
Abstract
Adaptor associated kinase 1 (AAK1), a member of the Ark1/Prk1 family of Ser/Thr kinases, is a specific key kinase regulating Thr156 phosphorylation at the μ2 subunit of the adapter complex-2 (AP-2) protein. Due to their important biological functions, AAK1 systems have been validated in clinics for neuropathic pain therapy, and are being explored as potential therapeutic targets for diseases caused by various viruses such as Hepatitis C (HCV), Dengue, Ebola, and COVID-19 viruses and for amyotrophic lateral sclerosis (ALS). Centreing on the advances of drug discovery programs in this field up to 2023, AAK1 inhibitors are discussed from the aspects of the structure-based rational molecular design, pharmacology, toxicology and synthetic routes for the compounds of interest in this review. The aim is to provide the medicinal chemistry community with up-to-date information and to accelerate the drug discovery programs in the field of AAK1 small molecule inhibitors.
Introduction
Diseases of the central nervous system, such as neuropathic pain, Alzheimer’s disease (AD), and Parkinson’s disease (PD), seriously affect human health and social development, and have become serious public health problems around the world. Among them, Alzheimer’s disease and Parkinson’s disease are characterised by progressive degeneration of specific neurons in the brain, which currently affect many people in ChinaCitation1. However, at present, the treatment of central nervous system diseases is still a major medical problem in the world. Adaptor Associated Kinase 1 (AAK1) plays an important role in the treatment of nervous system diseases. AAK1, also known as AP-2-associated protein kinase 1, belongs to the Numb-associated kinase (NAK) familyCitation2,Citation3. It is a highly conserved Ser/Thr protein kinaseCitation4,Citation5. AAK1 was first identified in α-adaptin protein-interacting partners associated with phosphorylating the μ2 subunit of AP-2 at Thr156 and playing a regulatory role in the process of CMECitation6,Citation7. The reason why AAK1 can play an important role in nervous system diseases is that AAK1 participates in many nervous system-related signal pathways, such as NDR and CDK signal pathways. Therefore, AAK1 is associated with many human diseases, such as neuropathic pain, ADCitation8, PDCitation9, amyotrophic lateral sclerosis (ALS)Citation10 and so on. At present, several AAK1 inhibitors have entered the clinical stage, such as BMS-986176/LX-9211 for diabetic peripheral neuropathic pain (NCT04455633) and post-herpetic neuralgia (NCT04662281)Citation11.
It is worth noting that with the outbreak of COVID-19 in recent years, AAK1i has shown significant clinical efficacy in the treatment of COVID-19Citation12–14. Increasing evidence shows that drugs such as Baricitinib, Erlotinib, Sunitinib, and Fedratinib on the market have the ability to target AAK1 activity and thus play a role in blocking virus endocytosisCitation15. The main mechanism is that AAK1 can phosphorylate AP2M1, which is very important for the virus to induce clathrin-mediated endocytic pathway into cells. And in order to reduce virus invasion, the activity of the AAKl is inhibited and phosphorylated AP2M1 is down-regulated in AAKli. Long before the treatment of coronavirus, Neveu et al. proposed that AAK1 inhibitors have the effect of hepatitis C virus (HCV) in 2012Citation16. Subsequently, it was clearly stated in the US patent filed by Einav et al. that AAK1 inhibitors can destroy the assembly of HCV and the replication of HIV-1Citation17. However, the anti-viral effect of AAK1 seems to have been ignored by scientists, and its related research has not been reported in the literature. Until the outbreak of COVID-19 in 2019, researchers actively explored various mechanisms of antiviral treatment, and paid more attention to the anti-viral effect of AAK1 inhibitors.
Over the past several decades, research involving AAK1 has proliferated, as evidenced by the large numbers of scientific publications on the topic. Literature using the SciFinder® search engine and PubMed website with “AAK1” or “adaptor associated kinase 1” as the key word provide all references for further analysis. These references were further analysed by publication year. The result reveals that the publication (papers and patents) number increased from 19 in 2005 to 429 in 2023. Then the search was conducted again using “inhibitor” to exclude those references focuses on pure biology without involving any AAK1 inhibitors. The references obtained above were then further categorised using the therapeutic area as criteria. Therapeutic applications of AAK1 inhibitors that are being identified or validated mainly focus on pain and neurological diseasesCitation18, and anti-viral compoundsCitation19. The outbreak of the COVID-19 epidemic in recent years significantly increased the awareness of AAK1 protein and contributed to the hike in publication number. Recent advances in treating sequelae of COVID-19 infection suggest AAK1 could become one of the potential therapeutic targets to treat COVID-19Citation12–14. drug discovery/drug discovery fields, a number of articles have been published in different major journals. However, there are few articles about AAK1 inhibitorsCitation20,Citation21. In terms of drug discovery targeting AAK1 protein with small molecule inhibitors, relevant work has been carried out in this research group for several years. In addition, a literature review of AAK1 inhibitors was initiated last summer to formulate a review of the field with a focus on the medicinal chemistry aspect. This updated summary of accomplishments in the field could ultimately be helpful to the drug discovery community And a detailed analysis of AAK1 inhibitors from biochemical concepts to design strategies has been conducted, with the aim of formulating a field review focusing on medicinal chemistry. It is worth believing that the latest summary of achievements in this field may eventually help the drug discovery community. And the hope is that it will serve as a quick reference guide for the scientific community and inspire new ideas to accelerate drug discovery in this field.
Structural and biological function of AAK1
Structure of AAK1
AAK1 is a Ser/Thr protein kinase that belongs to the numb-associated kinase (NAK) family. This family also includes serine/threonine kinase 16/myristoylated and palmitoylated serine/threonine kinase 1 (STK16/MPSK1), Cyclin G associated kinase (GAK), and bone morphogenic protein BMP-2 inducible kinase (BIKE)Citation22,Citation23. And AAK1 is composed of 868 amino acids, with a highly conserved kinase domain (about 267 amino acids) at the N-terminal, α-aptamer interaction domain (AID) at the C-terminal, and a glutamine (28%) -proline (18%) -alanine (13%) QPA-rich region (residues 312–630) in the middle region ()Citation3,Citation7. Moreover, a long form of AAK1 (AAK1L) containing an extended C-terminal has been identifiedCitation24. This kind of AAK1 encodes an additional grid protein binding domain (CBD2), which consists of several low-affinity interaction motifs. However, although AAK1L CBD2 can directly bind to grid protein, its overexpression impairs the endocytosis of transferrin. Moreover, the fact that AAK1 plays a role in multiple steps of the endosome pathway has also been confirmed.
Figure 1. Schematic diagram of the AAK1 kinase domain and domain organisation comparison of the human NAK family.
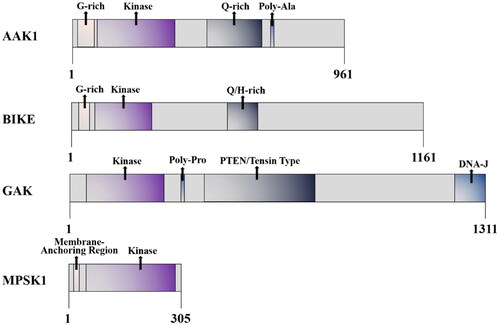
In the NAK family, kinase domains are all close to the amino terminal and highly similar, but they have their own characteristics in other regional domainsCitation4. This fact highlights the substrate specificity and the different physiological functions of each family member. For example, NAK is related to a wide range of cell functions. In addition to the previously-mentioned functions of binding to clathrin and phosphorylating the AP-2 μ2 subunit, NAK promotes Notch activation that regulates signal transduction between Notch cells. AAK1 is also a substrate for NDR1/2 phosphorylation that has been shown to control dendrite morphogenesis in developing mammalian neuronsCitation25. However, it has been reported that the AAK1 complex can be knockdown by RNAi approaches. In this study, it is demonstrated that AAK1 silencing affects the expression of epithelial-mesenchymal plasticity (EMP) markers and greatly improves the sensitivity of cancer cells to targeted therapyCitation26. This underscores the importance that AAK1 may also have in the treatments of cancers and other diseases.
BIKE is closely related to AAKl in structure, which is related to Numb. It plays a role in osteoblast differentiation and has recently been identified as a clathrin-coated vesicle-associated proteinCitation27,Citation28. BIKE also negatively regulates polyploidy and megakaryocyte differentiation in acute megakaryocytic leukaemia by interacting with cyclin-dependent kinase 2 (CDK2) and promoting mitosisCitation29. GAK is a known association partner of cyclin G and CDK5, some of its known functions are shared with AAKl. GAK is necessary for reticulin transport, and mediates the binding with plasma membrane and trans-Golgi network. GAK is necessary for maintaining centrosome maturation and progress through mitosisCitation30. GAK has been reported to be involved in chronic kidney disease (CDK) and has been discussed as a potential drug target for the treatment of viral infections due to its involvement in the entry and replication of multiple virusesCitation31. MPSK1 is the most distantly related member of the AAK1 family, and its physiological function is still poorly understood. It is known to be a Golgi-related kinase, which plays an uncertain role in regulating secretion in the constitutive secretion pathway of the trans-Golgi networkCitation32. MPSK1 is also related to mammary gland development in miceCitation33.
Kuai et al. found that K252a, a natural compound containing indolocarbazole functionalities (tryptophan sugar compounds), enhances the signal transduction and neurogenesis of Neuregulin-1 (Nrg1)/ErbB4-dependent neurotrophic factor by integrating chemical genomics and proteomics. However, the deletion of AAK1 as the related target of K252a affects the transport and expression level of ErbB4, which provides evidence for AAK1 in Nrg1-mediated neurotrophic factor signal transductionCitation34. In 2016, Sorrell et al. successfully resolved the eutectic structure of the AAK1/K252a complexCitation3 (). Subsequently this structure has provided a strong rationale for the design and optimisation of AAK1 inhibitors. Small molecule inhibitors K252a with a near-atomic resolution of 1.95 Å. They crystallised in space group P212121, wherein each asymmetric unit (chain A and B) has two AAK1 kinase domains (). In chain A, the C-terminal forms a part of the helix, extending outward from the kinase domain into the solvent channel of the crystal complex. Through gel filtration and crystallography, they showed that AAK1 chiefly exists in monomeric form rather than dimeric, with a typical bi-lobal catalytic domain structure. The details of AAK1’s crystal structure include the localisation of the activated C-terminal helix (ASCH), and the position of zinc atoms present at high concentration in the crystallisation solution. All are shown in relation to the symmetry-related molecule (Sym 1).
Figure 2. (A) The chemical structure of K252a. (B) Interaction of K252a with AAK1 (protein data bank (PDB) ID: 4WSQ). Inhibitors are shown in stick representation with yellow carbon atoms. Key interacting residues are shown and labelled.
