxanthone in combination with doxorubicin on b-cell lymphoma cells and its mechanism of action through molecular docking. Curr Ther Res Clin Exp. 2020;92:100576.
- Park HK, Lee JE, Lim J, Jo DE, Park SA, Suh PG, Kang B. Combination treatment with doxorubicin and gamitrinib synergistically augments anticancer activity through enhanced activation of bim. BMC Cancer. 2014;14(1):431.
- Evan GI, Vousden K. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411(6835):342–348.
- Brooks R. Cell cycle commitment and the origins of cell cycle variability. Front Cell Dev Biol. 2021;9:698066.
- Cohen G. Caspases: the executioners of apoptosis. Biochem J. 1997;326 (Pt 1) (Pt 1):1–16.
- Reed J, Jurgensmeier J, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366(1-2):127–137.
- Vogel C, Marcotte E. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13(4):227–232.
- Pollman MJ, Naumovski L, Gibbons G. Endothelial cell apoptosis in capillary network remodeling. J Cell Physiol. 1999;178(3):359–370.
- Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel N. Inhibition of vegf receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106(11):1311–1319.
- Orellana EA, Kasinski AL. Sulforhodamine b (srb) assay in cell culture to investigate cell proliferation. Bio Protoc. 2016;6(21):1984–1984.
- Ciftci H, Sever B, Bayrak N, Yıldız M, Yıldırım H, Tateishi H, Otsuka M, Fujita M, TuYuN AF. In vitro cytotoxicity evaluation of plastoquinone analogues against colorectal and breast cancers along with in silico insights. Pharmaceuticals. 2022;15(10):1266.
- Peña-Morán OA, Villarreal ML, Álvarez-Berber L, Meneses-Acosta A, Rodríguez-López V. Cytotoxicity, post-treatment recovery, and selectivity analysis of naturally occurring podophyllotoxins from bursera fagaroides var. Fagaroides on breast cancer cell lines. Molecules. 2016;21(8):1013.
- Braga CB, Kido LA, Lima EN, Lamas CA, Cagnon VH, Ornelas C, Pilli RA. Enhancing the anticancer activity and selectivity of goniothalamin using ph-sensitive acetalated dextran (ac-dex) nanoparticles: a promising platform for delivery of natural compounds. ACS Biomater Sci Eng. 2020;6(5):2929–2942.
- Abdelaal MR, Soror SH, Elnagar MR, Haffez H. Revealing the potential application of ec-synthetic retinoid analogues in anticancer therapy. Molecules. 2021;26(2):506.
- Abdelaal MR, Ibrahim E, Elnagar MR, Soror SH, Haffez H. Augmented therapeutic potential of ec-synthetic retinoids in caco-2 cancer cells using an in vitro approach. Int J Mol Sci. 2022;23(16):9442.
- Haffez H, Osman S, Ebrahim HY, Hassan Z. Growth inhibition and apoptotic effect of pine extract and abietic acid on mcf-7 breast cancer cells via alteration of multiple gene expressions using in vitro approach. Molecules. 2022;27(1):293.
- Khedr MA, Abu-Zied KM, Zaghary WA, Aly AS, Shouman DN, Haffez H. Novel thienopyrimidine analogues as potential metabotropic glutamate receptors inhibitors and anticancer activity: synthesis, in-vitro, in-silico, and SAR approaches. Bioorg Chem. 2021;109:104729.
- Lo KKW, Lee TKM, Lau JSY, Poon WL, Cheng SH. Luminescent biological probes derived from ruthenium (II) estradiol polypyridine complexes. Inorg Chem. 2008;47(1):200–208.
- Böhm I, Schild H. Apoptosis: The complex scenario for a silent cell death. Mol Imaging Biol. 2003;5(1):2–14.
- Su M, Li Y, Chung HY, Ye W. 2β-(isobutyryloxy) florilenalin, a sesquiterpene lactone isolated from the medicinal plant centipeda minima, induces apoptosis in human nasopharyngeal carcinoma cne cells. Molecules. 2009;14(6):2135–2146.
- Corripio-Miyar Y, MacLeod CL, Mair I, Mellanby RJ, Moore BD, McNeilly TN. Self-adjuvanting calcium-phosphate-coated microcrystal-based vaccines induce pyroptosis in human and livestock immune cells. Vaccines (Basel). 2023;11(7):1229.
