
Abstract
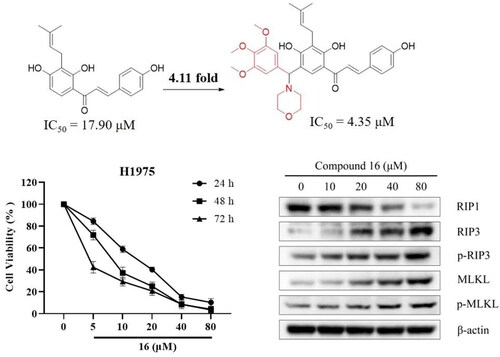
In this study, seventeen isobavachalcone (IBC) derivatives (1–17) were synthesised, and evaluated for their cytotoxic activity against three human lung cancer cell lines. Among these derivatives, compound 16 displayed the most potent cytotoxic activity against H1975 and A549 cells, with IC50 values of 4.35 and 14.21 μM, respectively. Compared with IBC, compound 16 exhibited up to 4.11-fold enhancement of cytotoxic activity on human non-small cell lung cancer H1975 cells. In addition, we found that compound 16 suppressed H1975 cells via inducing apoptosis and necroptosis. The initial mechanism of compound 16 induced cell death in H1975 cells involves the increasing of Bax/Bcl-2 ratio and Cyt C protein level, down-regulating of Akt protein level, and cleaving caspase-9 and -3 induced apoptosis; the up-regulation of RIP3, p-RIP3, MLKL, and p-MLKL levels induced necroptosis. Moreover, compound 16 also caused mitochondrial dysfunction, thereby decreasing cellular ATP levels, and resulting in excessive reactive oxygen species (ROS) accumulation.
Graphical Abstract
Introduction
Lung cancer is one of the most common causes of cancer morbidity and mortality worldwideCitation1. Non-small cell lung cancer (NSCLC) is the most frequent subtype of lung cancer, accounts for approximately 80–85% of all lung cancersCitation2. Commonly, NSCLC is not detected until the disease has progressed to an advanced stateCitation3. Up to now, surgery, radiation, chemotherapy, molecularly targeted therapy, and immunotherapy are used to treat NSCLCCitation4. Chemotherapy or tyrosine kinase inhibitors (TKIs) improve the median survival of patients with advanced NSCLC, but the overall survival remains poorCitation5,Citation6. However, with the progress of chemotherapy, TKIs do not have a good efficacy in the epidermal growth factor receptor (EGFR) mutation type of NSCLC, and it is inevitable that acquired drug resistance will occur, resulting in chemotherapy failureCitation7.
Programmed cell death (PCD), including apoptosis, autophagy, necroptosis, ferroptosis, pyrotosis, etc., refers to the form of cell death that can be regulated by various molecules or signals, which is different from necrosisCitation8,Citation9. Many studies showed that the discovery of small-molecule compounds targeting different forms of PCD has been emerging as a new strategy for many types of human cancersCitation10–14. Recently, we reported that isobavachalcone (IBC) induces multiple PCD in human triple-negative breast cancer MDA-MB-231 cells, including necroptosisCitation15.
Necroptosis, a type of PCD was identified in 2005, which is distinct from classical apoptosis and necrosisCitation16. Until now, many necroptosis inducers have been emerging as new anti-cancer agents. Shikonin induces necroptosis in glioma cells via promoting RIP1/RIP3 necrosome formation and ROS overproductionCitation17. Matrine induces necroptosis in cholangiocarcinoma cells by enhancing RIP3 expression and the following RIP3/MLKL/ROS signalling pathwayCitation18. LGH00168 induces necroptosis in human lung cancer A549 cells by the ROS-mediated ER stress and NF-κB inhibitionCitation19. 2-Methoxy-6-acetyl-7-methyljuglone induces necroptosis in lung cancer cells by targeting RIP1 and ROS in a TNFα-independent mannerCitation20. ZZW-115 induces necroptosis in hepatocellular carcinoma cells via concomitant mitochondrial metabolism failure that triggers lower ATP productionCitation21. However, the anti-cancer properties of IBC and its analogs in necroptosis of H1975 cells are not completely understood.
