
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Liver cancer exhibits a high degree of heterogeneity and involves intricate mechanisms. Recent research has revealed the significant role of histone lysine methylation and acetylation in the epigenetic regulation of liver cancer development. In this study, five inhibitors capable of targeting both histone lysine methyltransferase nuclear receptor-binding SET domain 2 (NSD2) and histone deacetylase 2 (HDAC2) were identified using a structure-based virtual screening approach. Notably, DT-NH-1 displayed a potent inhibition of NSD2 (IC50 = 0.08 ± 0.03 μM) and HDAC2 (IC50 = 5.24 ± 0.87 nM). DT-NH-1 also demonstrated a strong anti-proliferative activity against various liver cancer cell lines, particularly HepG2 cells, and exhibited a high level of biological safety. In an experimental xenograft model involving HepG2 cells, DT-NH-1 showed a significant reduction in tumour growth. Consequently, these findings indicate that DT-NH-1 will be a promising lead compound for the treatment of liver cancer with epigenetic dual-target inhibitors.
Introduction
Liver cancer is the second most common cause of cancer-related deaths worldwide due to its invasiveness and lack of effective treatment.Citation1–3 Targeted therapy has become a new approach to cancer treatment because of its high specificity and low side effects.Citation4 However, the majority of the current drugs developed for single-target cannot fully consider the essential characteristics of cancer with multiple pathogeneses, pathological links, and genetic correlations.Citation5,Citation6 Although drug combination therapy and fixed-dose combinations (FDCs) therapy are utilised as alternative approaches to achieve clinical efficacy, they are associated with a greater risk of drug-drug interactions, unpredictable pharmacokinetics, and poor safety.Citation7,Citation8 The ideal multi-target drugs can avoid drug resistance caused by single-target mutations and better balance the relationship among multiple pathological factors of the disease. In addition, multi-target drugs have exhibited a great potential in expanding the scope of treatment and improving patient compliance.Citation9,Citation10 Therefore, it is imperative to research and develop multitarget drugs, especially dual-targeting drugs, for the treatment of liver cancer.Citation11
Histone lysine deacetylases (HDACs), as the most important histone post-translational modifiers, promote a closed chromatin conformation by removing acetyl groups from ε-amino group of histone lysine residues.Citation12 To date, four classes of human HDAC isoenzymes have been identified: class I (HDAC1, 2, 3, and 8), class II (HDAC4, 5, 6, 7, 9, and 10), and class IV (HDAC11) including Zn2+-dependent enzymes that adopt an arginase deacetylase fold, whereas class III (Sirtuins) are NAD+-dependent enzymes that adopt an unrelated fold.Citation13 Among them, compared with other categories, HDAC1, 2, and 3 are not only expressed in the nucleus, but also widely expressed, and they may play the most important role in tumorigenesis.Citation14–16 In particular, clinical studies have demonstrated that serum HDAC2 level is higher in patients with liver failure than that in healthy individual people.Citation17 In addition, Kim et al. found that the mechanism of HDAC2 upregulation in a large number of patients with hepatocellular carcinoma (HCC) may be that epidermal growth factor (EGF) induces the transcriptional activation of HDAC2 in hepatoma cells activation of through CK2α/Akt signalling pathway.Citation18 HDAC2 knockdown increases the function of tumour suppressor p53, thereby inhibiting proliferation and inducing cellular senescence.Citation19,Citation20 Moreover, recent studies have shown that inhibition of HDAC2 can regulate the acetylation of PD-L1 to improve human immunity.Citation21 HDAC2 siRNA encapsulated in lipid nanoparticles was also found to inhibit the growth of HCC cells.Citation22,Citation23 A growing body of evidence has highlighted that HDAC2 plays a key role in the tumorigenesis of liver cancer by regulating hepatocyte death and cell cycle components.Citation24,Citation25 Therefore, the development of compounds that selectively inhibit HDAC2 is a very promising strategy for liver cancer treatment.
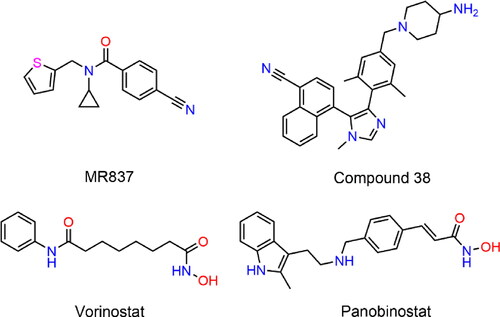
Liver cancer is a heterogeneous disease with different molecular and phenotypic characteristics, and it typically involves mutations in two or more carcinogenic pathways.Citation26 As an important class of epigenetic modifiers, histone lysine methyltransferase (HKMTase) nuclear receptor binding SET domain 2 (NSD2) specifically dimethylates H3K36 to form and maintain chromatin structure, regulate gene transcription and participate in the epithelial-mesenchymal transition (EMT).Citation27,Citation28 NSD2 is a multi-domain protein, containing two proline-tryptophin-tryptophin-proline (PWWP) domains and a SET domain that can catalyse methylation.Citation29 The NSD2 has been proven to be overexpressed in HCC cells, and it played an oncogene role.Citation30,Citation31 NSD2 overexpression resulted in a global increase in H3K36me2 expression level and it acted as a potent coactivator to mediate NF-κB signalling pathway to promote tumour growth, while NSD2 knockdown inhibited cancer cell proliferation.Citation32–36 Importantly, Qiu et al. demonstrated that NSD2 promoted the proliferation, migration, and invasion of HCC cells by activating mTORC1 signalling pathway, and determined that NSD2 could be a promising therapeutic target for HCC.Citation37 The recent development of small-molecule inhibitors targeting NSD2 has emerged as a very promising strategy to treat cancer progression and metastasis.Citation38 In 2021, Allali-Hassan et al. discovered a chemical antagonist MR837 that could specifically target the aromatic cage in the NSD2-PWWP1 domain by virtual screening, and successfully proposed a new method to target NSD2.Citation39 Subsequently, based on the cocrystal structure of the NSD3-PWWP1 domain chemical probe BI-9321 and MR837, Li et al. optimised a new small-molecule compound 38 targeting NSD2-PWWP1, and biological evaluation indicated that the compound partially inhibited the proliferation of tumour cells.Citation40 Therefore, the number and types of NSD inhibitors are relatively limited, and the development of small molecule inhibitors is still in its infancy.Citation41 In addition, the recent Food and Drug Administration (FDA) approval of Tazemetostat, a lysine methyltransferase (PKMT) inhibitor, for epithelioid sarcoma and follicular lymphoma further strengthens the therapeutic relevance and drug availability of the NSD2 target.Citation42 Therefore, additional research is urgently required to develop NSD2 inhibitors with high cellular potency or in vivo efficacy.
