
Abstract
In the present work, we report a new series of potent SARS-CoV-2 Main Protease (Mpro) inhibitors based on maleimide derivatives. The inhibitory activities were tested in an enzymatic assay using recombinant Mpro (3CL Protease from coronavirus SARS-CoV-2). Within the set of new Mpro inhibitors, 6e demonstrated the highest activity in the enzymatic assay with an IC50 value of 8.52 ± 0.44 µM. The IC50 value for Nirmatrelvir (PF-07321332, used as a reference) was 0.84 ± 0.37 µM. The cytotoxic properties were determined in the MTT assay using MRC-5 and HEK-293 cell lines. In the course of the investigation, we found that the newly obtained maleimide derivatives are not substantially cytotoxic (IC50 values for most compounds were above 200 µM).
Introduction
The global COVID-19 pandemic, caused by the novel coronavirus SARS-CoV-2, has had a profound and lasting impact on public health, economies, and societies worldwideCitation1. Since its emergence in late 2019, scientists have been relentlessly pursuing effective treatment strategies to mitigate the severity of the disease and reduce its mortality rate. This pursuit has led to dynamic and rapidly evolving therapeutic approaches, ranging from repurposed existing drugs to novel therapies specifically designed to combat the virus and its associated complicationsCitation2. In the early stages of the pandemic, efforts primarily focused on repurposing existing antiviral medications and investigating their potential efficacy against SARS-CoV-2. However, as our understanding of the virus and its interaction with the host immune system deepened, new therapeutic targets emerged.
Proteases play a crucial role in the replication and survival of viruses, including the SARS-CoV-2 virusCitation3. Among the various proteases encoded by the SARS-CoV-2 genome, the Main Protease (Mpro), also known as 3 C-like protease (3CLpro), is of particular interestCitation4. Understanding Mpro’s function and structure has been instrumental in developing potential treatments for COVID-19. SARS-CoV-2 Mpro is an enzyme responsible for processing viral polyproteins, ultimately facilitating the production of functional viral proteinsCitation5. It cleaves the polyproteins at specific sites, releasing individual proteins that are vital for viral replication. The SARS-CoV-2 Mpro exhibits a tripartite framework: domain I (encompassing residues 8–101), domain II (composed of residues 102–184) and domain III (residues 201–303). Domain I and domain II adopt a structural motif reminiscent of the antiparallel β-barrel structure commonly encountered in trypsin-like serine proteases, and domain III consists of five α-helices, forming an antiparallel conglomerate connected to the domain by a long loop region composed of residues 185–200Citation6. The active site of SARS-COV-2 Mpro is located in a cleft between domains I and II, holding a histidine/cysteine catalytic dyad (His41 and Cys145)Citation7,Citation8.
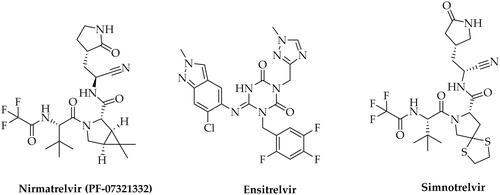
Inhibiting Mpro’s activity has been identified as one of the strategies (along with inhibition of SARS-CoV-2 RdRp - RNA dependent RNA polymerase) for treating COVID-19, as disrupting the virus’s ability to replicate can help control the infection. Currently, several small-molecule antivirals targeting Mpro, as well as SARS-CoV-2 RdRp, have been approved for clinical use ()Citation9. Notably, the Food and Drug Administration (FDA) has granted clinical approval for three small-molecule antivirals: Remdesivir, Molnupiravir (SARS-CoV-2 RdRp inhibitors), and Paxlovid, the latter being a combination of Nirmatrelvir (PF-07321332, Mpro inhibitor) and Ritonavir (an antiretroviral medication used along with other medications to treat HIV/AIDS)Citation10–12. Other pharmaceuticals whose mechanism of action assumes Mpro inhibition are Ensitrelvir (approved by the Ministry of Health, Labour, and Welfare in Japan)Citation13 or Xiannuoxin endorsed by the China National Medical Products Administration, being a combination of Simnotrelvir (Mpro inhibitor) and RitonavirCitation14. Unfortunately, these drugs exhibit certain limitations, including suboptimal efficacy, potential toxicity, and imperfect pharmacokinetic (PK) profiles. Furthermore, considering the emergence of drug-resistant variants to the presently approved drugs, the search for new active substances becomes particularly importantCitation9.
