
Abstract
Abnormal accumulation of branched-chain amino acids (BCAAs) can lead to metabolic diseases and cancers. Branched-chain α-keto acid dehydrogenase kinase (BCKDK) is a key negative regulator of BCAA catabolism, and targeting BCKDK provides a promising therapeutic approach for diseases caused by BCAA accumulation. Here, we screened PPHN and POAB as novel putative allosteric inhibitors by integrating allosteric binding site prediction, large-scale ligand database virtual screening, and bioactivity evaluation assays. Both of them showed a high binding affinity to BCKDK, with Kd values of 3.9 μM and 1.86 μM, respectively. In vivo experiments, the inhibitors demonstrated superior kinase inhibitory activity and notable antiproliferative and proapoptotic effects on diverse cancer cells. Finally, bulk RNA-seq analysis revealed that PPHN and POAB suppressed cell growth through a range of signalling pathways. Taken together, our findings highlight two novel BCKDK inhibitors as potent therapeutic candidates for metabolic diseases and cancers associated with BCAA dysfunctional metabolism.
Introduction
BCAAs are essential amino acids composed of leucine, isoleucine, and valineCitation1–3. They play pivotal roles in various life activities, including protein synthesis, lipogenesis, gluconeogenesis, neurotransmitter synthesis, and energy supportCitation4–8. As shown in , the metabolism of BCAAs involves two significant processesCitation4,Citation9,Citation10. In the first reaction, a reversible reaction, BCAAs are catalysed by branched-chain aminotransferase (BCAT) to generate branched-chain α-keto acids (BCKAs). In the second reaction, an irreversible reaction, oxidative decarboxylation of BCKAs is catabolized by the BCKA dehydrogenase complex (BCKDC) to produce branched-chain acyl-CoA intermediates, which contribute to subsequent catabolic pathways. The activity of BCKDC is regulated dynamically in the form of phosphorylation and dephosphorylation of BCKA dehydrogenase E1 subunit alpha (BCKDHA, also known as E1α), an important component of BCKDCCitation11,Citation12. BCKDHA can be phosphorylated and inactivated by BCKA dehydrogenase kinase (BCKDK)Citation1, while mitochondrial phosphatase 2 C (PP2Cm) can dephosphorylate and activate BCKDHACitation13. Previous studies focused on the regulated elements, such as BCAT and BCKDK, as drug targets for the treatment of BCAA metabolic diseasesCitation14,Citation15.
Figure 1. BCAA metabolism-related pathways reveal the potential to target BCKDK for the development of inhibitors to treat diseases caused by BCAA accumulation. This figure shows the detailed process of BCAA metabolism. BCAAs enter the cell through large amino acid transporters (LATs) on the cell membrane, and their catabolism consists of two main steps. The first step is reversible transamination catalysed by branched-chain aminotransferases (BCATs), which include cytoplasmic BCATc and mitochondrial BCATm. BCAT transfers the α-amino group of BCAAs to the amino receptor, usually α-ketoglutarate (α-KG), resulting in the production of glutamate (Glu) and the corresponding branched-chain-α-keto acids (BCKAs), such as α-keto-β-methylvalerate (KMV), α-ketoisovalerate (KIV), and α-ketoisocaproate (KIC). The second step is the irreversible oxidative decarboxylation of BCKA, catalysed by branched-α-keto acid dehydrogenase (BCKDH) in mitochondria. This process generates the corresponding branched-chain acyl coenzyme A derivative (R-CoA), which enters the TCA cycle to provide energy to the organism. BCKDH comprises three subunits: E1, E2, and E3. When BCKDK (BDK) acts on the LBD structural domain of the E2 subunit of BCKDH, it phosphorylates the E1 α subunit, leading to the inactivation of BCKDH. Conversely, PP2Cm (BDP) plays a regulatory role in dephosphorylation and activation. Imbalances in the BCAA metabolic pathway are associated with the development of various diseases. Furthermore, the cross-talk between BCKDK of BCAA catabolism and the MAPK pathway promotes tumorigenesis in colorectal cancer. Additionally, leucine accumulation activates the mTORC1 signalling pathway, promoting protein and nucleic acid synthesis and contributing to tumour development. Therefore, screening for high-affinity and high-specificity BCKDK inhibitors could provide a research basis for the design and development of drugs to treat various diseases triggered by BCAA accumulation and upregulation of BCKDK.
Abnormal accumulation of BCAAs in circulation has been reported to be closely related to the occurrence and development of metabolic disordersCitation16, diverse cancersCitation17, and nervous system diseasesCitation18–22. For example, high levels of BCAAs and BCKAs have been reported to be associated with insulin resistanceCitation23–29, obesityCitation30, type 2 diabetesCitation31,Citation32, and heart diseasesCitation33–36. In addition, the overactivation of some signalling pathways, such as mTOR, by BCAAs can lead to the occurrence of cancersCitation37–40. It has been demonstrated that the inhibition of BCKDK can reduce BCAA and BCKA concentrations in plasmaCitation41,Citation42. Previous studies also found that BCKDK can promote colorectal carcinogenesis through activation of the MEK/ERK (MAPK) signalling pathwayCitation43, suggesting a relationship between BCKDK metabolism and carcinogenesis. Therefore, BCKDK inhibitors are great candidates to reset the irregular metabolism of BCAAs and achieve metabolic disease and cancer treatmentCitation15.
In recent studies, several BCKDK allosteric inhibitors have been found by high-throughput screenings and structure-based designCitation44,Citation45. Of these compounds, 3,6-dichlorobenzo [b] thiophene-2-carboxylic acid (BT2) exhibits the greatest efficacy against BCKDK and is commonly employed as a BCKDK inhibitor. It can change the conformation of BCKDK and reduce the phosphorylation of BCKDHA by binding to the N-terminal domain in a unique allosteric site of BCKDK. The inhibitors can reduce the concentration of BCAAs in vivo by sensitising BCKDC. However, the limited size of the BT2 binding site constrains the optimisation of its structural design for a compound with improved activity. Additionally, BT2 not only inhibits BCKDK but is also a potent and selective Mcl-1 inhibitorCitation46. Novel BCKDK inhibitors with higher specificity and activity are urgently needed for pharmacological applications.
In this study, we aimed to identify and evaluate novel putative allosteric inhibitors to develop new therapeutic compounds for metabolic diseases and cancers caused by BCAA accumulation. Allosteric binding site prediction, virtual screening, and various bioactivity evaluation assays were conducted, which resulted in two novel inhibitors, PPHN and POAB. These inhibitors exhibit favourable binding affinity and significant kinase inhibitory activity towards BCKDK. The compounds were also found to inhibit cell proliferation and promote apoptosis in various cancer cell lines and affect BCAA metabolism and signalling pathways. This study provides a research basis for further structural optimisation of lead compounds and the development of treatments for diseases caused by BCAA accumulation.