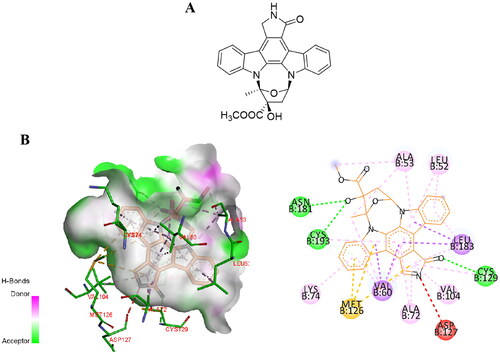
In AAK1, hydrogen bonding residues Gln203 interact with the N-terminal of ASCH and the αE/αF loop on Ile239. Residues 204–208 in AAK1 (and equivalent residues in BIKE) form a short turn through intramolecular interactions to provide greater conformational change. In addition, the kinase Ala-Pro-Glu (APE) motif (Glu229 in AAK1) forms a salt bridge that stabilises the C-terminal loop. The residues Tyr225 and Arg226 before the APE sequence in AAK1 help to lock the activated fragment in a certain position. AAK1 is a constitutively active kinase because Met94 of α-C helix, Tyr106 of β4, and His174 and Phe195 of HRD and DFG motifs constitute the regulatory spine (R spine) that keep it aligned. In addition, there is a flexible phosphate-binding loop at the ATP site in AAK1, called the “P ring”, which is located in Ala53-Ala59 of AAK1. Adding a glycine at the X2 position in the sequence G-X1-G-X2-φ-G allows this sequence to become a di-glycine motif that changes the conformation of the loop to adapt to various inhibitor scaffoldsCitation3.
Biological function of AAK1
AAK1 is implicated in multiple cellular pathways. For example, AAK1 participates in the NF-κB signalling pathway. AAK1 mediates IKBα to combine with inhibitors and forms a trimer p50-p65-IKBα complex, which becomes phosphorylated (). Under catalysis by Skp1-cullin 1-F-box (SCF) E3 ligase complex (SCF-E3 ubiquitinase), IKBα becomes ubiquitinated and degraded by a protease. The activated NF-κB is transferred to the endosome where it combines with its related DNA motifs to induce transcription of the target gene. This leads to degradation of target cellular proteins, including signal transducers, cell cycle regulators, and transcription factors. Recruitment of the target gene produces widespread immune and inflammatory reactions (). Lian et al. explained that miR-671-5P transmitted by MenSCs through extracellular vesicles (EVs) directly degrades AAK1. This results in inhibition of the activation of NF-κB and the signalling pathway mediated by AAK1Citation35. In addition, Abhinand et al. reported that AAK1-AP2M1 regulates vascular endothelial growth factor (VEGFR) by endocytosisCitation36. The combination of VEGFA-165 and VEGFR2 activates other pathways such as RAS-MAPK, PI3K-AKT-mTOR, PLC-PKC, JAK-STAT, NF-κB, p38MAPK-FAK and WAK. The interaction between these pathways contributes to the proliferation, migration and survival of endothelial cells and the formation of tubes involved in angiogenesisCitation37.
Figure 3. AAK1 medicates NF-κB and NDR1/2 signalling pathway. Left, AAK1 accelerates the degradation of IKBα, which leads to the activation of trans-acting factor p50/p65 and the transcription of pro-inflammatory cytokines, such as IL-6, TNF-αand IL-1β. Right, the Ser635 phosphorylation of AAK1 is regulated by phosphorylated NDR1/2, which was controlled by the upstream kinases MST1/2 and MAP4Ks. Phosphorylated AAK1 inhibits the growth of dendritic branches.
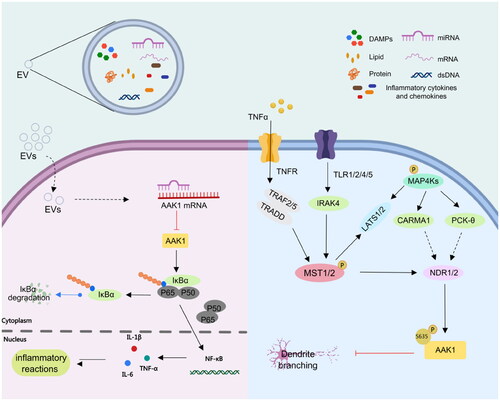
In addition, AAK1, as the substrate of NDR1/2 kinase, a key NDR family kinase in dendrite regulation, plays an important role in dendrite branching and dendritic spine growthCitation25. The phosphorylation of NDR1/2 is regulated by an upstream gene involving phosphorylated MST1/2 and MAP4Ks ()Citation21,Citation38. NDR1/2 is intimately involved in the process of polarised dendrite growth in an evolutionary conserved pathway from yeast to mammals. It is known to control growth and morphology in worm, fly, and mammal. Moreover, it is clear that NDR1/2 phosphorylation of AAK1 at Ser635, inhibits the growth of dendrite branching and arborisation (for a recent review see Ref. Citation21). AAK1 is not only implicated in pathways linked to neurodevelopment but also is involved in the regulation of the Notch pathway.
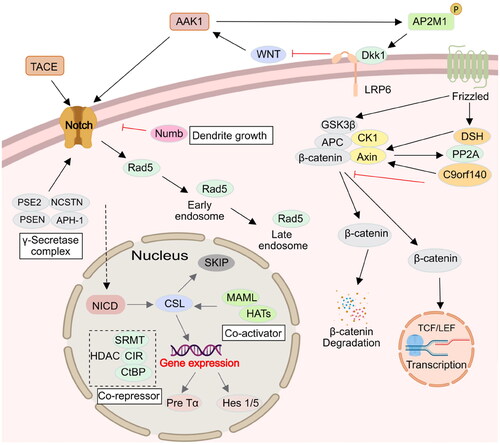
The Notch signalling pathway widely exists in vertebrates and invertebrates and is highly conserved in evolution. It regulates the differentiation and development of cells, tissues, and organs through the interaction between adjacent cells, and is related to many human diseasesCitation39. It is reported that γ-Secretase processing of Notch is essential in mono-ubiquitination and clathrin mediated endocytosis (CME). In addition, Eps15 contains many isoforms, such as Esp15b and its related protein Eps15R, which have been confirmed to participate in reticulin-mediated endocytosis, and the protein containing UIM is known to interact with the ubiquitination receptor, and might thus be involved in Notch endocytosis complex. Gupta-Rossi et al. confirmed that AAK1 acts as an adaptor for a Notch interaction with components of the clathrin-mediated pathway Eps15bCitation40. Transfected AAK1 increases the localisation of activated Notch cell types to produce Rab5-positive endocytic vesicles. On the other hand, AAK1 depletion or overexpression of Numb, an inhibitor of the pathway, interferes with this vesicle localisation and distribution. These results suggest that after ligand-induced activation of Notch, the membrane-tethered form is directed to different endocytic pathways leading to distinct fates. AAK1 also activates the WNT pathway and promotes the synthesis and transmission of the regulating factor Rab5 in early and late stage endosomes. In addition, AAK1 is activated by Numb. In turn, Notch receptors are inhibited by AAK1. This indicates that there is a dynamic balance between AAK1 and the Notch receptorCitation21 (). WNT promotes the endocytosis of the WNT receptor (LRP6) in a reticulin fibre meshwork. It activates AAK1 and phosphorylates the Thr156 site of AP2M1. However, the abundance of LRP6, which is also an inhibitor of WNT, thus forms a negative feedback loop driven by AAK1Citation38. In addition, AAK1i stabilises β-catenin and activates the β-catenin-dependent WNT signal pathway that accelerates the transcription of the target geneCitation41,Citation42 ().
Figure 4. AAK1 participates in CDK16 and Nrg1 signalling pathway. Left, the phosphorylation of AAK1 Thr389 was controlled by CDK16, which was involved in the regulation of neuronal synaptic transmission. Right, AAK1 inhibits the Nrg1/Erbb4-dependent neurotrophic factor signal transduction. Inhibition of AAK1 results in the sustained activation of downstream signalling by ErbB4, leading to enhanced Nrg1-mediated neurogenesis.
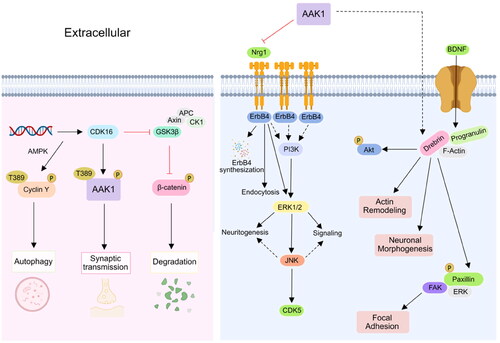
AAK1 also plays a role in the cyclic AMP dependent CDK16 signal pathway that regulates the phosphorylation of PRC1 in lung cancer cells. As a substrate of CDK16, AAK1 also plays a potential role in neuron and brain diseases. The abundance of MiR-125b-5p is known to affect the activity of CDK16, thus indirectly inhibiting cell degradation and promoting autophagy. CDK16 promotes synaptic transmission by phosphorylating the Thr389 site of AAK1 ()Citation43. AAK1 also inhibits the Nrg1/ErbB4-dependent neurotrophic factor signal transduction. By activating ERk1/2, the archetypical kinase enhances neurogenesis and signal transduction. At the same time, other ErbB receptors may also be susceptible to inhibition by AAK1Citation34. AAK1 regulates brain-derived neurotrophic factor (BDNF) that enhances the interaction between paxillin and drebrin may promote neuronal morphogenesis and actin remodellingCitation44.
Figure 5. AAK1 is involved in Notch and WNT signalling pathway. Firstly, AAK1 mediates the interaction of Notch and Eps15b, which accelerates the Notch pathway. Secondly, AAK1 negatively regulates WNT signalling by promoting CME of LRP6. Meanwhile, WNT activates AAK1-activated AP2M1 phosphorylation to promote endocytosis.
AAK1 and human diseases
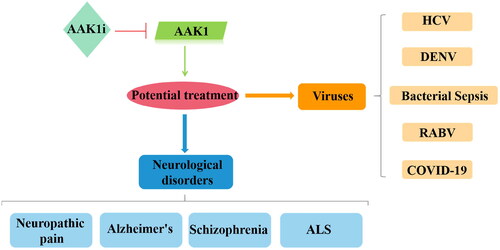
Due to the important biological functions of AAK1, studies of AAK1 as a potential therapeutic target have been vigorous and fruitful over the past few decades. Indeed, intervention in AAK1 phosphorylation has been shown useful in clinical trials for the treatment of neurological diseases such as neurological pain, AD, Schizophrenia, and ALS. Intervention in AAK1 catalysis has also been explored for potential therapies in the treatment of viral diseases including HCV, dengue virus and ebola virus (DEVN), bacterial sepsis, rabies virus (RABV), and COVID-19 ().
Figure 6. Relevance of AAK1 protein and therapies for human diseases. When AAK1 kinase is inhibited, it has a potential therapeutic effect, which is associated with neurological disorders and antiviral activity.
AAK1 and neurological diseases
Because endocytosis is closely related to many pathophysiological functions of the nervous system, and AAK1 closely regulates the steps endocytosis, we hypothesise that AAK1 may affect cellular function by participating in the pathological development of some nervous system diseases. Scientists have provided many studies to examine this point.