Figure 1. Chemical structures of SARS-CoV-2 Mpro inhibitors approved for clinical use for the treatment of COVID-19.
In this article, we present research on the design, synthesis, and biological evaluation of new SARS-CoV-2 Mpro inhibitors based on the maleimide framework. The inspiration for the design of the above-mentioned derivatives was the research paper published in 2020 by Prof. Marcin Drąg from Wroclaw University of Science and Technology (Poland), presenting the structures of potential substrates and substrates preferences of the SARS-CoV-2 Mpro enzymeCitation15. Based on the reported structures and data, we proposed new dipeptide, tripeptide, and tetrapeptide derivatives containing a maleimide pharmacophore. As one of the possible mechanisms of action for the designed compounds leading to the inhibition of SARS-CoV-2 Mpro activity, we assume the participation of maleimide pharmacophore in the Michael addition reaction between the -SH group of the crucial cysteine residue (Cys145) in the enzyme active site and the α,β-unsaturated bond of the maleimide moiety. This hypothesis was supported by our initial molecular docking calculations, which indicated that after docking maleimide moieties of designed compounds they occupy the region in close distance to the Cys145 residue (4.44 Å).
Molecular docking
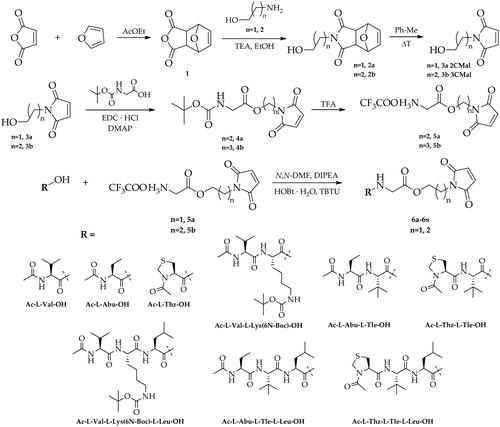
Initially, to verify that the newly designed compounds 6a-s (Scheme 1) can bind to the SARS-CoV-2 Mpro enzyme’s active site effectively, molecular docking studies were performed. The X-ray structure of SARS-CoV-2 Mpro was retrieved from the Protein Data Bank (code: 6Y2E) and properly prepared for docking calculations.
Scheme 1. Synthesis of dipeptide, tripeptide and tetrapeptide derivatives of 1–(2-hydroxyethyl)-1H-pyrrole-2,5-dione and 1–(3-hydroxypropyl)-1H-pyrrole-2,5-dione (6a-6s).
The calculated results for the proposed structures 6a-s indicated that they could, at least theoretically, efficiently bind to the active site of the SARS-CoV-2 Mpro enzyme. The free binding energies of designed compounds to the molecular target were at a satisfactory level in a range of −5.9 to −7.2 kcal/mol (). As it can be seen in for representative compound 6e, maleimide moiety occupies the region within a close distance to Cys145, which jointly with His41, forms a catalytic dyad within S1’ pocket. In the course of our research, we found that the distance between the maleimide group of compound 6e (which demonstrated the highest SARS-CoV-2 Mpro inhibitory activity) and Cys145 is 4.44 Å () suggesting the possible Michael addition reaction between the -SH group of cysteine and the α,β-unsaturated bond of the maleimide moiety. In addition, the N atom of the maleimide group may interact with Glu166 residue in the S1 binding pocket. A similar arrangement in the SARS-CoV-2 Mpro active site was observed for other newly designed derivatives. Furthermore, we detected that the peptide chains (P1-P4) of potential SARS-CoV-2 Mpro inhibitors 6a-6s are well accommodated in the cavity defined by several amino acid residues, which form the enzyme’s active site. For example, P1 residue (Glu) of compound 6e interacts with His41 of the S1’ binding pocket while the P2 residue (L-Thz) of compound 6e interacts with several amino acid residues (Thr25, Thr26, Cys44, and Asn142).