Results
Novel allosteric sites of BCKDK
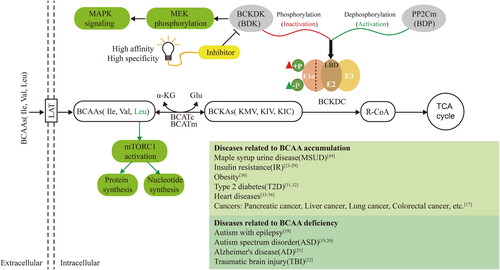
As illustrated in , BCKDK is a drug target for the treatment of metabolic diseases caused by BCAA accumulation. We thus utilised the CavityPlus web server to systematically identify allosteric sites of BCKDK. illustrates the fifteen potential ligand binding sites predicted employing the Cavity submodule of the web server, utilising the crystal structure of BCKDK (PDB ID: 3TZ0). Among these, site 4 was within the orthosteric site of BCKDK, while site 2 had been previously reported as an allosteric site for BCKDKCitation44,Citation45. Subsequently, with site 4 set as the orthosteric site, the allosteric properties of the remaining 14 sites were predicted with the CorrSite submodule of CavityPlus. The summarised results of the Cavity and CorrSite analyses are presented in , highlighting that site 1, 2, 3, and 5 exhibited better druggability. Notably, site 2 displayed the strongest DrugScore but exhibited a smaller surface area and volume, potentially limiting its accommodation of compounds with larger molecular weights. Moreover, it displayed the lowest allosteric score. In contrast to site 2, site 5 demonstrates good druggability and the highest allosteric score. Furthermore, site 5 is situated diagonally opposite to the orthosteric site (as depicted in ). The cavity associated with site 5 originates from the interior of the protein, extending towards the protein surface. This cavity offers the appropriate spatial dimensions, making it favourable for binding compounds with larger molecular weights and facilitating structural modifications. Based on these findings, we selected site 5 as the target allosteric site for BCKDK (as shown in ) for subsequent structure-based virtual screening.
Figure 2. The virtual screening process for BCKDK inhibitors. The screening of BCKDK inhibitors consists of the following major parts: binding site prediction, structure-based virtual screening and bioactivity evaluation. (A–C) The results of allosteric site prediction based on the crystal structure of BCKDK. (A) Fifteen ligand binding sites were predicted by the CavityPlus server based on the crystal structure of BCKDK (PDB ID: 3TZ0). (B) The following sites are displayed: Site 4 represents the orthosteric site, Site 2 corresponds to the allosteric site reported in the literature, and Site 5 is a novel allosteric site selected in this study. (C) The orthosteric site contains the ADP structure shown in orange stick, while the targeted allosteric site (with key amino acid residues shown in green stick) is the focus of this study. (D) The specific screening steps for BCKDK inhibitors after determining the ligand binding site: Step 1: Screening 11.4 million compounds from the VirtualFlow Real Library, which contains 1.4 billion compounds, using custom ligand screening conditions. The predicted novel allosteric site (Site 5) was defined as the docking site (site space information: centre_x = 5.945, centre_y = -26.304, centre_z = 17.208, size_x/y/z = 22.4). Step 2: Employing the rigid docking software Qvina2 and Smina, 11.4 million compounds were docked into the target site. The top 25,000 compounds with the highest docking scores of each software were retained, resulting in a primary screening database of 47,702 compounds after eliminating duplicates. Step 3: Setting the amino acid residues within 8 Å of the binding site as flexible residues, the compounds in the primary screening database were docked using the platform’s own flexible docking software, Vina. The top 200 compounds were screened to form the fine screening database. Step 4: Hermite software was used to predict the ADMET properties of the compounds in the fine screening database. Based on the prediction of ADMET properties of the compounds through the Hermite Platform (https://hermite.dp.tech, DP Technology), compounds with high solubility, favourable bioavailability, and low toxicity were selected. Furthermore, considering the key interactions between the compounds and key amino acid residues in the receptor binding site, seven compounds that exhibited both hydrogen bonding and hydrophobic interactions were identified as theoretical hit compounds (hits). Step 5: Biological activity evaluation assays were performed on the hits, leading to the identification of three lead compounds. (E) The interactions between the seven hits and the key amino acid residues within the targeted site were examined. The compound structures are shown as yellow sticks, while the key amino acid residues within the site are indicated by green sticks. All these compounds can form hydrogen bond interactions with ARG371 or THR41 residues and occupy the hydrophobic cavity composed of TYR349, MET172, LEU46, and other crucial amino acid residues.
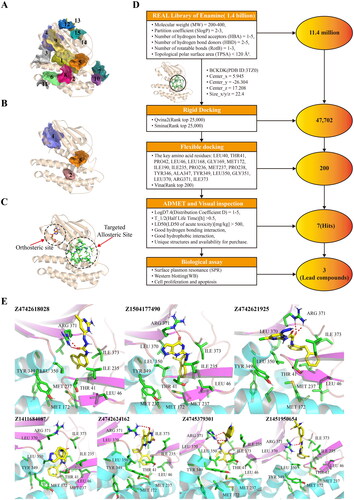
Table 1. Summary of the results of Cavity and CorrSite.
Structure-based virtual screening
Allosteric site 5 was targeted through structure-based virtual screening, as depicted in the flowchart in . For molecular docking, we utilised VirtualFlow, an ultralarge and open-source drug discovery platform. First, we selected 11.5 million compounds as the original ligand library from the REAL Library database, applying the Lipinski rule of fiveCitation47,Citation48 and considering the ligand library size. Second, 50,000 compounds were selected by performing rigid docking. Third, after removing duplicates, 47,702 compounds were screened using flexible docking, and the top 200 compounds were selected for subsequent analysis. Finally, we applied ADMET filters and assessed the ligand-receptor interactions of these compounds to identify seven theoretical hit compounds (hits) that met the following criteria: (1) satisfactory ADMET parameters, (2) plausible interactions, (3) both hydrophobic and hydrogen bonding interactions, (4) unique structures, and (5) availability for purchase from vendors. Table S1 summarises the basic information of these seven hits, including the Enamine ID, chemical structure, molecular weight, docking score, and a few key ADMET results. As shown in , all seven compounds can form 1–2 hydrogen bonds with residues ARG 371 or THR 41 and are located in the hydrophobic cavity formed by residues TYR 349, MET 172, LEU 46, and other crucial amino acid residues of the site.
Candidate lead compounds
We expressed and purified the recombinant BCKDK-His6-tag protein using the E. coli expression system. The successful expression and purification of the protein were confirmed by SDS–PAGE results shown in . Furthermore, the seven hit compounds were procured and subjected to surface plasmon resonance (SPR) assays to determine their binding affinity. The dissociation equilibrium constant (Kd) values for the seven hits obtained from the steady-state affinity model in the SPR results are presented in Table S1. Among them, Compounds 1, 3, and 4 exhibited favourable binding affinity towards BCKDK, with Kd values of 12.99 μM, 3.9 μM, and 1.86 μM, respectively. Based on their promising binding affinity, Compounds 1, 3, and 4 were identified as lead compounds, and were abbreviated as PCAB, PPHN, and POAB, respectively. The SPR steady-state affinity fitting curves of these three lead compounds are shown in . illustrates the chemical structure, International Union of Pure and Applied Chemistry (IUPAC) nomenclature, abbreviation, Enamine ID, and Kd values of the lead compounds.
Figure 3. Lead compounds were screened using surface plasmon resonance (SPR) experiments and kinase activity assessment. (A) SDS–PAGE analysis of recombinant BCKDK-His6-tag protein, with a band size of the target protein equal to 47 kDa. (B) Fitting results obtained from surface plasmon resonance (SPR) experiments for the lead compounds. Dashed lines indicate the positions of Kd values. (C) Structure, custom abbreviated name, Enamine ID, and Kd values of each lead compound. In the compound structures, the red group represents the portion that forms hydrogen bonding interactions with key amino acid residues at the binding site, the blue group represents the segment that occupies the hydrophobic cavity of the site, and the black group serves as a linker. (D) The proliferation inhibition rate of 293T cells by PCAB, PPHN, POAB and BT2 at different concentrations from 0.05 μM to 500 μM. (E) Western blotting results after treating 293T cells with the compounds at a concentration of 40 μM for 48 hours. (F) Quantitative results based on the graph in (D), indicating that both compounds PPHN and POAB significantly inhibited the phosphorylation of BCKDHA by BCKDK, with a stronger inhibitory effect compared to that of BT2.