Neuropathic pain, which is associated with poor quality of life, has huge unmet clinical needs. However, due to a variety of side effects, limited scope of treatment and unsatisfactory treatment, only a few new painkillers have entered the market in the past decade. Therefore, new drugs with higher efficacy and fewer adverse reactions are needed to treat neuropathic pain. In this regard, AAK1 is a new feasible target for the treatment of neuropathic pain, and it has been confirmed for the first time in mouse knockout phenotype. Kostich et al. reported AAK1 as a novel target for the treatment of neuropathic painCitation45. And it found that AAK1 knockout mice showed a significantly reduced response to persistent pain in the formalin test and had no mechanical ectopic pain within 3 weeks after spinal nerve ligation. In addition, a potent small molecule inhibitor of AAK1 (LP-935509, brain to plasma (B/P) ratio >2) was also identified. It could also reduce chronic contractile injury (CCI) in a rat model of diabetic peripheral neuropathy and reduction of pain responses in a rat streptozotocin modelCitation45. Moreover, Luo et al. found that BMS986176/LX9211 possesses good efficacy for pain remediation in two rodent neuropathic pain models. With the completion of phase I clinical studies, this effort in humans will continue. Louis et al. developed a potent and selective AAK1 radio-ligand, [3H]BMT-046091, and observed the distribution of AAK1 binding sites in the brain and spinal cord of rodents and cynomolgus monkeysCitation46. The results showed that AAK1 is widely expressed in the brain and spinal cord, with the highest expression level in the forebrain. In addition, orally bioavailable LP-935509 could be found to occupy the AAK1 binding site in a dose-dependent manner. And the target occupancy of LP-935509 was correlated with its anti-diabetic pain effectCitation46. In 2022, Chen et al. demonstrated that miRNA-384-3p attenuated sevoflurane-induced neuronal apoptosis and memory impairment. By proposing that this compound inhibits the expression of AAK1, the effect of mitigating sevoflurane-induced nerve damage in paediatric clinical surgery was demonstratedCitation47.
Apart from intensifying painful neuronal responses, AAK1 has been shown to promote amyloid β (Aβ)-induced damage in AD through the intervention of the AP2M1 adapter. AAK1 co-locates with endosomal and presynaptic protein markers, but it is wrongly identified as an aggregate containing mutant SOD1 and neurofilament protein in ALS, which complex inhibit the activity of AAK1. In 2018, Fu et al. used Aβ1-42 to establish a mouse AD model and used the Morris water maze test to verify that the periodic changes in AAK1, AP-2, and Rab-5 (early intranuclear marker) expression were closely related to the decline of cognitive ability associated with ADCitation8.
AAK1 and viral diseases
Viruses, capable of infecting all organisms from bacteria, plants, animals to humans, are a major source of infectious disease. Viral infections have caused huge burdens and inflicted damage to human health and economic activities through the ages. AAK1 was generally considered as a feasible biological target for the treatment of neuropathic pain, but recently, scientists have found that AAK1 may also be a potential anti-viral target. In the review by Huang, the role of the Numb-associated kinases family in anti-virus therapy was also mentioned, including AAK1 in April 2023Citation48. Recently, scientists have found that inhibiting AAK1 can potentially regulate endocytosis, thereby inhibiting virus invasion into cells ().
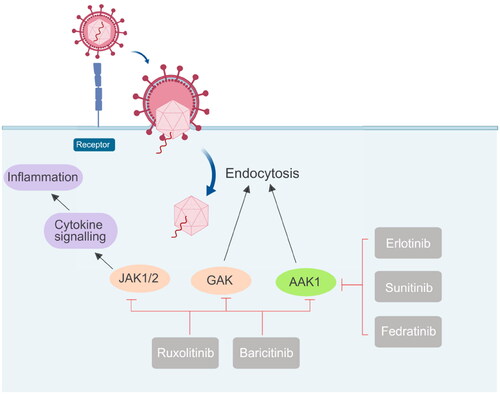
Figure 7. Different drugs currently on the market that act on JAK1/2, GAK, and AAK1 kinases to prevent virus invasion, mainly to inhibit the endocytosis of the virus.
Sunitinib and Erlotinib are new multi-targeted tyrosine kinase inhibitors, which were first approved for the treatment of cancer. The dissociation constants (Kd) of Sunitinib and Erlotinib for binding AAK1 to GAK are 11 nmol L−1 and 3.1 nmol L−1, respectivelyCitation49. In 2015, further studies confirmed that AAK1 and GAK were the key regulators of HCV entry into cells, and the anti-viral effects of Sunitinib and Erlotinib might contribute by mechanisms involving the inhibition of AAK1 and GAKCitation50. In 2017, Einav’s group reported that the roles of AP-1 and AP-2 complexes, as well as AAK1 and GAK, in the entry, assembly, and release of Flaviviridae family members are significant. It is further confirmed that these host factors represented a broad-spectrum of anti-inflammatory agents that were potential targets for anti-viral therapy. In addition, compared to direct-acting anti-viral drugs, administering the Sunitinib/Erlotinib combination could potentially provide greater resistance against DENV. This combination limited DENV infection in vitro and reduced the related viremia, morbidity, and mortality in a mouse model. It was also found that the combination of tyrosine kinase inhibitors blocked the entry and late stages of the life cycle of four DENV serotypes and multiple virusesCitation51.
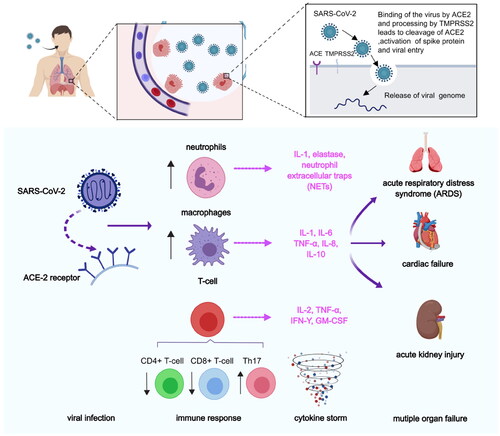
Angiotensin-converting enzyme 2 (ACE2) is the primary receptor for the COVID-19. It was recently reported that inhibition of the ACE2 receptor provides a new therapeutic direction in fighting COVID-19 and reducing its mortalityCitation52. In addition, inhibition of the transmembrane protease, serine 2 (TMPRSS2) and cathepsin L (CTSL), of the endocytosis regulator AAK1, and of the pro-inflammatory cytokines- IL-6, IL-1β, TNF-α, and IFN-γ, all significantly reduce mortality and morbidity associated with COVID-19 infection (). Also, Akbarzadeh-Khiavi et al. summarised two possible mechanisms of cell entry of SARS-CoV-2Citation53. The first is that the virus binds to the ACE-2 receptor outside the plasma membrane, and the endocytosis process occurs by assembling clathrin and coat proteins on the inner side of the plasma membrane in the form of a vesicle. The second mechanism presupposes that SARS-CoV-2 directly uses clathrin-dependent endocytosis to enter host cells. Following viral entry, viral RNA is released into the cytoplasmic space to initiate the production of structural and non-structural viral proteins. Finally, virions of coronaviruses are formed by budding into the endoplasmic reticulum-Golgi intermediate compartment and released as new virions. AAK1 may affect the assembly and entry of the SARSCoV-2 virus through the operation of one of these two possible mechanisms. Later studies by Ou et al. showed that endocytosis is the preferred pathway for SARS-CoV-2 to enter host cellsCitation54. In addition, The Kar et al. paper also suggests a new method for the treatment of COVID-19. Kar et al. analysed the inhibitory potential of 605 plant compounds, selected from Indian medicinal plants with reported antiviral and anti-inflammatory activities, against the receptor-binding domain of spike proteins of the SARS-CoV-2 wild-type and the variants of concern, including variants B.1.1.7 (Alpha), B.1.351 (Beta), P0.1 (Gamma), B.1.617.2 (Delta), and B.1.1.529 (Omicron)Citation55. On the other hand, they found taraxerol antagonises the human ACE2, CTSL, and TNF-α. β-amyrin antagonises the human TMPRSS2. Cynaroside antagonises the human AAK1 and IL-1β. Friedelin antagonises the human IL-6 and IFN-γ.
Figure 8. Pathological process after SARS-CoV-2 infection. First, SARS-CoV entered into lung epithelial cells, by binding of the virus to the ACE-2 receptor via spike proteins expressed on the surface of the virus. After the viruses proliferated in large numbers, the viruses can be recognised by neutrophils, macrophages, or T-cells. The inflammatory responses were triggered and the danger signalling molecules such as certain cytokines (e.g. IL-1, IL-8, etc.) were producted. In addition, immune response were elicited by reducing the number of CD4+ T-cells and CD8+ T-cells and increasing the number of Th17 cells.
Janus kinase (JAK) is a family of intracellular, non-receptor tyrosine kinases that transduce cytokine-mediated signals via the JAK-STAT pathway. Baricitinib, an effective and selective JAK inhibitor, is currently used to treat rheumatoid arthritis (RA). However, recently Baricitinib has been considered as a potential therapeutic drug for COVID-19 because of its anti-inflammatory and anti-viral activities, which may prevent the abnormal production of pro-inflammatory cytokines commonly observed in COVID-19 patients. In addition, Baricitinib also inhibits the virus from entering the target cells by binding AP2-related protein kinase 1 (AAK1). In 2020, Lo et al. reported for the first time the favourable clinical course and outcome of an RA patient who had been treated with Baricitinib for more than one year and who developed COVID-19. The patient received supplemental oxygenation, in addition to lopinavir/ritonavir, hydroxychloroquine, while continuing to use Baricitinib. After recovery, the treatment was judged to be successful. She is currently apyretic with an O2 saturation 95% in ambient air. While his family received the same treatment except for Baricitinib, his condition deteriorated rapidly, and he died of respiratory failure a few days laterCitation56. Although the clinical data here are only preliminary, they provide strong support for further study of the use of Baricitinib in the treatment of COVID-19. When the pandemic ended, a large number of investigators employed artificial intelligence (AI) algorithms to independently predict and evaluate in vitro pharmacology and in vivo pharmacokinetics of Baricitinib in related leukocyte subsets. Through analysis of reported biochemical and cellular results, this series of numerous case studies confirmed the anti-cytokine signal transduction behaviour and reported the antiviral effect of BaricitinibCitation52. This treatment may be related to the clinical and radiological recovery, the rapid decline of SARS-CoV-2 viral load, drop in inflammatory markers and IL-6 levelsCitation51. In a series of clinical I/II randomised controlled studies in 2021, González et al. confirmed that Baricitinib could prevent respiratory insufficiency in tumour and hematological disease patients with COVID-19Citation57. Also, the drug Ruxolitinib has displayed clinical effectiveness in COVID-19 patientsCitation58. The above results prove that effective anti-viral drugs can be found from other existing drugs. Because the proteins in the host have complex functions and various mechanisms of action, they cannot be used for treatment rashly. Moreover, considering the toxicity problem, the in vitro experiment can not be applied to the in vivo experiment.
