
Abstract
Anthraquinones have attracted considerable interest in the realm of cancer treatment owing to their potent anticancer properties. This study evaluates the potential of a series of new anthraquinone derivatives as anticancer agents for non-small-cell lung cancer (NSCLC). The compounds were subjected to a range of tests to assess their cytotoxic and apoptotic properties, ability to inhibit colony formation, pro-DNA damage functions, and capacity to inhibit the activity of tyrosine kinase proteins (PTKs). Based on the research findings, it has been discovered that most active derivatives (i84, i87, and i90) possess a substantial capability to impede the viability of NSCLC while having mostly a negligible effect on the human kidney cell line. Moreover, the anthraquinones displayed pro-apoptotic and genotoxic attributes while blocking the phosphorylation of multiple PTKs. Collectively, our findings indicate that these derivatives may demonstrate promising potential as effective anticancer agents for lung cancer treatment.
GRAPHICAL ABSTRACT
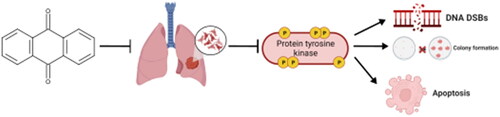
Introduction
Cancer is a significant health problem globally. It is a complex disease characterised by uncontrolled cell growth and proliferation, often resulting in the formation of malignant tumours. There are many types of cancer, each with unique characteristics and treatment challenges. This disease can affect people of all ages and demographics, and its incidence is expected to continue to rise due to factors such as ageing of populations and lifestyle factors such as smoking, poor diet, and lack of physical activity. One of the most common types of cancer worldwide and the leading cause of cancer-related deaths globally is lung cancer. There are two primary types of lung cancer: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), with NSCLC accounting for about 85% of all casesCitation1.
Anthraquinones are a class of naturally occurring organic compounds that have garnered significant attention in the field of cancer therapy due to their potent anticancer activity. These compounds are widely distributed in the plant, fungal, and lichen kingdoms and exhibit a diverse range of biological activities, including anti-inflammatory, antimicrobial, and antitumour propertiesCitation2,Citation3. Extensive research has been conducted to explore the anticancer potential of anthraquinones, and numerous studies have reported their ability to arrest cell cycle progression, induce apoptosis, and inhibit cell proliferation in various cancer cell linesCitation4–7. Mechanistically, anthraquinones have been shown to exert their anticancer effects via several mechanisms, including the inhibition of topoisomerase activity, the induction of oxidative stress, and the modulation of signalling pathways involved in cell survival and proliferationCitation8–10. Among the well-known anthraquinones, emodin (1,3,8-trihydroxy-6-methyl-anthraquinone), a strong inhibitor of the protein tyrosine kinase (PTK) p56lck, stands out for its potent anticancer activity against various cancer types, including breast, lung, prostate, and liver cancerCitation11,Citation12. Emodin induces apoptosis in cancer cells and exhibits anti-metastatic activity by suppressing the migration and invasion of cancer cellsCitation12,Citation13.
PTKs are a group of enzymes that play a crucial role in the regulation of several cellular processes, including cell growth, differentiation, and survivalCitation14. The dysregulation of PTKs has been implicated in the development and progression of various types of cancer. As a result, PTKs have emerged as an important target for the development of anticancer therapiesCitation15. For example, imatinib is a protein tyrosine kinase inhibitor (PTKI) approved for the treatment of chronic myeloid leukaemia and gastrointestinal stromal tumoursCitation16. Imatinib targets kinases such as c-kit and BCR-ABL, effectively killing cancer cells and inducing remissionCitation17. Another example of PTKI is dasatinib, which targets multiple PTKs, including BCR-ABL, SRC, and c-KITCitation18. Dasatinib has shown promising results in the treatment of several types of cancer, including chronic myeloid leukaemia, acute lymphoblastic leukaemia, and non-small cell lung cancerCitation19,Citation20. In addition to imatinib and dasatinib, numerous other PTKIs are currently undergoing development or clinical trials for the treatment of various types of cancer. This underscores the significance of compounds that act on this target in the field of cancer research and therapy.
This study aimed to assess the molecular pharmacology of 15 anthraquinone derivatives, () to determine their potential as anticancer agents against lung cancer cell lines. These compounds were previously obtained, purified, and physiochemically characterised; however, their molecular mechanism of action was unknownCitation21. Given the involvement of PTKs and the structural formulas of anthraquinones, we evaluated these compounds to determine their ability to inhibit phosphorylation of this enzyme class.
Table 1. Chemical structures of 9,10-dioxoanthracenyldithiocarbamates. 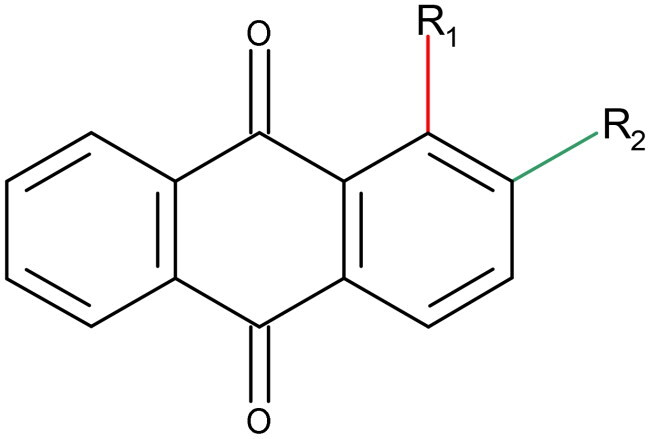
Results
Cytotoxic activity of anthraquinones
In this study, we assessed the antiproliferative activity of anthraquinones against human non-small cell lung cancer cell lines, namely A549, H226, and H460 and non-malignant human embryonic kidney cell line HEK293. Cisplatin and etoposide served as reference compounds. The cells were exposed to various concentrations of the compounds (0.5–100 µM) for 72 h and analysed using the MTT assay. The IC50 concentration that inhibits 50% growth of cells was used to express the results (). Most of the compounds showed no activity, having an IC50 value greater than 50 μM; however, select compounds, specifically i64, i68, i79, i82 and i89, displayed moderate activity, while i84, i87, and i90 showcased pronounced efficacy. Interestingly, compounds i84, i87, and i90 manifested exceptional cytotoxicity against the A549 cell line. Their IC50 values observed a pattern: H460 > H226 > A549, and they outperformed the benchmark compound, cisplatin. Notably, i84, i89, and i90 exhibited diminished impacts on the HEK293 cell line, hinting at their potential cancer cell specificity. Expanding our scope, we also evaluated the three most potent compounds against normal bronchial epithelial cells, NHBE2594. The results suggest that these compounds display comparable activity to that observed in A549 cells and demonstrate lower cytotoxicity than etoposide. These results demonstrate the potential utility of anthraquinones as effective cytotoxic agents for non-small cell lung cancer.