IBC is a chalcone bearing prenyl group that was first isolated from the seed of Psoralea corylifolia L. in 1968Citation22. Many research findings revealed that chalcones could be served as a common scaffold (e.g. xanthohumol, panduretin A, and licochalcones) for anti-cancer agent research and developmentCitation23–26. To improve the anti-cancer activity of IBC, in this study seventeen IBC derivatives were synthesised and investigated cytotoxicity against three lung cancer cell lines. Furthermore, we demonstrated that the mechanism action of compound 16 induces apoptosis and necroptosis in H1975 cells.
Results
Semisynthesis of isobavachalcone derivatives
As shown in Scheme 1, three series of IBC derivatives (1–17) were designed and synthesised by using IBC as the starting material. Compounds 1–5 were synthesised from IBC through Williamson reaction in the presence of Na2CO3 in DMF. An aldehyde group was substituted on IBC afforded intermediate 6. Next, compound 6 was further reacted with aminoguanidine bicarbonate in the presence of catalytic amounts of acetic acid to provide compound 7. Finally, the target compounds with the structures of 8–17 were synthesised by the Mannich reaction of IBC with appropriately substituted benzaldehyde and morpholine in MeCN.
Scheme 1. Synthetic route for IBC derivatives. (i) DMF, CH3I or benzyl chloride, Na2CO3, 65 °C, 10–12 h. (ii) 25%NaOH, CHCl3, 30 °C, 9 h. (ii) Acetic acid, aminoguanidine carbonate, 50 °C, 5 h. (iv) MeCN, aldehydes, morpholine, 80 °C, 12 h.

In vitro cytotoxic activity of IBC derivatives
Compounds 1–17 were initially evaluated for their cytotoxic activities against the three human lung cancer cell lines (H1975, A549, and PC9) using the MTT assay (). Among these, compounds 7 and 16 showed excellent cytotoxicity against the human lung cancer (H1975 and A549) cell lines, with IC50 values of 4.35–15.04 μM. Compared to IBC, compound 16 displayed 4.11- and 1.53-fold enhancement of the cytotoxic effects on H1975 and A549 cells. We observed that both 2-OH and 4′-OH substituted IBC derivatives were inactive at 50 μM. Compounds 6 and 7, which were substituted with the aldehyde group and aminoguanidine at C-5 position, showed better cytotoxic activity against the H1975 cells than the parent compound. In addition, compounds 10, 13, 16, and 17, Mannich base substituted IBC derivatives, had potent cytotoxic activity against H1975 cells, with IC50 values of 6.76, 7.39, 4.35, and 7.01 μM, respectively. In addition, compound 16 exhibited relatively low cytotoxic activity against normal mouse hepatocytes Aml-12 cells, with IC50 values of 10.09 μM, indicating its selective inhibition (SI) to cancer cells ().
Table 1. Cytotoxic activity of compounds 1–17 and IBC (IC50, μM, 72 h).
Table 2. Cytotoxic activity of compound 16 on Aml-12 cells for 72 h (IC50, μM).
Compound 16 inhibited cell viability against human lung cancer cells
As shown in , compound 16 inhibited cell viability against the three human lung cancer (H1975, A549, and PC9) cell lines. These results also indicated that H1975 cells were more sensitive to cell growth inhibition by compound 16. Similar to the MTT assays, the colony-formation assays showed that treatment with compound 16 (0, 1, 2, and 4 μM) inhibited cell growth in a concentration-dependent manner, with the colony formation rates of 100, 51.34, 27.70, and 14.87%, respectively ().
Figure 1. Inhibitory effect of compound 16 on cell viability in human lung cancer cells. (A) The cell viabilities of three human lung cancer (H1975, A549, and PC9) cells treated with various concentrations (0, 5, 10, 20, 40, and 80 μM) of 16 for 24, 48, and 72 h. (B) Effect of compound 16 on the ability of cells to form colonies in H1975 cells using a colony-formation assay. (C) Quantification of the colony-formation assay. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the control.