Table 1. The compounds-SARS-CoV-2 Mpro enzyme interactions for selected Mpro inhibitors (and distances [Å]) identified using BIOVIA software.
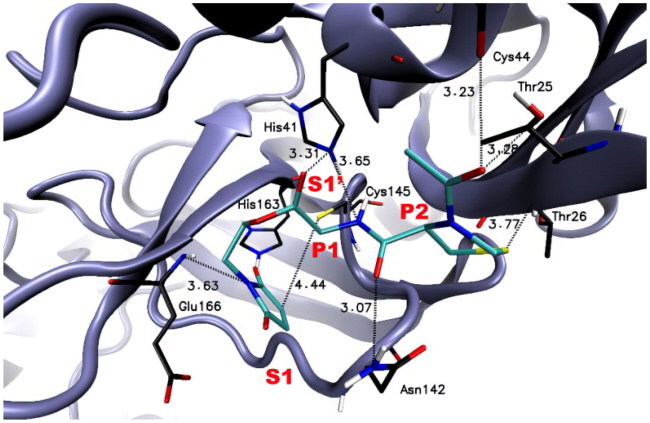
Furthermore, additional electrostatic interactions between the amino acids at the P1 and P2 positions of compound 6e and other amino acid residues of the enzyme’s binding pockets (e.g., His163; His164; Leu141; Met165; Met49; Leu27; Thr45; Ser46; Thr24) were detected using BIOVIA, Dassault Systèmes, Discovery Studio Visualiser software (). These interactions may be crucial in the binding process of compound 6e. All of the detected interactions such as conventional hydrogen bonds, carbon hydrogen bonds, π-alkyl, or van der Waals interactions may potentially stabilise the compound-enzyme complex and provide favourable binding and promising inhibitory activity of designed compounds.
Chemistry
The newly designed compounds 6a-6s based on dipeptide, tripeptide, and tetrapeptide derivatives containing a maleimide pharmacophore were synthesised according to the route shown in Scheme 1 and Scheme 2.
Scheme 2. Synthesis of trifluoroacetate derivatives 6g’, 6h’, 6m’, and 6n’.
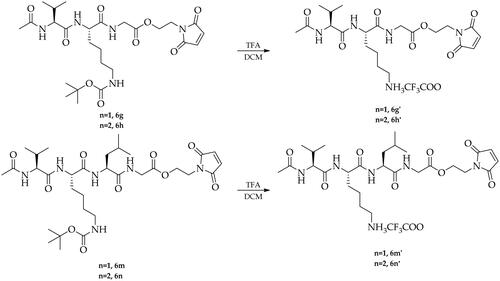
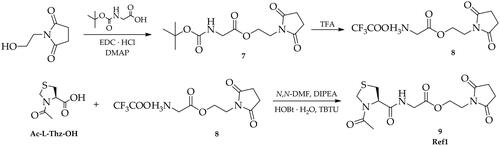
The first step of the synthesis involved the preparation of trifluoroacetate derivatives of 2–(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl glycine (5a) and 3–(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)propyl glycine (5b). For this purpose, maleic anhydride was reacted in a Diels-Alder cycloaddition reaction with furan leading to the bicyclic derivative 1, which was then treated with 2-aminoethan-1-ol or 3-aminopropan-1-ol in the presence of triethylamine to give 2–(2-hydroxyethyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (2a) and 2–(3-hydroxypropyl)-3a,4,7,7a-tetrahydro-1H-4,7-epoxyisoindole-1,3(2H)-dione (2b). The next step included the preparation of maleimide derivatives 3a and 3b by heating the compounds 2a and 2b in toluene as a result of a reverse Diels-Alder reaction, which were then coupled with tert-butoxycarbonylglycine in the presence of EDC·HCl and DMAP. Next, the obtained compounds (4a, 4b) were treated with trifluoroacetic acid (TFA) to remove the tert-butoxycarbonyl protective group (Boc) leading to the formation of trifluoroacetate salt derivatives (5a, 5b). The next step of the synthesis included a series of coupling reactions of the obtained trifluoroacetate derivatives of 5a and 5b with appropriate N-protected amino acids, dipeptides, and tripeptides using HOBt and TBTU as coupling reagents in the presence of DIPEA resulting in the formation of the maleimide derivatives (6a-6s). Finally, derivatives containing a tert-butoxycarbonyl protective group (6 g, 6h, 6 m, 6n) were transformed to the corresponding trifluoroacetate salts (6g’, 6h’, 6m’, 6n’) by treatment with TFA (Scheme 2). Furthermore, to investigate the mechanism of inhibition for designed new Mpro inhibitors, the reference compound- Ac-l-Thz-Gly-2CSucc (Ref1) was obtained in the same way starting from the commercially available 1–(2-hydroxyethyl)pyrrolidine-2,5-dione (Scheme 3).