Table 2. The in vitro growth inhibitory activity of anthraquinones was assessed by determining their IC50 values (± SD, µM), representing the compound concentration required to inhibit 50% of cell growth. The values reported are the means of three independent experiments, with etoposide and cisplatin used as references.
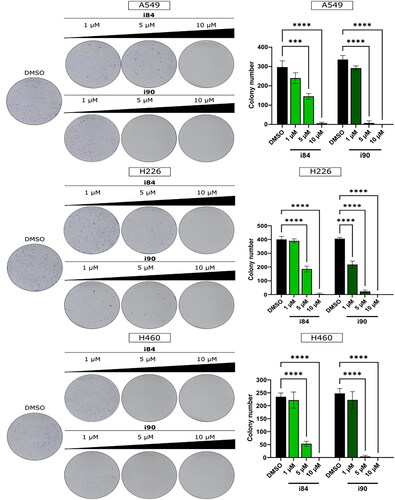
To assess the antiproliferative nature of compounds, the i84 and i90 were selected due to their lower toxicity levels. The colony formation assay was employed to test these compounds, and the results displayed a dose-dependent reduction in the number of colonies in all the tested cell lines, as shown in . This indicates that treatment with i84 and compounds effectively reduced the adhesion-independent proliferation of the tested cell lines.
Figure 1. The ability of NSCLC cells to form colonies is impeded by anthraquinones. A visual representation of the colony formation assay is displayed on the left, and the quantitative results are presented on the right. The data shown in the panels represent the mean ± SEM of three independent experiments. ***p < 0.0001 and ****p < 0.00001 (two-way ANOVA and post hoc Dunnett’s test).
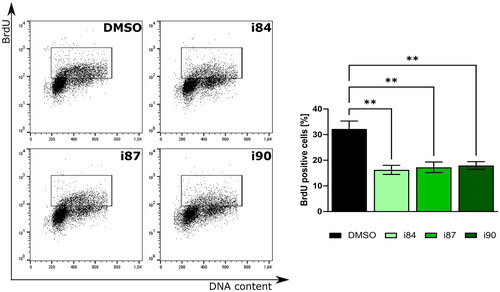
To further underscore the antiproliferative effects of anthraquinone derivatives, we performed the bromodeoxyuridine (BrdU) incorporation assay on the A549 cell line. reveals that upon treatment with equitoxic doses of these compounds, there was a notable 2-fold reduction in BrdU-positive cells. Collectively, these results underscore the promise of anthraquinone derivatives as potential inhibitors of in vitro cancer cell proliferation.
Figure 2. Analysis of BrdU incorporation after treatment of A549 cells with anthraquinones. DNA staining results are depicted in representative histograms with associated statistical evaluations. Graph bars present the quantification data, with error bars indicating the mean ± SD from three independent experiments (two-way ANOVA test).
Anthraquinones induce DNA damage and apoptosis
Various studies have reported a correlation between the cytotoxicity of anthraquinones and their potential to induce DNA damage, including DNA double-strand breaks (DSBs)Citation22,Citation23. One of the markers for DSBs is the phosphorylation of histone variant H2AX on Serine 139, resulting in the formation of nuclear fociCitation24. To determine the extent of DNA damage caused by the anthraquinone compounds, A549 cells were used, as compounds exhibit the highest cytotoxic activity towards this cell line. As shown in , all compounds caused a significant increase in DNA DSBs in NSCLC cells, as evidenced by the accumulation of γ-H2AX fraction (p < 0.01).
Figure 3. Anthraquinones induce DNA DSBs. (a) Representative high-resolution laser scanning confocal images showing the γ-H2AX after treatment A549 cell lines. For negative and positive controls, DMSO and etoposide (ETP) were utilised, respectively. Nuclei were counterstained with DAPI. Scale bars = 10 μm. (b) Representative histograms showing induction γ-H2AX in the A549 cell line at 24 h of treatment. (c) The quantification of the analysis is presented on a bar graph. **p < 0.001, ****p < 0.00001 (two-way ANOVA and post hoc Dunnett’s test).