Prior to investigating the inhibitory effect of the three lead compounds on BCKDK in vivo, we assessed the concentration of the administered compounds by employing the CCK-8 kit. Specifically, the effects of three lead compounds and a positive control compound, BT2, on the viability of 293 T cells were assayed within a concentration range of 0.05 to 500 μM. By fitting the curves as presented in , a dose-dependent response towards cell proliferation of 293 T was observed for these four compounds. The IC50 values for PCAB, PPHN, POAB, and BT2 on 293 T cells were calculated to be 10.17 μM, 10.17 μM, 15.6 μM, and 247.4 μM, respectively. Hence, a concentration that induces a marginally greater inhibitory effect than that corresponding to the IC50 value of the lead compounds, which is 40 μM, was selected for administration in the cellular experiments.
To examine the inhibitory effect of the three lead compounds on BCKDK in vivo, we conducted Western blotting experiments using the 293 T cell line and measured the quantity of phosphorylated BCKDHA, a downstream substrate of BCKDK. The results depicted in demonstrate that all three lead compounds inhibited the function of BCKDK at a concentration of 40 μM. Notably, both PPHN and POAB exhibited a significant inhibitory effect, as evidenced by the pBCKDHA/BCKDHA ratio, which surpassed that of the positive control BT2 ().
The lead compounds affect tumour cell proliferation and apoptosis
Due to its potential as a therapeutic target for BCAAs-associated diseases, BCKDK has garnered significant attentionCitation15. Consequently, we investigated the effects of inhibiting BCKDK through PCAB, PPHN, and POAB on the proliferation and apoptosis of the following distinct cell lines: human embryonic kidney 293 T cells, human lung cancer A549 cells, and human colorectal cancer HCT116 cells.
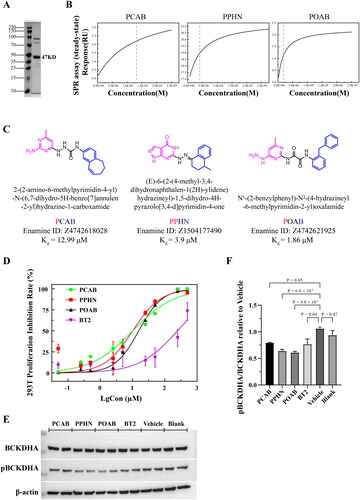
The CCK-8 assay results, displayed in , indicate the effect of PCAB, PPHN, POAB, and BT2 on cell proliferation at a concentration of 40 μM. As depicted in the figure, both PPHN and POAB showed significant inhibitory effects on the proliferation of all three cell types, and PPHN exhibited the best inhibitory effect, inhibiting almost 87% of 293 T cells, 80% of A549 cells, and 90% of HCT116 cells at 72 h, while POAB inhibited 85% of 293 T cells and 55% of A549 and HCT116 cells. PCAB, on the other hand, inhibited 75% of 293 T cells but showed no significant inhibitory effect on the proliferation of A549 and HCT116 cells. The literature reports that the positive control compound BT2 has an inhibitory effect on HCT116 tumour cells at 100–250 μMCitation40. Our results demonstrate that PPHN and POAB showed superior inhibition of the proliferation of 293 T, A549, and HCT116 cells compared to BT2 cells when the dosage was relatively low.
Figure 4. Effect of lead compounds on cell growth of normal 293 T cells and tumour cells (A549 and HCT116). (A) Cell viability results of different compounds (40 μM) measured using the CCK-8 assay after 24 hours, 48 hours, and 72 hours of treatment. All three lead compounds inhibited the proliferation of normal 293T cells, and compounds PPHN and POAB also inhibited the proliferation of A549 and HCT116 tumour cells. (B) Apoptosis analysis of different compounds (40 μM) on different cells, as measured using flow cytometry. Q1: Necrotic cells, Q2: Late apoptotic cells, Q3: Early apoptotic cells, Q4: Live cells. (C) Quantitative statistics of the apoptotic cells (Q2 + Q3) in the graph from (B). The compound PPHN demonstrated significant proapoptotic effects on all three cell types.
presents the results of the apoptosis assay, revealing that PPHN significantly promoted the apoptosis of all three types of cells at a concentration of 40 μM, with the percentage of apoptotic cells being approximately 2%, 4%, and 8%, respectively. POAB significantly promoted the apoptosis of HCT116 cells, with the percentage of apoptotic cells being approximately 7%. In contrast, the control compound BT2 caused no significant effect on the three cell types.
In summary, these results show that PPHN and POAB exert promising effects on antitumor cell proliferation and protumor cell apoptosis, suggesting their potential therapeutic applications in tumour treatment.
The underlying mechanisms of lead compounds
In our previous experiments, we observed significant antiproliferative and proapoptotic effects of PPHN and POAB on 293 T, A549, and HCT116 cell lines. To unravel the underlying mechanism, we conducted a bulk RNA-seq analysis to assess the effects of PPHN and POAB on the transcriptome of these three cell lines.
To investigate the impact of inhibition by BCKDK on BCAA metabolism, we conducted a transcriptome-level analysis of the expression differences of 38 genes involved in BCAA metabolism across various groups (). The results indicate that the majority of genes related to BCAA metabolism were affected. Particularly in HCT116 cells, the expression of ACAA2, ACAT2 and HADH was significantly downregulated. Furthermore, BCAT1 was significantly upregulated in 293 T and A549 cells, while ACADM and DLD showed significant upregulation across all three cell lines. Conversely, BCAT2 and ACAD5 were significantly downregulated in 293 T and A549 cells. It is worth mentioning that the aminotransferases BCAT1 and BCAT2 exhibited contrasting alterations in this context, potentially attributable to variances in their tissue distribution and cellular localizationCitation1,Citation17. Notably, ACAA2, ACAT2, HADH, ACADM, and ACAD5 are also involved in fatty acid metabolism, playing vital roles in energy generation through beta-oxidation, while DLD is crucial in the tricarboxylic acid (TCA) cycle. Consequently, the observed differential expression patterns suggest potential disruptions in BCAA metabolism, fatty acid metabolism, and overall energy production in the respective cell lines.
Figure 5. Comprehensive analysis of the results obtained from the bulk RNA-seq assay. (A) Heatmap illustrating the expression levels of some differentially expressed genes (DEGs) involved in BCAA metabolism. These genes were identified as being impacted by PPHN and POAB treatment across three distinct cell types at the transcriptome level. (B) Bubble plot highlighting the pathways that were specifically affected by PPHN and POAB and selected from the top 40 signalling pathways enriched with DEGs in Supplementary Figure 1. The analysis was performed using an adjusted p value cut-off of 0.05. The DEGs were derived from 293T, A549, and HCT116 cell lines treated with PPHN and POAB. The plot was divided into two columns representing downregulation and upregulation. The colour bar indicates the adjusted p value of the pathway, calculated using clusterProfiler. These results offer valuable insights into the molecular changes induced by PPHN and POAB, shedding light on their impact on cellular processes and potential therapeutic implications.
