
Abstract
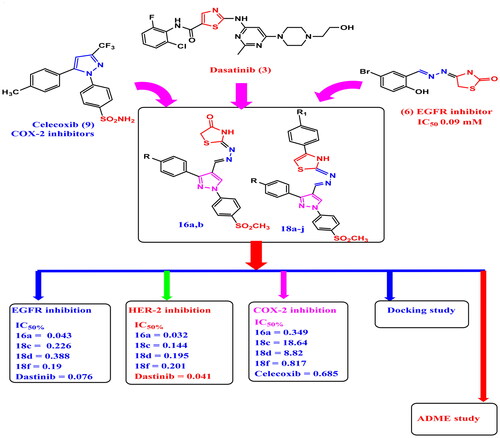
Two new series of pyrazolyl-thiazolidinone/thiazole derivatives 16a–b and 18a–j were synthesised, merging the scaffolds of celecoxib and dasatinib. Compounds 16a, 16b and 18f inhibit COX-2 with S.I. 134.6, 26.08 and 42.13 respectively (celecoxib S.I. = 24.09). Compounds 16a, 16b, 18c, 18d and 18f inhibit MCF-7 with IC50 = 0.73–6.25 μM (dasatinib IC50 = 7.99 μM) and (doxorubicin IC50 = 3.1 μM) and inhibit A549 with IC50 = 1.64–14.3 μM (dasatinib IC50 = 11.8 μM and doxorubicin IC50 = 2.42 μM) with S.I. (F180/MCF7) of 33.15, 7.13, 18.72, 13.25 and 8.28 respectively higher than dasatinib (4.03) and doxorubicin (3.02) and S.I. (F180/A549) of 14.75, 12.96, 4.16, 7.07 and 18.88 respectively higher than that of dasatinib (S.I. = 2.72) and doxorubicin (S.I = 3.88). Derivatives 16a, 18c, 18d, 18f inhibit EGFR and HER-2 IC50 for EGFR of 0.043, 0.226, 0.388, 0.19 μM respectively and for HER-2 of 0.032, 0.144, 0.195, 0.201 μM respectively.
GRAPHICAL ABSTRACT
Introduction
Combination therapy, a strategy for treatment modality that merges two or more therapeutic agents, is an important issue in combating cancerCitation1. Combination of anti-cancer drugs enhances efficacy compared to the mono-therapy manner because it targets key pathways in a characteristically synergistic or an additive behaviourCitation2. However, the wide use of combined drugs was faced by some restrictions including the high cost, drug toxicity and drug-drug interactionsCitation3. These problems can be overcome with the use of multi-target anticancer agentsCitation4. Since cancer is a compacted and multiple genes involved disease, drugs that act at multiple targets can improve efficacy and lower drug resistance and are considered as the future of worthy anticancer drug developmentCitation5. One of the most efficient and known manner in cancer therapy, is targeting protein kinases due to their vital role in regulation of various cellular signal transductions and activities, such as, proliferation, differentiation, apoptosis, metabolism and transcriptionCitation6–8.
EGFR is the highly expressed-cell surface-tyrosine kinase receptor that upon activation (phosphorylation) it becomes responsible for the cancer cells’ proliferation causing apoptosis prevention, metastasis, and invasionCitation9,Citation10. Specifically, EGFR and HER-2 are overexpressed in 20–40% of human solid tumours (such as lung, breast, gastric, ovarian and oral cancers) and are associated with increased metastasis and poor prognosisCitation11,Citation12. Recently, there is an increasing interest in developing dual/multi-kinase inhibitors for cancer treatmentCitation13. HER-2 is unique within the EGFR family in that it does not have known ligand and cannot form ligand-dependent homodimerCitation14–16. Thus, to initiate downstream signalling, it must form heterodimer with other EGFR proteins once their specific ligands have boundCitation17,Citation18.
Female breast cancer (BC) has surpassed lung cancer as the most commonly spread cancer with about 2.3 million new cases (11.7% of total cases) worldwideCitation19,Citation20.
There are various small molecules acting as EGFR inhibitors that are now available in clinics; including either reversible inhibitors like erlotinib (1), or irreversible inhibitors as dacomitinib (2) Citation21,Citation22. In addition, the thiazole motif is a rational building block in medicinal chemistry for designing and synthesising multiple bioactive anti-cancer agentsCitation23. Different studies investigated that many thiazole analogs exhibited very potent antitumor activity targeting specific pathways such as; inhibition of microtubule polymerisation as well as suppression of various molecular targets such as receptor tyrosine kinases (RTKs), non-receptor tyrosine kinases (nRTKs), phosphatidylinositol-3-kinases (PI3Ks), serine/threonine kinases (STKs)Citation24–26.
Dasatinib (3) and dabrafenib (4) are examples for thiazole containing drugs acting as selective protein kinase inhibitorsCitation27,Citation28. Several molecular docking studies proved that the nitrogen and sulphur atoms of thiazole ring form H-bonds with various amino acid residues in the active sites of different protein kinases including EGFRCitation29. In addition, EGFR–ERK pathway participates in many inflammatory conditions as it was reported that tri (2-chloroethyl) phosphate (TCEP) causes inflammatory reactions in HepG2 cells by activating EGFR, which can be faced by EGFR inhibitionCitation30. EGFR activation was also participated in respiratory inflammation with respiratory syncytial virus (RSV) and Non-Typeable Haemophilus influenzae (NTHi)Citation31–33. Furthermore, different thiazole derivatives were reported as effective EGFR kinase inhibitors, pyrazolyl-thiazoles (5a), (5b) exhibited marked EGFR inhibitory activity with IC50 = 0.06, 0.18 μM respectively. It also showed significant in vitro cytotoxicity against MCF-7 with IC50 = 0.07, 0.09 μM respectively comparing to erlotinib (IC50 = 0.02 μM)Citation34,Citation35. It was reported that, hydrazone linker inhibit EGFR and has high cytotoxic effects against MCF-7Citation36.
Additionally, the hydrazonyl-thiazole derivative (6) showed potent activity against the MCF-7 cell line and EGFR inhibitory activity (IC50 = 0.09 μM) compared to erlotinibCitation37. Pyrazolyl-thiazolidinone skeleton with hydrazone linkage (7) was reported as potent anticancer agent inducing strong apoptosis and cell cycle arrest in addition to effective EGFR suppression with nanomolar IC50 valueCitation38,Citation39. Furthermore, thiazole and thiazolidine pharmacophores occupy an important place in the field of anticancer chemotherapy targeting different proteinsCitation40–42. Pyrazolyl-thiazole derivative (8) demonstrated promising cytotoxicity through its potent dual inhibitory activity against HER-2 and EGFR with IC50 of 0.013 μM and 0.009 μM against HER-2 and EGFR respectively, in a comparable way to lapatinibCitation43.
On the other hand, inflammation acts as a defensive mechanism for the body, but it is also associated with pathological disorders, like atherosclerosis, cancers, infections and rheumatoid arthritisCitation44–46. Most of the clinically employed non-steroidal anti-inflammatory drugs (NSAIDs) are known to inhibit the two isoforms of cyclooxygenase (COX) enzyme: COX-1 (constitutive form) and COX-2 (inducible form)Citation47,Citation48. There is an evidence to suggest a role for COX-2 in the process of breast cancer developmentCitation49–52. Celecoxib (9), a vicinal diaryl pyrazole, a member of coxibs family, inhibit COX-2 isozyme selectively, thus, reducing the risk of gastric ulcers and cardiovascular disordersCitation53–55.
Fentiazac (10a), sudoxicam (10b), and meloxicam (10c) are another non-steroidal anti-inflammatory candidates containing thiazole moiety. Meloxicam (10c) is relatively selective for COX-2 at lower doses with increased cardiovascular safety than that of celecoxibCitation56.
It was reported that selective COX-2 inhibitors represent a potentially useful class of anticancer drugs so the use of COX-2 inhibitors has been associated with reduced breast cancer riskCitation57. Furthermore, several studies have suggested a reduced risk of recurrence and death from breast cancer in women who have used non-steroidal anti-inflammatory drugs (NSAIDs) after diagnosisCitation58,Citation59. also, non-steroidal anti-inflammatory drugs (NSAIDs) are used as an auxiliary therapy with chemotherapeutics in order to cure pain caused by cancer or its treatmentCitation57. Celecoxib (9) promotes apoptosis in MCF-7 cells via raising production of the pro-apoptotic protein Bax and activating caspase-7Citation60. In medicinal chemistry, a drug hybridisation is an approach of combining two or more pharmacophores into a single molecule. Therefore, a molecule with different pharmacophores will have different actions and is useful in cancer treatmentCitation61. Based on the aforementioned data and as a continuation of our research interest in the scope of synthesis and biological evaluation of new hybrid anti-inflammatory and anti-cancer agents, here in, we designed and synthesised compounds containing the Y-shaped structure of COX-2 selective coxibs with their bioactive pharmacophore (SO2CH3) which is important for COX-2 selectivity. Also, thiazole and thiazolidinone moieties were incorporated which were known with their anti-cancer activities (EGFR and HER-2 inhibition) utilising hydrazone moiety acting as a linker between the thiazolidinone/thiazole scaffold tail of dasatinib and vicinal diaryl pyrazole scaffold of celecoxib in order to inhibit both EGFR and HER-2 enzymes and suppress the carcinogenic progress. Consequently, we describe the synthesis, in vitro evaluation as COX-1/COX-2 inhibitors, in vitro anti-cancer activity on MCF7, A549, F180 cell lines, cell cycle analysis, apoptosis, mechanistic study of EGFR & HER-2 enzymes inhibition, docking and SwissADME studies for new two series of thiazole/thiazolidinone linked to pyrazole scaffold through hydrazone bridge as celecoxib/dasatinib analogs. The target derivatives 16a,b & 18a-j are hybrid molecules, their design was derivatised from lead compounds dasatinib (3), celecoxib (9), meloxicam (10c) and previously reported EGFR inhibitors (5a,b), (6), (7) and HER-2/EGFR inhibitor Pyrazolyl-thiazole (8) in which some modifications in the structure of these compounds were carried out to trigger COX-2/EGFR/HER-2 selective inhibition as: (i) thiazole moiety from dasatinib (3) as EGFR inhibitor and Pyrazolyl-thiazole derivative (8) as HER-2 inhibitor was maintained in target compounds 18a–j (ii) thiazolidinone moiety of both EGFR inhibitors (6) and (7) was maintained in the target final compounds 16a,b (iii) pyrazolyl-hydrazone linker of the EGFR inhibitor (6) and pyrazole moiety of HER-2 inhibitor (8) was maintained in both the target series 16a,b & 18a–j. (iv) Y-shape structure, central vicinal diaryl pyrazole of celecoxib and SO2CH3 moiety which is important pharmacophore for COX-2 inhibition were maintained in all designed compounds 16a,b & 18a–j. iv) Finally, thiazole moiety of NSAIDs fentiazac (10a), sudoxicam (10b) and meloxicam (10c) was maintained in the target compounds 18a–j ( and ).
Figure 1. Structure of reversible EGFR inhibitor Erlotinib (1), irreversible EGFR inhibitor Dacamitinib (2), Dasatinib (3), Dabrafenib (4) EGFR inhibitor Pyrazolyl-thiazole derivatives (5a), (5b), EGFR inhibitor hydrazonoyl-thiazole derivative (6), EGFR inhibitor pyrazolyl-thiazolidinone derivative (7), HER2 and EGFR inhibitor Pyrazolyl-thiazole (8), selective COX-2 inhibitors Celecoxib (9), thiazole containing NSAID Fentiazac (10a), thiazole containing NSAID Sudoxicam (10b) and Meloxicam (10c).