AAK1 inhibitors for the treatment of neuropathic pain
AAK1 inhibitors were first used to treat neuropathic pain, and several inhibitors have entered clinical research. The interaction mode between inhibitors and AAK1 kinase can be realised by X-ray crystal diffraction, which will be beneficial to further structural optimisation. Herein the latest research progresses on different categories and different chemical scaffolds of small molecular AAK1 inhibitors were summarised with the hope to provide a reference for the treatment of pain and to accelerate the drug discovery in this field.
Imidazo[1,2-b]pyridazine and pyrazolo[1,5-a]pyrimidine derivatives
Imidazo[1,2-b]pyridazines
The general structure for the class of compounds depicted by 1 () contains a common fragment, immidazo[1,2-b]pyridazine. The compounds in this group have been reported in patent reviews, and this article summarises them only briefly.
Figure 9. The chemical structures of patented AAK1 inhibitors with imidazo[1,2-b]pyridazine.
![Figure 9. The chemical structures of patented AAK1 inhibitors with imidazo[1,2-b]pyridazine.](/cms/asset/969606db-9353-403e-8a67-3c7bf6155b80/ienz_a_2279906_f0009_c.jpg)
This class of compounds was mainly pursued by Lexicon pharmaceuticals and Bristol Myers Squibb (BMS). Lexicon pharmaceuticals applied for two patents in 2013 (WO2013/134219, WO2013/134336), of which WO2013/134336 belongs to the patent utility category for medical use in the treatment of painCitation59,Citation60. LP-922761 (compound 2, ) is one of the effective means of relieving neuropathic pain. However, for compound 2, oral administration still did not relief the pain. In this kind of structure, most of the active compounds carry a piperazinyl or pyrrolidinyl moiety at position 6, with the second nitrogen preferentially being derivatized as a carbamate moiety. At position 3, various aryl groups were introduced. Here a 2-methoxy-pyridinyl functionality often produces an active inhibitor (compound 3 and 4, ). Unlike the derivatives disclosed by Lexicon pharmaceuticals, alkylamino, cycloalkylamino and phenylamino groups at position C8 of the core structure are claimed in the BMS patent (WO2015/026574)Citation61. The patent declaration by BMS identifies seven imidazo[1,2-b]pyridazine compounds for which potent binding and inhibition of AAK1 were observed (IC50 < 10 nM). The disadvantage is that only the analogue without structural modification at position C8 (compound 5, ) was tested and disclosed in this patent. At a dose of 60 mg/kg, this compound was less active than Gabapentin, when dosed at the equivalent 200 mg/kg (Gabapentin: The corresponding inhibition fraction at 200 mg/kg administered subcutaneously is 73%).
Pyrazolo[1,5-a]pyrimidine
Compared to Imidazo[1,2-b]pyridazines, this section focuses on compounds based on the imidazo[1,2-b]pyridazine scaffold in which the bridging nitrogen atom moves around the scaffold. The general structure is based on compound 6 (see ). Compounds in this group have been reported in patent reviews that this article only briefly summarises.
Figure 10. The chemical structures of patented AAK1 inhibitors with Pyrazolo[1,5-a]pyrimidine. EC50: median effective concentration, the concentration of drugs that causes 50% of the organisms to produce some toxic effect; TC50: median toxic dose, the drug dose that causes poisoning in half of the experimental animals.
![Figure 10. The chemical structures of patented AAK1 inhibitors with Pyrazolo[1,5-a]pyrimidine. EC50: median effective concentration, the concentration of drugs that causes 50% of the organisms to produce some toxic effect; TC50: median toxic dose, the drug dose that causes poisoning in half of the experimental animals.](/cms/asset/800fca82-a804-4a9d-b594-e6d071649366/ienz_a_2279906_f0010_c.jpg)
Lexicon pharmaceuticals proposed a series of pyrazolo[1,5-a]pyrimidine AAK1 inhibitors in the patent (WO2013/134228). Eighteen representative compounds that exhibited IC50 values of less than 10 nM were disclosed in the biochemical and cellular patentCitation62. In this compound group, a piperazinyl or pyrrolidinyl functionality at position 6 and a (hetero)aryl at position 3 are the most common substituents. Among them, compound 7, 8 and 9 () have the best inhibitory activity as demonstrated in a preclinical mouse model of neuropathic pain (the mouse formalin paw test). Compounds 7 and 8 are as active at a dose of 30 mg/kg (sc), as compared with Gabapentin at 200 mg/kg (sc). Compound 9 had good activity by oral administration (at 30 mg/kg sc), indicating good oral bioavailability, and 9 has also been reported in several reports in the biochemical literature.
Compound 10 (also known as LP-935509, ) is an effective, orally available, widely distributed and highly permeable AAK1 inhibitorCitation44. Compound 10 inhibits the phosphorylation of μ2 protein (IC50 = 3.3 ± 0.7 nM, Cell IC50 = 2.8 ± 0.4 nM). In addition, 10 is competitive with ATP binding, having a Ki value of 0.9 nM. It relieves neuropathic pain in various preclinical models of mice and rats, such as the CCI and streptozotocin (STZ) models. Unfortunately, it has no effect on acute pain. In short, compounds 10 is a promising candidates worthy of further study.
Lexicon pharmaceutical corporation synthesised compound 11 (), and reported it to be a potentially important AAK1 inhibitor in the patent (WO2015/142714)Citation63. The 3-methyloxetan-3-yl-4–(3-(2-methoxypyridin-3-yl)pyrazolo[1,5-a]pyrimidin-5-yl)piperazine-1-carboxylate showed potent activities with IC50 values of 0.9 nM in radio-active enzymatic and 4.7 nM in cell-based AAK1 assays. This compound demonstrated activity in both the formation assay and single nucleotide polymorphism SNP assay (also known as the Chung model). Bi et al. disclosed a number of analogues for AAK1 inhibitors based on pyrazolo[1,5-a]pyrimidine scaffolds with a carbon linker at position 5 in the patent (WO2015/035117)Citation64. In vivo data are not discussed. Compound 11 was revealed to have certain antiviral utility in a separate patent application (WO2021/216454)Citation65. The base compound was evaluated in three different in vitro cell models. Over the course of testing the efficacy in preventing infection by CoV-229E strain in MRC-5 cells, compound 11 exhibited good safety and low toxicity compared with the other three positive control drugs (EC50 > 10 μM; TC50 > 10 μM). In Huh‑7 cells, compound 11 and Sunitinib malate were used to test the anti-coronavirus activity of the OC43 strain of seasonal human oronavirus (HCoV). Results were characterised by a different anti-viral activity measure (by cytopathic effect, CPE) and cell viability was characterised by uptake of neutral red dye. The experimental results showed that compound 11 had similar antiviral outcomes to the positive control, but its cytotoxicity was much less. In addition, compound 11 appeared useful for reducing the viral load of HCoV-OC43 in H292 cells.
In addition, the effectiveness of a series of substituted compounds built upon the Pyrrolo[2,3-b]pyridine scaffold were summarised in a patent reviewCitation20. The derivatives include: pyrrolo[2,1-f][1,2,4]triazine analogues, quinolines, and benzimidazoles. Then, the AAK1 inhibitors in the literature are discussed from the aspects of structure and rational design. Full considerations are given to the pharmacology and toxicology of the compounds of interest.
Aryl amides
After screening their in-house compound library, Hartz et al. identified compound 12 (IC50 = 69 nM, ) as a potent lead compound with AAK1 inhibitory activity based on its desirable drug-like propertiesCitation66. This compound had octanol: water partition coefficient cLogP = 0.93, topological polar surface area (tPSA) = 90 Å2, ligand efficiency (LE) = 0.47, and lipophilic ligand efficiency (LLE) = 6.2. Then, the structural modifications of compound 12 are focused on enhancing the initial inhibitory activity of AAK1.
Figure 11. Structures of the AAK1 inhibitors with the aryl amide scaffold or its analogs selected from the references.
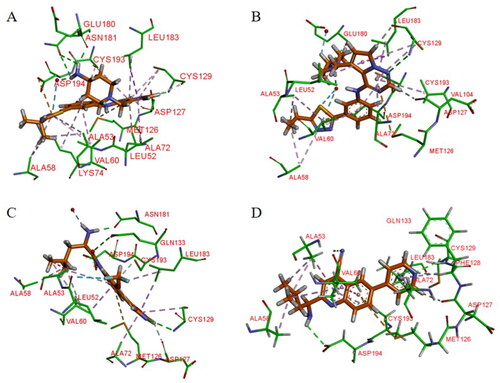
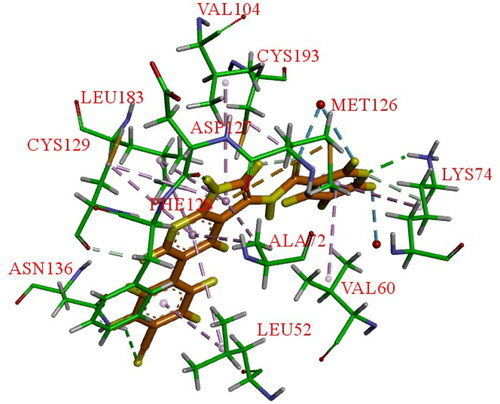
Hartz et al. synthesised a series of AAK1 inhibitors based on 12 by attaching the different alkyl groups to the amide side capping groupCitation66. The introduction of a carbon chain with different lengths influences the activity of AAK1, and its activity will increase with the lengthening of the chain. By extending the alkyl side chain beyond the isopropyl group, the activity of AAK1 increased. Moreover, the addition of branches, such as analogues based on D-isoleucine, was found to lead to further improvements of inhibition. In addition, the substitution of the D-isoleucine-derived amide (13, IC50 = 31 ± 7 nM, ) by a D-leucine group (14, IC50 = 12 ± 2 nM, ) produced better efficacy. This substitution provided a lesson which may be drawn relating the size of side chains of the probe with respect to access to the ATP binding site of the kinase. Hartz et al. generated the X-ray crystal structure of AAK1 which showed the binding mode of compound 14 with 2.65 Å resolutionCitation66. From this structure, it can be found that the oxazole ring nitrogen forms a single hydrogen bond with the backbone NH of Cys129 within the kinase hinge region (see ). At the same time, the X-ray crystal structure of 14 also demonstrated that a key hydrophobic interaction occurs between the isopropyl group and a small lipophilic pocket in the P-loop region of the kinase, which could improve the potency and selectivity of the kinase. Further investigations revealed that replacing the methoxy group with a hydrogen results in a 13-fold reduction in potency. Hartz et al. attributed this to the loss of a hydrophobic contact between the Leu52 and Ala53 on the P-loop above and Leu183 belowCitation66. However, the side chain of D-leucine-capped amide seems to exhibit optimal hydrogen bonding interactions through the amide and amine nitrogens and an appropriate alkyl side chain length that promotes a stabilising bridging effect between the N-terminal domain (via the P-loop) and C-terminal domain (via the DFG motif) of the AAK1 kinase (). Based on the strength of the interaction between the hydrogen bond and kinase hinge region formed by oxazole nitrogen in 14, they considered using similarly substituted heterocyclics to optimise such interactions. Of all the compounds tested, only pyrazole heterocyclic analog (i.e. 15, IC50 = 15 ± 4 nM, ) had similar activity.