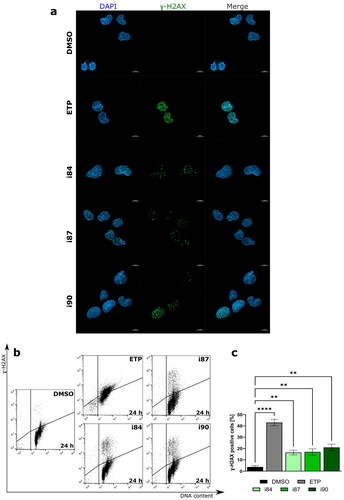
DNA damage triggers checkpoint mechanisms, which arrest the cell cycle to allow time for DNA repair. If the damage is too severe, the cells undergo apoptosis, which is the most common form of cell death. Therefore, the pro-apoptotic properties of anthraquinones were examined using flow cytometry (). A549 cells were treated with IC90 concentration of anthraquinone derivatives for 24 h and stained with 7-AAD and Annexin V-FITC, which has a high affinity to phosphatidylserine. Dual staining enabled the differentiation between live cells (Annexin V-FITC(-)/7-AAD(-)), early-phase apoptotic cells (Annexin V-FITC(+)/7-AAD(-)), late-phase apoptotic cells (Annexin V-FITC(+)/7-AAD(+)), and necrotic cells (Annexin V-FITC(-)/7-AAD(+)).
Figure 4. Flow cytometric analysis of A549 cell line after 24 h of treatment with anthraquinone compounds, using Annexin V-FITC/7-AAD. For negative and positive controls, DMSO and etoposide (ETP) were utilised, respectively. (a) Representative dot plots. (b) The quantitation of data is presented on a bar graph. **p < 0.001, ***p < 0.0001, and ****p < 0.00001 (two-way ANOVA and post hoc Dunnett’s test).
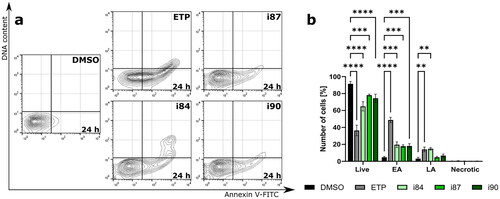
Treatment with anthraquinone derivatives resulted in a significant decline in the number of live cells, while the proportion of Annexin V-positive cells increased (). This indicates phosphatidylserine externalisation and ongoing apoptotic cell death. Compounds i84 and i90 exhibited the most pronounced pro-apoptotic activity among all compounds with a 4.3-fold and 3.1-fold increase in apoptosis, respectively, compared to the control. Moreover, all compounds showed a constant number of necrotic fractions under the same conditions (). The 7-AAD dye can only enter cells with compromised cell membrane integrity, indicating late-stage apoptosis or necrosis. These findings confirmed that the investigated anthraquinones trigger apoptotic rather than necrotic cell death in NSCLC cell lines.
Anthraquinones inhibit multiple tyrosine kinases’ phosphorylation
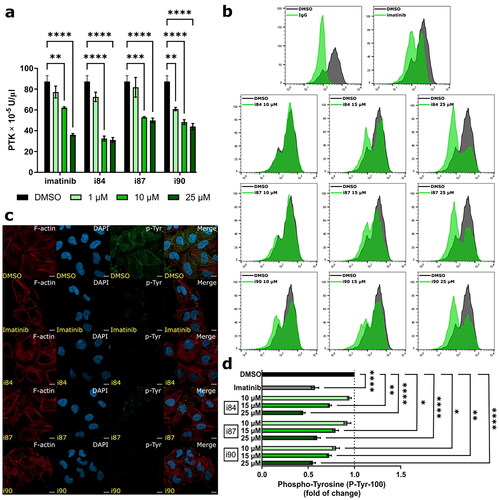
Numerous data have reported that anthraquinones can inhibit the phosphorylation of diverse protein tyrosine kinases (PTKs), such as emodin or chrysophanol (1,8-dihydroxy-3-methylanthrone)Citation25. Therefore, we investigated the inhibitory activity of compounds against PTKs using an enzyme-linked immunosorbent assay (ELISA), considering the role of PTKs and the structural formulas of anthraquinones. Imatinib, a selective PTK inhibitor used in medical treatment, was employed as a reference. As shown in , all tested compounds exhibited a concentration-dependent decrease in PTK activity. Compound i84 showed the most potent inhibitory activity against PTK and was more effective at a 1 μM concentration than imatinib. To further assess thehosphoy of tested compounds to suppress phosphorylation of PTK, high serum-supported A549 cells were treated with increasing concentrations of compounds and analysed by flow cytometry. Representative histograms and corresponding quantification are presented in . All the compounds decreased the phosphorylation of p-Tyr in a concentration-dependent manner. At a 25 μM concentration, i84 and i90 significantly reduced protein tyrosine phosphorylation, resulting in an almost 2-fold decrease in the p-Tyr expression compared to the vehicle. Moreover, both compounds exhibited higher activity than imatinib. Furthermore, A549 cells treated with compounds at 25 μM were analysed using confocal microscopy (). Within a 4-h treatment with i84 or i90, a remarkable decrease in p-Tyr staining was observed. I84 displayed similar activity to imatinib, whereas no apparent changes were observed for i87. These results are consistent with those obtained in the ELISA and flow cytometry tests.
Figure 5. Analysis of changes in phosphorylation of protein tyrosine kinases after compounds treatment. DMSO and imatinib were used as negative and positive controls, respectively. (a) In vitro determination of PTK activity using A549 extracts determined by ELISA. (b) Representative histograms of compounds-treated A549 cells after intracellular hosphor-flow cytometry. (c) Representative microscopy images presenting immunofluorescence of microtubule and p-Tyr foci in A549 cells at 4 h of treatment. The microtubules are depicted in red, p-Tyr in green, and the nucleus is stained with DAPI (blue). Scale bars = 10 μm. (d) Quantification of intracellular hosphor-flow cytometry. *p < 0.01, **p < 0.001, ***p < 0.0001, ****p < 0.00001 vs. vehicle (one-way ANOVA and post hoc Dunnett’s test).
The inhibitory activity of i84 and i90 against particular PTKs was investigated. A549 cells were treated with the tested compounds for 6 h and analysed using a human RTK phosphorylation Ab array, which can screen the expression of 71 different RTKs simultaneously (). The relative phosphorylation levels of all investigated target molecules were presented in .