Scheme 3. Synthesis of Ac-l-Thz-Gly-2CSucc derivative (Ref1).
Biological evaluation
In order to verify our hypothesis and experimentally confirm the results obtained in the molecular modelling studies, we determined the ability of the newly synthesised maleimide derivatives to inhibit the function of SARS-CoV-2 Mpro. For this purpose, we conducted an enzyme assay using commercially available recombinant 3CL Protease from coronavirus SARS-CoV-2 (Sigma-Aldrich SAE0172) according to the modified procedure described in the literatureCitation15. The summarised results of the enzymatic assay are collected in .
Table 2. The inhibition of SARS-CoV-2 Mpro enzyme activity.
Table 3. MTT cell viability assay results (human lung fibroblasts MRC-5 and human kidney epithelial cells HEK-293).
The received data showed that all derivatives effectively inhibited SARS-CoV-2 Mpro enzyme in the low micromolar concentration range from 8.52 to 27.28 µM. In the course of the research, we found that the highest inhibitory activity was exhibited by the compound 6e containing Ac-l-Thz-Gly- scaffold in its structure (IC50 = 8.52 ± 0.44 µM). The showed above analysis that the influence of the length and type of the peptide scaffolds did not significantly affect the effectiveness of inhibition (keeping it at a constantly high level) which can explain the very similar predicted free binding energy values of the inhibitor-enzyme complexes calculated in molecular docking studies. These observations may indicate that a pharmacophore based on maleimide derivatives is mainly responsible for the high potency of the tested compounds, which, according to our assumptions, may undergo Michael addition reaction between the α,β-unsaturated bond of the maleimide moiety and -SH group of cysteine in the catalytic site of SARS-CoV-2 Mpro. In this case, the peptide core of the inhibitors ensures a good arrangement of the molecules in the catalytic site, stabilising the inhibitor-enzyme complexes through numerous electrostatic interactions. To investigate the impact of α,β-unsaturated bonds presence on Mpro inhibition, and to confirm the proposed inhibition mechanism involving the Michael addition reaction, we performed the synthesis and activity studies for succinimide derivative Ref1. In the course of our studies, we found that succinimide analogue has no inhibitory effects on the SARS-CoV-2 Mpro enzyme even at 400 μM concentration. The above studies suggest that the presence of α,β-unsaturated bonds in the structure of the designed new inhibitors is crucial for effectively inhibiting the enzyme’s function and, as assumed, the mechanism of inhibition may involve the 1,4 addition reaction occurring between a Michael donor (-SH nucleophile) and a Michael acceptor (α,β-unsaturated carbonyl group). Additionally, we evaluated the type of inhibition for compound 6e (chosen as the representative compound for the series). For this reason, KM and Vmax of the SARS-CoV-2 Mpro activity were measured under different concentrations of inhibitor (0–50 µM) and substrate (5–80 µM). The reaction kinetics studies and analysis of the Lineweaver–Burk plot (Figure S74, ESI) indicated a mixed type of inhibition for compound 6e. In this case, the reversibility of the enzymatic reaction may be related to the retro-Michael reactionCitation16,Citation17 when the Michael adduct (thiosuccinimide derivative) is regenerated back to maleimide moiety. The reversibility of this reaction is common due to the presence of thiolate moiety as a good leaving group.