Figure 12. X-ray crystal structure of AAK1 in complex with compound 14 (PDB ID: 7LVH), 16 (PDB ID: 7LVI), 17 (PDB ID: 7RJ8) and 18 (PDB ID: 7RJ7). The compounds are rendered as a stick models with orange carbon atoms. Hydrogen bonds are shown as purple dashed lines.
Subsequently, a structural constraint was introduced to the amide side chain of D-leucine derivatives, in which the flexible isobutyl group was tied back within a six-membered (piperidine) ring. It was also substituted with isopropyl at the 3-position. These modifications improved the potency and imparted a more rigid structure. Ultimately, compound 16 (IC50 = 10 ± 3 nM, ) was selected, which had a potency equivalent to 14.
It is unfortunate that 16 had no follow-up progress to report due to its metabolic instability in mice. Eventually, a SAR analysis of substituents on the central benzene ring showed that 17 (IC50 = 12 nM, ) is more effective than compound 14. Following a different line of investigation, they found compound 17 inhibited AAK1 activity by a more direct measurement of the phosphorylation of μ2. Using a Western blot analysis of the μ2 subunit and showed 82% reduction of μ2 phosphorylation following acute administration of compound 17, vs 90% for control. The EC50 of inhibition of μ2 phosphorylation in vivo was determined to be 8.3 μM, resulting in an ED50 of 6 mg/kg. Kinase selectivity profiling of compound 17 against a panel of 219 kinases at 1 μM showed the compound had good overall kinase selectivity. By comparing their pharmacokinetic (PK) parameters their compound’s (14, 15, 17) pharmacokinetic parameters in male G57BL6 mice, compound 17 was identified as the excellent inhibitor.
Due to its ability to form hydrogen bonds within the ATP binding site at the hinge region (see ), compound 17 becomes a potentially ATP-competitive small molecule kinase inhibitor. Compound 17 (later named BMS-911172) demonstrated the following favourable pharmacologic properties: a clogP = 1.9, tPSA = 90 Å2, LE = 0.45 and LLE = 6.0. The aqueous solubility of 17 at pH 1 and pH 7.4 was 5.85 mg/mL and 0.60 mg/mL, respectively. An increasing volume of in vivo measurements found that 17 significantly reduces phase II hind paw flinching and licking/biting in the formalin pain model. In the CCI model, compound 17 (60 mg/kg, sc) significantly reduced hyperalgesia, with the maximal reversal increased by 24% (thermal) and 13% (mechanical) with respect to the positive comparator gabapentin.
Subsequently, new work was reported in 2022 in which novel compounds possessing quinazoline and quinoline structures that differed from compound 17. Hartz continued to modify 17 to obtain compounds with better activity. The new compounds transferred the amide of 17 back to the neighbouring phenyl ring to form a bicyclic benzene-pyridine structures, then modified the synthesis to produce a series of fused pyridine-benzene (quinoline) heterocycles and bicyclic pyridine compounds. Among them, a selective potent AAK1 kinase inhibitor 18 () was discovered, which improved enzyme and cellular potency compared to 14. In vitro testing in a HEK cell culture showed that 18 is the most potent compound with an IC50 value of 19 nM. Compound 18 also presented favourable metabolic stabilityCitation67. Compound 18 had high permeability across membranes, which allowed it to penetrate into the central nervous system (CNS). Its LE value is 0.47 and LLE value is 5.4. Compound 18 also showed good overall selectivity in a kinome panel screening (using 403 kinases). Because of its potent capacity to inhibit the phosphorylation of μ2 in mouse brain, it was also regarded as a likely candidate for safe testing in human subjects because drug exposures can be kept low and still be effective. Therefore, 18 seemed to be a candidate potential for clinical development or further preclinical research.
Bis(hetero)aryl ethers
Luo et al. first reported a series compounds containing a bis(hetero)aryl scaffold as inhibitors of AAK1Citation11. These compounds against AAK1 were tested and their activities were mostly in the Nano molar range. In addition, the oxazole-phenyl compound 19 was screened () as the lead against AAK1 using a high-throughput screening campaign. And it could be found that compound (20, ) to be optimal after conducting an extensive SAR study of compounds bearing biaryl and amino-amide chainsCitation11. In the CCI model, 20 reduced the nociceptive response induced by phase II formalin in rats. This is a reaction known to be closely associated with the phosphorylation level of μ2. In addition, chemical studies confirmed that the key hydrogen-bonding interaction occurred between the oxazole head group of compound 20 and the AAK1 hinge binding site. When other heterocyclic substituted oxazoles were found to be effective inhibitors, only pyridyl (21, IC50 = 15 nM, ) was well tolerated and its efficacy was equivalent to 20. When the amide group was substituted by other groups (such as an ether group), the potency decreases, presumably due to the destruction of the hydrogen-bond-linkage between the amide NH and AAK1. However, the potency of 22 (IC50 = 49 nM, ) that carried additional fluorocarbon groups was still within the acceptable range. Lu made a full SAR study on the ether derivative 22. Encouragingly, compound 22 is more effective, and its IC50 is 49 nM. For this reason, compound 22 appeared to be a promising lead compound.
Figure 13. Structures of the AAK1 inhibitors with the Bis(hetero)aryl ethers or their analogs selected from Ref. Citation11, Citation68.
CYP3A4 (Cytochrome P4503A4 enzyme) is one of the members of the cytochrome P450 oxidase family that is the major enzyme for the detoxification of bile acids. Eliminating CYP3A4 inhibition is a problem that is often considered in drug research and development. It mainly exists in the small intestine and liver, and can oxidise foreign organic small molecules, such as toxins or drugs, so that they can be excreted. CYP3A4 enzyme inactivates many drugs, so before testing the drugs effect in vivo it is frequently necessary to inhibit CYP3A4. Compounds that show excellent potency for inhibiting AAK1 (23, IC50 = 0.6 nM and Cell IC50 = 2.9 nM, ) may also have CYP3A4 inhibition problems. In the course of in vivo testing, Luo and his group discovered that compound 23 was susceptible to CYP3A4 inactivation.
Subsequently, Luo found that adding a methyl group beside the chiral amine (in 21, ), or placing it next to a pyridyl nitrogen (24, 25, ) avoids CYP3A4 inhibition. This is a major discovery. In DPNP model testing, compound 26 () was shown to be worthy of attention because of its good permeability across the CNS and the high drug in the brain vs drug in plasma ratio (b/p ratio = 11). Unfortunately, 26 may be susceptible to some problems such as oral exposure and CV liabilities. Moreover, the lack of a good safety profile prevented it from entering clinical trials. This prompted medicinal chemists to further optimise the core in order to identify more active allosteric inhibitors. To solve the potential problem of CV liabilities, 2-pyridyl, and 3-pyridyl analogues were synthesised and a series of inhibitors were obtained. Among these, representative compound 27 () not only possesses good cell potency (IC50 = 2.0 nM) but also shows good metabolic stability and AR relaxation of 42%. Compound 27 showed good kinase selectivity in our internal 249 kinase panels. In the CCI model, the pain response of 27 was reversed by 97% at the dose of 1 mg/kg. However, the lowest effective dose is 0.3 mg/kg. At effective dose levels, compound 27 was less likely to produce sports injuries than gabapentin.
A potent and selective radio ligand of the [3H]BMT-046091 is applied to determine AAK1 distribution and target engagement. An in vitro [3H]BMT-046091 binding autoradiography assay determined the spinal cord occupancy rate of compound 27 to enable a better understanding of the relationship between AAK1 target engagement and the anti-neuropathic pain effects. The results showed good target binding in the spinal cord. In the diabetic peripheral neuropathic pain (DPNP) rat model, oral administration of 27 significantly reduces mechanical abnormal pain, and the inhibition rate of pain response is more than 60% at the dose of 1 mg/kg. Compound 27 (i.e. BMS-986176, renamed as LX-9211 by Lexicon) had entered into clinical phase II research studies and is currently aimed at delivering palliative relief to patients with diabetic peripheral neuropathic pain and patients with postherpetic neuralgiaCitation11.
Subsequently, 28 and 29 () were tested head-to-head with 27. Both of them showed higher potency against AAK1 and were effective in CCI and DPNP rat pain models. Unfortunately, a degree of hepatotoxicity and other CV liabilities at high doses prevented these two compounds from further developmentCitation68. Although 28 and 29 still have some problems, the results of this study provide a new scaffold for further developing novel AAK1 inhibitors.
AAK1 inhibitor for inhibiting virus
AAK1 is a potential antiviral biological target. Up to now, no AAK1 inhibitor has been approved for antiviral indications. In this section, the structure and antiviral activity of AAK1 inhibitors with antiviral effect are systematically summarised, which provides a reference for the subsequent research on developing antiviral drugs targeting AAK1.
Repurposing of other kinase inhibitors
Sunitinib
Sunitinib (30, ) was approved by the FDA in 2006 for the treatment of gastrointestinal stromal tumours (GIST) and renal cell carcinoma (RCC). However, 30 maintains a nanomolar affinity for AAK1 and had been widely used to study the role of AAK1 in virus infection (see ). Einav’s research group found that 30 showed broad-spectrum anti-viral activity against a variety of virusesCitation51. The EC50 values for most viruses were below 1 μM, and the CC50 values were all greater than 10 μM. (The CC50 (for cytotoxic concentration) value indicates the toxicity to normal cells). The higher of value means better safety.
Figure 14. Chemical structures of approved drugs 30–32 and their derivatives 33–35.
Table 1. Anti-viral activity of 30Citation51.
Baricitinib
Baricitinib (31, ) was approved by the FDA for the treatment of rheumatoid arthritis in 2018. As early as 2016, Sorrell et al. found that 31 had a strong inhibitory effect on AAK1Citation3. The results of isothermal titration calorimetry (ITC) showed that the Kd value of 31 on AAK1 reached 17.2 nM. Recently, the Stebbing team analysed and calculated the inhibitor drugs on the market using BenevolentAI’s unique database and artificial intelligence algorithmCitation52. In the literature, it is recommended that 31 be tested to block the process of virus infection, because of an anticipated property to interfere with endocytic viral entry in lung cells. Later, the in vitro pharmacology and in vivo pharmacokinetics of 31 in relevant white blood cell subset were evaluated. It was found to inhibit the signal transduction of cytokines associated with the novel coronavirus infection. Compound 31, predicted by AI, weas verified to have a positive biochemical inhibition on human NAKs family members, and its Kd values on AAK1 was determined to be 8.2 nM at the same time. There are currently twenty-six clinical studies underway to examine the role of 31 for the treatment of novel coronaviruses, utilising searches of the FDA clinical database. According to the FDA’s official website, five of them have completed phase II/III clinical trials (NCT04421027, NCT04358614, NCT04362943, NCT04401579, and NCT04640168). The combination of 31 and the anti-viral drug Remdesivir may be an appropriate combination therapy strategy. Investigators have been concerned about the risk of adverse effects such as immunosuppression, secondary infection, and thrombosis using this combination regimen. Thankfully, clinical studies have shown fewer acceptable adverse effects.