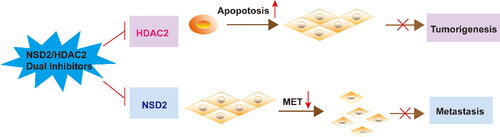
To date, it has become a consensus that inhibitors developed against HDAC induce cell cycle arrest, differentiation, and apoptosis by preventing aberrant HDAC deacetylation in cancer patients.Citation43 Some HDAC inhibitors that inhibit HDAC2 have been approved by the FDA, such as Vorinostat and Panobinostat.Citation44,Citation45 However, clinical results indicated that the therapeutic efficacy of these drugs is unsatisfactory, and patients still experience tumour invasion and metastasis.Citation46 Recent date demonstrated that NSD2 is involved in tumour cell invasion and metastasis through EMT.Citation47 Moreover, studies have found that NSD2 is closely associated with histone methyltransferase EZH2, and EZH2 mediates trimethylation of H3K27 (H3K27me3) to further upregulate NSD2 expression level.Citation28 The combined treatment of human acute myeloid leukaemia (AML) cells with EZH2 inhibitor 3-deazaneplanocin A and Panobinostat caused more depletion of EZH2 and inhibition of trimethylation of lysine 27 on histone H3 than treatment with each drug alone, and significantly improved the survival rate of immunodeficient mice with leukemia.Citation48 Hence, it is imperative to design NSD2/HDAC2 dual-targeting small-molecule compounds to prevent tumorigenesis and inhibit tumour metastasis to improve the efficacy of liver cancer treatment (Scheme 1).
Scheme 1. Mechanistic diagram of NSD2/HDAC2 inhibitors in the treatment of HCC.
With the recent advances in the understanding of tumour molecular biology, molecular targets for treatment have been identified, such as tumour antigens, growth factors, transcription factors, and epigenetic modifiers.Citation49,Citation50 Small-molecule drugs targeting tumours have noticeable advantages of reaching intracellular targets and greater drug exposure in the tumour microenvironment. Therefore, there is an urgent need to develop small molecules to modulate the immune system and treat diverse type of cancer.Citation51 From discovery of bioactive compounds to corresponding biological verification, computer-aided drug design (CADD) has remarkably accelerated the process of drug research and reduced the overall cost.Citation52 The virtual screening method based on structure is robust and efficient, and it is one of the most promising in silico techniques.Citation53–55 Through virtual screening and functional verification, Ucar et al. and Thiyagarajan et al. identified the lead compound of dual-targeting inhibitors of focal adhesion kinase (FAK) and other factors (insulin-like growth factor 1 and ribosomal S6 kinase 1).Citation56,Citation57 Moreover, the TAOK1 and MAP4K5 dual inhibitor compounds identified using a structure-based virtual screening approach arrested cancer cells at G0/G1 phase.Citation58
Simultaneous targeting of HDAC2/NSD2 by a small-molecule compound is a promising cancer treatment that has rarely been reported, making them an attractive strategy for the development of drugs to treat liver cancer. In the present study, the first NSD2/HDAC2 dual-targeted inhibitor (DT-NH-1) was successfully identified using structure-based virtual screening. Subsequently, DT-NH-1 exhibited encouraging in vitro enzymatic inhibitory activity, anti-cell proliferation activity, and biosafety. Finally, it was found to display significant antitumor efficacy in a HepG2 xenograft mouse model. In summary, DT-NH-1 is a promising dual-targeting inhibitor in the treatment of cancer.
Materials and methods
Materials
Four HCC cell lines (HepG2, HuH-1, SK-HEP-1, and Bel-7405) and human normal liver L-02 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured in a Dulbecco’s modified Eagle’s medium (DMEM; Gibco, New Nork, USA), supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% foetal calf serum, and maintained in a humidified incubator with 5% CO2 and 95% air at 37 °C. All compounds were purchased from WuXi AppTec (Shanghai, China). NSD2 and HDAC2 proteins were obtained from Abcam (Cambridge, UK).
Establishment of a structure-based pharmacophore model for NSD2
Pharmacophore models based on validated protein structures have been widely utilised in the drug design and development.Citation59,Citation60 The X-ray crystal structure of NSD2 (PDB ID: 7VLN) was obtained from Protein Data Bank (PDB). The protein preparation tool in the Molecular Operating Environment program (MOE, Chemical Computing Group Inc, Montreal, Quebec, Canada) was used to pretreat the protein structure of NSD2 by removing water molecules and adding hydrogen atoms. Then, the energy was minimised through Amber14: EHT force field. The representative pharmacophore features in NSD2 active site, including hydrogen bond acceptor (Acc), hydrogen bond donor (Don), aromatic centre (Aro), and hydrophobic (Hyd) features, were selected by the pharmacophore query editor of MOE.
Validation of the pharmacophore model
To validate the reliability of the NSD2 model described above, the Güner–Henry (GH) scoring method was utilised as described previously.Citation61 A test database consisted of 1981 inactive molecules from the Enhanced Database of Useful Decoys (DUDe) and 19 reported active molecules.Citation38,Citation39,Citation41 The proposed model was subsequently used as a three-dimensional (3D) query to filter the test database using the pharmacophore search protocol available in MOE. Finally, statistical parameters such as total hits (Ht), yield of actives (%), ratio of actives (%), the goodness of hit score (GH), and enrichment factor (E) were calculated. The E value is proportional to the ability of pharmacophore model to identify active compounds. A GH score greater than 0.7 represents a higher reliability of the model. The GH score was calculated based on the following formula:Citation62
Docking-based virtual screening
Molecular docking is a computational process that simulates the interaction between a ligand and a target protein, aiming to predict the binding affinity and the specific binding orientation (pose) of the two molecules. This is achieved through the calculation of various physicochemical parameters.Citation63 To reduce computational effort and obtain the optimal conformation, the semi-flexible docking method was employed, which was to set the target protein to be rigid and the ligand compound to be flexible. The energy minimisation principle of MOE was followed to convert 35,000 molecules from the obtained ligand database into 3D compounds. The crystal structures of NSD2 (PDB ID: 7VLN) were utilised as a screening template, and the docking score of MR837 was used as a threshold to screen compounds in the database for subsequent filtering of HDAC2 targeted compounds. The crystal structures of HDAC proteins co-crystallized by the FDA-approved drug Vorinostat (PDB ID: 4LXZ) were obtained from the PDB. The two proteins were imported into the MOE program and prepared by removing water, adding hydrogen atoms, calculating partial charges, and performing energy minimisation in Amber14: EHT force field.