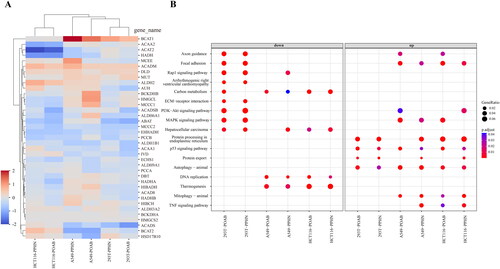
To clarify the fundamental molecular mechanisms of these compounds within the distinct contexts of HCT116, A549, and 293 T cells, KEGG pathway enrichment was utilised to analyse the deeper implications of the gene expression changes. Figure S1 illustrates the top 40 signalling pathways enriched with the differentially expressed genes, while presents the specific pathways consistently affected by both compounds. The results suggest that the compounds’ antiproliferative and proapoptotic effects may be linked to the downregulation of specific pathways in 293 T cells, including axon guidance, focal adhesion, Rap1 signalling pathway, PI3K-Akt signalling pathway, ECM-receptor interaction, arrhythmogenic right ventricular cardiomyopathy, MAPK signalling pathway, and hepatocellular carcinoma, as well as DNA replication and thermogenesis in cancer cells. Notably, the MAPK and PI3K-Akt (PI3K/Akt/mTOR) signalling pathways align with the signalling pathways associated with BCAA metabolism, as previously outlined in the Introduction. These pathways are known to play critical roles in cell growth and survival, and their downregulation can lead to decreased cell proliferation and increased apoptosis. Interestingly, the observed proapoptotic effects of the compounds may also be linked to the upregulation of pathways involved in regulating cellular stress responses, including protein processing in the endoplasmic reticulum, protein export, p53 signalling pathway, and autophagy-animal in all three cell types. These pathways are responsible for detecting and responding to cellular stress, and their upregulation suggests that the compounds induce stress in the cells, ultimately leading to apoptosis. The bulk RNA-seq findings offer significant insights into the potential mechanisms that underlie the observed impacts of the compounds on cell proliferation and apoptosis.
Revealing the possible allosteric effects of lead compounds binding to BCKDK through molecular dynamics simulations
To elucidate the potential mechanisms underlying the allosteric inhibition of BCKDK by lead compounds, we designed six systems of lead compounds targeting the allosteric sites of BCKDK studied in this paper with or without ADP binding to the orthosteric site. Molecular dynamics simulations lasting 100 ns were conducted for each system. It is noteworthy that the temporal progression of root-mean-square deviation (RMSD) values for the proteins and ligands of each system, as depicted in , indicates that the latest system attains convergence to stability at approximately 80 ns. The total binding free energies (ΔGtotal) afford valuable insights into the affinities of the lead compounds for the binding site in the presence or absence of ADP. Strikingly, PPHN and POAB had heightened binding affinities compared to PCAB, consistently aligning with experimental findings. The enhanced binding affinity of the lead compounds to the allosteric site in the absence of ADP binding at the orthosteric site is noteworthy.
Figure 6. Molecular dynamics simulations between lead compounds and BCKDK in the presence and absence of ADP. (A) Time evolution of RMSD values of protein/ligand and total binding free energy for the six protein-ligand interaction systems. (B) RMSF values and B-factor scores during molecular dynamics simulations. The numerical labels in the figure correspond to the positions of the residues in the original crystal structure (PDB ID: 3TZ0). (C) The structures of the proteins are coloured according to their B-factor, and B-factor putty superimposed with cartoons is presented. Warmer colours (red) denote higher flexibility, and cooler colours (blue) indicate lower flexibility. (D) The conformation of PPHN and POAB within the allosteric site, along with their hydrogen bonding interactions with crucial amino acid residues, are shown during the steady state of the MD simulation. The compounds are depicted as green sticks to signify the absence of ADP, while yellow sticks represent the presence of ADP. The protein structure is illustrated in cartoon mode in cyan, while the essential amino acid residues are depicted as white sticks, and the yellow dashed lines indicate hydrogen bonds.
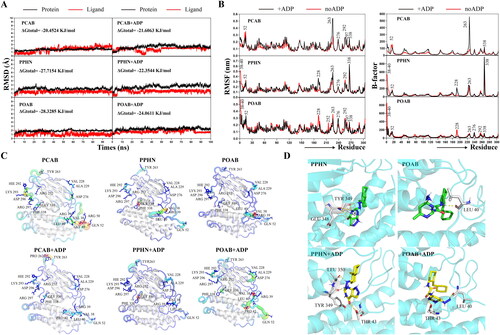
The flexibility of residues during simulations is revealed through the analysis of root-mean-square fluctuation (RMSF) results and B-factor scores, as shown in . The structure of the protein is characterised by B-factor scores, as elucidated in . It is evident that the interaction of lead compounds with the protein induces significant conformational changes, including the orthosteric site where residues LYS 293, ARG 252, ARG 297, GLY 339 and PHE 338 are located and the substrate binding site where residues VAL 38, LEU 40, GLN 52 and VAL 228 are located. This may be a potential mechanism by which the lead compounds exert an allosteric inhibitory effect. The steady-state conformation of PPHN and POAB bound to BCKDK is depicted in in both the presence and absence of ADP. The distinct conformations within the allosteric site are revealed. Notably, hydrogen bonding interactions with key amino acid residues, particularly residues GLU 348 and TYR 349, as well as residue LEU 40, emerge as crucial factors supporting the binding of PPHN and POAB, respectively. Additionally, upon the engagement of ADP at the orthosteric site, PPHN exhibits changes in interactions with residues THR 43, TYR 349 and LEU 350, while POAB shows increased interactions with residue THR 43. These findings reveal the potential allosteric effects induced by lead compounds upon their interaction with target proteins, providing an interpretation that aligns with experimental observations.
Consequently, understanding this mechanism establishes a fundamental foundation for future endeavours in compound screening, wherein molecular dynamics simulations can be employed to screen more promising BCKDK inhibitors.
Discussion
In this study, we have developed a novel allosteric site in BCKDK, which is located in the C-terminal domain and opposite to the orthosteric site and was predicted by the CavityPlus server. This site is different from another well-studied allosteric site discovered by Shih-Chia Tso et al.Citation44. The new site exhibits a higher allosteric score and a larger space, making it a promising target for developing BCKDK allosteric inhibitors and more amenable for structural optimisation. Instead, BT2 possesses a smaller ligand binding space and is surrounded by many helices, which limits the optimisation of inhibitors. This may explain why BT2 has not yet entered preclinical studies.
The abnormal accumulation of branched-chain amino acids (BCAAs) has been linked to various metabolic disorders and cancersCitation16,Citation17. BCKDK, a crucial negative regulator of BCAAs, can be inhibited by phenyl butyric acid (PB), a BCKDK inhibitor, resulting in reduced BCAA accumulation in patients with moderate MSUDCitation49. Additionally, PB can impede the direct phosphorylation of MEK by BCKDK, thereby decreasing the activation of the MAPK signalling pathway and, consequently, tumorigenicityCitation43. Furthermore, BT2 exhibited superior BCKDK inhibition efficacy than PB but has demonstrated growth inhibition of colorectal cancer cells at a higher dosageCitation40. Therefore, the identification of more potent BCKDK inhibitors is crucial for the therapeutic management of tumours characterised by the abnormal accumulation of BCAAs or the aberrant activation of the MAPK signalling pathway. In this study, we discovered two novel putative allosteric inhibitors, PPHN and POAB, with strong binding affinity and kinase inhibitory activity of BCKDK, by conducting surface plasmon resonance (SPR) and western blotting assays. At a concentration of 40 μM, these compounds significantly inhibited BCKDK phosphorylation on the downstream substrate BCKDHA. Additionally, these compounds exhibited noteworthy antiproliferative and proapoptotic effects on 293 T, A549 and HCT116 cell lines. These findings suggest that these two compounds hold promise as BCKDK inhibitors for use in tumour treatment. However, it is worth noting that although both PPHN and POAB showed comparable inhibitory effects on BCKDHA phosphorylation, PPHN exhibited a more pronounced inhibitory effect than POAB in cellular activity assay experiments. This finding suggests that PPHN may have an additional impact on other enzymes. Therefore, further investigations are required to determine whether these compounds selectively target BCKDK.