Figure 6. Analysis of changes in phosphorylation of protein tyrosine kinases after compounds treatment. (a) Human PTK overview. Green and red points refer to non-receptor and receptor PTKs, respectively. (b) Proteome profiler analysis of A549 cells after treatment with compounds. DMSO and imatinib were used as negative and positive controls, respectively. (c) Template presenting the location of tyrosine kinase antibody. (d) Profiling changes in PTKs after treatment with anthraquinones or imatinib with respect to the vehicle. Non-receptor and receptor PTKs are presented on the left and right panels, respectively.
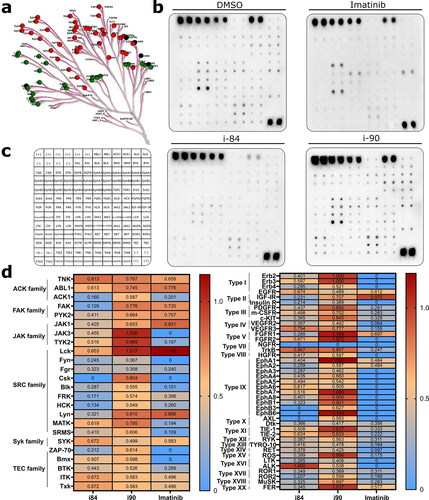
The study findings revealed that i90 exhibited partial inhibition of kinase phosphorylation, with thirteen protein tyrosine kinases appearing to be largely unaffected (). Notably, NGFR kinase was fully inhibited, while Fyn, Frg, and ROR1 kinases demonstrated a 2.8-fold decrease in activity compared to the control group, indicating significant inhibition by i90. On the other hand, i84 downregulated all investigated p-PTKs except ALK and exhibited higher potency towards PTKs than i90 (). However, i84 was less active than imatinib, indicating its potential selectivity.
It is noteworthy to mention that the A549 cell line exhibited higher basal expression of certain protein tyrosine kinases, indicating potential implications in cancer progression. Notably, non-receptor kinases from the ACK1, FAK, and SRC families, including FRK, Csk, and Lyn, as well as receptor kinases from types II (IGF-IR, Insulin-R), X (AXL, Dtk), XIV (EphA2), and XV (ROS) were found to be upregulatedCitation26. These findings suggest that these kinases may play crucial roles in cancer development and require further investigation. I84 significantly downregulated the expression of all those kinases, suggesting that inhibiting these kinases may have a significant impact on non-small cell lung cancer development and warrant further investigation (). Additionally, most of the SRC family kinases were significantly inhibited by i84, indicating that its may act more efficiently on these particular proteins (). Further research in this area is necessary and will be conducted in future studies.
Molecular docking studies
In addition to the abovementioned biological tests, a series of docking simulations were performed using the Autodock Vina software. The program utilised an empirical scoring function, reminiscent of the X-score function, to quantitatively assess the ligand-receptor binding affinity. Four ligands were subjected to this analysis: i87, Bosutinib, Dasatinib, and Imatinib. Notably, Bosutinib, Dasatinib, and Imatinib are established for their efficacy against PTK targets and were thus used as reference ligands in this study.
From the human RTK phosphorylation Ab array, compound i84 emerged as the most potent candidate, prompting its selection for docking studies. Kinases that exhibited the most inhibition in the test, and for which a PDB structure was available, were investigated. Specifically, we looked at those kinases with a petite cavity bordered by alpha helices on one side and beta sheets on the other.
Of all the simulations performed, we shortlisted the top 10 based on the scoring function. Following a visual assessment, the best results were chosen based on spatial arrangement and docking score (). The findings revealed that i84 had docking scores superior to those of Dasatinib and Bosutinib, but generally inferior to Imatinib. However, the scores across all ligands had a close range, with a maximum difference of 2.8 kcal/mol. These results suggest that i84 could potentially be a potent inhibitor of the investigated PTKs.
Table 3. Ligand–protein energies calculated with the Autodock Vina program.
Discussion
Anthraquinones have been recognised as cancer growth inhibitors targeting topoisomerases, telomerase, matrix metalloproteinases, and protein kinaseCitation2,Citation27. These molecular targets are critical for providing necessary cell survival signals. Several drugs based on an anthraquinone scaffold, such as doxorubicin, epirubicin, pixantrone, valrubicin, and mitoxantrone, are clinically employed for treating various types of cancersCitation23. To further explore the mechanism of anthraquinones in modulating cancer cell growth, the cellular and molecular effects of newly developed anthraquinones were studied in vitro. Results suggest that these newly formulated anthraquinones show promising potential in regulating cancer growth by directly targeting multiple signalling pathways.
Among the examined anthraquinone derivatives, compounds i84, i87 and i90 were found to be the most active, which effectively inhibited the growth of NSCLC cells and inhibited the formation of colonies in cell cultures. Moreover, our studies revealed that these compounds induced DNA DSBs, likely leading to apoptotic cell death. Anthraquinones are generally known to act as DNA intercalators, causing DNA single- and double-strand breaks, which can cause apoptosisCitation28. Interestingly, none of the analysed compounds possessed the ability to bind DNA (data not shown), indicating that their mechanism of action is distinct from that of the typical compounds from this chemical group. Notably, further studies revealed that the compounds tested had a strong inhibitory effect on phosphorylation of multiple PTKs, among which compound i84 was the most active according to our data.
It is worth noting that i84 has been shown to significantly decrease the activity of almost all the studied SRC kinases. SRC kinases are a family of non-receptor tyrosine kinases which play a role in signal transduction, cell growth and survival, and angiogenesis pathwaysCitation29. Dysregulation and increased activation of SRC kinases have been associated with the progression of several types of cancer, making them attractive targets for cancer treatment therapiesCitation30. Consequently, the development of small molecules as inhibitors for SRC kinases presents a viable strategy for targeting them. These inhibitors bind to the ATP-binding site of the kinase and effectively hinder its activity. Certain compounds, including anthraquinones like anthraquinone-2-carboxylic acid and flavonoids such as scutellarein, have demonstrated strong inhibitory effects against SRC kinasesCitation31,Citation32. By blocking SRC kinases, these substances effectively suppress the inflammatory response and impede the activation of NF-κB.