Enzymatic assays for HDAC2 and NSD2-PWWP1
In vitro HDAC2 inhibition experiments were carried out as described previously.Citation64 In the 96-well plate, 10 μL of HDAC2 enzyme solution was first added, and 50 μL of inhibitor compounds with different concentrations were mixed. After incubating the mixture at 37 °C for 5 min, 40 μL of the fluorescent substrate Boc-Lys (acetyl) -AMC was added to the mixture. The plates were incubated at 37 °C for an additional 0.5 h. Finally, the developer (100 μL) containing trypsin and TSA was injected, and the fluorescence intensity at wavelengths of 390 and 460 nm was measured 20 min later by a microplate reader (BioTek Cytation 5; Agilent Technologies, Inc., Santa Clara, CA, USA).
The enzymatic activity of NSD2-PWWP1 was determined by a previously described homogeneous time-resolved fluorescence (HTRF) assay.Citation40 First, different concentrations of inhibitor compounds were prepared and dissolved in dimethyl sulfoxide (DMSO). Subsequently, the mixture of 5 μL of NSD2-PWWP1 (4 nM) dilution buffer and 5 μL of substrate polypeptide dilution buffer was added to 96-well plates according to the manufacturer’s protocol. Then, 5 μL of the final 1:1000 dilution of the inhibitor compound was sucked into the mixture, and 5 μL of the Tb/d2 mixture was added to each well. The 96-well plates were incubated at room temperature for 3 h, and the fluorescence intensity was recorded using a microplate reader (BioTek Cytation 5) at test wavelengths of 665 and 620 nm. All experiments were repeated three times under the same conditions.
Molecular dynamics (MD) simulation
GROMACS 2021.5 was employed to perform the MD simulations of NSD2-hit complex and HDAC2-hit complexes. The topology files for the NSD2 and HDAC2 proteins were created using GROMACS, utilising the AMBER99SB-ILDN force field. The topology file for the compound of interest (hit) was generated through the Acpype Server (www.bio2byte.be) and employed GAFF force field. NSD2-hit complex was placed in the centre of a cubic box with each side length of 71 Å, and HDAC2-hit complex was placed in the centre of a cubic box with each side length of 82 Å. TIP3P water model was added into the boxes to solvate the complex systems, and appropriate Na+ and Cl- ions were also added to neutralise the complex systems. Thereafter, the two complex systems were subjected to energy minimisation using the steepest descent method for 5000 steps. Furthermore, the two systems were gradually heated from 0 to 300 K using the V-rescale thermostat in a 1-ns NVT simulation to equilibrate systems. Subsequently, 1-ns NPT simulation was performed to relax systems (pressure = 1 atm) using the Parrinello-Rahman barostat. Finally, 50 ns MD was carried out with the integration step of 1 fs, and hydrogen-bonds were constrained using the linear constraint solver (LINCS) algorithm. Trajectory data were recorded every 10 ps. The root-mean-square deviation (RMSD) calculation was conducted using the gmx_rms module, the root-mean-square fluctuation (RMSF) calculation was performed using the gmx_rmsf module, and the secondary structure was evaluated via DSSP 4.0 software.
MTT assay
The effects of candidate compounds on cell proliferation were determined as previously reported by MTT assay.Citation65 Firstly, the most potent compound was selected by the inhibition rate of all compounds on the HepG2 cell line under the same condition (the same concentration), and then the IC50 value of the compound on more HCC cell lines was determined.Citation60,Citation66 The cells were seeded into 96-well culture plates at a density of 5 × 104 cells/well. After overnight incubation, cells were treated with various concentrations of the candidate compounds for 72 h. Each well was filled with 100 μL MTT (0.5 mg/mL) solution dissolved in phosphate-buffered saline (PBS), and the 96-well plates were placed at 37 °C for 4 h until a purple precipitate appeared. Then, 200 μL dimethyl sulfoxide (DMSO) was injected into the mixture and shaken gently for 10 min. The absorbance of the samples at 570 nm was measured using a Synergy 4 microplate reader (BioTek Instruments Inc., Winooski, VT, USA). Each set of experiments was performed three times in a parallel manner.
In vivo antitumor assay
To investigate the antitumor activity of DN-NH-1 in vivo, a human tumour xenograft model was established as described previously.Citation55 The animal experiments were approved by the Ethics Committee of China Pharmaceutical University (Nanjing, China). The specific pathogen-free grade 6-week-old male BALB/c nude mice were purchased from Changzhou Cavens Experimental Animal Limited Company (Changzhou, China). HepG2 cells (1 × 107 cells) were injected into the right subcutaneous space of mice. When the average tumour volume reached about 80–100 mm3, mice were randomly divided into 4 groups: vehicle-control group, low-dose treatment group (3 mg/kg), middle-dose treatment group (6 mg/kg), and high-dose treatment group (12 mg/kg). All mice were injected intraperitoneally. Subcutaneous tumour volume was measured every 3 days for a total of seven times during treatment and was calculated by the formula: (c × c × d)/2, where c and d represent the smallest and largest diameters of tumours, respectively ().
Figure 1. Structures of the representative NSD2 and HDAC inhibitors.
Results
Generation of NSD2 pharmacophore model
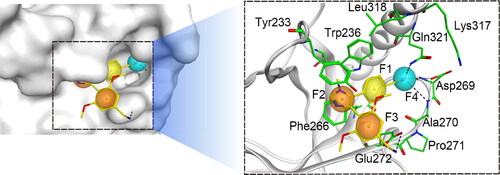
The retrieved X-ray crystal structures of NSD2 (PDB ID: 7VLN) co-crystallized with imidazole-5-ylbenzonitrile compounds were utilised to characterise the NSD2 pharmacophore model. PWWP1 is the reader domain of NSD2, which performs the function of preferentially combining with nucleosome containing H3K36me2 and maintaining a proper nuclear localisation, which depends on the conservative aromatic cage composed of Trp236, Tyr233, and Phe266. As illustrated in , the pockets of the aromatic cage can accommodate reasonably designed compounds (e.g., the cyclopropyl group of MR837). In this study, four important pharmacophore features were generated (F1-F4): feature F1 corresponds to hydrophobic amino acid residues Trp236 and Leu318, features F2 and F3 are both aromatic centres, corresponding to amino acids Tyr233 and Phe266 that are components of the aromatic pocket, and the F4 feature is a hydrogen bond receptor, forming hydrogen bond interactions with Gln321, Ala270, and Asp269. According to the above-mentioned four features, the NSD2 pharmacophore model was finally established for the subsequent molecular docking.
Figure 2. Details of the NSD2-based pharmacophore model features (F1: hydrophobic feature, F2 and F3: aromatic features, F4: hydrogen-bond acceptor feature).