Figure 2. Structural hyperdization of designed compounds from EGFR inhibitor Dasatinib (3), reported EGFR inhibitor Pyrazolyl-thiazole (5a,b), reported EGFR inhibitor Hydrazono-thiazole (6), reported EGFR inhibitor pyrazolyl-thiazolidinone, reported HER2 and EGFR inhibitor Pyrazolyl-thiazole (8), COX-2 inhibitors Celecoxib (9) and Meloxicam (10c).

Experimental
Chemistry
General
Melting points were determined on a Thomas-Hoover capillary apparatus and are uncorrected. Infra-red (IR) spectra were recorded as films on KBr plates using a Nicolet 550 Series II Magna FT-IR spectrometer. 1H NMR and 13C NMR spectra were measured on a Bruker 400 MHz NMR Spectrophotometer, Faculty of Pharmacy, Cairo University, Egypt in dimethyl sulfoxide (DMSO)-d6 with TMS as the internal standard, where J (coupling constant) values are estimated in Hertz (Hz). Mass spectra (MS) were recorded on a Water’s Micromass ZQ 4000 mass spectrometer using the electro-spray (ES) ionisation mode. Microanalyses were performed for C, H and N were carried out on Perkin-Elmer 2400 analyser (Perkin-Elmer, Norwalk, CT, USA) at the microanalytical unit of EL-Azhar University, Egypt. All compounds were within ± 0.4% of the theoretical values. Compounds 13b,d-eCitation62, 14b,d-eCitation62, 15b,d,eCitation63, 17a,bCitation64 were prepared according to previously reported procedures. (supplementary data S2)
Experimental procedures and spectral data for compound 14a,c
A mixture of hydrazone dervative (13a or c) (0.01 mole) and phosphorous oxy chloride (0.01 mole) in dimethyl formamide (10 ml) was heated under reflux for 24 h. The reaction mixture after cooling was poured into crushed ice and neutralised by 10% sodium carbonate solution, the formed percipitate was dried and crystallised from ethanol to give compounds 14a,c. Physical and spectral data are listed below:
1–(4-(Methylsulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazole-4-carbaldehyde (14a)
Yield 80%; white solid; m.p. 240–242 °C; IR (KBr): 3115 (C–H aromatic), 2982 (C–H aliphatic), 2700 (aldehydic-H), 1676 (C = O), 1515 (C = N), 1337, 1141 (SO2CH3) cm−1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.39 (s, 3H, CH3), 3.26 (s, 3H, SO2CH3), 7.33 (d, 2H, J = 7.6 Hz, 4-methylphenyl H-3, H-5), 7.84 (d, 2H, J = 7.6 Hz, 4-methylphenyl H-2, H-6), 8.11 (d, 2H, J = 8.0 Hz, 4-methanesulfonylphenyl H-3, H-5), 8.28 (d, 2H, J = 8.0 Hz 4- methanesulfonylphenyl H-2, H-6), 9.50 (s, 1H, pyrazole H-5), 10.01 (s, 1H, aldehydic-H); 13C NMR (DMSO-d6 100 MHz, δ ppm): 34.49, 43.98, 120.01, 123.20, 128.50, 128.93, 129.36, 139.53, 139.68, 142.42, 152.77, 156.76, 162.82, 185.21; MS m/z (ES+) 340.09 (M+) (100%). Anal. Calcd. for C18H16N2O3S: C, 63.51; H, 4.74; N, 8.23; Found; C, 63.22; H, 4.58; N, 8.55.
3–(4-isobutylphenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazole-4-carbaldehyde (14c)
Yield 71%; yellow solid; m.p. 233–235 °C; IR (KBr): 3115 (C–H aromatic), 2979 (C–H aliphatic), 2700 (aldehydic-H), 1649 (C = O), 1525 (C = N), 1302, 1139 (SO2CH3) cm−1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 0.89 (d, 6H, J = 6.56 Hz, isobutyl- 2CH3), 1.86 (septet, 1H, J = 6.68 Hz isobutyl- H), 2.50 (d, 2H, J = 1.52 Hz, isobutyl- CH2), 3.29 (s, 3H, SO2CH3), 7.30 (d, 2H, J = 8.0 Hz, 4-isobutylphenyl H-3, H-5), 7.85 (d, 2H, J = 8.0 Hz, 4-methylphenyl H-2, H-6), 8.11 (d, 2H, J = 8.8 Hz, 4-methanesulfonylphenyl H-3, H-5), 8.28 (d, 2H, J = 8.8 Hz 4- methanesulfonylphenyl H-2, H-6), 9.50 (s, 1H, pyrazole H-5), 10.01 (s, 1H, aldehydic-H); 13C NMR (DMSO-d6 100 MHz, δ ppm): 22.61, 30.05, 34.55, 44.83, 119.98, 123.21, 128.81, 128.97, 129.36, 129.62, 136.23, 139.68, 142.42, 143.17, 153.78, 185.19; MS m/z (ES+) 382.14 (M+) (100%). Anal. Calcd. for C21H22N2O3S: C, 65.95; H, 5.80; N, 7.32; Found; C, 66.14; H, 5.88; N, 7.22.
Experimental procedures and spectral data for compounds 15a,c
A mixture of pyrazole aldehyde (14a or c) (0.01 mole) and thiosemicarbazide (0.01 mole) in 30 ml ethanol 95% was heated under reflux for 10h. the formed precipitate was filtered, dried, and crystallised from ethanol to give compounds 15a,c. Physical and spectral data are listed below:
(E)-2-((1–(4-(methylsulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazol-4-yl)methylene)hydrazinecarbothioamide (15a)
Yield 85%; white solid; m.p. 212–214 °C; IR (KBr):3255, 3367 (NH2), 3128(NH), 3008 (C–H aromatic), 2974 (C–H aliphatic), 1597 (C = N), 1337, 1149 (SO2CH3) cm−1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.39 (s, 3H, CH3), 3.28 (s, 3H, SO2CH3), 7.33 (d, 2H, J = 7.6 Hz, 4-methylphenyl H-3, H-5), 7.58 (d, 2H, J = 7.6 Hz, 4-methylphenyl H-2, H-6), 7.75 (s, 1H, olefenic-H), 8.10 (d, 2H, J = 8.0 Hz, 4-methanesulfonylphenyl H-3, H-5), 8.15 (d, 2H, J = 8.0 Hz, 4- methanesulfonylphenyl H-2, H-6), 8.22 (s, 2H, NH2, D2O exchangeable), 9.31 (s, 1H, pyrazole H-5), 11.25 (s, 1H, NH, D2O exchangeable); MS m/z (ES+) 413.10 (M+) (100%). Anal. Calcd. for C19H19N5O2S2: C, 55.19; H, 4.63; N, 16.94; Found; C, 55.42; H, 4.48; N, 17.12.
(E)-2-((3–(4-isobutylphenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazinecarbothioamide (15c)
Yield 70%; white solid; m.p. 204–206 °C; IR (KBr): 3244, 3333 (NH2), 3182 (NH), 3028 (C–H aromatic), 2954 (C–H aliphatic), 1583 (C = N), 1346, 1145 (SO2CH3) cm−1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 0.89 (d, 6H, J = 6.8 Hz, isobutyl- 2CH3), 1.87 (septet, 1H, J = 6.8 Hz isobutyl-H), 2.51 (d, 2H, J = 1.52 Hz, isobutyl- CH2), 3.26 (s, 3H, SO2CH3), 7.30 (d, 2H, J = 7.6 Hz, 4-isobutylphenyl H-3, H-5), 7.59 (d, 2H, J = 7.6 Hz, 4- isobutylphenyl H-2, H-6), 8.08 (d, 2H, J = 8.8 Hz, 4-methanesulfonylphenyl H-3, H-5), 8.14 (d, 2H, J = 8.8 Hz 4- methanesulfonylphenyl H-2, H-6), 8.22 (s, 2H, NH2, D2O exchangeable), 9.27 (s, 1H, pyrazole H-5), 11.35 (s, 1H, NH, D2O exchangeable); MS m/z (ES+) 455.14 (M+) (100%). Anal. Calcd. for C22H25N5O2S2: C, 58.00; H, 5.53; N, 15.37; Found; C, 58.16; H, 5.66; N, 15.42.
Experimental procedures and spectral data for compounds 16a,b
An equimolecular mixture of thiosemicarbazone derivatives 15a or c (0.01 mole), ethyl chloroacetate (0.01 mole), and sodium acetate (0.012 mole) in ethanol 95% (40 ml) was heated under reflux for 24 h. The formed percipitate was crystallised from ethanol to give compounds 16a,b. Physical and spectral data are listed below:
Z)-2-((E)-((1–(4-(methylsulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazol-4-yl)methylene)hydrazono)thiazolidin-4-one (16a)
Yield 77%; white solid; m.p. 286 – 288 °C; IR (KBr): 3367 (NH), 3012 (C–H aromatic), 2970 (C–H aliphatic), 1643 (C = O), 1593 (C = N), 1400, 1145 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.38 (s, 3H, CH3), 3.18 (s, 3H, SO2CH3), 3.92 (s, 2H, CH2), 7.31 (d, 2H, J = 7.6 Hz, methylphenyl H-3, H-5), 7.76 (d, 2H, J = 7.6 Hz, methylphenyl H-2, H-6), 8.07 (d, 2H, J = 8.4 Hz methanesulfonylphenyl H-3, H-5), 8.04 8.25 (d, 2H, J = 8.4 Hz methanesulfonylphenyl H-2, H-6), 8.41 (s, 1H, olefenic-H), 9.10 (s, 1H, pyrazole H-5), 11.88 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 21.42, 33.71, 44.07, 49.07, 118.04, 119.50, 129.03, 129.25, 129.32, 129.61, 130.84, 138.86, 138.98, 142.74, 149.41, 152.99; MS m/z (ES+) 453.09 (M+) (100%). Anal. Calcd. For C21H19N5O3S2: C, 55.61; H, 4.22; N, 15.44; Found; C, 55.46; H, 4.38; N, 15.28.
(Z)-2-((E)-((3–(4-isobutylphenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazono)thiazolidin-4-one (16b)
Yield 66%; white solid; m.p. 216– 218 °C; IR (KBr): 3421 (NH), 3020 (C–H aromatic), 2954 (C–H aliphatic), 1639 (C = O), 1593 (C = N), 1404, 1145 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 0.89 (d, 6H, J = 6.8 Hz, isobutyl- 2CH3), 1.88 (septet, 1H, J = 6.8 Hz isobutyl-H), 3.18 (d, 2H, J = 1.52 Hz, isobutyl- CH2), 3.29(s, 3H, SO2CH3), 3.92 (s, 2H, CH2), 7.30 (d, 2H, J = 7.6 Hz, 4-isobutylphenyl H-3, H-5), 7.77 (d, 2H, J = 7.6 Hz, 4-isobutylphenyl H-2, H-6), 8.08 (d, 2H, J = 8.0 Hz, 4-methanesulfonylphenyl H-3, H-5), 8.26 (d, 2H, J = 8.0 Hz 4-methanesulfonylphenyl H-2, H-6), 8.43 (s, 1H, olefinic-H) 9.12 (s, 1H, pyrazole H-5), 11.91 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 22.60, 30.13, 33.66, 44.07, 44.88, 49.08, 56.33, 118.06, 119.47, 128.92, 129.32, 129.55, 129.60, 130.77, 138.98, 142.51, 142.75, 149.39, 155.08; MS m/z (ES+) 545.13 (M+) (100%). Anal. Calcd. For C24H25N5O3S2: C, 58.16; H, 5.08; N, 14.13; Found; C, 58.28; H, 5.18; N, 14.36.
Experimental procedures and spectral data for compounds 18a-j
An equimolecular mixture of carbazone derivatives 15a–e (0.01 mole), phenacyl bromide derivatives (17a or b) (0.01 mole) and pyridine (0.012 mole) in ethanol 95% (40 ml) was heated under reflux for 24 h. The reaction mixture was evaporated to dryness and the residue was crystallised from ethanol to give compounds 18a–j. Physical and spectral data are listed below:
(Z and E)-4–(4-bromophenyl)-2-((E)-((1–(4-(methylsulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazol-4-yl)methylene)hydrazono)-2,3-dihydrothiazole (18a)
Yield 81%; white solid; m.p. 296 – 298 °C; IR (KBr): 3348 (NH), 3070 (C–H aromatic), 2939 (C–H aliphatic), 1597 (C = N), 1342, 11465 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.39 (s, 3.6H, phenyl-CH3 of both E and Z isomers), 3.28 (s, 3.6H, SO2CH3 of both E and Z isomers), 7.34 (s, 1H, thiazole H-5 of E isomer), 7.35 (d, 2H, J = 7.6 Hz, 4-methylphenyl H-3, H-5 of E isomer), 7.59 (d, 2.4H, J = 7.6 Hz, methylphenyl H-2, H-6 of both E and Z isomers), 7.70 (d, 0.4H, J = 7.6 Hz, 4-methylphenyl H-3, H-5 of Z isomer),7.77 (s, 0.2H, thiazole H-5 of Z isomer), 7.79 (d, 2.4H, J = 7.6 Hz, bromophenyl H-3, H-5 of both E and Z isomers), 8.09 (d, 2.4H, J = 7.6 Hz bromophenyl H-2, H-6 of both E and Z isomers), 8.14 (d, 2.4H, J = 7.6 Hz 4-aminosulfonylphenyl H-3, H-5 of both E and Z isomers), 8.21 (s, 1.2H, olefinic-H of both E and Z isomers), 8.26 (m, 2.4H, 4-aminosulfonylphenyl H-2, H-6 of both E and Z isomers), 9.03 (s, 1H, pyrazole H-5 of Z isomer), 9.31 (s, 1H, pyrazole H-5 of E isomer), 11.37 (s, 1.2H, NH, D2O exchangeable of both E and Z isomers); 13C NMR (DMSO-d6 100 MHz, δ ppm): 10.08, 44.02, 119.34, 119.40, 123.99, 124.43, 127.73, 129.47, 129.72, 130.36, 131.10, 134.34, 138.56, 139.37, 140.15, 141.20,142.38, 142.58, 147.87, 150.39, 178.22; MS m/z (ES+) 591.04 (M+) (100%). Anal. Calcd. For C27H22BrN5O2S2: C, 54.73; H, 3.74; N, 11.82; Found; C, 54.62; H, 3.86; N, 11.48.
(Z)-2-((E)-((1–(4-(methylsulfonyl)phenyl)-3-(p-tolyl)-1H-pyrazol-4-yl)methylene)hydrazono)-4–(4-nitrophenyl)-2,3-dihydrothiazole (18b)
Yield 79%; white solid; m.p. 232–234 °C; IR (KBr): 3317 (NH), 3032 (C–H aromatic), 2934 (C–H aliphatic), 1597 (C = N), 1338, 1141 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 2.41 (s, 3H, phenyl-CH3), 3.29 (s, 3H, SO2CH3), 7.35 (d, 2H, J = 8.0 Hz, 4-methylphenyl H-3, H-5), 7.59 (d, 2H, J = 8.0 Hz, methylphenyl H-2, H-6), 7.71 (s, 1H, thiazole H-5), 8.07 (d, 2H, J = 7.6 Hz, nitrophenyl H-3, H-5), 8.09 (d, 2H, J = 7.6 Hz nitrophenyl H-2, H-6), 8.18 (s, 1H, olefinic-H), 8.26 (m, 4H, 4-aminosulfonylphenyl H-2, H-3, H-5, H-6), 9.05 (s, 1H, pyrazole H-5), NH not detected; 13C NMR (DMSO-d6 100 MHz, δ ppm): 21.41, 44.09, 108.90, 118.28, 119.34, 124.58, 126.79, 128.69, 128.88, 129.29, 129.41, 129.70, 135.31, 138.78, 138.81, 141.15, 142.88, 146.67, 148.93, 152.41, 168.83; MS m/z (ES+) 558.11 (M+) (100%). Anal. Calcd. For C27H22N6O4S2: C, 58.05; H, 3.97; N, 15.04; Found; C, 58.19; H, 3.72; N, 15.33.
(E)-4–(4-bromophenyl)-2-((E)-((3–(4-methoxyphenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-2,3-dihydrothiazole (18c)
Yield 74%; white solid; m.p. 288–290 °C; IR (KBr): 3348 (NH), 3070 (C–H aromatic), 2939 (C–H aliphatic), 1597 (C = N), 1342, 11465 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 3.29 (s, 3H, SO2CH3), 3.58 (s, 3H, phenyl-OCH3), 7.34 (s, 1H, thiazole H-5), 7.57 (d, 2H, J = 8.4 Hz, 4-methoxyphenyl H-3, H-5), 7.69 (d, 2H, J = 8.4 Hz, methoxyphenyl H-2, H-6), 7.79 (d, 2H, J = 8.4 Hz, bromophenyl H-3, H-5), 7.91 (d, 2H, J = 8.4 Hz bromophenyl H-2, H-6), 8.09 (d, 2H, J = 8.8 Hz 4-aminosulfonylphenyl H-3, H-5), 8.14 (s, 1H, olefinic-H), 8.29 (d, 2H, J = 8.8 Hz 4-aminosulfonylphenyl H-2, H-6), 9.10 (s, 1H, pyrazole H-5), 11.39 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 43.58, 44.11, 105.00, 118.65, 118.98, 119.41, 125.97, 128.00, 129.33, 139.69, 130.19, 131.66, 131.97, 133.90, 134.29, 138.98, 139.16, 142.78, 149.74, 151.58, 168.65; MS m/z (ES+) 607.03 (M+) (100%). Anal. Calcd. For C27H22BrN5O3S2: C, 53.29; H, 3.64; N, 11.51; Found; C, 53.48; H, 3.52; N, 11.66.
(E and Z)-2-((E)-((3–(4-methoxyphenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-4–(4-nitrophenyl)-2,3-dihydrothiazole (18d)
Yield 67%; white solid; m.p. 264–266 °C; IR (KBr): 3309 (NH), 3035 (C–H aromatic), 2927 (C–H aliphatic), 1597 (C = N), 1346, 1149 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 3.29 (m, 4.5H, SO2CH3 of both E and Z isomers), 3.59 (m, 4.5H, phenyl-OCH3 of both E and Z isomers), 7.66 (d, 1H, J = 8.0 Hz, 4-methoxyphenyl H-3, H-5 of Z isomer), 7.69 (d, 2H, J = 8.0 Hz, 4-methoxyphenyl H-3, H-5 of E isomer), 7.70 (s, 1H, thiazole H-5 of E isomer), 7.81 (s, 0.5H, thiazole H-5 of Z isomer),7.83 (d, 1H, J = 8.0 Hz, methoxyphenyl H-2, H-6 of Z isomer), 7.90 (d, 2H, J = 8.0 Hz, methoxyphenyl H-2, H-6 of E isomer), 8.09 (m, 6H, nitrophenyl H-2, H-3, H-4, H-6 of both E and Z isomers), 8.17 (s, 0.5H, olefinic-H of Zisomer), 8.20 (s, 1H, olefinic-H of E isomer), 8.26 (m, 6H, 4-aminosulfonylphenyl H-2, H-3, H-4, H-6 of both E and Z isomers), 9.12 (s, 1H, pyrazole H-5 of E isomer), 9.37 (s, 0.5H, pyrazole H-5 of Z isomer), 11.37 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 43.59, 44.06, 109.05, 118.56, 119.25, 119.42, 124.57, 126.80, 129.35, 129.46, 129.68, 130.21, 131.67, 131.93, 134.31, 134.56, 134.66, 139.03, 141.15, 142.77, 168.97; MS m/z (ES+) 574.11 (M+) (100%). Anal. Calcd. For C27H22N6O5S2: C, 56.43; H, 3.86; N, 14.63; Found; C, 56.70; H, 3.62; N, 14.88.
(E)-4–(4-bromophenyl)-2-((E)-((3–(4-isobutylphenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-2,3-dihydrothiazole (18e)
Yield 81%; white solid; m.p. 269–271 °C; IR (KBr): 3244 (NH), 3020 (C–H aromatic), 2954 (C–H aliphatic), 1573 (C = N), 1350, 1145 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 0.91 (d, 6H, J = 6.8 Hz, isobutyl- 2CH3), 1.91 (septet, 1H, J = 6.8 Hz isobutyl-H), 2.54 (d, 2H, J = 1.52 Hz, isobutyl- CH2), 3.28 (s, 3H, SO2CH3), 7.32 (d, 2H, J = 7.6 Hz, 4-isobutylphenyl H-3, H-5) 7.38 (s, 1H, thiazole H-5), 7.58 (d, 2H, J = 7.6 Hz, 4-isobutylphenyl H-2, H-6), 7.70 (d, 2H, J = 8.0 Hz, 4–4-bromophenyl H-3, H-5), 7.79 (d, 2H, J = 8.0 Hz 4-bromophenyl H-2, H-6), 8.07 (d, 2H, J = 8.8 Hz 4-methanesulfonylphenyl H-3, H-5), 8.26 (s, 1H, olefinic-H), 8.28 (d, 2H, J = 8.8 Hz 4-methanesulfonylphenyl H-2, H-6), 9.05 (s, 1H, pyrazole H-5), 12.00 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 22.65, 30.17, 44.10, 44.87, 104.84, 118.37, 118.63, 119.12, 119.33, 120.97, 128.03, 128.47, 128.65, 128.80, 129.32, 129.70, 129.87, 132.01, 135.23, 138.71, 142.90, 152.46, 168.58; MS m/z (ES+) 633.09 (M+) (100%). Anal. Calcd. For C30H28BrN5O2S2: C, 56.78; H, 4.45; N, 11.04; Found; C, 56.92; H, 4.64; N, 11.19.
(E)-2-((E)-((3–(4-isobutylphenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-4–(4-nitrophenyl)-2,3-dihydrothiazole (18f)
Yield 80%; white solid; m.p. 286–288 °C; IR (KBr): 3209 (NH), 3024 (C–H aromatic), 2912 (C–H aliphatic), 1581 (C = N), 1342, 1145 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 1H NMR (DMSO-d6, 400 MHz, δ ppm): 0.92 (d, 6H, J = 6.8 Hz, isobutyl-2CH3), 1.91 (septet, 1H, J = 6.8 Hz isobutyl-H), 2.56 (d, 2H, J = 1.52 Hz, isobutyl- CH2), 3.29 (s, 3H, SO2CH3), 7.32 (d, 2H, J = 7.6 Hz, 4-isobutylphenyl H-3, H-5), 7.70 (d, 2H, J = 7.6 Hz, 4-isobutylphenyl H-2, H-6), 7.78 (s, 1H, thiazole H-5), 8.10 (m, 4H, 4-nitrophenyl H-2, H-3, H-5, H-6), 8.19 (s, 1H, olefinic-H), 8.26 (m, 4H, 4-methanesulfonylphenyl H-2, H-3, H-5, H-6), 9.06 (s, 1H, pyrazole H-5), 12.11 (s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 22.65, 30.16, 44.09, 44.87, 108.86, 118.27, 119.04, 119.30, 124.56, 126.80, 128.44, 128.76, 128.80, 129.29, 129.67, 138.77, 141.15, 142.42, 142.88, 146.66, 148.94, 152.47, 168.80; MS m/z (ES+) 600.16 (M+) (100%). Anal. Calcd. For C30H28N6O4S2: C, 59.98; H, 4.70; N, 13.99; Found; C, 60.18; H, 4.88; N, 14.25.