On the other hand, i84 also inhibited phosphorylation of several receptor tyrosine kinases (RTKs), such as IGF-IR, Insulin-R, NGFR, EphB3, EphB6 or AXL. Receptor kinases are transmembrane proteins that transmit signals from the extracellular environment into the cell, influencing various cellular processes including cell growth, survival, and proliferationCitation33. Over the past twenty years, various molecules targeting RTKs have been employed as primary or secondary therapeutic agents in numerous cancer typesCitation34. Despite the promising clinical benefits that approved kinase inhibitors have provided to an array of cancer patients, these drugs are unfortunately not curative, merely delaying tumour progression. Advanced tumours in particular are able to craft escape routes to sidestep inhibiting target proteins, leading to cytostatic drug resistanceCitation35. One potential way to combat multidrug resistance is to simultaneously target multiple kinases, as this can improve inhibiting cancer cell growth.
Targeting multiple PTKs has been shown to offer multiple benefits compared to targeting just one. The dysregulated activity of different PTKs in cancer cells renders them insensitive to treatments aimed at a single PTK. As such, targeting multiple PTKs provides increased chances of inducing cell death and preventing treatment resistanceCitation36. Furthermore, this approach may lead to a broader anticancer effect and improved treatment outcomes. This is because PTKs are often involved in interconnected signalling pathways, and simultaneous inhibition of multiple PTKs can disrupt these pathways at various points, resulting in stronger and more effective anticancer responsesCitation36. Thus, targeting multiple PTKs is likely to lead to improved anticancer activity and greater treatment efficacy.
Molecular docking has emerged as an instrumental tool in supporting biological investigations. In our studies, it was observed that compound i84 demonstrates a notable affinity towards various non-receptor and receptor kinases. This binding behaviour is reminiscent of established chemotherapeutic agents, underscoring the potential therapeutic value of i84. To gain a deeper understanding of the nuances and specificities of the binding interactions, we suggest that future research should leverage cutting-edge molecular modelling techniques. These techniques, especially thermodynamic analyses, can shed light on the stability and energetics of the binding process, offering insights beyond what traditional docking can provide.
Furthermore, while molecular docking provides an initial glimpse into potential binding interactions, a comprehensive understanding requires experimental validation. Notably, the RTK membrane assay, commonly employed in these studies, offers only semi-quantitative data. This limitation highlights the necessity for more definitive analytical techniques. We propose that subsequent investigations integrate enzymatic assays. Such assays would not only validate the binding events but also help pinpoint the exact concentrations required to inhibit specific kinases, thereby enhancing the precision and applicability of the findings.
Considering the extensive role of PTKs in the regulation of a variety of cellular functions, targeting these enzymes with compounds such as anthraquinones may provide promising compounds that can exert avenues for the development of novel therapeutic strategies for the treatment of cancer. Their anticancer activity through multiple mechanisms could prove advantageous in tackling cancer cells that are resistant to traditional chemotherapeutic treatment. Additionally, due to the unique properties of anthraquinones, it could be possible to design a more precise and effective strategy for the treatment of this devastating disease. Further investigation is required to explore the potential of anthraquinones as effective treatments for cancer.
Methods
Compounds
Compounds were synthesised accordingly to the protocol described earlierCitation21. Etoposide, cisplatin, and imatinib were purchased from Sigma-Aldrich.
Cell culture
A549, H226, H460, and HEK293 cell lines were obtained from the American Type Culture Collection. All cell lines were cultured in a controlled environment held at a temperature of 37 °C, with a 5% CO2 atmosphere. Furthermore, regular screenings for Mycoplasma contamination were diligently conducted. A549, H226, and H460 cells were cultured in the RPMI-1640 medium (Corning), while the HEK293 cell line was cultured in DMEM (Corning). Each medium was supplemented with 10% foetal bovine serum (Corning), 2 mM L-glutamine (Corning), and antibiotics (penicillin 62.6 μg/ml and streptomycin 40 μg/ml).
Cell viability
MTT (3,(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was employed to assess cell viability. The cells were first seeded into 96-well plates and exposed to the investigated compounds at varying concentrations (0 to 50 µM) for 72 h. Etoposide and cisplatin served as references. After the treatment period, the cells were incubated with the MTT solution (0.4 mg/ml PBSx1) for three hours at 37 °C. Then, the medium was removed, and formazan crystals were dissolved in 100 μl of DMSO. Subsequently, the absorbance was quantified at 540 nm using an ASYS UVM340 microplate reader (Biochrom Ltd.). The experiment was performed independently in triplicate to ensure the reliability and validity of the results.
Colony forming assay
To assess the clonogenic potential of A549, H226 and H460 cells were seeded at a density of 400 cells per well into 6-well plates. The cells were subsequently exposed to different concentrations of the investigated carbazole derivatives for 24 h. Following treatment, the cells were washed and cultured for 8 days. Methanol was employed to fix the cells, which were then subjected to 0.5% crystal violet staining. Visible colonies were counted using ImageJ software, and cell viability was determined concerning the control. Colony visualisation and counting were facilitated by ImageJ 1.53n software.