The effectiveness of a large-scale screening library in reducing the false-positive rate and improving the quality of lead compounds has already been establishedCitation50. Such high-quality leads show the potential to lower dose delivery, minimise off-target effects, and improve ADME/T propertiesCitation51. In this research, we applied VirtualFlow, a large-scale open-source drug discovery platform, to conduct molecular docking-based virtual screening. Initially, 11.4 million compounds from its own Real Library database were built as a large-scale ligand library, and 720 CPU cores were used for parallel computing. This approach has increased the calculation speed and improved the efficiency of this virtual screening. In the present study, we found two specific BCKDK inhibitors, PPHN and POAB, based on the inhibition of BCKDK phosphorylation on the downstream substrate BCKDHA, as well as the strong binding affinity and kinase inhibitory activity of BCKDK, by conducting SPR and Western blotting assays. Furthermore, these two BCKDK inhibitors, PPHN and POAB, exert superior pharmacological antiproliferative and proapoptotic effects on 293 T, A549 and HCT116 cancer cells.
Through utilising bulk RNA-seq experiments, we elucidated the crucial role of BCKDK in linking BCAA metabolism and cancer-related pathways. The antiproliferative and proapoptotic effects resulting from the inhibition of BCKDK by PPHN and POAB can be linked to the disruption of genes associated with BCAA metabolism and the impaired lipid metabolism and energy production of cells. Because BCAA metabolism has been reported to be associated with lipid metabolismCitation52–54 and energy productionCitation10. Furthermore, KEGG enrichment analysis revealed the downregulation of intracellular signalling pathways, including MAPK and PI3K-Akt (PI3K/Akt/mTOR) in 293 T, and DNA replication in A549 and HCT116. In addition, upregulation of autophagy was observed across all combinations of cell lines and compounds, which may be related to the antiproliferative and proapoptotic behaviours observed in CCK-8 and apoptosis assays. The mTOR pathway is a nutrient-sensing signalling pathway that can be activated by insulin and BCAAsCitation55–58, thereby promoting protein synthesis for cell growth. Consequently, the role of BCKDK as a hub connecting the BCAA metabolic pathway with cancer-associated signalling pathways underscores the criticality of targeting BCKDK in the treatment of BCAA-accumulated cancers.
However, it is essential to consider that different tissues and organs may exhibit varying expression levels of BCKDK due to the regulation requirements of BCAACitation59. For instance, in the malignant stage of advanced tumours or in tissues such as skeletal muscle, which require BCAAs for anabolism, the metabolic consumption of BCAAs can impair protein anabolism, leading to cancer cachexia and muscle atrophyCitation60,Citation61. Hence, subsequent studies will require a deeper understanding of the complex mechanisms by which BCKDK inhibitors function in different tissues, organs and disease stages.
To advance our understanding, further research should investigate the mechanism by which the identified inhibitors affect the structural changes in BCKDK, optimise their structures for improved antitumor drugs targeting BCKDK, and explore the potential of these compounds in vivo, such as in animal models of metabolic diseases and cancers caused by BCAA accumulation.
Methods
Predicting novel allosteric sites for BCKDK
The CavityPlus serverCitation62 was used to predict potential allosteric sites for BCKDK. First, all possible ligand-binding sites in the entire BCKDK protein were identified by the CavityCitation63,Citation64 submodule, and the orthosteric site was determined by combining the information of predicted cavities and available literatureCitation44. Next, based on the parameter setting of “not excluding the orthosteric site”, the allosteric properties of all of the above predicted cavities were scored using the CorrSiteCitation65,Citation66 submodule. Then, sites with a score greater than 0.6 were marked by one star as potential allosteric sites, and the highest one was marked by two stars and designated as a novel allosteric site of BCKDK for subsequent virtual screening.
Virtual screening
The VirtualFlowCitation51,Citation67,Citation68 platform was used to discover novel drug candidates by targeting the BCKDK allosteric site and carrying out virtual screening. A custom ligand library that included 11.4 million compounds was built from its REAL Library database through the following filtering criteria: molecular weight (MW) = 200–400, partition coefficient (SlogP) = 2–3, number of hydrogen bond acceptors (HBA) = 1–5, number of hydrogen bond donors (HBD) = 2–5, number of rotatable bonds (RotB) = 1–3 and topological polar surface area (TPSA) < 120 Å2. In the rigid docking step, Qvina02 and Smina (built-in docking programs in VirtualFlow) were used to perform docking and scoring. Based on the scoring ranking, the top 25,000 compounds of each procedure were selected. After duplicates were eliminated, the remaining 47,702 compounds were used to construct the initial screening database. In the flexible docking step, the residues within 8 Å from the centre of the allosteric site were defined as the key amino acid residues and set as flexible amino acids in the configuration file of Vina (built-in docking programs in VirtualFlow). The top 200 compounds ranked by flexible docking scores were selected as the fine screening database. Next, the ADMET properties of these 200 compounds were predicted by using the Hermite Platform (https://hermite.dp.tech, DP Technology), and compounds with high toxicity and poor solubility were excluded. PyMOL (The PyMOL Molecular Graphics System, version 2.4.1) was then used to observe the interactions between these compounds and the key amino acid residues in the site, and compounds with good hydrogen bonding interactions and hydrophobic interactions were reserved. Finally, the structural similarity of compounds and their commercial availability were considered, and seven candidate hits were selected and purchased from TargetMol (TargetMol, Boston, USA).
Expression and purification of BCKDK
The human BCKDK sequence (Gene ID: 10295, UniProt KB: O14874) was used to generate the pQLink-MBP-3C-BCKDK-His6-tag expression vector. The recombinant protein was expressed in Rosetta competent cells at 20 °C for 20 h. After expression, cell pellets were harvested and lysed in 5 volumes (w/v) of lysis buffer (25 mM HEPES, 300 mM NaCl, 15% glycerol, 1 mM PMSF, DNAse, 1 mM MgCl2, pH 8.0), and Ni-NTA affinity chromatography was used for the purification of recombinant protein. After the samples were washed 10 times, the Ni-NTA bead volume of buffer A (50 mM HEPES, 500 mM NaCl, 1 mM PMSF, 0.5 mM EDTA, 1 mM β-mercaptoethanol, pH 8.0) with 5 mM and 30 mM imidazole recombinant protein was digested with HRV 3C protease on the column. Then, through elution with a 3-fold column volume of buffer B (50 mM HEPES, 300 mM NaCl, 1 mM PMSF, 0.5 mM EDTA, 1 mM β-mercaptoethanol, pH 8.0) containing 500 mM imidazole, BCKDK-His6-tag was purified on an AKTA system (AKTA purifier) by equilibration and stored in buffer C (50 mM HEPES, 250 mM NaCl, 250 mM KCl, 300 mM arginine, 2 mM MgCl2, 20 mM β-mercaptoethanol, pH 8.0).