Validation of the pharmacophore model
A database of 1981 inactive and 19 active molecules for the pharmacophore model validation was used for screening. The Güner–Henry (GH) scoring method assessed the specificity and accuracy of the structure-based model. The value of the enrichment factor (E) in is 95, and 18 active hits were identified from 20 molecules, demonstrating that the pharmacophore model is more capable of recognising active molecules. Moreover, a GH score of 0.91 (greater than 0.7) was obtained, indicating an excellent ability to distinguish between active and inactive compounds.
Table 1. The validation of the pharmacophore model by the GH scoring method.
Virtual screening of HDAC2/NSD2 dual-targeting inhibitors
In the present study, the steps of multi-step virtual screening are depicted in . Firstly, a two-dimensional (2D) small-molecule database containing 35,000 compounds was established, and each compound was replaced with a 3D structure based on the principle of energy minimisation. The above-validated NSD2 pharmacophore model features were used as the screening constraints to obtain 207 hit compounds matching with all pharmacophore features. The reliability of the docking method was verified in NSD2 and HDAC2 before molecular docking. The original ligand was utilised as a template, and the docking ligand was redocked flexibly to the active sites of NSD2 and HDAC2 by the Triangle Matcher placement method. The results revealed that the poses of the original and docking ligands were almost identical (). Subsequently, these 207 compounds were further screened by docking with the active sites of NSD2 and HDAC2, respectively. The docking-binding free energy can be used to predict the binding affinity between each hit compound and the active site of the target. A lower value indicates a stronger binding affinity. MR837 was selected as a positive control for NSD2 molecular docking with a docking score of −10.51 kcal/mol. The compounds were screened for the active site connected to NSD2, and the docking score of each hit was calculated. The reasonable docking score threshold of < −10.51 kcal/mol was set as the screening criterion (), and 37 compounds were obtained from 207 compounds. Subsequently, a high-resolution crystal structure of HDAC2 (PDB ID: 4LXZ) was obtained, and based on docking scores below −12 kcal/mol, 19 compounds were achieved from 207 compounds for HDAC2 docking screening. As shown in , based on docking scores below < −10.51 and −12 kcal/mol, five potential dual-targeting NSD2/HDAC2 candidate compounds (referred to as DT-NH-1–5) were ultimately obtained that met both scoring thresholds. Five screened compounds were finally purchased for subsequent biological experiments ().
Figure 3. Identification of hit compounds targeting NSD2 and HDAC2 through a comprehensive strategy of structure-based virtual screening and biological validation. (A) Workflow of the combinatorial screening and biological validation process of NSD2/HDAC2 dual-targeting inhibitors. (B) The docking validation of NSD2 before molecular docking (yellow indicates docking pose, and cyan indicates original pose). (C) The docking validation of HDAC2 before molecular docking (yellow indicates docking pose, and cyan indicates original pose). (D) Identification of five hit compounds with lower binding energies than both NSD2 (-10.51 kcal/mol) and HDAC2 (-12 kcal/mol).
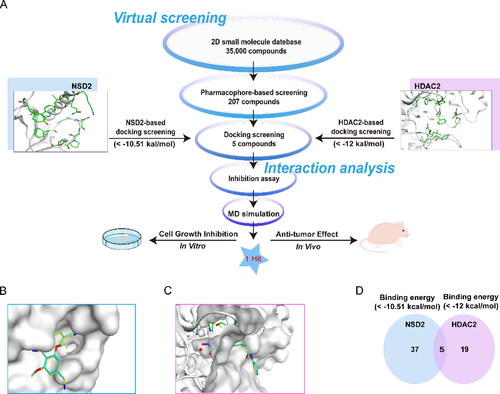
Figure 4. The chemical structures of five hit compounds.
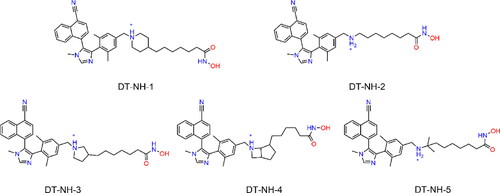
Table 2. The docking scores and enzymatic inhibitory activities of 5 hit compounds.
Interaction analysis
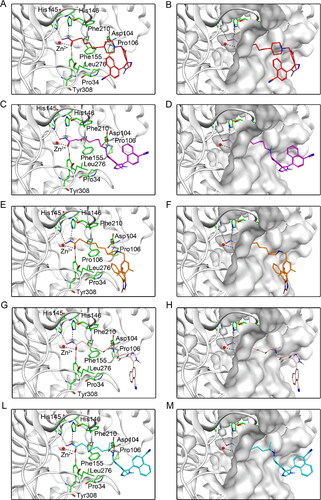
The potential five compounds were sequentially docked into NSD2 and HDAC2 proteins to assist in studying their binding modes. shows the interaction between five hit compounds and NSD2 proteins. The nitrogen atoms of all the compounds not only formed stable hydrogen bonds with Gln321, Ala270, Asp269, and Glu272, but also provided additional hydrogen bonds with Lys317 by the oxygen atoms at the end of the fatty chain. In addition, each compound exhibited hydrophobic interactions with hydrophobic amino acids such as Tyr233, Trp236, and Phe266 (). Furthermore, five potential compounds that matched well with the active pockets of NSD2 were identified (). Next, the binding modes of the five compounds to the HDAC2 protein are illustrated in . Each hit compound contained an N-Hydroxyformamide group, which not only formed important ionic bonds with positively charged zinc ions, but also exhibited hydrogen bond interactions with His145, His146, and Tyr308. In addition, the fatty chains of each compound showed hydrophobic interactions with Phe155 and Phe210. These potent interactions are consistent with the fit results of each compound with the HDAC2 surface active pocket, as displayed in . Therefore, the docking results of the hit compounds with NSD2 and HDAC2 indicated that DT-NH-1–5 could simultaneously interact with the key residues of the active pockets of both NSD2 and HDAC2, which might lead to the occurrence of dual inhibitory effects on NSD2 and HDAC2.
Figure 5. Predicted docking poses of five hit compounds at the NSD2 active site. Compounds in (A), (C), (E), (G), and (L) are represented by different colours (red for DT-NH-1, purple for DT-NH-2, brown for DT-NH-3, light pink for DT-NH-4, and cyan for DT-NH-5). (B), (D), (F), (H), and (M) are their corresponding surface maps. The coded green bars represent the active amino acid residues of NSD2. Hydrogen bonds are indicated by dashed black lines.