BrdU assay
To identify DNA synthesis, A549 cells were exposed to a solution of 20 μM BrdU (5-bromo-2′-deoxyuridine) from Sigma Aldrich for an hour before the treatment concluded. Subsequently, the cell samples were collected using a trypsin solution and then preserved in 75% ethanol at −20 °C for an extended period, often overnight. After rehydration with PBS for 10 min, the samples underwent denaturation using 2 M HCl for 45 min at room temperature. To neutralise the suspension, a 0.1 M sodium tetraborate solution with a pH of 8.5 was applied for 10 min at room temperature. Blocking was performed using 1% w/v bovine serum albumin (BSA) in PBS for 30 min at room temperature. Following the blocking step, the samples were subjected to a rat anti-BrdU antibody (diluted 1:100) from Abcam (#ab6326) and incubated for an hour at 37 °C. Subsequently, a goat anti-rat conjugated antibody (diluted 1:200) from Abcam (#ab150157) was applied and incubated for 30 min at 37 °C. For DNA staining, a solution containing 20 μg/μl of propidium iodide (PI) and 100 μg/μl of RnaseA in PBS was used for 20 min at room temperature.
DNA damage
In this study, DNA damage was assessed by subjecting treated cells to a multi-step protocol. First, the cells were harvested and fixed in 75% ethanol, followed by overnight storage at −20 °C or longer. Upon rehydration with PBS, the cells were permeabilized in 0.2% Triton X-100 in PBS for 15 min at room temperature. To detect DNA damage, the samples were blocked with 2% BSA in PBS and incubated with Alexa488-conjugated mouse anti-γH2AX (Ser139) antibody (dilution of 1:100, BioLegend, #613406). The DNA was subsequently stained with 7-AAD (Sigma Aldrich) and 100 μg/μl RnaseA in PBS for 20 min at room temperature. As a reference, etoposide was utilised.
Proapoptotic properties
After treatment with the investigated compounds and etoposide, A549 cells were harvested through trypsinization, rinsed twice with PBS, and subjected to Annexin V FITC conjugate staining (Thermo Fisher Scientific, #A13199) as per the manufacturer’s instructions.
Universal tyrosine kinase assay
The Universal Tyrosine Kinase Assay Kit (#MK410; TaKaRa) was utilised to evaluate the activity of PTK in the A549 cell extract, following the manufacturer’s instructions. Initially, A549 cells were collected with extraction buffer and centrifuged at 10000 g for 10 min at 4 °C. Each sample was mixed with 40 mM ATP-2Na solution and incubated for 30 min at 37 °C. Subsequently, the sample solution was removed, and the wells were washed four times with washing solution. The wells were then blocked with blocking solution for 30 min at 37 °C, and anti-phosphotyrosine (PY20)-HRP solution was added to each well, followed by incubation for 30 min at 37 °C. After washing the wells four times, HRP substrate solution was added, and the wells were incubated for 20 min at 37 °C. The reaction was stopped using a stop solution, and the absorbance at 450 nm was measured with an ASYS UVM340 microplate reader (Biochrom Ltd.). The activity of PTK was calculated using the PTK standard curve provided in the kit.
Phospho-flow cytometry assay
For hosphor-flow cytometry, cells were seeded onto tissue culture plates and allowed to adhere overnight. The complete medium was replaced with a medium containing 1% FBS for 12 h before stimulation. Subsequently, the cells were pre-treated with compounds for 45 min, washed, and treated with compounds in 20% FBS for an additional 75 min. Cells were then detached with 1 mM EDTA in PBS, washed 2–3 times with PBS, and collected by centrifugation at 1100 rpm for 5 min at 4 °C. Next, cells were fixed with 4% PFA in PBS for 15 min at 4 °C and centrifuged at 1100 rpm for 5 min at 4 °C. After fixation, cells were permeabilised by gradually adding 4.5 ml of ice-cold 100% methanol to the pre-cooled cells in 0.5 ml of PBS, with gentle vortexing, to achieve a final concentration of 90% methanol. The samples were then incubated for 45 min on ice. Cells were washed with incubation buffer (0.5% BSA in PBS), centrifuged at 1100 rpm for 5 min at 4 °C, and blocked with blocking buffer at room temperature for 30 min. Next, cells were incubated with anti-p-Tyr-100 antibody (1:1500 dilution; #9411; Cell Signalling) or IgG Isotype control (1 μg/mL dilution; ab37355; Abcam) antibodies at room temperature for 1 h, followed by incubation with anti-mouse IgG cross-adsorbed secondary antibody conjugated to DyLight 488 (1:200 dilution; #SA5-10166; Thermo Fisher Scientific) for 30 min at room temperature. Cells were then washed twice with PBS-T and stained with 7-AAD and Rnase A (50 μg/mL) in the dark at room temperature for 15 min. Finally, we analysed the samples using flow cytometry (Guava EasyCyte 8 cell sorter, Merck Millipore) and FlowJo software v10. Each experiment was repeated independently three times.
Immunofluorescence
The cells were seeded on coverslips and allowed to attach overnight. Following this, cells treated with drugs were washed with PBS, fixed using 4% paraformaldehyde (Sigma-Aldrich) in PBS for 10 min at room temperature, and permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) in PBS for 15 min. The cells were then washed twice with PBS, blocked using 3% BSA in PBS for 1 h at room temperature, and incubated with primary antibodies diluted in 3% BSA in PBS-T (PBS containing 0.1% (v/v) Tween-20 (Sigma-Aldrich)) for 1 h at 37 °C. The primary antibodies used were mouse anti-phospho-tyrosine (P-Tyr-100), (#9411, Cell Signalling) at a dilution of 1:500 or Alexa488-conjugated mouse anti-γH2AX (Ser139) antibody at a dilution of 1:250 (#613406, BioLegend). The slides were washed with PBS-T and then incubated with the secondary antibody and/or Alexa Fluor 647 phalloidin (A22287, Thermo Fisher Scientific) in 3% BSA in PBS-T for 1 h at 37 °C. The secondary antibody used was a goat anti-mouse IgG antibody conjugated to Alexa Fluor 488 at a dilution of 1:200 (#SA5-10166, Invitrogen). After the secondary incubation, the slides were washed and stained with 0.5 µg/ml DAPI. The images were acquired using an LSM 800 inverted laser scanning confocal microscope (Carl Zeiss) equipped with an Airyscan detector and a × 63 1.4 NA Plan Apochromat objective (Carl Zeiss). The microscopy analysis was performed using equipment funded by the Foster Foundation (USA).