- Saad MH, El-Moselhy TF, S E-D N, Mehany AB, Belal A, Abourehab MA, Tawfik HO, El-Hamamsy MH. Discovery of new symmetrical and asymmetrical nitrile-containing 1, 4-dihydropyridine derivatives as dual kinases and p-glycoprotein inhibitors: synthesis, in vitro assays, and in silico studies. J Enzyme Inhib Med Chem. 2022;37(1):2489–2511.
- Hassan A, Mubarak FA, Shehadi IA, Mosallam AM, Temairk H, Badr M, Abdelmonsef AH. Design and biological evaluation of 3-substituted quinazoline-2, 4 (1 h, 3 h)-dione derivatives as dual c-met/vegfr-2-tk inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):2189578.
- Elkady H, El-Dardir OA, Elwan A, Taghour MS, Mahdy HA, Dahab MA, Elkaeed EB, Alsfouk BA, Ibrahim IM, Husein DZ, et al. Synthesis, biological evaluation and computer-aided discovery of new thiazolidine-2, 4-dione derivatives as potential antitumor vegfr-2 inhibitors. RSC Adv. 2023;13(40):27801–27827.
- Abdallah AE, Mabrouk RR, Al Ward MMS, Eissa SI, Elkaeed EB, Mehany AB, Abo-Saif MA, El-Feky OA, Alesawy MS, El-Zahabi M. Synthesis, biological evaluation, and molecular docking of new series of antitumor and apoptosis inducers designed as vegfr-2 inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):573–591.
- Abdallah AE, Mabrouk RR, Elnagar MR, Farrag AM, Kalaba MH, Sharaf MH, El-Fakharany EM, Bakhotmah DA, Elkaeed EB, Al Ward M. New series of vegfr-2 inhibitors and apoptosis enhancers: design, synthesis and biological evaluation. Drug Des Devel Ther. 2022; 16:587–606.
- Livak KJ, Schmittgen T. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-Delta Delta C(T)). Method. Methods. 2001;25(4):402–408.
- Zhou Y, Li L, Mao C, Zhou D. Astragaloside iv ameliorates spinal cord injury through controlling ferroptosis in H2O2-damaged pc12 cells in vitro. Ann Transl Med. 2022;10 (21):1176–1176.
- Fu J, Zhao J, Zhang H, Fan X, Geng W, Qiao S. Microrna‑451a prevents cutaneous squamous cell carcinoma progression via the 3‑phosphoinositide‑dependent protein kinase‑1‑mediated pi3k/akt signaling pathway. Exp Ther Med. 2021;21(2):1–1.
- Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (moe): application of qsar and molecular docking to drug discovery. Curr Top Med Chem. 2008;8(18):1555–1572.
- Scholz C, Knorr S, Hamacher K, Schmidt B. Docktite- a highly versatile step-by-step workflow for covalent docking and virtual screening in the molecular operating environment. J Chem Inf Model. 2015;55(2):398–406.
- El Hassab MA, Ibrahim TM, Al-Rashood ST, Alharbi A, Eskandrani RO, Eldehna W. In silico identification of novel sars-cov-2 2′-o-methyltransferase (nsp16) inhibitors: structure-based virtual screening, molecular dynamics simulation and mm-pbsa approaches. J Enzyme Inhib Med Chem. 2021;36(1):727–736.
- Hassab MA, Fares M, Amin MK, Al-Rashood ST, Alharbi A, Eskandrani RO, Alkahtani HM, Eldehna W. Toward the identification of potential α-ketoamide covalent inhibitors for sars-cov-2 main protease: fragment-based drug design and mm-pbsa calculations. Processes. 2021;9(6):1004.
- Schüttelkopf AW, Van Aalten D. Prodrg: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–1363.
- El Hassab MA, Ibrahim TM, Shoun AA, Al-Rashood ST, Alkahtani HM, Alharbi A, Eskandrani RO, Eldehna W. In silico identification of potential sars cov-2 2′-o-methyltransferase inhibitor: fragment-based screening approach and mm-pbsa calculations. RSC Adv. 2021;11(26):16026–16033.