(E and Z)-4–(4-bromophenyl)-2-((E)-((1–(4-(methylsulfonyl)phenyl)-3–(4-nitrophenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-2,3-dihydrothiazole (18 g)
Yield 66%; white solid; m.p. 217–219 °C; IR (KBr): 3309 (NH), 3066 (C–H aromatic), 2962 (C–H aliphatic), 1593 (C = N), 1342, 1149 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 3.29 (m, 4H, SO2CH3 of both E and Z isomers), 7.38 (s, 1H, thiazole H-5 of E isomer), 7.58 (d, 2H, J = 8.4 Hz, 4-bromophenyl H-3, H-5 of E isomer), 7.68 (d, 1H, J = 8.0 Hz, 4-bromophenyl H-3, H-5 of Z isomer), 7.73 (m, 3H, 4-bromophenyl H-2, H-6 of both E and Z isomers), 7.79 (m, 3H, 4-nitrophenyl H-3, H-5 of both E and Z isomers), 7.83 (s, 0.4 H, thiazole H-5 of Z isomer), 8.08 (m, 3H, nitrophenyl H-2, H-6 of both E and Z isomer), 8.13 (s, 1H, olefinic-H of E isomer), 8.15 (m, 3H, methanesulfonylphenyl H-3, H-5 of both E and Z isomers), 8.20 (s, 0.4H, olefinic-H of Z isomer), 8.25 (m, 3H, 4-aminosulfonylphenyl H-2, H-6 of both E and Z isomers), 9.08 (s, 1H, pyrazole H-5 of E isomer), 9.33 (s, 0.4H, pyrazole H-5 of Z isomer), NH is not detected; 13C NMR (DMSO-d6 100 MHz, δ ppm): 44.07, 104.93, 118.48, 118.79, 119.21, 119.40, 120.96, 122.67, 128.00, 129.04, 129.33, 129.45, 129.62, 130.66, 131.09, 131.36, 131.64, 132.00, 132.26, 134.35, 134.56, 134.69, 138.96, 139.11, 142.73, 142.76, 149.73, 150.98, 168.53; MS m/z (ES+) 622.01 (M+) (100%). Anal. Calcd. For C26H19BrN6O4S2: C, 50.08; H, 3.07; N, 13.48; Found; C, 50.23; H, 3.23; N, 13.75.
(E)-2-((E)-((1–(4-(methylsulfonyl)phenyl)-3–(4-nitrophenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-4–(4-nitrophenyl)-2,3-dihydrothiazole (18h)
Yield 64%; white solid; m.p. 228–230 °C; IR (KBr): 3317 (NH), 3106 (C–H aromatic), 2977 (C–H aliphatic), 1589 (C = N), 1342, 1149 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 3.28 (s, 3H, SO2CH3), 7.37 (s, 1H, thiazole H-5), 7.45 (d, 2H, J = 8.8 Hz 4-nitrophenyl H-3, H-5), 7.60 (d, 2H, J = 8.8 Hz 4-nitrophenyl H-2, H-6), 7.82 (d, 2H, J = 8.0 Hz 4-nitrophenyl H-3, H-5), 7.87 (d, 2H, J = 8.0 Hz, 4-nitrophenyl H-2, H-6), 8.07 (d, 2H, J = 8.4 Hz 4-methanesulfonylphenyl H-3, H-5), 8.16 (s, 1H, olefenic H), 8.25 (d, 2H, J = 8.4 Hz 4-methanesulfonylphenyl H-2, H-6), 9.08 (s, 1H, pyrazole H-5), 11.99 (s, 1H, NH D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 44.05, 109.02, 118.90, 119.35, 124.12, 124.56, 126.78, 129.36, 129.73, 130.61, 134.35, 134.58, 138.58, 139.40, 141.08, 147.89, 148.91, 149.76, 150.40, 178.26; MS m/z (ES+) 589.08 (M+) (100%). Anal. Calcd. For C26H19N7O6S2: C, 52.96; H, 3.25; N, 16.63; Found; C, 52.87; H, 3.36; N, 16.62.
(E)-4–(4-bromophenyl)-2-((E)-((3–(4-bromophenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-2,3-dihydrothiazole (18i)
Yield 64%; white solid; m.p. 228–230 °C; IR (KBr): 3329 (NH), 3070 (C–H aromatic), 2920 (C–H aliphatic), 1593 (C = N), 1365, 1145 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 3.28 (s, 3H, SO2CH3), 7.60 (d, 2H, J = 8.4 Hz 4-bromophenyl H-3, H-5), 7.69 (s, 1H, thiazole H-5), 7.86 (d, 2H, J = 8.4 Hz 4-bromophenyl H-2, H-6), 8.06 (d, 2H, J = 8.0 Hz 4-bromophenyl H-3, H-5), 8.10 (d, 2H, J = 8.0 Hz, 4-bromophenyl H-2, H-6), 8.17 (s, 1H, olefenic H), 8.24 (d, 2H, J = 8.4 Hz 4-methanesulfonylphenyl H-3, H-5), 8.26 (d, 2H, J = 8.4 Hz 4-methanesulfonylphenyl H-2, H-6), 9.08 (s, 1H, pyrazole H-5), 12.11(s, 1H, NH D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 44.07, 108.95, 118.37, 119.37, 124.56, 126.78, 128.96, 129.06, 129.32, 129.54, 129.72, 130.83, 131.26, 134.96, 138.94, 142.73, 146.66, 148.91, 150.94, 168.75; MS m/z (ES+) 654.93 (M+) (100%). Anal. Calcd. For C26H19Br2N5O2S2: C, 47.50; H, 2.91; N, 10.65; Found; C, 47.82; H, 3.19; N, 10.90.
(E)-2-((E)-((3–(4-bromophenyl)-1–(4-(methylsulfonyl)phenyl)-1H-pyrazol-4-yl)methylene)hydrazono)-4–(4-nitrophenyl)-2,3-dihydrothiazole (18j)
Yield 64%; white solid; m.p. 228 – 230 °C; IR (KBr): 3317 (NH), 3106 (C–H aromatic), 2977 (C–H aliphatic), 1589 (C = N), 1342, 1149 (SO2CH3) cm1; 1H NMR (DMSO-d6, 400 MHz, δ ppm): 3.28 (s, 3H, SO2CH3), 7.38 (s, 1H, thiazole H-5), 7.55 (d, 2H, J = 8.4 Hz 4-bromophenyl H-3, H-5), 7.58 (d, 2H, J = 8.4 Hz 4-bromophenyl H-2, H-6), 7.78 (d, 2H, J = 8.4 Hz 4-nitrophenyl H-3, H-5), 7.87 (d, 2H, J = 8.4 Hz, 4-nitrophenyl H-2, H-6), 8.08 (d, 2H, J = 8.8 Hz 4-methanesulfonylphenyl H-3, H-5), 8.18 (s, 1H, olefenic H), 8.25 (d, 2H, J = 8.8 Hz 4-methanesulfonylphenyl H-2, H-6), 9.07 (s, 1H, pyrazole H-5), 11.98 (s, 1H, NH D2O exchangeable); 13C NMR (DMSO-d6 100 MHz, δ ppm): 44.08, 104.94, 118.47, 119.38, 120.96, 128.00, 128.96, 129.06, 129.32, 129.53, 129.63, 130.83, 131.08, 131.99, 134.00, 134.61, 138.92, 142.75, 150.92, 168.48; MS m/z (ES+) 622.01 (M+) (100%). Anal. Calcd. For C26H19BrN6O4S2: C, 50.08; H, 3.07; N, 13.48; Found; C, 50.28; H, 3.26; N, 13.21.
Biological evaluation
In vitro anti-inflammatory activity
The in vitro COX-1/COX-2 inhibition was determined using COX-1 inhibitor screening Kit-K548 (Biovision, S. Milpitas Blvd., Milpitas, CA 95035 USA) and COX-2 inhibitor screening Kit-K547 (Biovision, S. Milpitas Blvd., Milpitas, CA 95035 USA). The reported procedure utilised in the biological assays was conducted herein were performed as reported earlier. The potency of the tested compounds was determined as the concentration causing 50% enzyme inhibition (IC50). In addition, the COX-2 selectivity indexes (S.I values) which are calculated as IC50 (COX-1)/IC50 (COX-2) were carried out and compared with that of the standard drugs celecoxib & indomethacin.
In vitro cytotoxicity screening and IC50 determination
3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) cell viability assay was done in all the three cancer cell lines MCF-7, A549 and F180. In brief, cells were seeded at 1 × 104 density per well in 96-well plate, in the next day cell were treated with a single dose of 10 µM from each compound. The wells without treatment were used as a control. After 48 h, media was removed and fresh media which contains 0.5 mg/mL thiazolyl blue tetrazolium bromide reagent (Sigma-Aldrich- St. Louis, MO, USA, M2128) was added. The cells where incubated for 3 h at 37 °C and 5% CO2, then DMSO (Sigma-Aldrich- St. Louis, MO, USA) was added to solubilise the formed formazan crystals and was taken at 570 nm using Varioskan Flash (Thermo Fisher Scientific, Waltham, MA, USA) microplate spectrophotometer. The half-maximal inhibitory concentration (IC50) of the most potent compounds was assessed using dose-response curve (GraphPad Prism software, version 8.00, Graph Pad software, Inc)Citation65,Citation66.
Cell cycle analysis
The effect of compounds 18d on cell growth was determined using flow cytometry according to the procedure reported earlier. Compounds 18d were added on breast MCF7 cells with (IC50 = 14, 16, 15, 9 μM respectively) for 24h. Rinsed breast cells with phosphate buffer saline (PBS), centrifuged, 70% ethanol was added, cells were washed with PBS and suspended with 0.1 mg/mL RNase. Moreover, cells were marked with 40 mg/mL propidium iodide (PI), and cell cycle distribution was determined using a FACS Calibur flow cytometer (Becton Dickinson) ().
Table 1. Cell cycle analysis of compounds 18d and control for breast MCF7 cells.
Determination of apoptosis using Annexin-V
The apoptosis of compounds 18d was determined using Annexin-V-FITC method reported earlier. Where compounds 18d were added on MCF7 breast cells at their IC50 = 14, 16, 15, 9 μM respectively, cells were incubated for 24 h, collected, rinsed twice with PBS and marked with mixture of fluorescein isothiocyanate (FITC), Annexin-V- and PI (propidium iodide) are left for 30 min at room temperature in the dark. analysis was carried out using FACS Calibur flow cytometer (BD Biosciences, San Jose, CA, USA) ().
Table 2. Apoptosis of compounds 18d and control against breast MCF7 cell line.
EGFR Kinase inhibitory activity
EGFR-WT(Catalog # 40321), EGFR(L858R) (Catalog # 40324), EGFR (T790M/L858R) (Catalog # 40322) Kinase Assay Kits were used to assess EGFR kinase inhibitory activity of compounds 16a, 18c, 18d, 18f according to the reported procedureCitation67: Thaw ATP, Poly (Glu:Tyr 4:1) and PTK substrate (10 mg/ml) in 5× Kinase Buffer 1. (Optional) To achieve a 10 mM concentration, add DTT to 5× Kinase Buffer 1 (e.g. add 10 l of 1 M DTT to 1 ml of 5× Kinase Buffer 1). The master mixture (25 l per well) as follows: 6 l (5× Kinase Buffer 1), 1 l of ATP (500 M), 1 l of the PTK substrate Poly (Glu:Tyr 4:1) (10 mg/ml), and 17 l of water are divided among N wells. Pour 25 l into each well. Add 5 l of the “Test Inhibitor” well’s inhibitor solution. Add 5 l of the identical solution devoid of an inhibitor to the “Positive Control” and “Blank” samples (Inhibitor buffer). By combining 2400 ml of water with 600 ml of 5× Kinase Buffer 1, you can make 3 ml of 1x Kinase Buffer 1. For 100 reactions, 3 ml of 1× Kinase Buffer 1 are sufficient. The "Blank" wells should have 20 l of 1× Kinase Buffer 1 added to them. On ice, thaw the EGFR enzyme. When the tube containing the enzyme initially defrosts, quickly spin it to recover its entire contents. Determine how much EGFR is needed for the test, then diluted the enzyme with 1× Kinase Buffer 1 to 1 ng/l. At −80 °C, aliquot the remaining undiluted enzyme. Note: The freeze-thaw cycle is sensitive to the EGFR enzyme. Reduce the number of freeze-thaw cycles. Reusing thawed aliquots or diluted enzyme is not advised. Add 20 l of diluted EGFR enzyme to the “Positive Control” and “Test Inhibitor Control” wells to start the reaction. Incubate for 40 min at 30 °C. Defrost the Kinase-Glo Max solution. 50 l of the Kinase-Glo Max reagent should be added to each well following the 40 min reaction. Plate should be covered with aluminium foil and incubated for 15 min at room temperature. Use the microplate reader to measure luminescence.
HER-2 inhibitory activity
HER-2 inhibitory activity was determined using Kinase-Glo MAX (Promega, #V6071) (Catalog # 40230). IC50 of compounds 16a, 18c, 18d, 18f were determined in MCF7 cells according to the following procedureCitation68, Thaw 5× Kinase Buffer 1, ATP and PTK substrate Poly (Glu:Tyr 4:1) (10 mg/ml). (Optional: If desired, add DTT to 5× Kinase Buffer 1 to make a 10 mM concentration; e.g. add 10 µl of 1 M DTT to 1 ml 5× Kinase Buffer 1). Prepare the master mixture (25 µl per well): N wells × (6 μl 5× Kinase Buffer 1 + 1 µl ATP (500 µM) + 1 µl PTK substrate Poly (Glu:Tyr 4:1) (10 mg/ml)+ 17 μl water). Add 25 μl to every well. Add 5 μl of Inhibitor solution of each well labelled as “Test Inhibitor”. For the “Positive Control” and “Blank”, add 5 μl of the same solution without inhibitor (Inhibitor buffer). Prepare 3 ml of 1× kinase Buffer 1 by mixing 600 µl of 5× Kinase Buffer 1 with 2400 µl water. 3 ml of 1× Kinase Buffer 1 is sufficient for 100 reactions. To the wells designated as “Blank”, add 20 μl of 1× kinase Buffer 1. Thaw HER-2 enzyme on ice. Upon first thaw, briefly spin tube containing enzyme to recover full content of the tube. Calculate the amount of HER-2 required for the assay and dilute enzyme to 7 ng/µl with 1× Kinase Buffer 1.
Molecular docking study
The docking study was out using the Molecular Operating Environment (MOE) program (2014) and all docking studies were carried out using enzyme downloaded from the Protein Data Bank (PDB). The enzymes included in the study were COX-2, EGFR, HER-2 enzymes. The crystal structure of the reference drug celecoxib bound at the COX-2 active site obtained from protein data bank at Research Collaboration for Structural Bioinformatics (RSCB) protein data bank COX-2 enzyme [PDB] (entry 3LN1)Citation69, EGFR enzyme (PDB, ID: 1M17)Citation70. Her-2 enzyme (PDB, ID:3PP0)Citation71. The preparation of the target derivatives for docking was done via their 3D structure built by MOE. Before docking, 3D protonation of the structures, running conformational analysis using systemic search, selecting the least energetic conformer and applying the same docking protocol used with ligand were done.
Pharmacokinetics and ADME studies
ADME studies were carried out using the Swiss ADME website according to previously reported methodCitation72. ADME studies were used to determine physicochemical parametersCitation73. The most active compounds 16a, 16b, 18c, 18d, 18f to determine drug likeness and medicinal chemistry of the synthesised compounds and correlates activity with physicochemical parameters.
Statistical analysis
All statistical analyses were performed using Graph Pad Prism 7 for Mac OS X (Intuitive Software for Science, San Diego, CA, USA). Differences between experimental groups were compared using analysis of variance (ANOVA) followed by Tukey’s test (p < 0.05).
Results and discussion
Chemistry
The general reactions used for the preparation of the final target pyrazolyl-thiazolidinone/thiazole hybrids 16a, b & 18a–j are outlined in Scheme 1. Hydrazones 13b,d–eCitation62, pyrazole aldehyde derivatives 14b,d–eCitation62, thiosemicarbazone derivatives 15b,d–eCitation63 and 4-substituted phenacyl bromide derivatives 17a,bCitation64 were synthesised according to previously reported procedure. The nucleophilic addition reaction of 4-methanesulfonylphenylhydrazine hydrochloride 12 to 4-substituted acetophenone derivatives 11a,c in ethanol resulted in formation of hydrazone derivatives 13a,c which further cyclized into pyrazole aldehydes 14a,c utilising Vilsimier Hack reaction conditions with the appearance of characteristic pyrazole H-5 and aldehydic protons at 9.5, 10.01 ppm in H1NMR respectively. Pyrazole thiosemicarbazone intermediates 15a,c were synthesised through condensation of pyrazole aldehydes 14a,c with thiothemicarbazide in ethanol with the appearance of D2O exchangeable NH2, NH at 8.22, 11.25–11.35 ppm in H1NMR respectively on the expense of the aldehydic protons peaks. The target pyrazolyl-thiazolidinones 16a,b were synthesised by cyclisation of thiosemicarbazides 15a,c with ethyl chloroacetate with the appearance of singlet peak at 3.92 ppm corresponding to aliphatic CH2 moiety of thiazolidinone ring in H1NMR and appearance of sharp intense peak at 1643, 1639 cm−1 corresponding to carbonyl group of thiazolidinone ring in I.R. while cyclisation of the intermediate thiosemicarbazone 15a–e with phenacyl bromide derivatives 17a,b gave the target pyrazolyl-thiazole derivatives 18a–j with the appearance of additional aromatic protons in H1NMR corresponding to aryl derivatives coming from the utilised phenacyl bromide derivatives and in addition of appearance of singlet peak at 7.34–7.78 ppm due to thiazole H-5. It was worthy to note that pyrazolyl-thiazole final compounds 18a, d, g showed two geometrical isomers at different ratios around hydrazone double bond due to restricted rotation and that was clear in H1NMR spectrum of these derivatives as the ratios of (E and Z) isomers were predicted by comparing the intensity of pyrazole H-5 in H1NMR spectrum for the two isomers as these ratios were 1:0.2 & 1:0.5 & 1:0.4 respectively. The existence of two geometrical isomers for these derivatives was further confirmed with 2D NOESY H1NMR.
Scheme 1. Reagents and conditions: (a) ethanol 95% reflux 12 h, (b) POCl3/DMF reflux 24 h, (c) thiosemicarbazide, ethanol 95%, reflux 10 h, (d) ethanol 95%, sod. acetate, reflux, 24 h, (e) ethanol 95%, pyridine, reflux, 24 h.