RTK phosphorylation array
A549 cells were serum-starved for 12 h before treatment with 20 μM of tested compounds for 45 min. After washing, the cells were treated with 20% FBS for an additional 75 min. Lysis buffer was used to collect cell samples, which were subsequently analysed using the RayBio Human Phospho Array Kit (#AAH-PRTK-1–4; RayBiotech) according to the manufacturer’s instructions. In brief, RTK array membranes were blocked with a blocking Cocktail buffer for 1 h, followed by incubation with 300 μg of protein from experimental samples for 5 h. After extensive washing, membranes were incubated with biotinylated antibodies overnight at 4 °C. Then, membranes were washed and incubated with HRP-conjugated streptavidin for 2 h at room temperature. The unbound HRP antibody was washed out, and each membrane array was detected by chemiluminescence using a ChemiDoc Imaging System (Bio-Rad). Signal intensities were measured using densitometric analysis with ImageLab and normalised using the positive control spotted on the membrane. Each experiment was repeated independently three times.
Molecular docking
The selection of kinases for docking receptors was selected by their PDB structures’ availability and the RTK phosphorylation array results. A distinguishing feature observed among PDB structures was a petite cavity bordered by alpha helices on one side and beta sheets on the opposite side. From the initial pool, 54 of the 71 structures displayed this characteristic. The remaining either lacked this feature or were devoid of an accompanying PDB for the kinase in question. Molecular docking studies were conducted using the PDB structures for IGF-IR (2OJ9), Insulin R (5E1S), EphB3 (5L6P), AXL (5U6B), ROR2 (4GT4), ACK1 (5ZXB), FAK (2JKK), Csk (1BYG), FRK (2MRK), and ZAP-70 (1U59). For the ligands Bosutinib, Dasitinib, and Imatinib, their structures were sourced directly from their respective PDBs, where they were found to be bound to specific kinases. I84 is a new molecule; therefore, its structure was built and optimised using the Biovia softwareCitation37. The grid size in docking was set to 30 Å × 30 Å × 30 Å, with its centre aligned to the aforementioned kinase cavity, recognised as the inhibition site. All docking simulations were executed using Vina AutoDockCitation38. Prior to simulation, input files underwent necessary conversion to match the software’s format requirements.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 9 software. Uniform levels of significance were employed throughout the manuscript, wherein the following symbols were used to indicate p values: ns (p > 0.01), * (p < 0.01), ** (p < 0.001), *** (p < 0.0001), and **** (p < 0.00001).
Author contributions
Conceptualisation: M.O., N.M; methodology: M.O., N.M., M.S., V.Z.; validation: M.O., N.M.; formal analysis: M.O. N.M.; writing—original draft preparation: M.O., N.M.; graphical conceptualisation: M.O., N.M.; writing—review and editing: M.O., N.M.
Acknowledgements
We extend our gratitude to Jakub Jurasz from the Gdańsk University of Technology for his invaluable assistance in conducting molecular docking, which significantly enhanced the depth and quality of our research.
The financial support to the maintenance of research facilities used in these studies from Gdańsk University of Technology by the DEC–2/2021/IDUB/V0.6/Si grant under the SILICIUM SUPPORTING CORE R&D FACILITIES – “Excellence Initiative – Research University” program is gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The datasets presented in the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Thandra KC, Barsouk A, Saginala K, Aluru JS, Barsouk A. Epidemiology of lung cancer. Contemp. Oncol. 2021;25(1):45.
- Siddamurthi S, Gutti G, Jana S, Kumar A, Singh SK. Anthraquinone: a promising scaffold for the discovery and development of therapeutic agents in cancer therapy. Future Med Chem. 2020;12(11):1037–1069.
- Gaya E, Fernández-Brime S, Vargas R, Lachlan RF, Gueidan C, Ramírez-Mejía M, Lutzoni F. The adaptive radiation of lichen-forming Teloschistaceae is associated with sunscreening pigments and a bark-to-rock substrate shift. Proc Natl Acad Sci U S A. 2015;112(37):11600–11605.
- Kim HS, Lee YS, Kim DK. Doxorubicin exerts cytotoxic effects through cell cycle arrest and Fas-mediated cell death. Pharmacology. 2009;84(5):300–309.
- Tian W, Li J, Su Z, Lan F, Li Z, Liang D, Wang C, Li D, Hou H. Novel anthraquinone compounds induce cancer cell death through paraptosis. ACS Med Chem Lett. 2019;10(5):732–736.
- Park SH, Lee J, Kang MA, Jang KY, Kim JR. Mitoxantrone induces apoptosis in osteosarcoma cells through regulation of the Akt/FOXO3 pathway. Oncol. Lett. 2018;15(6):9687.
- Levitzki A. Tyrosine kinases as targets for cancer therapy. Eur J Cancer. 2002;38 Suppl 5:S11–S18.
- Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF. Topoisomerase Iiβ–mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67(18):8839–8846.
- Ren L, Li Z, Dai C, Zhao D, Wang Y, Ma C, et al. Chrysophanol inhibits proliferation and induces apoptosis through NF-κB/cyclin D1 and NF-κB/Bcl-2 signaling cascade in breast cancer cell lines. Mol. Med. Rep. 2018;17(3):4376–4382.