Next, we also investigated the effect of compounds on human lung fibroblasts (MRC-5) and human kidney epithelial cells (HEK-293) to evaluate whether newly obtained derivatives induced changes in cell viability (Ebselen as promising SARS-CoV-2 Mpro inhibitorCitation18 was used as a reference). Our data indicated that all analysed compounds induced lowering of MRC-5 cell viability at a higher concentration than Ebselen (IC50 = 94 ± 8 μM for Ebselen, 145 ± 21 μM for Ac-l-Thz-Gly-3CMal 6f, and higher than 200 μM for other compounds) (). Interestingly, more variable results were observed in the HEK-293 cell line. However, Ebselen IC50 was similar to MRC-5 (IC50 = 137 ± 36 μM); part of the analysed compounds 6a-6f, 6i, 6j, and 6 l lowers cell viability at a lower concentration than Ebselen in comparison to MRC-5 cell line. Other compounds’ IC50 values were higher than 200 μM (see ). Overall, the conducted studies showed that the obtained compounds based on maleimide derivatives show low cytotoxicity and are very promising for further research.
Materials and methods
Molecular modelling
Ligands preparation
Prior to docking procedures, the potential inhibitors were prepared using Portable HyperChem 8.0.7 Release (Hypercube, Inc., Gainesville, FL, USACitation19). Each ligand was optimised using a MM + force fieldCitation20 and the Polak–Ribière conjugate gradient algorithm (terminating at a gradient of 0.05 kcal mol−1 Å −1).
Protein preparation
The X-ray structures of the SARS-CoV-2 Mpro enzyme used for molecular modelling studies was taken from the Protein Databank (Protein Data Bank accession code: 6Y2E). The docking analysis was carried out after standard preparation procedures including the removal of co-crystalized ligands and water molecules, and the addition of hydrogen atoms and Gasteiger charges to each atom.
Molecular docking
Docking calculations of the optimised ligands to the prepared structure of SARS-CoV-2 Mpro enzyme were carried out with Autodock Vina 1.1.2 software (The Molecular Graphic Laboratory, The Scripps Research Institute, La Jolla, CA, USA)Citation21 with exhaustiveness, num_modes, and energy_range parameters set as 8, 30, and 10, respectively. For all of the docking studies, a grid box size was centred on the sulphur atom of Cys145, and the size of the grid box was 30 Å x 30 Å x 30 Å (x = −13.504, y = −30.226, z = 3.454). Then, the best poses for a particular ligand were visually inspected. Illustrations of the 3D models were generated using VMD 1.9 (University of Illinois at Urbana – Champaign, Urbana, IL, USA)Citation22. Identification of the compounds-SARS-CoV-2 Mpro enzyme interactions was performed using Discovery Studio Visualiser v20. 1. 0. 19295 (BIOVIA, Dassault Systèmes, San Diego, CA, USA)Citation23.
Biological assays
The inhibition of SARS-CoV-2 Mpro enzyme activity
The SARS-CoV-2 Mpro activity was measured fluorometrically at 37 °C, according to literature with slight modificationsCitation8,Citation15. Briefly, 0.15 µg of Mpro from coronavirus SARS-CoV-2 (Sigma-Aldrich SAE0172) was incubated with different concentrations of analysed compounds (0, 5, 10, 17.5, 25, 33, 50 and 100 µM) resuspended in DMSO (maximal concentration of DMSO −1%, up to 7% DMSO did not affect the assay) in buffer (20 mM Tris-HCl, 100 mM NaCl, 1 mM EDTA, and 1 mM DTT, pH 7.3). Next, the mixture (100 µL of the final volume) was shaken at 37 °C for 10 min. After incubation, 20 µL of the substrate (Dabcyl-KTSAVLQSGFRKM-Glu(Edans)-NH2, Biosyntan, Germany) was added (final concentration − 20 µM), and enzymatic activity was measured in Synergy H1 Hybrid Multi-Mode microplate reader (BioTek, USA) at 340/490 nm (excitation/emission). Each data point was replicated 3 times.
Determination of the SARS-CoV-2 Mpro inhibition type
To determine the type of inhibition, KM and Vmax of the SARS-CoV-2 Mpro activity were measured under different concentrations of compound 6e (0, 0.1, 1, 5, 10, 25, and 50 µM) and substrate (5, 10, 20, 40, and 80 µM). Briefly, 0.15 µg of Mpro from coronavirus SARS-CoV-2 (Sigma-Aldrich SAE0172) was incubated with different concentrations of the analysed compound in the buffer. Next, the mixture was shaken at 37 °C for 10 min. After incubation, different substrate concentrations were added (final volume 100 µL), and enzymatic activity was measured in Synergy H1 Hybrid Multi-Mode microplate reader (BioTek, USA) at 340/490 nm (excitation/emission). The nonlinear regression analysis (Michaelis-Menten enzyme kinetics) was performed using the GraphPad Prism 8.3.0 software. Each data point was replicated 3 times.