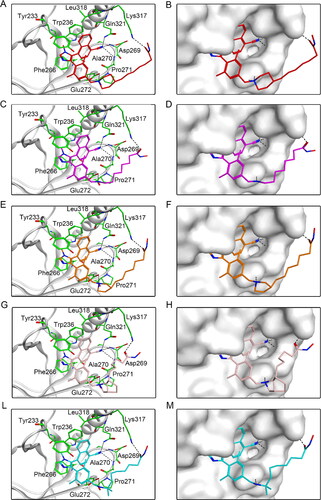
Figure 6. Predicted docking poses of five hit compounds at the HDAC2 active site. Compounds in (A), (C), (E), (G), and (L) are represented by different colours (red for DT-NH-1, purple for DT-NH-2, brown for DT-NH-3, light pink for DT-NH-4, and cyan for DT-NH-5). (B), (D), (F), (H), and (M) are their corresponding surface maps. The coded green bars represent the active amino acid residues of HDAC2. Hydrogen and ionic bonds are indicated by dashed black lines.
Enzymatic inhibitory assay of NSD2 and HDAC2
Five potential dual-targeting NSD2/HDAC2 candidate compounds were obtained through virtual screening and interaction analysis. Their enzymatic inhibitory activities against NSD2 and HDAC2 were measured using homogeneous time-resolved fluorescence (HTRF) technology and fluorescence signal-based methods, respectively. The reported NSD2 inhibitor MR837 and the approved HDAC inhibitor Vorinostat were utilised as positive controls. As shown in , the half maximal inhibitory concentration (IC50) of the five hit compounds for NSD2 in the range of 0.08–0.64 μM was significantly smaller than that of MR837 (17.13 ± 1.95 μM). The reliability of feature screening based on the pharmacophore model was also confirmed. In addition, apart from DT-NH-3, the other four dual-targeting compounds exhibited superior inhibitory effects on HDAC2 versus Vorinostat. Notably, the enzymatic inhibitory activity of DT-NH-1 was optimal, with 214.1- and 1.8-fold inhibition of NSD2 and HDAC2 proteins compared with the controls MR837 and Vorinostat, respectively.
MD Simulation
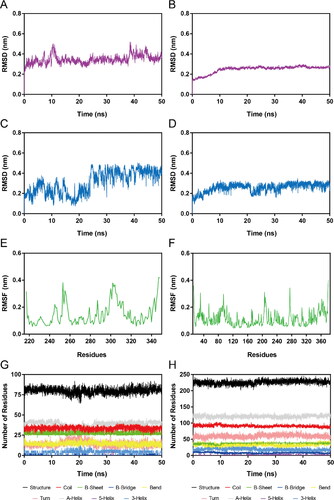
To further evaluate the time-dependent binding stability of NSD2-DT-NH-1 and HDAC2-DT-NH-1 complexes, 50 ns MD simulations were carried out. The RMSD generally represents the system stability during MD simulation. The lower RMSD indicated a greater stability. The RMSDs of the NSD2-DT-NH-1 and HDAC2-DT-NH-1 complexes are respectively illustrated in . The RMSDs of the hit in the NSD2-DT-NH-1 and HDAC2-DT-NH-1 complexes are respectively depicted in . It was found that the RMSDs of the two complexes were relatively stable at about 0.38 and 0.26 nm, respectively. The RMSDs of the hit in the two complex systems were also stabilised at about 0.42 and 0.28 nm, respectively. These results indicated the appropriate structural stability of the hit and the two complexes during the MD simulation, and the hit could simultaneously bind stably with NSD2 and HDAC2. Furthermore, RMSF is generally utilised to descript the structural adaptability of each protein residue. The lower RMSF presents less flexibility, which is mainly related to the interactions between protein and ligand. As illustrated in , RMSF values of all residues within NSD2 and HDAC2 were lower than 0.4 nm, suggesting the appropriate stability of protein residues and the promising binding affinity between the two proteins and the DT-NH-1 during the MD simulation. Finally, to determine whether the secondary structure of the NSD2 and HDAC2 was stable during the MD simulation, DSSP analysis was conducted. As depicted in , no noticeable alterations were found in secondary structural elements of NSD2 and HDAC2 proteins, suggesting the proper structural stability of the two proteins. Collectively, these MD simulation results suggested that the DT-NH-1 could simultaneously bind stably to NSD2 and HDAC2 with promising binding stability. Thus, the DT-NH-1 could potentially serve as a potent dual-target inhibitors for NSD2 and HDAC2.
Figure 7. MD simulation of DT-NH-1 in complex with NSD2 and HDAC2. (A), (B) RMSD of NSD2-DT-NH-1 and HDAC2-DT-NH-1 complexes. (C), (D) RMSD of DT-NH-1 in NSD2-DT-NH-1 and HDAC2-DT-NH-1 complexes. (E), (F) RMSF of Cα atoms of NSD2 residues and HDAC2 residues respectively in NSD2-DT-NH-1 and HDAC2-DT-NH-1 complexes. (G), (H) The secondary structural analysis of DT-NH-1 in complex with NSD2 and HDAC2.
In vitro anti-proliferative assay
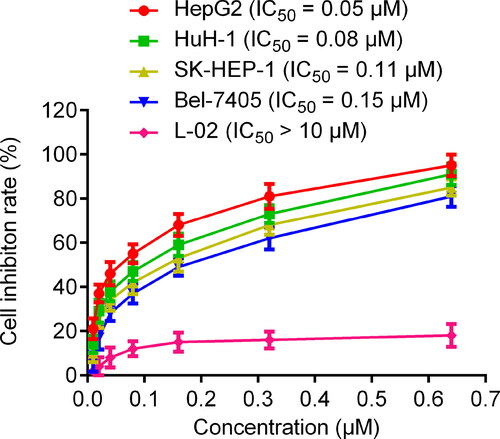
To evaluate the inhibitory effects of five hit compounds on liver cancer cells, MTT assay was carried out. As shown in , all hit compounds exhibited stronger inhibitory effects on HepG2 cells than MR837 and Vorinostat at a concentration of 0.6 μM. Among them, DT-NH-1 was evaluated as the strongest inhibitor with > 90% inhibition of HepG2 cells. Considering the need to evaluate the biosafety of DT-NH-1 and its inhibitory effects on multiple liver cancer cells, the effects of DT-NH-1 on normal human liver cell lines and four liver cancer cell lines (HepG2, HuH-1, SK-HEP-1, and Bel-7405) were further assessed. As illustrated in , DT-NH-1 exhibited an excellent biosafety for normal human liver cells (IC50 > 10 μM), while DT-NH-1 showed strong inhibitory effects on four HCC cell lines (IC50 = 0.05–0.15 μM), especially on HepG2 cells (IC50 = 0.05 μM). Importantly, the selectivity index (SI) of DT-NH-1, which is the IC50 value of a compound on normal cells divided by the IC50 value of cancer cells.Citation67, ranged from 66.7 to 200, confirming the high selectivity and excellent biosafety of DT-NH-1. The above-mentioned results of in vitro anti-proliferation experiments indicated that DT-NH-1 in this study has promising anti-tumour potential, which lays the foundation for the future dual-targeted anti-tumour drug design. Therefore, we will further investigate the anti-tumour effects of DT-NH-1 on HepG2 xenograft in nude mice.