2D NOESY H1NMR of compounds 18a, d, g showed a correlation between the olifenic proton of each geometrical isomer and the pyrazole H-5 of it and that indicating the presence of two geometrical isomers (E & Z) with the previously mentioned ratios and explained the peak duplication of H1NMR spectra of compounds 18a, d, g ((a–c) of see supplementary data S2).
Biological evaluation
In vitro anti-inflammatory activity
COX-1 inhibitor screening Kit-K548 (Biovision, S. Milpitas Blvd., Milpitas, CA 95035 USA) and COX-2 inhibitor screening Kit-K547 (Biovision, S. Milpitas Blvd., Milpitas, CA 95035 USA) were used. The potency of the screened compounds was estimated as the concentration causing 50% enzyme inhibition (). Thiazole containing compounds 16a–b and 18a–j showed inhibitory activity COX-2 isozyme inhibitory activities (IC50 = 0.349–27.76 μM range) in comparison with celecoxib (IC50 = 0.685 μM). The selectivity index range of thiazole containing compounds were (S.I. = 1.6–34.6) in comparison with celecoxib (S.I. = 24.09). From the above results, it can be concluded that compounds 16a, 16b, 18f have outstanding COX-2 selectivity indexes of 134.6, 26.08–49.38 respectively when compared to celecoxib (the COX-2 selective reference drug) (COX-1 IC50 = 16.5 μM, COX-2 IC50 = 0.685 μM and S.I. = 24.09). (Supplementary data S1)
Table 3. In vitro COX-1 and COX-2 inhibitory activity of thiazole 16a,b, 18a–j and reference drugs indomethacin, celecoxib.
In vitro cytotoxicity screening and IC50 determination
MCF7 human breast adenocarcinoma and A549 small lung cancer and F180 fibro blast cell lines (purchased from Sigma-Aldrich (Labco), were used to evaluate the anti-cancer activity of the newly synthesised thiazole containing compounds 16a,b and 18a-j. IC50 of compounds 16a,b and 18a–j was determined using the MTT cell viability assay (; ). Compounds 16a,b and 18a–j showed good inhibitory activity against MCF-7 with IC50 = 0.73–36.0 μM compared to dasatinib (IC50 = 7.99 μM) and doxorubicin (IC50 = 3.10 μM) (MCF-7). Componnd 16a showed excellent inhibitory activity (IC50 = 0.73 μM) which was more potent than both standard drugs dasatinib (IC50 = 7.99 μM) and doxorubicin IC50 = 3.10 μM (MCF-7). On the other hand, the tested compounds 16a,b and 18a–j showed moderate inhibitory activity against A549 (small lung cancer cell line) with IC50 = 1.25–68.7 μM compared to dastinib with IC50 = 11.8 μM and doxorubicin IC50 = 2.42 μM (A549). Similarly compound 16a showed excelent inhibitory activity of IC50 = 1.64 μM more potent than both standard drugs dasatinib (IC50 = 11.8 μM and doxorubicin IC50 = 2.42 μM) (A549). Morever the effect of thiazole containing compounds 16a,b & 18a–j on normal cell line (F180) showed safety of such compounds against normal cell line. Further more the selectivity index of thiazole containing compound were calculated. The selectivity index (F180/MCF7) of compounds 16a, 16b, 18c, 18d and 18f were S.I = 33.15, 7.13, 18.72, 13.25 and 8.28 respectivly that higher than selectivity index of the reference drugs dastinib (4.03) and doxorubicin (3.02). The selectivity index (F180/A549) of compounds 16a, 16b, 18c, 18d and 18f were 14.75, 12.96, 4.16, 7.07 and 18.88 respectivly which were higher than standard drugs dastinib (S.I = 2.72) and doxorubicin (S.I = 3.88). (Supplementary data S1)
Figure 3. (a) 2D NOESY H1NMR of compounds 18a showing correlation between the pyrazole H-5 and olefinic proton of each geometrical isomer, (b) 2D NOESY H1NMR of compounds 18d showing correlation between the pyrazole H-5 and olefinic proton of each geometrical isomer, (c) 2D NOESY H1NMR of compounds 18g showing correlation between the pyrazole H-5 and olefinic proton of each geometrical isomer.