Cell culture
Cell lines were obtained from the American Type Culture Collection (ATCC). A human lung fibroblast cell line (MRC-5, ATCC, CCL-171) was cultured in Eagle’s Minimum Essential Medium (cat no. P04-08058, PanBiotech, Germany) supplemented with 10% foetal bovine serum (FBS) and 1% of penicillin/streptomycin (P/S). A human epithelial kidney cell line (HEK-293, ATCC, CRL-1573) was cultured in Dulbecco’s Modified Eagle’s Medium (cat no. P04-03596, PanBiotech, Germany) supplemented with 10% FBS and 1% of penicillin/streptomycin (P/S). All cells were cultured under standard conditions (37 °C in a humidified atmosphere of 5% CO2). Cells were maintained in a 75 cm2 tissue culture flask, and the medium was replaced every 48 h. When confluent cells were detached with a trypsin-EDTA solution and subcultured into a newer flask.
MTT viability assay
MTT assay was used to determine cell viability. Cells were seeded in 96 plates at a density of 2 × 104 cells per well for MRC-5 and 4 × 103 cells per well for HEK-293. After 24 h, the medium was changed to a medium not supplemented with FBS, and cells were treated with different concentrations of analysed compounds (0, 31.25, 62.5, 125, 250, 500 µM resuspended in DMSO (maximal concentration of DMSO − 0.1% for 500 µM and 0.006% for 31.25 µM). After 72 h incubation the media was supplemented with water-soluble tetrazolium salt [3–(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (final concentration 0.45 mg/mL). Next, microplates were incubated at 37 °C in 5% CO2 for 2 h. After incubation, the medium was removed, and formazan crystals were diluted in 100 µL of DMSO. After 15 min, cell viability was assessed by measuring absorbance at 540 nm (reference 630 nm) using Synergy H1 Hybrid Multi-Mode microplate reader (BioTek, USA). Viability was determined as a percentage of control.
Conclusion
In the present work, we described our research on molecular modelling, synthesis, and biological evaluation of a new series of potent SARS-CoV-2 Mpro inhibitors based on peptidomimetic maleimide derivatives. Enzymatic assay performed using recombinant Mpro (3CL Protease from coronavirus SARS-CoV-2) indicated that all of the newly synthesised inhibitors 6a-s were able to effectively inhibit the action of Mpro. Among them, the highest inhibitory activity was exhibited by compound 6e with an IC50 value of 8.52 ± 0.44 µM. The IC50 value for Nirmatrelvir (used as a reference) was 0.84 ± 0.37 µM. The analysis showed that the influence of the length and type of the peptide scaffolds did not significantly affect the effectiveness of inhibition suggesting that a pharmacophore based on maleimide derivatives is mainly responsible for the potency of tested compounds. Based on the results of molecular docking studies and experimental data obtained for succinimide derivative Ref1, we postulate that newly designed compounds may undergo a Michael addition reaction between the α,β-unsaturated bond of the maleimide moiety and -SH group of cysteine in the catalytic site of SARS-CoV-2 Mpro suggesting this type of inhibition mechanism. On the other hand, the reaction kinetics studies performed for compound 6e and analysis of the Lineweaver–Burk plot indicated a mixed type of inhibition. Its reversibility may be related to the retro-Michael reaction when the Michael adduct (thiosuccinimide derivative) is regenerated back to maleimide moiety. In the cell line experiments using the human lung fibroblast (MRC-5) and human epithelial kidney (HEK-293) cell lines, we determined the initial safety profile of the obtained compounds. We found that the newly obtained maleimide derivatives are safe, do not show any cytotoxic properties (IC50 values for most compounds were above 200 µM), and could be promising candidates for further preclinical investigations.