Figure 8. Inhibitory effects of five hits, MR837 and Vorinostat on HepG2 cells at a concentration of 0.6 μM. The results are representative of three independent experiments and are expressed as mean ± SD.
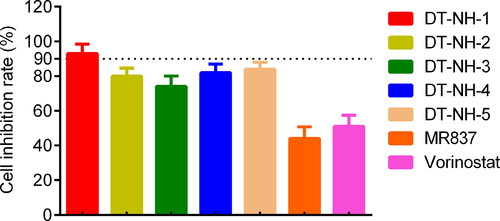
Figure 9. Inhibitory effects of DT-NH-1 on normal liver cell lines and four liver cancer cell lines. The results are representative of three independent experiments and are expressed as mean ± SD.
In vivo antitumor efficacy of DT-NH-1
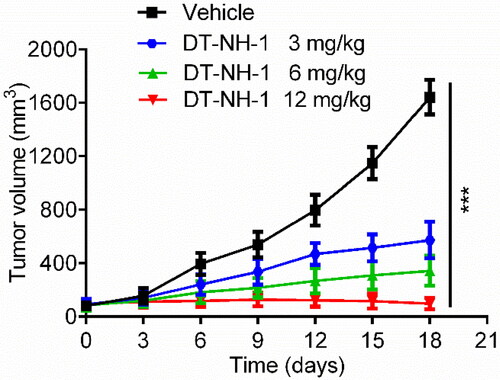
The in vivo antitumor efficacy of DT-NH-1 was evaluated based on the inhibitory effects of the target protein and the anti-cell proliferation potency in vitro. A xenograft model of BALB/c nude mice was first successfully established, carrying the HepG2 cell line, and vehicle and different doses of DT-NH-1 were then intraperitoneally injected, respectively. Compared with the vehicle-control group, the tumour growth rate of the DT-NH-1 treated group was significantly reduced in a dose-dependent manner, and DT-NH-1 had the optimal anti-tumour effect at the dose of 12 mg/kg (). The above-mentioned findings demonstrated that DT-NH-1 exerted a strong anticancer effect in vivo.
Figure 10. The change in tumour volume with varying concentrations of DT-NH-1. The results are presented as mean ± SD, n = 5. ***p < 0.001 mean significant difference versus the vehicle group.
Conclusions
Genomic changes and epigenetic modifications jointly promote the progression and metastasis of cancer, and immune checkpoint inhibitors have been widely studied in cancer treatment. However, the severe reality of drug resistance in patients and the urgent need to develop new targets have attracted scholars’ attention in the research of epigenetic modifications-related cancer treatment targets. Moreover, due to the strong efficacy and satisfactory safety, designing drugs with dual-targeting combined effects is a promising strategy for treating cancer. The NSD2 and HDAC2 play a crucial role in the cancer cell proliferation and cancer metastasis. In the present study, five novel NSD2/HDAC2 dual-targeting small-molecule compounds were identified using a comprehensive virtual screening scheme. Particularly, enzymatic inhibitory experiments confirmed that compound DT-NH-1 has a strong inhibitory effect on NSD2 (IC50 = 0.08 ± 0.03 μM) and HDAC2 (IC50 = 5.24 ± 0.87 μM). Moreover, DT-NH-1 exhibited a potent antiproliferative activity against various liver cancer cell lines, particularly HepG2 cells, and exhibited a high level of biological safety according to the results of in vitro cell proliferation experiments. Afterwards, the HepG2 cell line, which was the most sensitive to DT-NH-1, was selected to established a xenograft model for in vivo evaluation experiments. The results revealed that DT-NH-1 significantly inhibited tumour growth in mice. In summary, designing small-molecule compounds that simultaneously target NSD2 and HDAC2 has become a new therapeutic strategy for hindering tumour progression. In addition, our laboratory is currently conducting further optimisation research on DT-NH-1.
Ethical statement
All experimental protocols were reviewed and approved by the Animal Ethics Committee of China Pharmaceutical University.
Author contributions
GB, QZ and XJ contributed to the conception of the study. YW, XJ and MMN performed experiments and acquired data. YW, JC, MMN, and YY performed statistical analysis. XJ and YW contributed significantly to manuscript preparation. All authors had contributed and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Liao S-H, Su T-H, Jeng Y-M, Liang P-C, Chen D-S, Chen C-H, Kao J-H. Clinical manifestations and outcomes of patients with sarcomatoid hepatocellular carcinoma. Hepatology. 2019;69(1):209–221.
- Niu L, Liu L, Yang S, Ren J, Lai PBS, Chen GG. New insights into sorafenib resistance in hepatocellular carcinoma: responsible mechanisms and promising strategies. Biochim Biophys Acta Rev Cancer. 2017;1868(2):564–570.
- Li J-j, Liang Q, Sun G-c. Traditional Chinese medicine for prevention and treatment of hepatocellular carcinoma: a focus on epithelial-mesenchymal transition. J Integr Med. 2021;19(6):469–477.
- Cidon EU. Systemic treatment of hepatocellular carcinoma: past, present and future. World J Hepatol. 2017;9(18):797–807.
- Leon R, Garcia AG, Marco-Contelles J. Recent advances in the multitarget-directed ligands approach for the treatment of Alzheimer’s disease. Med Res Rev. 2013;33(1):139–189.
- Liao M, Zhang J, Wang G, Wang L, Liu J, Ouyang L, Liu B. Small-molecule drug discovery in triple negative breast cancer: current situation and future directions. J Med Chem. 2021;64(5):2382–2418.
- Pourkavoos N. Unique risks, benefits, and challenges of developing drug-drug combination products in a pharmaceutical industrial setting. combprodther. 2012;2(1):2.
- Thakur A, Tawa GJ, Henderson MJ, Danchik C, Liu S, Shah P, Wang AQ, Dunn G, Kabir M, Padilha EC, et al. Design, Synthesis, and biological evaluation of quinazolin-4-one-based hydroxamic acids as dual PI3K/HDAC inhibitors. J Med Chem. 2020;63(8):4256–4292.
- Zhang W, Pei J, Lai L. Computational multitarget drug design. J Chem Inf Model. 2017;57(3):403–412.
- Ramsay RR, Popovic-Nikolic MR, Nikolic K, Uliassi E, Bolognesi ML. A perspective on multi-target drug discovery and design for complex diseases. Clin Transl Med. 2018;7(1):3.
- Liu X-J, Zhao H-C, Hou S-J, Zhang H-J, Cheng L, Yuan S, Zhang L-R, Song J, Zhang S-Y, Chen S-W, et al. Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg Chem. 2023;133:106425.