Table 4. In vitro MCF-7, A549, F180 inhibitory activity of thiazole containing derivatives 16a,b & 18a–j and reference drugs dasatinib & doxorubicin.
EGFR Kinase inhibitory activity
EGFR-WT (Catalog # 40321), EGFR(L858R) (Catalog # 40324), EGFR (T790M/L858R) (Catalog # 40322) Kinase Assay Kits were used to assess EGFR kinase inhibitory activity of compounds 16a, 18c, 18d and 18f. Five concentrations were used in this assay and dasatinib was used as a reference for the determination of IC50, in addition the selectivity indices were calculated for these derivatives in order to show their selectivity against single mutant EGFR (L858R) and double mutant EGFR (T790M/L858R) tyrosine kinases. The results showed that compounds 16a, 18c, 18d, and 18f induced improved inhibitory activity against EGFR with IC50 of 0.043, 0.226, 0.388, 0.19 μM respectively which was comparable to that of dasatinib (IC50 = 0.076 μM). Moreover, these promising derivatives (except 18c) showed excellent selectivity indices (2.45–6.12) compared to dasatinib (2.05) against single mutant EGFR (L858R). In addition, all these compounds (16a, 18c, 18d, and 18f) showed improved selectivity indices (1.38–4.43) compared to dasatinib (1.35) against double mutant EGFR (T790M/L858R) (). (Supplementary data S1)
Figure 4. IC50 of compounds 16a, b & 18a–j against of MCF-7, A549, F180 cell lines and reference drugs dasatinib & doxorubicin.