Analogs derived from clinical compound
In addition to the above-mentioned kinase inhibitors that have been marketed for clinical use, there are also some newly synthesised small molecule inhibitors that are being investigated in preclinical studies. For example, on the basis of Ruxolitinib (32, ) and 31, Lin et al. designed some small molecule analogues using a scaffold jumping strategy (compounds 33, 34, and 35 in )Citation58. Replacing the pyrazole ring in 31 with a trifluoromethyl substituted indole ring gave compound 33 with comparable potency for AAK1 (IC50 = 161 nM), JAK1 (IC50 = 2.49 nM), and JAK2 (IC50 = 0.95 nM). However, 33 showes much weaker inhibition against GAK (IC50 > 10 μM) than 31 displayed (IC50 = 1.35 μM), which indicates that the substitution of the pyrazole ring by a trifluoromethyl substituted indole ring reduces the modified inhibitor’s affinity towards GAK. The reason may be the introduction of a fused trifluoromethyl phenyl group produces a steric hindrance with Lys69 and Cys190 of GAK. Compared with 31, the selectivity of 33 for AAK1, JAK1, and JAK2 is better than for GAK. However, its selectivity may not be compatible with the anti-coronavirus effect. On the other hand, the improvement in the selectivity of 33, particularly against AAK and JAK, may be more useful for intervention in other diseases, such as autoimmune diseases, where GAK inhibition is not deemed to be useful. Increased selectivity reduces the risk of toxicity caused by less off-target binding. Sadly, compound 34 showed slightly decreased (3–6-fold) potency for binding to AAK1 and GAK but dramatically decreased (59–88-fold) potency for JAK1/2 compared to 32. Similar to 34, compound 35 was 87–144-fold less effective than 32 for binding to JAK1/2. Compared to 32, 35 showed comparable binding to GAK, but a 22-fold lower affinity for AAK1. Ultimately, compound 33 warranted further study in more advanced preclinical studies to fully understand its potential as an effective anti-viral therapy.
Pyrrolo[2,3-b]pyridines
This section focuses on AAK1 inhibitors bearing a pyrrolo[2,3-b]pyridine scaffoldCitation69. The general structure of pyrrolo[2,3-b]pyridines-based analogues is shown in compound 36 ().
Figure 15. Structures of the AAK1 inhibitors based on the pyrrolo[2,3-b]pyridine scaffold or its analogs as selected from the references.
![Figure 15. Structures of the AAK1 inhibitors based on the pyrrolo[2,3-b]pyridine scaffold or its analogs as selected from the references.](/cms/asset/113eda2e-c1e3-49be-82d1-e42680ad2161/ienz_a_2279906_f0015_c.jpg)
Using the DiscoverX binding assay format, Verdonck et al. tested an unsubstituted pyrrolo[2,3-b]pyridine scaffold and a 7-aza-indole derivative (37, ) as novel AAK1i candidates with nanomolar potency (Kd = 53 nM)Citation69. And a series of 577 structurally diverse compounds were screened across a panel of 203 protein kinases. The results of binding-displacement assays show that compound 37 was 3-fold more selective for AAK1 over GAK, 8-fold more selective for AAK1 over BMP2K, and 22-fold more selective for AAK1 over STK16. In the crystal structure of compound 37 bound to AAK1 to 20 Å resolution, the nitrogen atoms of the pyrrolo[2,3-b]pyridine moiety form two hydrogen bonds to the peptide backbone of residues Asp127 and Cys129 that are located at the kinase hinge region. The 4-cyanophenyl moiety is oriented towards the solvent with the phenyl ring directly sandwiched between the backbone of Gly132 of the hinge and Leu52 of β1 in the N-lobe. The nitrogen of the cyano moiety forms a polar interaction with the side-chain of Asn136 (). Therefore, it is a promising lead that can be further optimised.
Figure 16. X-ray crystal structure of AAK1 in complex with compound 37 (PDB ID: 5L4Q). The compound is rendered as a stick model with orange carbon atoms. Hydrogen bonds are shown as purple dashed lines.
When the 4-cyanophenyl derivative in 37 was replaced with a 3,4-dimethoxy-phenyl functionality, compound 39 () was resulted. 39 showed stronger binding affinity for AAK1 and also demonstrated the most effective anti-DENV activity (EC50 = 1.64 μM, EC90 = 7.46 μM). Verdonck et al. carefully studied the structure-activity relationship (SAR) of the above derivativesCitation69. Then, the inhibition effect was examined after the replacement of the 3-pyridyl group by a number of (hetero) aromatics and cycloaliphatic groups. These compounds demonstrated a 100-fold reduced affinity for AAK1 IC50 values ranged between 0.1–0.6 μM and antiviral activity relative to compound 39 was diminished, indicating the important role of 3-pyridyl moiety for biological activity. Furthermore, comparative inhibition studies showed that the amide moiety is not necessary for AAK1 binding and can be substituted with other groups, such as ketone (40, ) or alkyne (41, ). Verdonck et al. synthesised an innovative series of substituted phenylacetylene derivatives and found that the introduction of a 3-pyridylacetylene (38, ) significantly improves the affinity of AAK1 (IC50 = 0.0042 μM), enhances anti-viral activity (EC50 = 0.72 μM), and exhibits only a moderate cytotoxic effect (CC50 = 17 μM)Citation65. Therefore, 38 appeared to be a candidate potential for clinical development or for further pre-clinical investigation.
In subsequent work, Verdonck et al. synthesised three different compounds based on the 7-aza-indole scaffold (compound 37)Citation69. Methylation of the pyrrole nitrogen on compound 39 produced compound 42 (). In addition, a pyrazolo[3,4-b]pyridine analogue (43, ), and a pyrrolo[2,3-b]pyrazine analogue (40, ) were also synthesised. Among these, compounds 42 and 43 showed low AAK1 affinity (> 800-fold and 100-fold loss in activity compared to compound 39). The significantly enhanced cytotoxicity (by 3-fold) of compound 44 was showed in Huh7 cells. Compounds 37, 38 and 39 were tested for their activity against the unrelated EBOV to evaluate the potential for broad-spectrum antiviral coverage. The biological assay showed that compound 38 possessed low nanomolar potency and excellent anti-EBOV (Ebola) activity with EC50 and EC90 values of 0.0428 μM and 1.49 μM, which its mechanism was to regulate the phosphorylation of AP2M1 via AAK1 inhibition. These assays demonstrated that 38 was an effective inhibitor regulating viral uptake in the treatment of a broad spectrum of viral agents. Unfortunately, the KINOMEScan assay (Discover X) showed that 38 was non-selective as an AAK1 inhibitor. In addition to AAK1, compound 38 targets multiple other kinases, which might contribute to its antiviral effects. Therefore, in the future, pharmacists can use compound 38 as a lead compound to optimise its structure and improve its selectivity.
Other AAK inhibitors
1H-indazole analogues
The base scaffold structure of compounds in the class is shown as 45 (). Well et al. developed a series of novel 3-acylaminoindazole derivatives as a small molecule chemical probe (SGC-AAK1-1, 48, ). Compound 45 bound with strong affinity for AAK1/BMP2K over other AAK1 family membersCitation70. Compound 48 was discovered to possess high affinity for both AAK1 and BMP2K with Ki values of 9.1 and 17 nM, respectively. The effect of compound 48 on WNT signalling was recently reported. Novel 3-acylaminoindazole analogues were designed and synthesised. These afforded a series of modified compounds that could be assayed for the strength of binding and their ability to inhibit phosphorylation by WNT and other family members. Scientists hope to improve the cell activity by changing the substituents on sulphonamides. On the other hand, different lengths of alkyl chains or alkylamines were used as the linker to explore the conformation effects of the target molecules. From these data, it was determined that a sulphonamide moiety appending at the 3-position of the aryl ring produced the greatest AAK1 inhibition. In addition, sulphonamides with small saturated rings (< 6 atoms) that was mono-substituted by short alkyl chains or alkylamines also increased the activity against AAK1Citation70. It is preferable to make modifications at the C3 position of the aryl ring than at the C4 position of the indazole scaffold. This allows a hydrogen-bonding network to become established in the ATP-binding pocket, which can be entered by 3-position groups. Therefore, the C3 position of the aryl ring is the key site for establishing a hydrogen bond with the ATP binding pocket. Then these become capable of donating a hydrogen bond and provide favourable interactions with the DFG motif aspartate residue (Asp198/194). Small acidic groups at these positions are not favoured due to charge repulsionCitation70. So the interaction between charges has an important impact on the structure of matter and molecular changes.
Figure 17. Structures of the AAK1 inhibitors with the 1H-indazole scaffold or its analogs selected from the references.
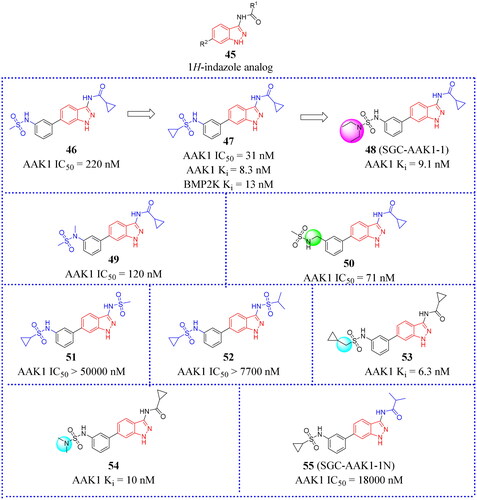
N-Methylation is a common method in the process of chemical compound design. Not surprisingly, methylation of the aryl 3-nitrogen (49, IC50 = 120 nM, ) or insertion of a methylene spacer between the sulphonamide and aryl ring (50, IC50 = 71 nM, ) increased potency for AAK1. By contrast, the substituent at the indazole 3-position appeared to form a polar interaction with the kinase hinge region that was sensitive to structural modifications. Sulphonamides with cyclopropyl substituents (47, IC50 = 31 nM, ) were selected for further SAR studies. Due to steric hindrance and charge interactions caused by the proximity of the sulphonamide oxygen atoms with nearby protein side chains, it was not advisable to replace amide with sulphonamide groups (eg. 51 and 52, )Citation70. Co-crystal structures reveal that the group attached to the 3-position amide exposes solvent at this position, which could explain the moderate tolerance of AAK1 for larger and bulkier substituents.