- Nemeikaite-Čeniene A, Sergediene E, Nivinskas H, Čenas N. Cytotoxicity of natural hydroxyanthraquinones: role of oxidative stress. Z Naturforsch C J Biosci. 2002;57(9-10):822–827.
- Zhang Q, Chen WW, Sun X, Qian D, Tang DD, Zhang LL, Li MY, Wang LY, Wu CJ, Peng W, et al. The versatile emodin: A natural easily acquired anthraquinone possesses promising anticancer properties against a variety of cancers. Int J Biol Sci. 2022;18(8):3498–3527.
- Hsu SC, Chung JG. Anticancer potential of emodin. Biomedicine (Taipei)). 2012;2(3):108–116.
- Jia X, Yu F, Wang J, Iwanowycz S, Saaoud F, Wang Y, Hu J, Wang Q, Fan D. Emodin suppresses pulmonary metastasis of breast cancer cells accompanied with decreased macrophage recruitment and M2 polarization in the lungs. Breast Cancer Res Treat. 2014;148(2):291–302.
- Kim M, Baek M, Kim DJ. Protein tyrosine signaling and its potential therapeutic implications in carcinogenesis. Curr. Pharm. Des. 2017;23(29):4226.
- Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol. 2020;13(1):1–23.
- Iqbal N, Iqbal N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother Res Pract. 2014;2014:357027–357029.
- Li S. Src-family kinases in the development and therapy of Philadelphia chromosome-positive chronic myeloid leukemia and acute lymphoblastic leukemia. Leuk Lymphoma. 2008;49(1):19–26.
- Shayani S. Dasatinib, a multikinase inhibitor: therapy, safety, and appropriate management of adverse events. Ther Drug Monit. 2010;32(6):680–687.
- Zhang M, Tian J, Wang R, Song M, Zhao R, Chen H, Liu K, Shim J-H, Zhu F, Dong Z, et al. Dasatinib inhibits lung cancer cell growth and patient derived tumor growth in mice by targeting LIMK1. Front Cell Dev Biol. 2020;8:556532.
- Senapati J, Sasaki K, Issa GC, Lipton JH, Radich JP, Jabbour E, Kantarjian HM. Management of chronic myeloid leukemia in 2023 – common ground and common sense. Blood Cancer J. 2023;13(1):58.
- Zvarych V, Stasevych M, Novikov V, Vovk M. Synthesis and study of antimicrobial activity of 2-dithiocarbamate-n-(9,10-dioxo-9,10-dihydroanthracenyl)acetamides. Biointerface Res. Appl. Chem. 2021;11(1):7725–7734.
- Chiang J-H, Yang J-S, Ma C-Y, Yang M-D, Huang H-Y, Hsia T-C, Kuo H-M, Wu P-P, Lee T-H, Chung J-G, et al. Danthron, an anthraquinone derivative, induces DNA damage and caspase cascades-mediated apoptosis in SNU-1 human gastric cancer cells through mitochondrial permeability transition pores and Bax-triggered pathways. Chem Res Toxicol. 2011;24(1):20–29.
- Malik MS, Alsantali RI, Jassas RS, Alsimaree AA, Syed R, Alsharif MA, Kalpana K, Morad M, Althagafi II, Ahmed SA, et al. Journey of anthraquinones as anticancer agents – a systematic review of recent literature. RSC Adv. 2021;11(57):35806–35827.
- Podhorecka M, Skladanowski A, Bozko P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucleic Acids. 2010;2010:1–9.
- Fouillaud M, Venkatachalam M, Girard-Valenciennes E, Caro Y, Dufossé L. Anthraquinones and derivatives from marine-derived fungi: structural diversity and selected biological activities. Mar Drugs. 2016;14(4):64.
- The Human Protein Atlas. https://www.proteinatlas.org/. Accessed May 30, 2023.
- Maciejewska N, Olszewski M, Jurasz J, Baginski M, Stasevych M, Zvarych V, Folini M, Zaffaroni N. Teloxantron inhibits the processivity of telomerase with preferential DNA damage on telomeres. Cell Death Dis. 2022;13(11):1005.
- Al-Otaibi JS, Teesdale Spittle P, El Gogary TM. Interaction of anthraquinone anti-cancer drugs with DNA:Experimental and computational quantum chemical study. J. Mol. Struct. 2017;1127:751–760.
- Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23(48):7906–7909.
- Zhang S, Yu D. Targeting Src family kinases in anti-cancer therapies: turning promise into triumph. Trends Pharmacol Sci. 2012;33(3):122–128.
- Sung NY, Kim MY, Cho JY. Scutellarein reduces inflammatory responses by inhibiting src kinase activity. Korean J Physiol Pharmacol. 2015;19(5):441–449.
- Park JG, Kim SC, Kim YH, Yang WS, Kim Y, Hong S, Kim K-H, Yoo BC, Kim SH, Kim J-H, et al. Anti-inflammatory and antinociceptive activities of anthraquinone-2-carboxylic acid. Mediators Inflamm. 2016;2016:1903849.
- Li E, Hristova K. Receptor tyrosine kinase transmembrane domains: Function, dimer structure and dimerization energetics. Cell Adh Migr. 2010;4(2):249–254.
- Cohen P, Cross D, Jänne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. 2021;20(7):551–569.
- Yang Y, Li S, Wang Y, Zhao Y, Li Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduct. Target Ther. 2022;7(1):1–36.
- Mohi MG, Boulton C, Gu T-L, Sternberg DW, Neuberg D, Griffin JD, Gilliland DG, Neel BG. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukaemia caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101(9):3130–3135.
- Discovery Studio Visualizer, v19. 1.0, BIOVIA. San Diego: Dassault Systems. 2018.
- Quiroga R, Villarreal MA. Vinardo. A scoring function based on Autodock Vina improves scoring, docking, and virtual screening. PloS One. 2016;11(5):e0155183.