Compound 16 induced apoptosis in H1975 cells
Following inhibition with compound 16 for 24 h, flow cytometry was used to detect cell death. As shown in , Annexin V/PI dual staining resulted in detected cell death rates of 14.7, 22.7, 34.3, and 54.9% for the H1975 cells exposed to increasing concentrations (10, 20, 40, and 80 μM) of compound 16, respectively. In addition, Calcein-AM and PI staining revealed significantly increased red fluorescence (death cells) in the H1975 cells by treated with the compound 16 ().
Figure 2. Compound 16 induced apoptosis in H1975 cells. (A) Flow cytometric analysis of cell death treated with different concentrations (10, 20, 40, and 80 μM) of 16 using Annexin V/PI dual staining. (B) H1975 cells were treated with 16 for 24 h, subjected to FITC and PI staining, and visualised using fluorescence microscopy. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the control.

Western blotting showed that compound 16 induced apoptosis in H1975 cells as confirmed by the increasing the Bax and Cyt C expression, decreasing the Bcl-2 and Akt protein levels, and cleaving caspase-9 and -3 (). Furthermore, the H1975 cells were pre-treated with z-VAD-FMK, a pan-caspase inhibitor. As shown in , we observed that the cell viability was 12.71% by treated with compound 16 alone and increased to 35.43% in combination with z-VAD-FMK (p < 0.01). These results indicated that compound 16 induced apoptosis in H1975 cells.
Figure 3. Compound 16 induced caspase-dependent apoptosis in H1975 cells. (A) Western blotting analysis of Bax, Bcl-2, Akt, and Cyt-C protein levels. (B) Western blotting analysis of caspase-9, and -3. (C) Cell viability following treated with compound 16 (40 μM) for 24 h alone or with pre-treatment with z-VAD (20 μM) as measured by MTT assay. **p < 0.01 compared with the control.

Compound 16 induced necroptosis in H1975 cells
We observed morphological features of typical necrosis of H1975 cells under electron microscopy after treated with 40 μM of compound 16 (). Compound 16 induced typical nuclear chromatin condensation, massive mitochondrial damage, and disruption of plasma membrane. Western blotting analysis showed that compound 16 increased the expression of RIP3, p-RIP3, MLKL, and p-MLKL protein levels (). Furthermore, the H1975 cells were pre-treated with Nec-1 (necroptosis inhibitor). As shown in , we observed that the cell viability was 14.82% by treated with compound 16 alone and increased to 25.33% in combination with Nec-1 (p < 0.05). Meanwhile, NSA (MLKL inhibitor) also had a protective effect on the compound 16 induced necroptosis in H1975 cells (p < 0.01). These results indicated that compound 16 induced necroptosis in H1975 cells via the regulation of the RIP3-MLKL signalling pathway.
Figure 4. Compound 16 induced necroptosis in H1975 cells. (A) Morphological characteristics of the control using electron microscopy. (B) Morphological characteristics of 16-treated H1975 cells showing necrosis. Red arrowheads indicated cell nuclear chromatin condensation, massive mitochondrial damage, and disruption of plasma membrane. (C) Western blotting analysis of RIP1, RIP3, p-RIP3, MLKL, and p-MLKL protein levels. (D) Cell viability following treated with compound 16 (40 μM) for 24 h alone or with pre-treatment with Nec-1 (20 μM) and NSA (20 μM) as measured by MTT assay. *p < 0.05 and **p < 0.01 compared with the control.

Effect of RIP3 knock-down on necroptosis of H1975 cells by siRNA
To explore the role of RIP3 in compound 16-induced necroptosis, we investigated the effect of RIP3 knock-down in H1975 cell viability using siRNA. As shown in , the siRNA1171 gradually silenced RIP3 expression, leading to a loss of Nec-1 protective effect in the compound 16 treated cells. These results suggested that compound 16 induced necroptosis in H1975 cells, which may be mimicked by up-regulation of RIP3.
Figure 5. Knock-down of RIP3 using siRNA protected against compound 16 induced cell death in H1975 cells. (A) The H1975 cells were transfected with RIP3 siRNA, and whole-cell lysates were subjected to western blot analysis. (B) After transfection with RIP3 siRNA1171, the cells were treated with compound 16 (40 μM) for 24 h, alone or with pre-treatment with Nec-1. The cell viability was measured by MTT assay. *p < 0.05 compared between the groups of compound 16 and 16 with Nec-1.