HER-2 inhibitory activity
HER-2 (Catalog # 40230) Kinase Assay Kit was used to assess HER-2 kinase inhibitory activity for compounds 16a, 18c, 18d and 18f. Five concentrations were used in this assay and dasatainib was used as a reference for the determination of IC50. The results showed that compounds 16a, 18c, 18d, and 18f induced improved inhibitory activity against HER-2 with IC50 of 0.032, 0.144, 0.195, 0.201 μM respectively which was comparable to that of dasatinib (IC50 = 0.041 μM) (). (Supplementary data S1).
Figure 5. IC50 values of compounds 16a, 18c, 18d and 18f compared to dasatinib on EGFR-WT, EGFR (L858R), EGFR (T790M/L858R).

Structure activity relationship (SAR) of in vitro anti-inflammatory and cytotoxic activities:
The relationship between the chemical structure of the new target compounds 16a,b &18a–i and their COX-2 inhibitory activity and cytotoxicity/selectivity was outlined in () in which the thiazolidinone derivatives 16a,b were more potent COX-2 inhibitors than the substituted phenyl-thiazole derivatives 18a–j. Among the substituted phenylthiazole derivatives 18a–j, compound 18f (R = isobutyl, R1 = NO2) showed the highest COX-2 inhibitory activity and selectivity.
Figure 6. IC50 values of compounds 16a, 18c, 18d, and 18f compared to that of dasatinib on HER-2.

As cytotoxic agents against MCF-7 and A549 cancer cell lines, the thiazolidinone derivative 16a (R = CH3) revealed the highest cytotoxic activity with excellent safety. Replacement of methyl group in compound 16a by isobutyl moiety in compound 16b resulted in a drop in cytotoxic activity and selectivity.
Expansion of the structure by replacement of the thiazolidinone ring by substituted phenyl-thiazole derivatives affected the cytotoxic activity as follow: (i) high cytotoxicity and selectivity against MCF7 cell line was associated with the substitution of the pyrazolo-phenyl ring by a methoxy group (R = OCH3) in compounds 18c,d. (ii) enhanced cytotoxicity and selectivity against the A549 cell line was detected when the thiazole-phenyl ring was substituted with a nitro group (R1 = NO2) in compounds 18d,f. (iii) reduction in cytotoxic activity against both cell line was observed when the pyrazolo-phenyl ring was substituted with either methyl, nitro or bromo groups (R = CH3, NO2, or Br) in compounds 18a, 18b, 18g–j.
Cell cycle analysis
Cell cycle analysis of compounds 18d was tested against breast cell line MCF-7 at their IC50 concentrations, cell cycle phases alterations were noticed upon compound 18d addition on breast MCF-7 cells (). It was noted that the percentage of cells upon adding compounds 18d at G0/G1 was decreased to 62.43% respectively, in comparison to the control which showed 59.61%. On the other hand, when treating breast cells MCF-7 with compounds 18d a dramatic increase in cells at the G2/M phase was occurred to (4.39%) than the control (13.11%). According to the displayed results, it can be concluded that compound 18d showed cell cycle arrest at the G2/M phase.
Figure 7. SAR of in vitro anti-inflammatory and cytotoxic activities of compounds 16a,b, and 18a–j.

Determination of apoptosis using annexin-V
Treating MCF-7 breast cells with compound 18d resulted in an increment of annexin-V positive apoptotic cells at the early and late apoptotic stages with 12.58% (early) and 27.45 (late) respectively, compared to control 0.55% (early) and 0.16% (late) respectively. Moreover, compounds 18d induced a percentage of necrosis 6.52 which were considerably higher than the control that showed only 1.76% necrosis ().
Figure 8. Compounds 18d effect on DNA-ploidy flow cytometric analysis of breast MCF-7 cells compared to negative control.

Molecular docking study
Molecular docking studies are used to predict protein-ligand interactions with different enzymes at the atomic level. In this study, molecular docking studies of thiazole containing derivatives 16a–b & 18a–j was done with different enzymes downloaded from protein data bank (PDB) such as COX-2 enzyme (PDB, ID: 3LN1), EGFR enzyme (PDB ID: 1M17), HER-2 enzyme (PDB, ID:3PP0).
Docking of thiazole containing derivatives with COX-2 enzyme
The target thiazole derivatives 16a–b and 18a–j were docked with COX-2 enzyme (PDB, ID; 3LN1). The native ligand celecoxib showed an energy score −4.9 Kcal/Mol and the selected compounds 16a,b, 18c, 18d and 18f showed energy score of −3.7, −3.6, −4.6, −3.9 and −3.5 Kcal/Mol respectively. The higher selectivity index of such compounds may attributed to the presence of thiazole or azo group or SO2CH3 moieties which are hydrogen bond acceptor and methane sulphonyl group which is COX-2 selective moiety in addition these derivatives contain NO2 and OCH3 moieties which can form hydrogen bond with amino acids of the target protein (Table 5 of see supplementary data S2, ).
Figure 9. The percentage of Annexin-V-FITC-positive staining in breast MCF-7 cells when treated with compounds 18d compared to negative control.

Docking of thiazole derivatives with EGFR enzyme
Thiazoles 16a–b and 18a–j and thiazole containing drug dasatinib were docked with EGFR enzyme (PDB, ID: 1M17). The thiazole containing drug dasatinib showed an energy score −4.5 Kcal/Mol and the selected compounds 16a, 16b, 18c, 18d and 18f showed energy score of −4.2, −4.3, −4.5, −4.1 and −4.3 Kcal/Mol respectively. The higher inhibitory activity of compounds 16a, 16b, 18c, 18d and 18f to EGFR enzyme may attribute to the presence thiazole moiety available for formation of hydrogen bond. They formed three or four H-bond interactions commonly with different amino acids (Lys721, Met742, Thr766) amino acids residues through SO2CH3 or thiazole ring or N = N moiety (Table 6 of see supplementary data S2, ).
Figure 10. (A) Visual representation (2D) of celecoxib docked with 3LN1 active site (B) Visual representation (2D) of compound 16a docked with 3LN1active site, (C) Visual representation (2D) of compound 16b docked with 3LN1 active site (D) Visual representation (2D) of compound 18c docked with 3LN1 active site (E) Visual representation (2D) of compound 18f docked with 3LN1 active site (F) Visual representation (2D) of compound 18d docked with 3LN1 active site.

Docking of thiazol containing derivatives with HER-2 enzyme
The synthesised thiazole containing derivatives 16a–b and 18a–j and thiazole containing drug dasatinib were docked with HER-2 enzyme (PDB, ID: 3PP0). The dasatinib showed an energy score −3. 8 Kcal/Mol and the selected compounds 16a, 16b, 18c, 18d and 18f showed energy score of −3.8, −3.7, −4.6, −3.6 and −3.9 Kcal/Mol respectively. They formed H bond interactions commonly with different amino acids (Gly729, Asp863, Thr798) amino acids residues through SO2CH3 or N = N or thiazole moieties (Table 7 of see supplementary data S2, ).
Figure 11. (A) Visual representation (2D) of compound 16a docked with 1M17 active site, (B) Visual representation (2D) of compound 16b docked with 1M17 active site (C) Visual representation (2D) of compound 18c docked with 1M17 active site. (D) Visual representation (2D) of compound 18f docked with 1M17 active site (E) Visual representation (2D) of compound 18d docked with 1M17 active site (F) Visual representation (2D) of dasatinib docked with 1M17 active site.

SwissADME studies
In drug development, bioavailability is a factor that must be considered in clinical applications. It is affected by the different physicochemical properties of compoundsCitation72.
The most active compounds 16a, 16b, 18c, 18d and 18f and the standard dasatinib were subjected to view physicochemical properties and drug likeness using the SwissADME site free web tool, developed by the Molecular Modelling Group of the Swiss Institute of BioinformaticsCitation72. ()
Figure 12. (A) Visual representation (2D) of 16a docked with HER-2 active site (3PP0) (B) Visual representation (2D) of compound 16b docked with HER-2 active site (3PP0), (C) Visual representation (2D) of compound 18c docked with HER-2 active site (3PP0), (D)Visual representation (2D) of compound 18d docked with HER-2 active site (3PP0),(E) Visual representation (2D) of compound 18f docked with HER-2 active site (3PP0) (E) Visual representation (2D) of compound dasatinib docked with HER-2 active site (3PP0).