Binding affinity detection assay
The binding affinity between seven hits and BCKDK-His6-tag was assessed using the surface plasmon resonance (SPR) technique. A Biacore T200 instrument and an NTA sensor chip were used. First, 350 mM EDTA solution was used as the regeneration solution on the F1 and F2 channels for surface condition adaptation, and the flow rate was set at 10 μL/min at 25 °C for 1 min. Then, the system was stabilised to output a smooth test baseline. The F2 channel was injected with 0.5 mM NiCl2 solution at a flow rate of 10 μL/min at 25 °C for 1 min. Then, the His-tagged BCKDK protein was captured at a concentration of 10 µg/ml on the F2 channel, and the flow rate was set to 10 μL/min at 25 °C for 3 min. The small molecules were dissolved in 1x PBS-P + 2% DMSO buffer and prepared into test solutions with concentrations of 50 μM, 25 μM, 12.5 μM, 6.25 μM, 3.125 μM, 1.5625 μM and 0.78125 μM. The dissociation of small molecules from BCKDK was recorded at a flow rate of 30 μL/min on the chip surface. After dissociation, the small molecules remaining on the NTA chip were eluted with a 350 mM EDTA solution as regeneration solution to release the binding sites of the receptor protein. After the test, the equilibrium dissociation constant Kd was analysed using the “Affinity” model in Biacore T200 evaluation software version 2.0.
Western blotting
Cells were spread into 6-well plates at a concentration of 2 x 105 cells/ml, 2 ml per well, two biological replicates and two technical replicates were performed. After incubating for 24 h, the medium was replaced with fresh medium containing 40 μM compound. After further incubation for 48 h, cell pellets were washed three times in cold PBS and resuspended in 50–150 µl of lysis solution (abs9116, Absin, China). Next, western blotting was performed according to the standard procedure, in which a PVDF membrane (Immobilon®-P, Millipore) was used for protein transfer and 5% non-fat dry milk (P1622, Applygen Technologies, Inc., Beijing, China) in TBST (20 mM Tris-HCl pH = 8.0, 150 mM NaCl, 0.1% Tween 20) was used as a block between incubations. The protein concentration of each sample was diluted to 2 µg/µl in the lysis buffer and the protein loading volume was performed as 18 µl. The specific antibodies used for this assay are described below: rabbit anti-BCKDHA (A303-790A-T, Bethyl, 1:10,000), rabbit anti-phospho-BCKDHA (A304-672A-T, Bethyl, 1:10,000), rabbit anti-β-actin (abs132001, Absin, 1:2500), and goat anti-rabbit IgG-HRP (abs20040, Absin, 1:10,000).
Cell Counting Kit-8 (CCK-8) assay
Cell Counting Kit-8 (CCK-8, DC101-02, DiNing, China) was used to evaluate the viability of 293 T, A549 and HCT116 cells after different treatments. In this study, the HCT116 cell line (RRID: CVCL_0291) was a gift from Chuanhui Han lab at Peking University, and this cell line was obtained from the Cell Resource Centre, Peking Union Medical College (PCRC). The 293 T cell line (ATCC CRL-3216, RRID: CVCL_0063) was a gift from Peng Du lab at Peking University. The A549 cell line was acquired from the Vector Core at CIBR (ATCC CCL-185, RRID: CVCL_0023). And the culture condition of all cell lines was consistently set to 37 °C, 5% CO2, and humidified air. Cells were seeded in 96-well plates at 3 x 104 cells/ml in 200 µl per well and three duplicate wells were set for each concentration. After culturing in an incubator of 5% CO2 and 37 °C for 24 h, the medium was replaced with a medium containing the target compound and proceeded to incubate for 48 h. Next, the medium was replaced with 100 µl fresh medium containing 10% CCK-8 reagent, the cells were incubated for 2–4 h in the incubator, and the absorbance was measured at 450 nm with a microplate reader (The Spark® multimode microplate reader, Tecan, Switzerland).
Apoptosis assay
Cells were seeded into 6-well plates at a concentration of 2 x 105 cells/ml and a volume of 2 ml per well. Three biological replications were set in this study. After cell adherence, the medium was replaced with fresh medium containing a 40 μM concentration of various compounds, and incubation was continued for 48 h. Then, an apoptosis assay was performed according to the standard procedure of the Annexin V-FITC/PI Apoptosis Kit (E-CK-A211, Elabscience, China). Analysis was performed with a BD LSR Fortessa flow cytometer (BD, USA), and data were processed with FlowJo software. The gating strategy for flow cytometry was defined as follows: Necrotic cells: FITC- PI+; Late apoptotic cells: FITC+ PI+; Early apoptotic cells: FITC+ PI-; Live cells: FITC- PI-.
Bulk RNA-seq
The cell treatment method was the same as that used for western blotting and apoptosis assays, except that 6-well plates were replaced with 12-well plates. Three biological replicates were set up for each group. The isolation of total RNA from cell pellets was performed utilising TRIzol Reagent (DP201-01, DiNing, China). RNA-seq libraries were subsequently prepared using the VAHTS® Universal V6 RNA-seq Library Prep Kit, and 100 bp paired-end reads were sequenced through DNBSEQ-2000RS (MGI). Human genomic sequences were obtained from Ensembl (GCA_000001405.28), and quality control was conducted using fastqc (0.11.9) software. Sequence alignment was performed using STAR (2.7.1a). The gene expression matrix was obtained by htseq (0.11.2), and differential expression analysis (DEA) was carried out utilising the R package DESeq2 (1.30.0). KEGG pathway enrichment analysis was conducted using clusterProfiler (4.6.2).
Molecular dynamics simulations
Using the crystal structure of BCKDK (PDB ID: 3TZ0) and the best docking model of lead compounds, systems containing and lacking ADP were constructed. The simulations were conducted using Amber software version 22 (Amber 2023, University of California, San Francisco; https://ambermd.org/). A gaff2 force field parameter was assigned to the ligand, and a ff19SB parameter was assigned to the protein. The complex was solvated using the OPC water model, and the system charge was neutralised by introducing CL ions. The systems were centred within a 10 × 10 × 10 Å truncated octahedron box, and periodic boundary conditions were applied. The process of minimisation involved three stages. In the first stage, the atoms of the protein and small molecules were restrained by 10 kcal/mol/Å2 force for 20,000 steps; in the second stage, the heavy atoms of the protein backbone and small molecules were restrained by a 10 kcal/mol/Å2 force for 20,000 steps; and finally, a stage without restraint was performed for 20,000 steps. Subsequently, the system was gradually heated to 300 K in the NVT ensemble while maintaining restraints on the protein and small-molecule atoms for 20 ps, utilising a Langevin thermostat during this period. In the equilibration phase, the Langevin thermostat was consistently employed for a duration of 200 ps while conducting equilibration in the NPT ensembles with constraints on the heavy atoms of the protein backbone and small molecules. Then, the NVT ensemble was used to control the volume and remove all constraints, allowing the system to continue equilibrating for 1 ns. Additionally, a production simulation lasting 100 ns was conducted in the NPT ensembles, with a temperature of 300 K and a pressure of 1 atm, without any constraints. To maintain the temperature and pressure, Langevin dynamics (with a 1.0 ps-1 collision frequency) and isotropic position scaling (relaxation time of 2 ps) methods were employed. The force constant utilised in the simulation was 10 kcal/mol/Å2. The particle mesh Ewald (PME) algorithm was employed to handle long-range electrostatic interactions, while the shake algorithm was utilised to constrain covalent bonds involving hydrogen atoms. The time step was set to 2 fs, and a cut-off value of 8 Å was established for nonbonded interactions. The binding free energy was calculated utilising MMGB/PBSA. The cpptraj module in AmberTools 22 was employed to conduct analyses on RMSD, RMSF, and B-factor. Structure visualisation was also performed using PyMOL software.