- Rodriguez Y, Hinz JM, Smerdon MJ. Accessing DNA damage in chromatin: preparing the chromatin landscape for base excision repair. DNA Repair. 2015;32:113–119.
- Hai Y, Christianson DW. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol. 2016;12(9):741–747.
- Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280(2):168–176.
- Schneider G, Kramer OH, Schmid RM, Saur D. Acetylation as a transcriptional control mechanism-HDACs and HATs in pancreatic ductal adenocarcinoma. J Gastrointest Cancer. 2011;42(2):85–92.
- Fritzsche FR, Weichert W, Röske A, Gekeler V, Beckers T, Stephan C, Jung K, Scholman K, Denkert C, Dietel M, et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer. 2008;8(1):381.
- Wang Y, Chen Q, Jiao F, Shi C, Pei M, Wang L, Gong Z. Histone deacetylase 2 regulates ULK1 mediated pyroptosis during acute liver failure by the K68 acetylation site. Cell Death Dis. 2021;12(1):55.
- Kim HS, Chang YG, Bae HJ, Eun JW, Shen Q, Park SJ, Shin WC, Lee EK, Park S, Ahn YM, et al. Oncogenic potential of CK2alpha and its regulatory role in EGF-induced HDAC2 expression in human liver cancer. Febs J. 2014;281(3):851–861.
- Harms KL, Chen X. Histone deacetylase 2 modulates p53 transcriptional activities through regulation of p53-DNA binding activity. Cancer Res. 2007;67(7):3145–3152.
- Gao B, Li X, Li S, Wang S, Wu J, Li J. Pan-cancer analysis identifies RNA helicase DDX1 as a prognostic marker. Phenomics. 2022;2(1):33–49.
- Han R, Ling C, Wang Y, Lu L. Enhancing HCC treatment: innovatively combining HDAC2 inhibitor with PD-1/PD-L1 inhibition. Cancer Cell Int. 2023;23(1):203.
- Lee Y-H, Seo D, Choi K-J, Andersen JB, Won M-A, Kitade M, Gómez-Quiroz LE, Judge AD, Marquardt JU, Raggi C, et al. Antitumor effects in Hepatocarcinoma of isoform-selective inhibition of HDAC2. Cancer Res. 2014;74(17):4752–4761.
- Mohammad NS, Nazli R, Zafar H, Fatima S. Effects of lipid based multiple micronutrients supplement on the birth outcome of underweight pre-eclamptic women: a randomized clinical trial. Pak J Med Sci. 2022;38(1):219–226.
- Ler SY, Leung CHW, Khin LW, Lu G-D, Salto-Tellez M, Hartman M, Iau PTC, Yap CT, Hooi SC. HDAC1 and HDAC2 independently predict mortality in hepatocellular carcinoma by a competing risk regression model in a Southeast Asian population. Oncol Rep. 2015;34(5):2238–2250.
- Noh JH, Jung KH, Kim JK, Eun JW, Bae HJ, Xie HJ, Chang YG, Kim MG, Park WS, Lee JY, et al. Aberrant regulation of HDAC2 mediates proliferation of hepatocellular carcinoma cells by deregulating expression of G1/S cell cycle proteins. PLoS One. 2011;6(11):e28103.
- Chen C-Y, Chen C-C, Chuang W-Y, Leu Y-L, Ueng S-H, Hsueh C, Yeh C-T, Wang T-H. Hydroxygenkwanin inhibits class I HDAC expression and synergistically enhances the antitumor activity of Sorafenib in liver cancer cells. Front Oncol. 2020;10:216.
- Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13(2):115–126.
- Chen R, Chen Y, Zhao W, Fang C, Zhou W, Yang X, Ji M. The role of methyltransferase NSD2 as a potential oncogene in human solid tumors. Onco Targets Ther. 2020;13:6837–6846.
- Topchu I, Pangeni RP, Bychkov I, Miller SA, Izumchenko E, Yu J, Golemis E, Karanicolas J, Boumber Y. The role of NSD1, NSD2, and NSD3 histone methyltransferases in solid tumors. Cell Mol Life Sci. 2022;79(6):285.
- Wang S, Yang H, Su M, Lian F, Cong Z, Wei R, Zhou Y, Li X, Zheng X, Li C, et al. 5-Aminonaphthalene derivatives as selective nonnucleoside nuclear receptor binding SET domain-protein 2 (NSD2) inhibitors for the treatment of multiple myeloma. Eur J Med Chem. 2021;222:113592.
- Gao B, Liu X, Li Z, Zhao L, Pan Y. Overexpression of EZH2/NSD2 histone methyltransferase axis predicts poor prognosis and accelerates tumor progression in triple-negative breast cancer. Front Oncol. 2020;10:600514.
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang H-e. C, Taylor JE, Hu M, Englund NP, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013;45(11):1386–1391.
- Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick A, et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 2014;28(1):198–201.
- Zhao L-H, Li Q, Huang Z-J, Sun M-X, Lu J-J, Zhang X-H, Li G, Wu F. Identification of histone methyltransferase NSD2 as an important oncogenic gene in colorectal cancer. Cell Death Dis. 2021;12(11):974.
- Song D, Lan J, Chen Y, Liu A, Wu Q, Zhao C, Feng Y, Wang J, Luo X, Cao Z, et al. NSD2 promotes tumor angiogenesis through methylating and activating STAT3 protein. Oncogene. 2021;40(16):2952–2967.
- Yang P, Guo L, Duan ZJ, Tepper CG, Xue L, Chen X, Kung H-J, Gao AC, Zou JX, Chen H-W, et al. Histone methyltransferase NSD2/MMSET mediates constitutive NF-kappaB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol Cell Biol. 2012;32(15):3121–3131.
- Dai J, Jiang L, Qiu L, Shao Y, Shi P, Li J. WHSC1 promotes cell proliferation, migration, and invasion in hepatocellular carcinoma by activating mTORC1 signaling. Onco Targets Ther. 2020;13:7033–7044.
- Zhang L, Zha X. Recent advances in nuclear receptor-binding SET domain 2 (NSD2) inhibitors: An update and perspectives. Eur J Med Chem. 2023;250:115232.
- Ferreira de Freitas R, Liu Y, Szewczyk MM, Mehta N, Li F, McLeod D, Zepeda-Velázquez C, Dilworth D, Hanley RP, Gibson E, et al. Discovery of small-molecule antagonists of the PWWP domain of NSD2. J Med Chem. 2021;64(3):1584–1592.
- Li N, Yang H, Liu K, Zhou L, Huang Y, Cao D, Li Y, Sun Y, Yu A, Du Z, et al. Structure-based discovery of a series of NSD2-PWWP1 inhibitors. J Med Chem. 2022;65(13):9459–9477.