In particular, the compliance of compounds to traditional Lipinski’s “rule of five” was calculated to determine if the compound can be orally active in humanCitation73. This simple rule states that orally active drug has no more than one violation of the following criteria: molecular weight (MW) less than 500 Da; no more than 10 hydrogen bond acceptors (HBA); no more than five hydrogen bond donors (HBD); and calculated octanol-water partition coefficient (ilogP) not greater than 5Citation73. Also, topological polar surface area (TPSA) and the number of rotatable bonds are other critical properties that have been linked to drug bioavailabilityCitation74. The reports suggested that compounds with a TPSA of more than 140 Å2 and more than ten rotatable bonds are thought to have low pharmacological flexibility and permeability, respectivelyCitation75.
The tested thiazolidinone derivatives 16a&b have M.wt. 453.54 g/mol and 491.02 g/mol, in order (lower than 500 g/mol). While the examined phenylthiazoles 18c,d,f have M.wt. range (574.63 − 608.53 g/mol) compared to the standard dasatinib (M.wt = 491.97)
All of the tested compounds have a number of hydrogen bond donor atoms of 1–2 and hydrogen bond acceptor atoms of 6–8 compared to 3 hydrogen bond donors and 7 hydrogen bond acceptors in the standard dasatinib. The number of rotatable bonds of the tested compounds ranged from 5–9 bonds, while dasatinib contains 8 bonds ().
Table 8. Physicochemical parameters of compounds 16a, 16b, 18c, 18d, 18f and dsastinib.
The molar refractivity (MR) value should be between 40 and 130 for good absorption and oral bioavailability. Acceptable molar refractivity values, in combination with the number of rotatable bonds, indicate that substances have adequate intestinal absorption and oral bioavailabilityCitation75. Only compound 16a showed accepted MR = 126.21 m3/mol, but the remaining designed compound’s MR values ranging from 140.63 to 167.57 m3/mol, compared to the standard dasatinib (MR = 133.62 m3/mol). This indicates that most of the proposed compounds (except compound 16a and the standard dasatinib) may have low gastrointestinal absorption and oral bioavailability (). Also, the proposed derivatives’ 16a,b and 18c TPSA values were found 139.46, 139.46, 138.32, respectively compared to TPSA value of dasatinib 134.75. However, compounds 18d and 18f showed TPSA values higher than the standard range 184.14 and 174.91, in order.
Table 9. Pharmacokinetics of compounds 16a, 16b, 18c, 18d, 18f and dsastinib.
It is worth mentioning that compounds with TPSA values lower than 60 Å2 can penetrate the BBB easilyCitation75. That’s why all the proposed derivatives and the standard dasatinib cannot penetrate the blood-brain barrier well, as evidenced by the BBB determination ().
In addition, the implicit log P (IlogP) n-octanol/water partition coefficients was known to play a critical role in medication absorption in the mouth, as well as facilitating drug interactions with their biological targetsCitation73. The estimated values of IlogP () were found to be less than five (3.15–4.47), as recommended by Lipinski’s rule of five and compared to dasatinib (3.15).
Table 10. Lipophilicity and water solubility of compounds 16a, 16b, 18c, 18d, 18f and dsastinib.
Moreover, the estimated aqueous solubility of the tested compounds Log S (ESOL) ranged from −4.84 to −7.03 compared to −4.84 of dasatinb, indicating their low aqueous solubility (soluble compounds have Log S < −4) ().
Furthermore, studying the inhibitory activity of proposed derivatives against a certain CYP isoform becomes a critical factorCitation75. shows the results of the inhibitory prediction for three CYP isoforms (CYP1A2, CYP2C9, and CYP2C19). While all of the target compounds and dasatinib were found not to inhibit CYP1A2. The thiazolidinone derivatives 16a,b and the standard dasatinib were anticipated to inhibit CYP2C19 and CYP2C9 enzymes which means they are lower exposed to inhibitory drug metabolism.
In conclusion, the thiazolidinone compounds 16a and 16b are the same as dasatinib in applying with Lipinski rule (having 0 violations) and having good bioavailability score 0.55, so they may be a drug candidate (). The phenylthiazole derivative 18c showed 1 violation with a good bioavailability score 0.55. While compounds 18d&f indicated 3 violations of lipiniski’s rule and a low bioavailability score 0.17 indicating their low oral-drug likeness.
Table 11. Drug likeness of compounds 16a, 16b, 18c, 18d, 18f and dsastinib.
Conclusion
New pyrazolyl thiazole derivatives 16a–b and 18a–j were synthesised, merging the scaffolds of both celecoxib and dasatinib to be of expected anti-inflammatory and anti-breast cancer activities and was tested for anti-inflammatory, anti-proliferative activities, cell cycle arrest (). Where derivatives 16a, 16b and 18f were of the highest COX-2 selectivity indexes in 134.6, 26.08 and 42.13 respectively compared to celecoxib S.I. = 24.09). In addition, compounds 16a, 16b, 18c, 18d and 18f were found to inhibit MCF-7 with IC50 = 0.73–6.25 μM compared to dasatinib (IC50 = 7.99 μM) and doxorubicin (IC50 = 3.1 μM). On the other hand, derivatives 16a, 16b, 18c, 18d and 18f showed good inhibitory activity against A549 (small lung cancer cell line) with IC50 = 1.64 − 14.3 μM compared to to dasatinib with IC50 = 11.8 μM and doxorubicin IC50 = 2.42 μM (A549). The selectivity index (F180/MCF7) of compounds 16a, 16b, 18c, 18d and 18f were 33.15, 7.13, 18.72, 13.25 and 8.28 respectivly were higher than with reference drugs dasatinib (4.03) and doxorubicin (3.02) with selectivity index (F180/A549). The selectivity index (A549) of compounds 16a, 16b, 18c, 18d and 18f were 14.75, 12.96, 4.16, 7.07 and 18.88 respectivly that were higher than standard drugs dasatinib (S.I = 2.72) and doxorubicin (S.I = 3.88) and the physicochemical and pharmacokinetic properties of the most active compounds 16a, 16b, 18c, 18d and 18f were predicted by using the Swiss Adme tool. Compound 16a showed excellent inhibitory activity of IC50 = 0.73 μM more potent than both standred drugs to dasatinib (IC50 = 7.99 μM) and doxorubicin IC50 = 3.10 μM (MCF-7) and excellent inhibitory activity of IC50 = 1.64 μM more potent than both standred drugs dasatinib (IC50 = 11.8 μM and doxorubicin IC50 = 2.42 μM) (A549). Moreover, the most active compound 16a has good water solubility apply with Lipinski rule and have 0 violations and that qualifies it in the future to be a drug candidate.
Figure 14. Structure activity relationship of synthesised compounds 16a, b and 18a–j.

Figure 13. The physicochemical diagram for the oral bioavailiability of compounds 16a, 16b, 18c, 18d, 18f and dasatinib.