Supplemental Material
Download PDF (3.9 MB)Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Antonopoulou I, Sapountzaki E, Rova U, Christakopoulos P. Inhibition of the main protease of SARS-CoV-2 (Mpro) by repurposing/designing drug-like substances and utilizing nature’s toolbox of bioactive compounds. Comput Struct Biotechnol. 2022; 20:1306–1344.
- Katre SG, Asnani AJ, Pratyush K, Sakharkar NG, Bhope AG, Sawarkar KT, Nimbekar VS. Review on development of potential inhibitors of SARS-CoV-2 main protease (MPro). Futur J Pharm Sci. (1)2022; 8:36.
- Zhu W, Shyr Z, Lo DC, Zheng W. Viral proteases as targets for coronavirus disease 2019 drug development. J Pharmacol Exp Ther. 2021; 378(2):166–172.
- Parmar M, Thumar R, Patel B, Athar M, Jha PC, Patel D. Structural differences in 3C-like protease (Mpro) from SARS-CoV and SARS-CoV-2: molecular insights revealed by molecular dynamics simulations. Struct Chem. 2022; 23:1–18.
- Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. (5626)2003; 300:1763–1767.
- Zhang W, Lin SX. Search of novel small molecule inhibitors for the main protease of SARS-CoV-2. Viruses. 2023;15(2):580.
- Kneller DW, Phillips G, O'Neill HM, Jedrzejczak R, Stols L, Langan P, Joachimiak A, Coates L, Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat Commun. 2020; 11(1):3202.
- Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science. 2020; 368(6489):409–412.
- Huang C, Shuai H, Qiao J, Hou Y, Zeng R, Xia A, Xie L, Fang Z, Li Y, Yoon C, et al. A new generation Mpro inhibitor with potent activity against SARS-CoV-2 Omicron variants. Signal Transduct Target Ther. 2023; 8:128.
- US Food and Drug Administration (FDA) [Internet]. Available from: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment. [last accessed 10 October 2023].
- US Food and Drug Administration (FDA) [Internet]. Available from: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-additional-oral-antiviral-treatment-covid-19-certain. [last accessed 10 October 2023].
- US Food and Drug Administration (FDA) [Internet]. Available from: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-first-oral-antiviral-treatment-covid-19. [last accessed 10 October 2023].
- Ministry of Health, Labour and Welfare of Japan (MHW) [Internet]. Available from: https://www.mhlw.go.jp/stf/newpage_29320.html. [last accessed 10 October 2023].
- China National Medical Products Administration (NMPA) [Internet]. Available from: http://english.nmpa.gov.cn/2022-08/15/c_797867.htm. [last accessed 10 October 2023].
- Rut W, Groborz K, Zhang L, Sun X, Zmudzinski M, Pawlik B, Młynarski W, Hilgenfeld R, Drag M. Substrate specificity profiling of SARS-CoV-2 main protease enables design of activity-based probes for patient-sample imaging. BioRxiv 2020.03.07.981
- Weissman MR, Winger KT, Ghiassian S, Gobbo P, Workentin MS. Insights on the application of the retro michael-type addition on maleimide-functionalized gold nanoparticles in biology and nanomedicine. Bioconjug Chem. 2016;27(3):586–593.
- Dong L, Li C, Locuson C, Chen S, Qian MG. A two-step immunocapture LC/MS/MS assay for plasma stability and payload migration assessment of cysteine–maleimide-based antibody drug conjugates. Anal Chem. 2018;90(10):5989–5994.
- Liu W, Wang J, Wang S, Yue K, Hu Y, Liu X, Wang L, Wan S, Xu X. Discovery of new non-covalent and covalent inhibitors targeting SARS-CoV-2 papain-like protease and main protease. Bioorg Chem. 2023;140:106830.
- HyperChem(TM) Professional 7.51, Hypercube, Inc., 1115 NW 4th Street, Gainesville, Florida, USA. Available from: www.hyper.com.
- Hocquet A, Langgård M. An evaluation of the MM + force field. J Mol Model. (3)1998; 4:94–112.
- Trott O, Olson AJ. Autodock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. (2)2010; 31:455–461.
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. (1)1996; 14:33–38.
- Discovery Studio Visualizer, v20. 1. 0. p. 19295. San Diego: Dassault Systèmes.