Statistical analyses
The samples involved in this experiment were set up with at least three replicate groups. GraphPad Prism software was used for data analysis and graphing, data were described using the mean with SEM, and one-way ANOVA was used to analyse the differences between groups. p Values are represented as equalities according to each calculated value, and the smallest p value was reported as p < 0.001.
Authors contributions
Li Zhang and Quanjun Yang conceived, designed and supervised the project. Li Zhang, Quanjun Yang and Chunqiong Li wrote the manuscript. Chunqiong Li led all the experiments and data analyses with the help of Hao Meng on molecular dynamics simulations.
Supplemental Material
Download PDF (996 KB)Acknowledgements
We would like to thank Mrs Xinshuang Zhang for Bulk RNA-seq library construction and sequencing, Mr Xiangyu Zhou for Bulk RNA-seq data analyses, Prof. Yulong Li (Peking University) for distributing the donor plasmids containing the BCKDK sequence, Prof. Zhe Zhang and Dr. Yuefeng Jiang (Peking University) for giving instruction in the expression and purification of BCKDK, Wenjuan Zhang (National Center for Protein Sciences-Beijing, PHOENIX Center) for providing technical support during the SPR experiments and Mr Hao Meng in molecular dynamics simulations associated with during revision.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr. 1984;4(1):409–454.
- Davis TA, Fiorotto ML, Reeds PJ. Amino acid compositions of body and milk protein change during the suckling period in rats. J Nutr. 1993;123(5):947–956.
- Moura A, Savageau MA, Alves R. Relative amino acid composition signatures of organisms and environments. PLoS One. 2013;8(10):e77319.
- Adeva-Andany MM, López-Maside L, Donapetry-García C, Fernández-Fernández C, Sixto-Leal C. Enzymes involved in branched-chain amino acid metabolism in humans. Amino Acids. 2017;49(6):1005–1028.
- Supruniuk E, Żebrowska E, Chabowski A. Branched chain amino acids-friend or foe in the control of energy substrate turnover and insulin sensitivity? Crit Rev Food Sci Nutr. 2023;63(15):2559–2597.
- Drummond MJ, Glynn EL, Fry CS, Timmerman KL, Volpi E, Rasmussen BB. An increase in essential amino acid availability upregulates amino acid transporter expression in human skeletal muscle. Am J Physiol Endocrinol Metab. 2010;298(5):E1011–1018.
- Hyde R, Taylor PM, Hundal HS. Amino acid transporters: roles in amino acid sensing and signalling in animal cells. Biochem J. 2003;373(Pt 1):1–18.
- Yoshizawa F. New therapeutic strategy for amino acid medicine: notable functions of branched chain amino acids as biological regulators. J Pharmacol Sci. 2012;118(2):149–155.
- Brosnan ME, Lowry A, Wasi Y, Lowry M, Brosnan JT. Regional and subcellular distribution of enzymes of branched-chain amino acid metabolism in brains of normal and diabetic rats. Can J Physiol Pharmacol. 1985;63(10):1234–1238.
- Brautigam CA, Wynn RM, Chuang JL, Machius M, Tomchick DR, Chuang DT. Structural insight into interactions between dihydrolipoamide dehydrogenase (e3) and e3 binding protein of human pyruvate dehydrogenase complex. Structure. 2006;14(3):611–621.
- Zhao Y, Hawes J, Popov KM, Jaskiewicz J, Shimomura Y, Crabb DW, Harris RA. Site-directed mutagenesis of phosphorylation sites of the branched chain alpha-ketoacid dehydrogenase complex. J Biol Chem. 1994;269(28):18583–18587.
- Wynn RM, Kato M, Machius M, Chuang JL, Li J, Tomchick DR, Chuang DT. Molecular mechanism for regulation of the human mitochondrial branched-chain alpha-ketoacid dehydrogenase complex by phosphorylation. Structure. 2004;12(12):2185–2196.
- Lu G, Sun H, She P, Youn J-Y, Warburton S, Ping P, Vondriska TM, Cai H, Lynch CJ, Wang Y, et al. Protein phosphatase 2cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest. 2009;119(6):1678–1687.
- Qian L, Li N, Lu X-C, Xu M, Liu Y, Li K, Zhang Y, Hu K, Qi Y-T, Yao J, et al. Enhanced bcat1 activity and BCAA metabolism promotes RHOC activity in cancer progression. Nat Metab. 2023;5(7):1159–1173.
- East MP, Laitinen T, Asquith CRM. BCKDK: an emerging kinase target for metabolic diseases and cancer. Nat Rev Drug Discov. 2021;20(7):498–498.
- Du C, Liu W-J, Yang J, Zhao S-S, Liu H-X. The role of branched-chain amino acids and branched-chain α-keto acid dehydrogenase kinase in metabolic disorders. Front Nutr. 2022; 9:932670.
- Sivanand S, Vander Heiden MG. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell. 2020;37(2):147–156.
- Yoo HS, Shanmugalingam U, Smith PD. Potential roles of branched-chain amino acids in neurodegeneration. Nutrition. 2022; 103-104:111762.
- Novarino G, El-Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J, Silhavy JL, Kara M, Khalil RO, Ben-Omran T, et al. Mutations in bckd-kinase lead to a potentially treatable form of autism with epilepsy. Science. 2012;338(6105):394–397.
- Maynard TM, Manzini MC. Balancing act: maintaining amino acid levels in the autistic brain. Neuron. 2017;93(3):476–479.
- Polis B, Samson AO. Role of the metabolism of branched-chain amino acids in the development of Alzheimer’s disease and other metabolic disorders. Neural Regen Res. 2020;15(8):1460–1470.
- Cole JT, Mitala CM, Kundu S, Verma A, Elkind JA, Nissim I, Cohen AS. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc Natl Acad Sci U S A. 2010;107(1):366–371.
- Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. 2014;10(12):723–736.
- Zhou M, Shao J, Wu C-Y, Shu L, Dong W, Liu Y, Chen M, Wynn RM, Wang J, Wang J, et al. Targeting BCAA catabolism to treat obesity-associated insulin resistance. Diabetes. 2019;68(9):1730–1746.
- Chen T, Ni Y, Ma X, Bao Y, Liu J, Huang F, Hu C, Xie G, Zhao A, Jia W, et al. Branched-chain and aromatic amino acid profiles and diabetes risk in Chinese populations. Sci Rep. 2016;6(1):20594.
- Tai ES, Tan MLS, Stevens RD, Low YL, Muehlbauer MJ, Goh DLM, Ilkayeva OR, Wenner BR, Bain JR, Lee JJM, et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia. 2010;53(4):757–767.
- Biswas D, Dao KT, Mercer A, Cowie AM, Duffley L, El Hiani Y, Kienesberger PC, Pulinilkunnil T. Branched-chain ketoacid overload inhibits insulin action in the muscle. J Biol Chem. 2020;295(46):15597–15621.
- Chevalier S, Marliss EB, Morais JA, Lamarche M, Gougeon R. Whole-body protein anabolic response is resistant to the action of insulin in obese women. Am J Clin Nutr. 2005;82(2):355–365.
- Kadota Y, Toyoda T, Kitaura Y, Adams SH, Shimomura Y. Regulation of hepatic branched-chain α-ketoacid dehydrogenase complex in rats fed a high-fat diet. Obes Res Clin Pract. 2013;7(6):e439–444.
- She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab. 2007;293(6):E1552–1563.