Effect of compound 16 on mitochondrial function in H1975 cells
To explore whether compound 16 induced apoptosis and necroptosis are associated with mitochondrial function, we performed a flow cytometry with using the calcium indicator JC-1. As shown in , H1975 cells were treated with various concentrations (10, 20, 40, and 80 μM) of compound 16, increasing green fluorescence of JC-1 from 8.14 to 61.20% was observed (p < 0.05). Next, we observed that cellular ATP levels were gradually decreased with different concentrations of compound 16 (). Furthermore, intracellular ROS levels gradually significantly enhanced by 1.86, 2.21, 3.74, and 5.82% when treatment with various concentrations (10, 20, 40, and 80 μM) of compound 16, respectively ().
Figure 6. Effect of compound 16 on mitochondrial function in H1975 cells. (A) Mitochondrial membrane potential was assessed by JC-1 staining and flow cytometry. (B) Quantification of the green fluorescence. (C) Cellular ATP levels after treated with different concentrations (10, 20, 40, and 80 μM) of 16 for 6 h. (D) Intracellular ROS levels after treated with various concentrations of 16 for 6 h. (E) Intracellular ROS level analysis following treated with 16 with or pre-treatment with NAC (20 μM). (F) Cell viability following treated with 16 (40 μM) for 24 h alone or with pre-treatment with NAC as measured by MTT assay. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the control.

To further illuminate the relationship between ROS and cell death, the NAC (as ROS scavenger) was applied to analyse the intracellular ROS level. As shown in , we observed that the intracellular ROS level enhanced by 3.75% by treated with compound 16 (40 μM) alone, and the increasement decreased to 2.02% in combination with NAC (p < 0.01). In addition, NAC also had a protective effect on the compound 16 induced cell death in H1975 cells () (p < 0.01). These results indicated that compound 16 induced various forms of H1975 cell death that are associated with mitochondrial function.
Molecular docking of compound 16 on RIP3
In the present study, the binding mode of compound 16 on RIP3 was revealed based on molecular docking. As shown in , α,β-unsaturated carbonyl moiety formed a hydrogen bond with the Lys296 residue. Moreover, electrovalent bond and hydrogen bond were formed between the morpholine ring of compound 16 to occupy the Glu99 and Arg96 residue of RIP3, respectively.
Figure 7. Molecular docking model of compound 16 on RIP3 protein. (A) 3D-binding model of compound 16 with RIP3 according to molecular docking analysis by MOE. (B) 2D-docking model of compound 16 with RIP3.

Discussion
Important advancements in anti-NSCLC drugs research and development have been achieved over the past 20 years. The use of small molecule tyrosine kinase inhibitors (e.g. gefitinib and erlotinib) and immunotherapy has resulted in unprecedented survival benefits for selected NSCLC patientsCitation27–29. However, the overall cure and survival rates for NSCLC remain low, especially in metastatic NSCLC patientsCitation30. In this study, we found that a novel IBC derivative 16 induced apoptosis and necroptosis in H1975 cells.
IBC, a natural bioactive chalcone, was found in the seed of a traditional Chinese medicine P. corylifolia L. Several studies revealed that IBC could be served as a potential lead compound for anti-cancer agent discovery and developmentCitation15,Citation31–33. Therefore, to find more effective anti-lung cancer IBC derivatives, three series of new IBC derivatives modified on phenolic hydroxyls (2-OH and 4′-OH) or benzene ring (C-5) position were synthesised. Interestingly, our results revealed that the aldehyde group, aminoguanidine, and Mannich base substituted on C-5 position of IBC could be enhanced the cytotoxic activity in H1975 cells. Among these, compound 16 had the most potent cytotoxic activity against H1975 cells, with IC50 values of 4.35 μM. Relative to the strong cytotoxicity activity against H1975 cells, compound 16 exhibited relatively low toxicity in normal Aml-12 cells, with an SI value of 2.32.