- Tang H, Yu A, Xing L, Chen X, Ding H, Yang H, Song Z, Shi Q, Geng M, Huang X, et al. Structural modification and pharmacological evaluation of substituted quinoline-5,8-diones as potent NSD2 inhibitors. J Med Chem. 2023;66(2):1634–1651.
- Hanley RP, Nie DY, Tabor JR, Li F, Sobh A, Xu C, Barker NK, Dilworth D, Hajian T, Gibson E, et al. Discovery of a potent and selective targeted NSD2 degrader for the reduction of H3K36me2. J Am Chem Soc. 2023;145(14):8176–8188.
- Wan G, Feng Z, Zhang Q, Li X, Ran K, Feng H, Luo T, Zhou S, Su C, Wei W, et al. Design and synthesis of Fibroblast Growth Factor Receptor (FGFR) and Histone Deacetylase (HDAC) dual inhibitors for the treatment of cancer. J Med Chem. 2022;65(24):16541–16569.
- Kawamata N, Chen J, Koeffler HP. Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses translation of cyclin D1 in mantle cell lymphoma cells. Blood. 2007;110(7):2667–2673.
- Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the treatment of multiple myeloma. Clin Cancer Res. 2015;21(21):4767–4773.
- Liang X, Tang S, Liu X, Liu Y, Xu Q, Wang X, Saidahmatov A, Li C, Wang J, Zhou Y, et al. Discovery of Novel Pyrrolo[2,3-d]pyrimidine-based derivatives as potent JAK/HDAC dual inhibitors for the treatment of refractory solid tumors. J Med Chem. 2022;65(2):1243–1264.
- Ezponda T, Popovic R, Shah MY, Martinez-Garcia E, Zheng Y, Min D-J, Will C, Neri A, Kelleher NL, Yu J, et al. The histone methyltransferase MMSET/WHSC1 activates TWIST1 to promote an epithelial–mesenchymal transition and invasive properties of prostate cancer. Oncogene. 2012;32(23):2882–2890.
- Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114(13):2733–2743.
- Menderes G, Black J, Schwab CL, Santin AD. Immunotherapy and targeted therapy for cervical cancer: an update. Expert Rev Anticancer Ther. 2016;16(1):83–98.
- Gandhi J, Afridi A, Vatsia S, Joshi G, Joshi G, Kaplan SA, Smith NL, Khan SA. The molecular biology of prostate cancer: current understanding and clinical implications. Prostate Cancer Prostatic Dis. 2018;21(1):22–36.
- Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov. 2015;14(9):603–622.
- Macalino SJ, Gosu V, Hong S, Choi S. Role of computer-aided drug design in modern drug discovery. Arch Pharm Res. 2015;38(9):1686–1701.
- Maia EHB, Assis LC, de Oliveira TA, da Silva AM, Taranto AG. Structure-based virtual screening: from classical to artificial intelligence. Front Chem. 2020;8:343.
- Yang D‐S, Yang Y‐H, Zhou Y, Yu L‐L, Wang R‐H, Di B, Niu M‐M. A redox‐triggered bispecific supramolecular nanomedicine based on peptide self‐assembly for high‐efficacy and low‐toxic cancer therapy. Adv Funct Materials. 2019;30(4): 1904969.
- Zhou Y, Zou Y, Yang M, Mei S, Liu X, Han H, Zhang C-D, Niu M-M. Highly potent, selective, biostable, and cell-permeable cyclic d-peptide for dual-targeting therapy of lung cancer. J Am Chem Soc. 2022;144(16):7117–7128.
- Ucar DA, Kurenova E, Garrett TJ, Cance WG, Nyberg C, Cox A, Massoll N, Ostrov DA, Lawrence N, Sebti SM, et al. Disruption of the protein interaction between FAK and IGF-1R inhibits melanoma tumor growth. Cell Cycle. 2012;11(17):3250–3259.
- Thiyagarajan V, Lin SH, Chang YC, Weng CF. Identification of novel FAK and S6K1 dual inhibitors from natural compounds via ADMET screening and molecular docking. Biomed Pharmacother. 2016;80:52–62.
- Chao M-W, Lin TE, HuangFu W-C, Chang C-D, Tu H-J, Chen L-C, Yen S-C, Sung T-Y, Huang W-J, Yang C-R, et al. Identification of a dual TAOK1 and MAP4K5 inhibitor using a structure-based virtual screening approach. J Enzyme Inhib Med Chem. 2021;36(1):98–108.
- Opo F, Rahman MM, Ahammad F, Ahmed I, Bhuiyan MA, Asiri AM. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci Rep. 2021;11(1):4049.
- Zheng L, Ren R, Sun X, Zou Y, Shi Y, Di B, Niu M-M. Discovery of a Dual Tubulin and Poly(ADP-Ribose) polymerase-1 inhibitor by structure-based pharmacophore modeling, virtual screening, molecular docking, and biological evaluation. J Med Chem. 2021;64(21):15702–15715.
- Niu M, Wang F, Li F, Dong Y, Gu Y. Establishment of a screening protocol for identification of aminopeptidase N inhibitors. J Taiwan Inst Chem Eng. 2015;49:19–26.
- Zhou Y, Di B, Niu MM. Structure-based pharmacophore design and virtual screening for novel tubulin inhibitors with potential anticancer activity. Molecules. 2019;24(17):24.
- Fan J, Fu A, Zhang L. Progress in molecular docking. Quant Biol. 2019;7(2):83–89.
- Li X, Inks ES, Li X, Hou J, Chou CJ, Zhang J, Jiang Y, Zhang Y, Xu W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J Med Chem. 2014;57(8):3324–3341.
- Ding L-Y, Hou Y-C, Kuo I-Y, Hsu T-Y, Tsai T-C, Chang H-W, Hsu W-Y, Tsao C-C, Tian C-C, Wang P-S, et al. Epigenetic silencing of AATK in acinar to ductal metaplasia in murine model of pancreatic cancer. Clin Epigenetics. 2020;12(1):87.
- Gomha S, Salaheldin T, Hassaneen H, Abdel-Aziz H, Khedr M. Synthesis, characterization and molecular docking of novel bioactive thiazolyl-thiazole derivatives as promising cytotoxic antitumor drug. Molecules. 2015;21(1):E3.
- Ahlina FN, Nugraheni N, Salsabila IA, Haryanti S, Da’i M, Meiyanto E. Revealing the reversal effect of Galangal (Alpinia galanga L.) extract against oxidative stress in metastatic breast cancer cells and normal fibroblast cells intended as a co-chemotherapeutic and anti-ageing agent. Asian Pac J Cancer Prev. 2020;21(1):107–117.