- Yu D, Moore SC, Matthews CE, Xiang Y-B, Zhang X, Gao Y-T, Zheng W, Shu X-O. Plasma metabolomic profiles in association with type 2 diabetes risk and prevalence in Chinese adults. Metabolomics. 2016;12(1):3.
- Floegel A, Stefan N, Yu Z, Mühlenbruch K, Drogan D, Joost H-G, Fritsche A, Häring H-U, Hrabě de Angelis M, Peters A, et al. Identification of serum metabolites associated with risk of type 2 diabetes using a targeted metabolomic approach. Diabetes. 2013;62(2):639–648.
- Wang W, Zhang F, Xia Y, Zhao S, Yan W, Wang H, Lee Y, Li C, Zhang L, Lian K, et al. Defective branched chain amino acid catabolism contributes to cardiac dysfunction and remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol. 2016;311(5):H1160–h1169.
- Huang Y, Zhou M, Sun H, Wang Y. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res. 2011;90(2):220–223.
- Rhee EP, Gerszten RE. Metabolomics and cardiovascular biomarker discovery. Clin Chem. 2012;58(1):139–147.
- Bhattacharya S, Granger CB, Craig D, Haynes C, Bain J, Stevens RD, Hauser ER, Newgard CB, Kraus WE, Newby LK, et al. Validation of the association between a branched chain amino acid metabolite profile and extremes of coronary artery disease in patients referred for cardiac catheterization. Atherosclerosis. 2014;232(1):191–196.
- Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Fürnsinn C, Promintzer M, Anderwald C, et al. The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56(6):1600–1607.
- Ong PS, Wang LZ, Dai X, Tseng SH, Loo SJ, Sethi G. Judicious toggling of mTOR activity to combat insulin resistance and cancer: current evidence and perspectives. Front Pharmacol. 2016; 7:395.
- Katagiri R, Goto A, Nakagawa T, Nishiumi S, Kobayashi T, Hidaka A, Budhathoki S, Yamaji T, Sawada N, Shimazu T, et al. Increased levels of branched-chain amino acid associated with increased risk of pancreatic cancer in a prospective case-control study of a large cohort. Gastroenterology. 2018;155(5):1474–1482.e1471.
- Ericksen RE, Lim SL, McDonnell E, Shuen WH, Vadiveloo M, White PJ, Ding Z, Kwok R, Lee P, Radda GK, et al. Loss of BCAA catabolism during carcinogenesis enhances mtorc1 activity and promotes tumor development and progression. Cell Metab. 2019;29(5):1151–1165.e1156.
- Uddin GM, Zhang L, Shah S, Fukushima A, Wagg CS, Gopal K, Al Batran R, Pherwani S, Ho KL, Boisvenue J, et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc Diabetol. 2019;18(1):86.
- Oyarzabal A, Bravo-Alonso I, Sánchez-Aragó M, Rejas MT, Merinero B, García-Cazorla A, Artuch R, Ugarte M, Rodríguez-Pombo P. Mitochondrial response to the BCKDK-deficiency: some clues to understand the positive dietary response in this form of autism. Biochim Biophys Acta. 2016;1862(4):592–600.
- Xue P, Zeng F, Duan Q, Xiao J, Liu L, Yuan P, Fan L, Sun H, Malyarenko OS, Lu H, et al. BCKDK of BCAA catabolism cross-talking with the MAPK pathway promotes tumorigenesis of colorectal cancer. EBioMedicine. 2017; 20:50–60.
- Tso S-C, Qi X, Gui W-J, Chuang JL, Morlock LK, Wallace AL, Ahmed K, Laxman S, Campeau PM, Lee BH, et al. Structure-based design and mechanisms of allosteric inhibitors for mitochondrial branched-chain α-ketoacid dehydrogenase kinase. Proc Natl Acad Sci U S A. 2013;110(24):9728–9733.
- Tso S-C, Gui W-J, Wu C-Y, Chuang JL, Qi X, Skvora KJ, Dork K, Wallace AL, Morlock LK, Lee BH, et al. Benzothiophene carboxylate derivatives as novel allosteric inhibitors of branched-chain α-ketoacid dehydrogenase kinase. J Biol Chem. 2014;289(30):20583–20593.
- Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013;56(1):15–30.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1-3):3–26.
- Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341.
- Brunetti-Pierri N, Lanpher B, Erez A, Ananieva EA, Islam M, Marini JC, Sun Q, Yu C, Hegde M, Li J, et al. Phenylbutyrate therapy for maple syrup urine disease. Hum Mol Genet. 2011;20(4):631–640.
- Lyu J, Wang S, Balius TE, Singh I, Levit A, Moroz YS, O'Meara MJ, Che T, Algaa E, Tolmachova K, et al. Ultra-large library docking for discovering new chemotypes. Nature. 2019;566(7743):224–229.
- Gorgulla C, Boeszoermenyi A, Wang Z-F, Fischer PD, Coote PW, Padmanabha Das KM, Malets YS, Radchenko DS, Moroz YS, Scott DA, et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature. 2020;580(7805):663–668.
- Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15(5):606–614.
- Kujala UM, Peltonen M, Laine MK, Kaprio J, Heinonen OJ, Sundvall J, Eriksson JG, Jula A, Sarna S, Kainulainen H, et al. Branched-chain amino acid levels are related with surrogates of disturbed lipid metabolism among older men. Front Med. 2016; 3:57.
- Lee JH, Cho Y-R, Kim JH, Kim J, Nam HY, Kim SW, Son J. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp Mol Med. 2019;51(11):1–11.
- Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169(2):361–371.
- Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14(3):133–139.
- Kim D-H, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175.
- Kanazawa T, Taneike I, Akaishi R, Yoshizawa F, Furuya N, Fujimura S, Kadowaki M. Amino acids and insulin control autophagic proteolysis through different signaling pathways in relation to mTOR in isolated rat hepatocytes. J Biol Chem. 2004;279(9):8452–8459.
- Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406.
- Yoshizawa F. Regulation of protein synthesis by branched-chain amino acids in vivo. Biochem Biophys Res Commun. 2004;313(2):417–422.
- Eley HL, Russell ST, Tisdale MJ. Effect of branched-chain amino acids on muscle atrophy in cancer cachexia. Biochem J. 2007;407(1):113–120.
- Xu Y, Wang S, Hu Q, Gao S, Ma X, Zhang W, Shen Y, Chen F, Lai L, Pei J, et al. Cavityplus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018;46(W1):W374–w379.
- Yuan Y, Pei J, Lai L. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr Pharm Des. 2013;19(12):2326–2333.
- Yuan Y, Pei J, Lai L. Ligbuilder 2: a practical de novo drug design approach. J Chem Inf Model. 2011;51(5):1083–1091.
- Xie J, Wang S, Xu Y, Deng M, Lai L. Uncovering the dominant motion modes of allosteric regulation improves allosteric site prediction. J Chem Inf Model. 2022;62(1):187–195.
- Ma X, Meng H, Lai L. Motions of allosteric and orthosteric ligand-binding sites in proteins are highly correlated. J Chem Inf Model. 2016;56(9):1725–1733.
- Gorgulla C, Fackeldey K, Wagner G, et al. Accounting of receptor flexibility in ultra-large virtual screens with virtual flow using a grey wolf optimization method. Supercomput Front Innovat. 2020; 7:4–12.
- Gorgulla C, Çınaroğlu SS, Fischer PD, Fackeldey K, Wagner G, Arthanari H. Virtualflow ants-ultra-large virtual screenings with artificial intelligence driven docking algorithm based on ant colony optimization. Int J Mol Sci. 2021;22(11):5807.