Apoptosis is one of the main cell death in which chemo-therapeutic agents kill cancer cellsCitation13,Citation34. Our results showed that apoptosis was induced in human non-small cell lung cancer H1975 cells treated with compound 16 via regulating apoptosis-related proteins, including Bax, Bcl-2, Akt, and Cyt C. Many previous studies have reported that anti-cancer agents induced apoptosis via the activation of caspases, including caspase-3, -8, and -9Citation35,Citation36. Consistent with previous studies, we found that the treatment of compound 16 involves the increased in the Bax/Bcl-2 ratio, up-regulated Cyt C level, and cleaved of the caspase-9 and -3 with the induction of apoptosis. Thus, our results suggest that compound 16 induced apoptosis in H1975 cells in the caspase-dependent manner.
Necroptosis has been emerged as a potential therapeutic target for treating cancerCitation11,Citation37. In our previous studies, we found that IBC trigger various forms of death in human breast cancer MDA-MB-231 cells, including necroptosisCitation15. Here, we showed for the first time that compound 16 induced apoptosis and necroptosis in H1975 cells, whereas necroptosis might be more important for compound 16 triggered PCD. The state of RIP1 and RIP3 determines whether they function as biomarkers that trigger or inhibit PCDCitation38,Citation39. In the cell viability experiments, Nec-1 or NSA significantly suppressed the cell death induced by compound 16 (40 μM) in the H1975 cells. These data showed that compound 16 increased RIP3, p-RIP3, MLKL, and p-MLKL levels in a concentration-dependent manner. In the siRNA experiments, the siRNA1171 inhibited compound 16 induced cell death and caused loss of the protective effect of Nec-1. Here, we also used compound 16 for molecular docking with RIP3 to indirectly reflect the interaction of 16 with the target protein. The binding energy of compound 16 with RIP3 was −13.4 kcal/mol. The strong binding affinity of 16 with RIP3 supports the role of compound 16 as a new RIP3-dependent necroptosis inducer.
In addition, the down-regulation of RIP1 protein levels was observed after treated with compound 16 (20, 40, and 80 µM). RIP1 is an important upstream signalling protein in the cell death receptor that regulates both cell survival and PCDCitation40. RIP1 not only activates NF-κB against apoptosis but also induces death receptor-mediated apoptosis and necroptosisCitation41,Citation42. Corresponding to these references, our studies demonstrated that compound 16 not only induces necroptosis but also induces apoptosis in H1975 cells by treatment with 40 µM of compound 16. Therefore, the down-regulation of RIP1 expression can also be considered reasonable, and the molecular mechanism needs to be further studies.
Mitochondria is an important organelle that plays a central role in cellular ROS generation and energy productionCitation43,Citation44. In this study, compound 16 induced PCD was closely associated with similar changes in mitochondrial function, mainly a decline in mitochondrial membrane potential, a decrease in ATP level, and the generation and accumulation of ROS. High levels of intracellular ROS have been recognised as the driving forces, leading to PCDCitation45. When the H1975 cells were treated with NAC and compound 16, NAC significantly suppressed the increase of ROS level and cell death induced by compound 16. Therefore, our data suggest that compound 16 induced necroptosis in H1975 cells via the up-regulation of the RIP3-MLKL-ROS signalling pathway.
Conclusions
In summary, we synthesised seventeen IBC derivatives and investigated their in vitro anti-cancer activities. Among these derivatives, compound 16 exhibited the most potent cytotoxic activity against H1975 cells. Our further study showed that compound 16 induced PCD in H1975 cells by triggering apoptosis and necroptosis. Importantly, compound 16 induced necroptosis via up-regulating RIP3-MLKL signalling pathway and ROS accumulation. Therefore, compound 16 may be a potential anti-NSCLC agent that warrants further investigation.