To explore the effect of alkyl amine or longer chain hydrocarbon substituents on AAK1 activity, two additional 3-pacylaminoinda-zoles (53 and 54, )Citation70 were designed. Similar to compound 47, compounds 53 and 54 did show high inhibition for AAK1 at 1 μM in the split luciferase assay, inhibiting AAK1 by 93% and 94% for 54 and 55, respectively. Moreover, the TR-FRET binding-displacement assays showed that they are dual AAK1/BMP2K inhibitors with greater than 5-fold selectivity over STK16 and 50-fold selectivity over GAK. The only successful co-crystal structures were obtained with BMP2K-KD, while co-crystals of acylaminoindazoles bound to the AAK1-KD were not successfulCitation70. To enhance the utility of dual AAK1/BMP2K probes, Well et al. designed a series of effective inhibitors by incorporating other caps such as fluorinated aryl rings, alkyl chain capped with fluorine groups, small hydrocarbon chains and rings, and alkyl amines linked to the sulphonamideCitation70. Fifteen compounds were synthesised using different synthesis strategies to assemble the heterocycle caps. Among these, compound 48 () was purified and showed a high affinity of this series for AAK1 and BMP2K. 48 demonstrated at least 35-fold selectivity for BMP2K over GAK and 4–16-fold selectivity for BMP2K over STK16. The KINOMEscan (DiscoverX) assay against 403 wild type human protein kinases at 1 μM showed that compound 48 exhibited good binding affinity (Kd) of all kinases inhibited >80%. In addition, compound 48 was designated as a small molecule inhibitor probe that targets the ATP-binding site of AAK1 and BMP2K preferentially over other serine/threonine kinases. The complementary negative control analog compound 55 for this probe (also called SGC-AAK1-1N, ) was made commercially available, becoming the only chemical probe targeting AAK1 that is widely used in conjunction with its negative control. In short, the results provide a new scaffold for the further research of developing novel probe.
Conclusions and perspectives
As a specific key kinase of the NAKs family that regulates the phosphorylation of threonine at the 156th position of the μ2 subunit of AP-2 protein, AAK1 plays a crucial role in CME. And the phosphorylation of Thr156 plays an important role in the regulation of protein structure and function, such as affecting downstream signals, apoptosis, and differentiation. Further study of AAK1 plays an important role in exploring the physiological and pathological mechanisms in cells and promote the treatment and prevention of related diseases. Because AAK1 is expressed in both the membrane and cytoplasm of normal cells, its internalised substances come from the external environment, such as micronutrients, and even some viruses and bacteria. Therefore, there are some potential safety problems when developing small molecular inhibitors. However, AAK1i has already been proved to have certain activity in clinic, suggesting it will not cause irreversible harm to the human body. Nevertheless, since it is not clear whether interference of this endocytosis pathway to mitigate pathological and physiological conditions will eventually lead to adverse side effects, small molecule compounds should be designed with safety in mind. These objections may be an explanation for the limited drug discovery activity against AAK1. There are two main areas of interest in the development of AAK1 inhibitors, one for pain therapy and the other for viral infections. At present, only Lexicon and BMS pharmaceuticals are active in these two fields. Since few companies are active in AAK1 drug discovery, the limited structural diversity of AAK1 inhibitors has been explored. In the field of pain treatment, great progress has been made, and the existing compounds have reached Phase II clinical studies. On the virology front, the COVID-19 pandemic has significantly increased the scientific interests in AAK1 protein for treating virus infection. Consequently, the interest in AAK1 inhibitors development, as reflected in a number of publications, has seen a major hike in recent years. Despite the important role of the AAK1 protein in the clathrin-mediated endocytosis of virus invasion, more work is needed including studies on aspects of kinase selectivity, safety window, and anti-virus effectiveness. To date, no AAK1 inhibitor has been approved for therapeutic uses including for the treatment of emerging viral infections, such as Dengue, Ebola and novel coronaviruses.
Another issue that needs to be addressed is kinase selectivity. Among the existing various kinases, their ATP pockets are similar. It may lead to the development of small molecule compounds that act on other kinases and even cause off-target effects. Therefore, from the stand point of medicinal chemistry and drug discovery, the development of AAK1 inhibitors based on rational drug design is practical and productive. This is because the crystal structure of the AAK1 protein has been solved. Therefore, the structural characteristics and structure/activity relationships appear to be straightforward and easy to understand. In this paper, the up-to-date advances of AAK1i in medicinal chemistry are summarised with the hope of providing the scientific community with a quick knowledge reference to accelerate their drug discovery. A number of studies reported that AAK1i have shown efficacy in various preclinical animal models. Among all the animal models, the neuropathic pain model has been studied most extensively. On the other hand, more in-depth studies are required to understand the mechanism and the role of AAK1i in preventing or inhibiting virus entry into host cells. The greater breakthrough efforts in this area are required to facilitate identification of safe and effective mediators for potential anti-virus therapy against emerging infections, which is highly anticipated.
CRediT authorship contribution statement
Ying-Yui Yuan: Literature searching, Methodology, Resources, Data curation, Writing - original draft, Visualisation. Nian-Dong, Mao: Methodology, Resources, Data curation, Writing - original draft. Ji-Long Duan: Resources, Data curation. Hang Zhang: Graphs preparation. Carmen Garrido: Editing, Visualisation. Frédéric Lirussi: Editing, Visualisation. Yuan Gao: Supervision, Writing - review & editing, Visualisation. Tian Xie: Methodology, Conceptualisation. Xiang-Yang Ye: Conceptualisation, Writing - review & editing, Project administration.
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Funding
References
- Wasner G. Central pain syndromes. Curr Pain Headache Rep. 2010;14(6):489–496.
- Sorensen EB, Conner SD. AAK1 regulates Numb function at an early step in clathrin-mediated endocytosis. Traffic. 2008;9(10):1791–1800.
- Sorrell FJ, Szklarz M, Abdul Azeez KR, Elkins JM, Knapp S. Family-wide structural analysis of human numb-associated protein kinases. Structure. 2016;24(3):401–411.
- Smythe E, Ayscough KR. The Ark1/Prk1 family of protein kinases. Regulators of endocytosis and the actin skeleton. EMBO Rep. 2003;4(3):246–251.
- Takahashi T, Furuchi T, Naganuma A. Endocytic Ark/Prk kinases play a critical role in adriamycin resistance in both yeast and mammalian cells. Cancer Res. 2006;66(24):11932–11937.
- Conner SD, Schröter T, Schmid SL. AAK1-mediated micro2 phosphorylation is stimulated by assembled clathrin. Traffic. 2003;4(12):885–890.
- Conner SD, Schmid SL. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J Cell Biol. 2002;156(5):921–929.
- Fu X, Ke M, Yu W, Wang X, Xiao Q, Gu M, Lü Y. Periodic variation of AAK1 in an Aβ1-42-induced mouse model of Alzheimer’s disease. J Mol Neurosci. 2018;65(2):179–189.
- Latourelle JC, Pankratz N, Dumitriu A, Wilk JB, Goldwurm S, Pezzoli G, Mariani CB, DeStefano AL, Halter C, Gusella JF, et al. Genomewide association study for onset age in Parkinson disease. BMC Med Genet. 2009;10(1):98.
- Shi B, Conner SD, Liu J. Dysfunction of endocytic kinase AAK1 in ALS. Int J Mol Sci. 2014;15(12):22918–22932.
- Luo G, Chen L, Kostich WA, Hamman B, Allen J, Easton A, Bourin C, Gulianello M, Lippy J, Nara S, et al. Discovery of (S)-1-((2',6-Bis(difluoromethyl)-[2,4'-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine (BMS-986176/LX-9211): a highly selective, CNS penetrable, and orally active adaptor protein-2 associated kinase 1 inhibitor in clinical trials for the treatment of neuropathic pain. J Med Chem. 2022;65(6):4457–4480.
- Gil C, Ginex T, Maestro I, Nozal V, Barrado-Gil L, Cuesta-Geijo MÁ, Urquiza J, Ramírez D, Alonso C, Campillo NE, et al. COVID-19: drug targets and potential treatments. J Med Chem. 2020;63(21):12359–12386.
- Stebbing J, Phelan A, Griffin I, Tucker C, Oechsle O, Smith D, Richardson P. COVID-19: combining antiviral and anti-inflammatory treatments. Lancet Infect Dis. 2020;20(4):400–402.
- Cantini F, Niccoli L, Matarrese D, Nicastri E, Stobbione P, Goletti D. Baricitinib therapy in COVID-19: A pilot study on safety and clinical impact. J Infect. 2020;81(2):318–356.
- Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, Rawling M, Savory E, Stebbing J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020;395(10223):e30–e31.
- Neveu G, Barouch-Bentov R, Ziv-Av A, Gerber D, Jacob Y, Einav S. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog. 2012;8(8):e1002845.
- Einav S, Rina B-B, Neveu G, et al. Methods and composition for treating viral diseases. 2013, US2013086133.
- Krahn AI, Wells C, Drewry DH, Beitel LK, Durcan TM, Axtman AD. Defining the neural kinome: strategies and opportunities for small molecule drug discovery to target neurodegenerative diseases. ACS Chem Neurosci. 2020;11(13):1871–1886.
- Cossart P, Helenius A. Endocytosis of viruses and bacteria. Cold Spring Harb Perspect Biol. 2014;6(8):a016972–a016972.
- Martinez-Gualda B, Schols D, De Jonghe S. A patent review of adaptor associated kinase 1 (AAK1) inhibitors (2013-present). Expert Opin Ther Pat. 2021;31(10):911–936. 36.
- Xin X, Wang Y, Zhang L, Zhang D, Sha L, Zhu Z, Huang X, Mao W, Zhang J. Development and therapeutic potential of adaptor-associated kinase 1 inhibitorsin human multifaceted diseases. Eur J Med Chem. 2023;248:115102.
- Cope MJ, Yang S, Shang C, Drubin DG. Novel protein kinases Ark1p and Prk1passociate with and regulate the cortical actin cytoskeleton in budding yeast. J Cell Biol. 1999; 144(6):1203–1218.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934.
- Henderson DM, Conner SD. A novel AAK1 splice variant functions at multiple steps of the endocytic pathway. Mol Biol Cell. 2007;18(7):2698–2706.
- Ultanir SK, Hertz NT, Li G, Ge WP, Burlingame AL, Pleasure SJ, Shokat KM, Jan LY, Jan YN. Chemical genetic identification of NDR1/2 kinase substrates AAK1 and Rabin8 uncovers their roles in dendrite arborization and spine development. Neuron. 2012;73(6):1127–1142.
- Golkowski M, Lius A, Sapre T, Lau HT, Moreno T, Maly DJ, Ong SE. Multiplexed kinase interactome profiling quantifies cellular network activity and plasticity. Mol Cell. 2023;83(5):803–818.e8.
- Borner GH, Antrobus R, Hirst J, Bhumbra GS, Kozik P, Jackson LP, Sahlender DA, Robinson MS. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol. 2012;197(1):141–160.
- Krieger JR, Taylor P, Gajadhar AS, Guha A, Moran MF, McGlade CJ. Identification and selected reaction monitoring (SRM) quantification of endocytosis factors associated with Numb. Mol Cell Proteomics. 2013;12(2):499–514.
- Wang M, Zhang T, Zhang X, Jiang Z, Peng M, Huang Z. BMP2K dysregulation promotes abnormal megakaryopoiesis in acute megakaryoblastic leukemia. Cell Biosci. 2020;10(1):57.