Supplemental Material
Download Zip (4.8 MB)Acknowledgements
Prof. Dr. Wael A. A. Fadaly, Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Beni-Suef University would like to thank Dr. Ahmed Sami, Faculty of Graduate Studies for Advanced Sciences, Beni-Suef University for providing HPLC analysis for all final compounds 16a,b &18a–j.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B, Yeger H. Combination therapy in combating cancer systematic review: combination therapy in combating cancer background. Oncotarget. 2017;8(23):38022–38043.
- Yap TA, Omlin A, De Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol. 2013;31(12):1592–1605.
- Albain KS, Nag SM, Calderillo-Ruiz G, Jordaan JP, Llombart AC, Pluzanska A, Rolski J, Melemed AS, Reyes-Vidal JM, Sekhon JS, et al. Gemcitabine plus paclitaxel versus paclitaxel monotherapy in patients with metastatic breast cancer and prior anthracycline treatment. J Clin Oncol. 2008;26(24):3950–3957.
- Li X, Li X, Liu F, Li S, Shi D. Rational multitargeted drug design strategy from the perspective of a medicinal chemist. J Med Chem. 2021;64(15):10581–10605.
- Zhang W, Pei J, Lai L. Computational multitarget drug design. J Chem Inf Model. 2017;57(3):403–412.
- Lamtha T, Krobthong S, Yingchutrakul Y, Samutrtai P, Gerner C, Tabtimmai L, Choowongkomon K. A novel nanobody as therapeutics target for EGFR-positive colorectal cancer therapy: exploring the effects of the nanobody on SW480 cells using proteomics approach. Proteome Sci. 2022;20(1):9.
- Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181.
- Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313(5998):144–147.
- Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol. 2008;21 Suppl 2:S16–S22.
- Yin HL, Janmey PA. Phosphoinositide regulation of the actin cytoskeleton. Annu Rev Physiol. 2003;65(1):761–789.
- Kamath S, Buolamwini JK. Targeting EGFR and HER-2 receptor tyrosine kinases for cancer drug discovery and development. Med Res Rev. 2006;26(5):569–594.
- Ali A, Al-Jandan B, Suresh C. The importance of ctokeratins in the early detection of oral squamous cell carcinoma. J Oral Maxillofac Pathol. 2018;22(3):441.
- Raghavendra NM, Pingili D, Kadasi S, Mettu A, Prasad SVUM. Dual or multi-targeting inhibitors: the next generation anticancer agents. Eur J Med Chem. 2018;143:1277–1300.
- Hubbard SR, Mohammadi M, Schlessinger J. Autoregulatory mechanisms in protein-tyrosine kinases. J Biol Chem. 1998;273(20):11987–11990.
- Butti R, Das S, Gunasekaran VP, Yadav AS, Kumar D, Kundu GC. Receptor tyrosine kinases (RTKs) in breast cancer: signaling, therapeutic implications and challenges. Mol Cancer. 2018;17(1):34.
- Naishima NL, Faizan S, Raju RM, et al. Design, synthesis, analysis, evaluation of cytotoxicity against MCF-7 breast cancer cells, 3D QSAR studies and EGFR, HER2 inhibition studies on Novel Biginelli 1,4-dihydropyrimidines. J Mol Struct. 2023;1277:1–23.
- Mitri Z, Constantine T, O'Regan R. The HER2 receptor in breast cancer: pathophysiology, clinical use, and new advances in therapy. Chemother Res Pract. 2012;2012:743193–743197.
- Mohmmed EA, Ramadan SS, EL-Saiid AS, Shousha WG. Frequency and clinical features of over-expressed her2 in Egyptian breast cancer women patients. Egypt J Hosp Med. 2021;85(1):3431–3435.
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Williams CB, Phelps-Polirer K, Dingle IP, Williams CJ, Rhett MJ, Eblen ST, Armeson K, Hill EG, Yeh ES. HUNK phosphorylates EGFR to regulate breast cancer metastasis. Oncogene. 2020;39(5):1112–1124.
- Chang JW-C, Huang C-Y, Fang Y-F, Chang C-F, Yang C-T, Kuo C-HS, Hsu P-C, Wu C-E. Epidermal growth factor receptor tyrosine kinase inhibitors for non-small cell lung cancer harboring uncommon EGFR mutations: real-world data from Taiwan. Thorac Cancer. 2023;14(1):12–23.
- Zhang L, Deng X-S, Meng G-P, Zhang C, Liu C-C, Chen G-Z, Jiang X-L, Zhao Q-C, Hu C. Design, synthesis and biological evaluation of a novel series of indole-3- carboxamide derivatives for cancer treatment as EGFR inhibitors. LDDD. 2018;15(1):70–83.
- Djukic M, Fesatidou M, Xenikakis I, Geronikaki A, Angelova VT, Savic V, Pasic M, Krilovic B, Djukic D, Gobeljic B, et al. In vitro antioxidant activity of thiazolidinone derivatives of 1,3-thiazole and 1,3,4-thiadiazole. Chem Biol Interact. 2018;286:119–131.
- Ayati A, Emami S, Moghimi S, Foroumadi A. Thiazole in the targeted anticancer drug discovery. Future Med Chem. 2019;11(15):1929–1952.
- Gümüş M, Yakan M, Koca I. Recent advances of thiazole hybrids in biological applications. Future Med Chem. 2019;11(15):1979–1998.
- Sever B, Altıntop MD, Radwan MO, et al. Design, synthesis and biological evaluation of a new series of thiazolyl-pyrazolines as dual EGFR and HER2 inhibitors. Eur J Med Chem. 2019;182:1–17.
- Keating GM. Dasatinib: a review in chronic myeloid leukaemia and Ph + acute lymphoblastic leukaemia. Drugs. 2017;77(1):85–96.
- Puszkiel A, Noé G, Bellesoeur A, Kramkimel N, Paludetto M-N, Thomas-Schoemann A, Vidal M, Goldwasser F, Chatelut E, Blanchet B, et al. Clinical pharmacokinetics and pharmacodynamics of dabrafenib. Clin Pharmacokinet. 2019;58(4):451–467.
- Abdelsalam EA, Abd El-Hafeez AA, Eldehna WM, El Hassab MA, Marzouk HMM, Elaasser MM, Abou Taleb NA, Amin KM, Abdel-Aziz HA, Ghosh P, et al. Discovery of novel thiazolyl-pyrazolines as dual EGFR and VEGFR-2 inhibitors endowed with in vitro antitumor activity towards non-small lung cancer. J Enzyme Inhib Med Chem. 2022;37(1):2265–2282.
- Zhang Y, Zhang W, Hou J, Wang X, Zheng H, Xiong W, Yuan J. Combined effect of tris(2-chloroethyl)phosphate and benzo (a) pyrene on the release of IL-6 and IL-8 from HepG2 cells via the EGFR-ERK1/2 signaling pathway. RSC Adv. 2017;7(85):54281–54290.
- Xu X, Steere RR, Fedorchuk CA, Pang J, Lee J-Y, Lim JH, Xu H, Pan ZK, Maggirwar SB, Li J-D, et al. Activation of epidermal growth factor receptor is required for NTHi-induced NF-κB-dependent inflammation. PLOS One. 2011;6(11):e28216.
- Kalinowski A, Galen BT, Ueki IF, Sun Y, Mulenos A, Osafo-Addo A, Clark B, Joerns J, Liu W, Nadel JA, et al. Respiratory syncytial virus activates epidermal growth factor receptor to suppress interferon regulatory factor 1-dependent interferon-lambda and antiviral defense in airway epithelium. Mucosal Immunol. 2018;11(3):958–967.
- Huang B-R, Chen T-S, Bau D-T, Chuang I-C, Tsai C-F, Chang P-C, Lu D-Y. EGFR is a pivotal regulator of thrombin-mediated inflammation in primary human nucleus pulposus culture. Sci Rep. 2017;7(1):8578.
- Lv P-C, Li D-D, Li Q-S, Lu X, Xiao Z-P, Zhu H-L. Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives as EGFR TK inhibitors and potential anticancer agents. Bioorg Med Chem Lett. 2011;21(18):5374–5377.
- Wang H-H, Qiu K-M, Cui H-E, Yang Y-S, Xing M, Qiu X-Y, Bai L-F, Zhu H-L. Yin-Luo. Synthesis, molecular docking and evaluation of thiazolyl-pyrazoline derivatives containing benzodioxole as potential anticancer agents. Bioorg Med Chem. 2013;21(2):448–455.
- Al-Salem HS, Arifuzzaman M, Issa IS, Rahman AM. Isatin-hydrazones with multiple Receptor Tyrosine Kinases (RTKs) inhibitory activity and in-silico binding mechanism. Appl Sci. 2021;11:3746.
- Aiebchun T, Mahalapbutr P, Auepattanapong A, Khaikate O, Seetaha S, Tabtimmai L, Kuhakarn C, Choowongkomon K, Rungrotmongkol T. Identification of vinyl sulfone derivatives as EGFR tyrosine kinase inhibitor: in vitro and in silico studies. Molecules. 2021;26(8):2211.
- Abbas HAS, Abd El-Karim SS. Design, synthesis and anticervical cancer activity of new benzofuran–pyrazol-hydrazono- thiazolidin-4-one hybrids as potential EGFR inhibitors and apoptosis inducing agents. Bioorg Chem. 2019;89:103035.
- Abd El-Karim SS, Syam YM, El Kerdawy AM, Abdelghany TM. New thiazol-hydrazono-coumarin hybrids targeting human cervical cancer cells: synthesis, CDK2 inhibition, QSAR and molecular docking studies. Bioorg Chem. 2019;86:80–96.
- Thakral S, Saini D, Kumar A, Jain N, Jain S. A synthetic approach and molecular docking study of hybrids of quinazolin-4-ones and thiazolidin-4-ones as anticancer agents. Med Chem Res. 2017;26(8):1595–1604.
- Bhat M, Poojary B, Kalal BS, Gurubasavaraja Swamy PM, Kabilan S, Kumar V, Shruthi N, Alias Anand SA, Pai VR. Synthesis and evaluation of thiazolidinone-pyrazole conjugates as anticancer and antimicrobial agents. Future Med Chem. 2018;10(9):1017–1036.
- Lv P-C, Zhou C-F, Chen J, Liu P-G, Wang K-R, Mao W-J, Li H-Q, Yang Y, Xiong J, Zhu H-L, et al. Design, synthesis and biological evaluation of thiazolidinone derivatives as potential EGFR and HER-2 kinase inhibitors. Bioorg Med Chem. 2010;18(1):314–319.
- Fakhry MM, Mahmoud K, Nafie MS, Noor AO, Hareeri RH, Salama I, Kishk SM. Rational design, synthesis and biological evaluation of novel pyrazoline-based antiproliferative agents in MCF-7 cancer cells. Pharmaceuticals. 2022;15(10):1245–1273.
- Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, Golenbock D, Gresnigt MS, Heneka MT, Hoffman HM, et al. A guiding map for inflammation. Nat Immunol. 2017;18(8):826–831.
- Harris RE. Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology. 2009;17(2):55–67.
- Koki AT, Masferrer JL. Celecoxib: a specific COX-2 inhibitor with anticancer properties. Cancer Control. 2002;9(2 Suppl):28–35.
- Marchesi VT. From molecular analysis to medical practice: recent studies reveal new therapeutic targets for an old medicine. FASEB J. 1998;12(12):1061–1061.
- Abdellatif KRA, Abdelgawad MA, Labib MB, et al. Synthesis and biological evaluation of new diarylpyrazole and triarylimidazoline derivatives as selective COX-2 inhibitors. Arch Pharm. 2017;350:1–10.
- Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase-2 in cancer: a review. J Cell Physiol. 2019;234(5):5683–5699.
- Liu B, Qu L, Yan S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015;15(1):2–7.
- Johnson AJ, Hsu A-L, Lin H-P, Song X, Chen C-S. The cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2+-ATPases: A plausible link with its anti-tumour effect and cardiovascular risks. Biochem J. 2002;366(Pt 3):831–837.
- Harris RE, Casto BC, Harris ZM. Cyclooxygenase-2 and the inflammogenesis of breast cancer. WJCO. 2014;5(4):677–692.
- Abdellatif KRA, Abdelall EKA, Labib MB, Fadaly WAA, Zidan TH. Design, synthesis of celecoxib-tolmetin drug hybrids as selective and potent COX-2 inhibitors. Bioorg Chem. 2019;90:103029.
- Abdellatif KRA, Abdelgawad MA, Labib MB, Zidan TH. Synthesis, cyclooxygenase inhibition, anti-inflammatory evaluation and ulcerogenic liability of novel triarylpyrazoline derivatives as selective COX-2 inhibitors. Bioorg Med Chem Lett. 2015;25(24):5787–5791.
- Sharma JN, Jawad NM. Adverse effects of COX-2 inhibitors. ScientificWorldJournal. 2005;5:629–645.
- Asghar W, Jamali F. The effect of COX-2-selective meloxicam on the myocardial, vascular and renal risks: a systematic review. Inflammopharmacology. 2015;23(1):1–16.
- Fu S-L, Wu Y-L, Zhang Y-P, Qiao M-M, Chen Y. Anti-cancer effects of COX-2 inhibitors and their correlation with angiogenesis and invasion in gastric cancer. World J Gastroenterol. 2004;10(13):1971–1974.
- Abuo-Rahma GE-DAA, Abdel-Aziz M, Beshr EAM, Ali TFS. 1,2,4-Triazole/oxime hybrids as new strategy for nitric oxide donors: synthesis, anti-inflammatory, ulceroginicity and antiproliferative activities. Eur J Med Chem. 2014;71:185–198.
- Abdelhaleem EF, Kassab AE, El-Nassan HB, Khalil OM. Design, synthesis, and biological evaluation of new celecoxib analogs as apoptosis inducers and cyclooxygenase-2 inhibitors. Arch Pharm. 2022;355(11):e2200190.
- Jeong H-S, Choi HY, Lee E-R, Kim J-H, Jeon K, Lee H-J, Cho S-G. Involvement of caspase-9 in autophagy-mediated cell survival pathway. Biochim Biophys Acta – Mol Cell Res. 2011;1813(1):80–90.
- Abuo-Rahma GE-DAA, Abdel-Aziz M, Mourad MAE, Farag HH. Synthesis, anti-inflammatory activity and ulcerogenic liability of novel nitric oxide donating/chalcone hybrids. Bioorg Med Chem. 2012;20(1):195–206.
- Abdellatif KRA, Fadaly WAA, Elshaier YAMM, Ali WAM, Kamel GM. Non-acidic 1,3,4-trisubstituted-pyrazole derivatives as lonazolac analogs with promising COX-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg Chem. 2018;77:568–578.
- Abdellatif KRA, Fadaly WAA, Kamel GM, Elshaier YAMM, El-Magd MA. Design, synthesis, modeling studies and biological evaluation of thiazolidine derivatives containing pyrazole core as potential anti-diabetic PPAR-γ agonists and anti-inflammatory COX-2 selective inhibitors. Bioorg Chem. 2019;82:86–99.
- Nobuta T, Hirashima S-i, Tada N, Miura T, Itoh A. One-pot metal-free syntheses of acetophenones from styrenes through aerobic photo-oxidation and deiodination with iodine. Org Lett. 2011;13(10):2576–2579.
- Omar HA, Arafa E-SA, Salama SA, Arab HH, Wu C-H, Weng J-R. OSU-A9 inhibits angiogenesis in human umbilical vein endothelial cells via disrupting Akt-NF-κB and MAPK signaling pathways. Toxicol Appl Pharmacol. 2013;272(3):616–624.
- Nemr MTM, AboulMagd AM, Hassan HM, Hamed AA, Hamed MIA, Elsaadi MT. Design, synthesis and mechanistic study of new benzenesulfonamide derivatives as anticancer and antimicrobial agentsviacarbonic anhydrase IX inhibition. RSC Adv. 2021;11(42):26241–26257.
- Nakamura JL. The epidermal growth factor receptor in malignant gliomas: Pathogenesis and therapeutic implications. Expert Opin Ther Targets. 2007;11(4):463–472.
- Cruz-López O, Ner M, Nerín-Fonz F, et al. Design, synthesis, HER2 inhibition and anticancer evaluation of new substituted 1,5-dihydro-4,1-benzoxazepines. J Enzyme Inhib Med Chem. 2021;36:1553–1563.
- Wang JL, Limburg D, Graneto MJ, Springer J, Hamper JRB, Liao S, Pawlitz JL, Kurumbail RG, Maziasz T, Talley JJ, et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: the second clinical candidate having a shorter and favorable human half-life. Bioorg Med Chem Lett. 2010;20(23):7159–7163.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–46272.
- Aertgeerts K, Skene R, Yano J, Sang B-C, Zou H, Snell G, Jennings A, Iwamoto K, Habuka N, Hirokawa A, et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J Biol Chem. 2011;286(21):18756–18765.
- Daina A, Michielin O, Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17.
- Abdellatif KRA, Abdelall EKA, Labib MB, Fadaly WAA, Zidan TH. Synthesis of novel halogenated triarylpyrazoles as selective COX-2 inhibitors: Anti-inflammatory activity, histopatholgical profile and in-silico studies. Bioorg Chem. 2020;105:104418.
- Ibrahim ZY, Uzairu A, Shallangwa GA, Abech SE. Pharmacokinetic predictions and docking studies of substituted aryl amine-based triazolopyrimidine designed inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH). Futur J Pharm Sci. 2021;7(1):113.