Experimental section
General information
All reagents used in the synthesis were obtained commercially and used without further purification unless otherwise specified. The reaction was monitored by thin-layer chromatography (TLC) on silica gel plates and visualised using a combination of UV. Flash column chromatography was performed using silica gel (300–400 mesh) was purchased from Qingdao Haiyang Chemical Co. Ltd. (Qingdao, China). The 1H-NMR and 13C-NMR spectra were recorded using TMS as the internal standard on a Bruker Avance (Bruker, Billerica, MA, USA) instrument. Chemical shifts were reported in ppm (d). High-resolution mass spectrometry (HRMS) spectra were determined using a Thermo Scientific LTQ Orbitrap XL mass spectrometer (Bruker, Bremerhaven, Germany). IBC was purchased from Mansite Biological Co. Ltd. (Chengdu, China). Human lung cancer (H1795, A549, and PC9) cells and normal mouse hepatocytes Aml-12 cells were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). RPMI-1640 was purchased from Gibco (California, USA). Foetal bovine serum (FBS) was purchased from Sijiqing (Hangzhou, China). RIPA lysis buffer and dimethylsulphoxide (DMSO), Nec-1, z-VAD-FMK, NSA, and NAC were purchased from Sigma-Aldrich (St. Louis, MO, USA). BCA protein assay kit, Annexin V-FITC cell apoptosis detection kit, ATP assay kit, and ROS assay kit were purchased from Beyotime (Hangzhou, China). The JC-1 assay kit was purchased from BestBio (Shanghai, China). Anti-RIP1, anti-RIP3, anti-MLKL, anti-p-MLKL, and anti-caspase-3 antibodies were purchased from Abcam (Cambridge, MA, UK). Anti-Bax, anti-Bcl-2, anti-Akt, and β-Actin antibodies were purchased from Proteintech (Wuhan, China). Anti-Cyt-C, anti-caspase-3, and anti-p-RIP3 were purchased from Abclonal (Wuhan, China).
General procedure for the preparation of compounds 1–17
The general procedure for the preparation of 1–3
To a solution of IBC (178 mg, 0.55 mmol) in DMF (6 ml) were added 10% Na2CO3 solution (3 ml) and CH3I (0.084 ml, 1.65 mmol), and the mixture was stirred at 65 °C for 10 h. The reaction was quenched with water and extracted three times with ethyl acetate. Furthermore, the combined extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Finally, the crude residue was purified by silica gel column chromatography using petroleum/ethyl acetate (10:1) to afford pure compounds 1–3, respectively.
(E)-1-(2,4-Dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (1)
Yellow solid, yield 9.7%; 1H-NMR (300 MHz, DMSO-d6) δ 13.96 (1H, s, -OH), 10.59 (1H, s, -OH), 8.08 (1H, d, J = 8.9 Hz, H-β), 7.88 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.80 (2H, d, J = 5.7 Hz, H-α), 7.04 (2H, d, J = 8.8 Hz, H-5′), 6.50 (1H, d, J = 8.9 Hz, H-5), 5.18 (m, H-2″), 3.83 (3H, s, H-7′), 3.25 (2H, d, J = 6.8 Hz, H-1″), 1.73 (3H, s, H-5″), 1.62 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.73, 163.56, 162.36, 161.43, 143.62, 130.96, 130.96, 130.47, 129.95, 129.94, 127.29, 122.33, 118.48, 114.43, 114.43, 112.70, 107.34, 55.42, 25.49, 21.27, 17.71; ESI-HRMS (m/z): calcd. for C21H23O4 + [M + H] + 339.1591, found 339.1592.
(E)-1-(2-Hydroxy-4-methoxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl) prop-2-en-1-one (2)
Yellow solid, yield 22.0%; 1H-NMR (300 MHz, DMSO-d6) δ 13.81 (1H, s, -OH), 10.16 (1H, s, -OH), 8.25 (1H, d, J = 9.0 Hz, H-6), 7.81 (1H, d, J = 4.2 Hz, H-α), 7.80 (3H, d, J = 9.1 Hz, H-β, H-2′, H-6′), 6.86 (2H, d, J = 8.6 Hz, H-3′, H-5′), 6.69 (1H, d, J = 9.0 Hz, H-5), 5.13 (m, H-2″), 3.90 (3H, s, H-7), 3.27 (2H, d, J = 7.0 Hz, H-1″), 1.72 (3H, s, H-5″), 1.61 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 192.37, 162.91, 162.12, 160.41, 144.81, 131.41, 130.83, 130.43, 125.71, 122.04, 117.24, 115.89, 115.86, 114.04, 102.73, 56.06, 25.49, 21.25, 17.67; ESI-HRMS (m/z): calcd. for C21H23O4 + [M + H] + 339.1591, found 339.1591.