- Chaikuad A, Keates T, Vincke C, Kaufholz M, Zenn M, Zimmermann B, Gutiérrez C, Zhang RG, Hatzos-Skintges C, Joachimiak A, et al. Structure of cyclin G-associated kinase (GAK) trapped in different conformations using nanobodies. Biochem J. 2014;459(1):59–69.
- Tian X, Inoue K, Zhang Y, Wang Y, Sperati CJ, Pedigo CE, Zhao T, Yan M, Groener M, Moledina DG, et al. Inhibiting calpain 1 and 2 in cyclin G associated kinase-knockout mice mitigates podocyte injury. JCI Insight. 2020;5(22):e142740.
- In JG, Striz AC, Bernad A, Tuma PL. Serine/threonine kinase 16 and MAL2 regulate constitutive secretion of soluble cargo in hepatic cells. Biochem J. 2014;463(2):201–213.
- Stairs DB, Notarfrancesco KL, Chodosh LA. The serine/threonine kinase, Krct, affects endbud morphogenesis during murine mammary gland development. Transgenic Res. 2005;14(6):919–940.
- Kuai L, Ong SE, Madison JM, Wang X, Duvall JR, Lewis TA, Luce CJ, Conner SD, Pearlman DA, Wood JL, et al. AAK1 identified as an inhibitor of neuregulin-1/ErbB4-dependent neurotrophic factor signaling using integrative chemical genomics and proteomics. Chem Biol. 2011;18(7):891–906.
- Lian J, Zhu X, Du J, Huang B, Zhao F, Ma C, Guo R, Zhang Y, Ji L, Yahaya BH, et al. Extracellular vesicle-transmitted miR-671-5p alleviates lung inflammation and injury by regulating the AAK1/NF-κB axis. Mol Ther. 2023;31(5):1365–1382.
- Abhinand CS, Galipon J, Mori M, Ramesh P, Prasad TSK, Raju R, Sudhakaran PR, Tomita M. Temporal phosphoproteomic analysis of VEGF-A signaling in HUVECs: an insight into early signaling events associated with angiogenesis. J Cell Commun Signal. 2023;17(3):1067–1079.
- Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal. 2016;10(4):347–354.
- Agajanian MJ, Walker MP, Axtman AD, Ruela-de-Sousa RR, Serafin DS, Rabinowitz AD, Graham DM, Ryan MB, Tamir T, Nakamichi Y, et al. WNT activates the AAK1 kinase to promote clathrin-mediated endocytosis of LRP6 and establish a negative feedback loop. Cell Rep. 2019;26(1):79.e8–93.e8.
- MacGrogan D, Münch J, de la Pompa JL. Notch and interacting signalling pathways in cardiac development, disease, and regeneration. Nat Rev Cardiol. 2018;15(11):685–704.
- Gupta-Rossi N, Ortica S, Meas-Yedid V, Heuss S, Moretti J, Olivo-Marin JC, Israël A. The adaptor-associated kinase 1, AAK1, is a positive regulator of the Notch pathway. J Biol Chem. 2011;286(21):18720–18730.
- Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999.
- Yang WK, Peng YH, Li H, Lin HC, Lin YC, Lai TT, Suo H, Wang CH, Lin WH, Ou CY, et al. Nak regulates localization of clathrin sites in higher-order dendrites to promote local dendrite growth. Neuron. 2011;72(2):285–299.
- Shehata SN, Deak M, Collodet C, Spiegl SF, Geley S, Sumpton D, Sakamoto K. Identification of novel PCTAIRE-1/CDK16 substrates using a chemical genetic screen. Cell Signal. 2019;59:53–61.
- Chen C, Chu CH, Chu Y, Chang TY, Chen SW, Liang SY, Tsai YC, Chen BC, Tu HL, Cheng PL. Neuronal paxillin and drebrin mediate BDNF-induced force transduction and growth cone turning in a soft-tissue-like environment. Cell Rep. 2022;40(7):111188.
- Kostich W, Hamman BD, Li YW, Naidu S, Dandapani K, Feng J, Easton A, Bourin C, Baker K, Allen J, et al. Inhibition of AAK1 kinase as a novel therapeutic approach to treat neuropathic pain. J Pharmacol Exp Ther. 2016;358(3):371–386.
- Louis JV, Lu Y, Pieschl R, Tian Y, Hong Y, Dandapani K, Naidu S, Vikramadithyan RK, Dzierba C, Sarvasiddhi SK, et al. [3H]BMT-046091 a potent and selective radioligand to determine AAK1 distribution and target engagement. Neuropharmacology. 2017;118:167–174.
- Chen Y, Gao X, Pei H. miRNA-384-3p alleviates sevoflurane-induced nerve injury by inhibiting Aak1 kinase in neonatal rats. Brain Behav. 2022;12(7):e2556.
- Huang C, Ji C, Wang J. Current thoughts on cellular functions of numb-associated kinases. Mol Biol Rep. 2023;50(5):4645–4652.
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–132.
- Neveu G, Ziv-Av A, Barouch-Bentov R, Berkerman E, Mulholland J, Einav S. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J Virol. 2015;89(8):4387–4404.
- Bekerman E, Neveu G, Shulla A, Brannan J, Pu SY, Wang S, Xiao F, Barouch-Bentov R, Bakken RR, Mateo R, et al. Anticancer kinase inhibitors impair intracellular viral trafficking and exert broad-spectrum antiviral effects. J Clin Invest. 2017;127(4):1338–1352.
- Stebbing J, Krishnan V, de Bono S, Ottaviani S, Casalini G, Richardson PJ, Monteil V, Lauschke VM, Mirazimi A, Youhanna S, et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol Med. 2020;12(8):e12697.
- Akbarzadeh-Khiavi M, Torabi M, Rahbarnia L, Safary A. Baricitinib combination therapy: a narrative review of repurposed Janus kinase inhibitor against severe SARS-CoV-2 infection. Infection. 2022;50(2):295–308.
- Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J, et al. Characterization of spikeglycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun. 2020; 11(1):1620.
- Kar P, Saleh-E-In MM, Jaishee N, Anandraj A, Kormuth E, Vellingiri B, Angione C, Rahman PKSM, Pillay S, Sen A, et al. Computational profiling of natural compounds as promising inhibitors against the spike proteins of SARS-CoV-2 wild-type and the variants of concern, viral cell-entry process, and cytokine storm in COVID-19. J Cell Biochem. 2022;123(5):964–986. 75.
- Lo Caputo S, Corso G, Clerici M, Santantonio TA. Baricitinib: A chance to treat COVID-19? J Med Virol. 2020;92(11):2343–2344.
- Moreno-González G, Mussetti A, Albasanz-Puig A, Salvador I, Sureda A, Gudiol C, Salazar R, Marin M, Garcia M, Navarro V, et al. A Phase I/II Clinical Trial to evaluate the efficacy of baricitinib to prevent respiratory insufficiency progression in onco-hematological patients affected with COVID19: a structured summary of a study protocol for a randomised controlled trial. Trials. 2021;22(1):116.
- Lin Q, Li J, Wang Y, Zang J. Design, synthesis, and biological evaluation of novel ruxolitinib and baricitinib analogues for potential use against COVID-19. Chem Biol Drug Des. 2023;101(3):760–771.
- Bi YZ, Carson KG, Cianchetta G, Green MA, Kumi G, Liang Z, Liu YJ, Main A, Zhang YL, Zipp CG. Imidazo[1,2-b]pyridazine-based compounds compositions comprisingthem, and uses thereof. 2013, WO2013134219A1
- Lanthorn TH, Savelieva K, Zambrowicz B. Inhibition of adaptor associated kinase 1 for the treatment of pain. 2013, WO2013134336A2.
- Hartz RA, Ahuja VT, Rajamani R, Dzierba CD, Bronson JJ, Macor JE. Imidazopyridazine kinase inhibitors useful for treating a disease or disorder mediated by AAK1, such as Alzheimer’s disease, bipolar disorder, pain, and schizophrenia, WO2015026574A1. 2015.
- Bi YZ, Carson KG, Cianchetta G, Green MA, Kumi G, Liang Z, Liu YJ, Main A, Zhang YL, Zipp CG. pyrazolo[1,5-a]pyrimidine-based compounds as adaptor associated kinase 1 inhibitors, compositions comprising them, and methods of their use, WO2013134228A1. 2013.
- Bi YZ, Kumi G. Inhibitors of adapter associated kinase 1, compositions comprising them, and methods of their use. 2015, WO2015142714A1.
- Bi YZ, Gardyan MW, Green MG, Kumi G, Zhang YL. Pyrazolo[1,5-a]pyrimidine-based compounds, compositions comprising them, and methods of use. 2015, WO2015035117A1.
- Gopinathan S, Tyle P, Yang QM. 4-(3-(Pyridin-3-yl)pyrazolo[1,5-A]pyrimidin-5-yl)piperazine for use in the treatment of COV-229E OR COV-OC43 coronaviruses infections. 2021, WO2021216454A1.
- Hartz RA, Ahuja VT, Nara SJ, Kumar CMV, Brown JM, Bristow LJ, Rajamani R, Muckelbauer JK, Camac D, Kiefer SE, et al. Discovery, structure-activity relationships, and in vivo evaluation of novel aryl amides as brain penetrant adaptor protein 2-associated kinase 1 (AAK1) inhibitors for the treatment of neuropathic pain. J Med Chem. 2021;64(15):11090–11128.
- Hartz RA, Ahuja VT, Nara SJ, Kumar CMV, Manepalli R, Sarvasiddhi SK, Honkhambe S, Patankar V, Dasgupta B, Rajamani R, et al. Bicyclic heterocyclic replacement of an aryl amide leading to potent and kinase-selective adaptor protein 2-associated kinase 1 inhibitors. J Med Chem. 2022;65(5):4121–4155.
- Luo G, Chen L, Kostich WA, Hamman B, Allen J, Easton A, Bourin C, Gulianello M, Lippy J, Nara S, et al. Discovery and optimization of biaryl alkyl ethers as a novel class of highly selective, CNS-penetrable, and orally active adaptor protein-2-associated kinase 1 (AAK1) inhibitors for the potential treatment of neuropathic pain. J Med Chem. 2022;65(6):4534–4564.
- Verdonck S, Pu SY, Sorrell FJ, Elkins JM, Froeyen M, Gao LJ, Prugar LI, Dorosky DE, Brannan JM, Barouch-Bentov R, et al. Synthesis and structure-activity relationships of 3,5-disubstituted-pyrrolo[2,3-b]pyridines as inhibitors of adaptor-associated kinase 1 with antiviral activity. J Med Chem. 2019;62(12):5810–5831.
- Wells C, Couñago RM, Limas JC, Almeida TL, Cook JG, Drewry DH, Elkins JM, Gileadi O, Kapadia NR, Lorente-Macias A, et al. SGC-AAK1-1: a chemical probe targeting AAK1 and BMP2K. ACS Med Chem Lett. 2020;11(3):340–345.