(E)-1-(2-Hydroxy-4-methoxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-methoxyphenyl) prop-2-en-1-one (3)
Yellow solid, yield 44.6%; 1H-NMR (300 MHz, DMSO-d6) δ 13.76 (1H, s, -OH), 8.28 (1H, d, J = 9.0 Hz, H-6), 7.94 (1H, d, J = 15.8 Hz, H-β), 7.92 (2H, d, J = 7.8 Hz, H-2′, H-6′), 7.85 (1H, d, J = 15.3 Hz, H-α), 7.50 (2H, d, J = 8.1 Hz, H-3′, H-5′), 6.70 (1H, d, J = 9.0 Hz, H-5), 5.13 (m, H-2″), 3.91 (3H, s, H-7′), 3.83 (3H, s, H-7), 3.28 (2H, d, J = 7.2 Hz, H-1″), 1.73 (3H, s, H-5″), 1.62 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 192.37, 163.00, 162.12, 161.57, 144.30, 131.15, 130.86, 130.58, 127.22, 122.00, 118.37, 115.89, 114.44, 114.02, 102.78, 56.08, 55.42, 25.48, 21.24, 17.67; ESI-HRMS (m/z): calcd. for C22H25O4 + [M + H] +353.1747, found 353.1749.
The general procedure for the preparation of 4–5
To a solution of IBC (149 mg, 0.46 mmol) in DMF (6 ml) were added Na2CO3 (36 mg, 0.34 mmol) and benzyl chloride (0.3 ml, 2.61 mmol), and the mixture was stirred at 65 °C for 12 h. The reaction mixture was quenched with water and extracted three times with ethyl acetate. Furthermore, the combined extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Finally, the crude residue was purified by silica gel column chromatography using petroleum/ethyl acetate = 10:1 to afford pure compounds 4 and 5, respectively.
(E)-1-(4-(Benzyloxy)-2-hydroxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-(benzyl-oxy) phenyl)prop-2-en-1-one (4)
Yellow solid, yield 32.9%; 1H-NMR (300 MHz, DMSO-d6) δ 13.77 (1H, s, -OH), 8.26 (1H, d, J = 9.2 Hz, H-6), 7.94 (1H, d, J = 15.2 Hz, H-β), 7.91 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.84 (1H, d, J = 15.3 Hz, H-α), 7.41 (m, J = 8.1 Hz, H-9, H-10, H-11, H-12, H-13, H-9′, H-10′, H-11′, H-12′, H-13′), 7.13 (2H, d, J = 8.8 Hz, H-3′, H-5′), 6.78 (1H, d, J = 9.1 Hz, H-5), 5.23 (m, H-2″), 5.19 (2H, s, H-7), 3.30 (2H, d, J = 8.9 Hz, H-1″), 1.63 (3H, s, H-5″), 1.61 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 192.37, 162.24, 162.06, 160.65, 144.26, 136.68, 131.16, 130.85, 130.46, 128.48, 127.97, 127.82, 127.54, 121.96, 118.51, 116.26, 115.26, 114.08, 104.04, 69.75, 69.43, 25.49, 21.40, 17.65; ESI-HRMS (m/z): calcd. for C34H32O4Na+ [M + Na] + 527.2198, found 527.2195.
(E)-3-(4-(Benzyloxy)phenyl)-1-(2,4-dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl)prop-2-en-1-one (5)
Yellow solid, yield 13.6%; 1H-NMR (300 MHz, DMSO-d6) δ 13.83 (1H, s, -OH), 10.17 (1H, s, -OH), 8.23 (1H, d, J = 9.1 Hz, H-6), 7.86 (1H, d, J = 15.6 Hz, H-β), 7.80 (2H, d, J = 10.1 Hz, H-2′, H-6′), 7.75 (1H, d, J = 15.3 Hz, H-α), 7.41 (m, H-9′, H-10′, H-11′, H-12′, H-13′), 6.86 (2H, d, J = 8.5 Hz, H-3′, H-5′), 6.77 (1H, d, J = 9.1 Hz, H-5), 5.27 (2H, s, H-7′), 5.15 (m, H-2″), 3.32 (2H, d, J = 7.0 Hz, H-1″), 1.62 (3H, s, H-5″), 1.60(3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 192.37, 162.23, 161.95, 160.41, 144.83, 136.70, 131.40, 130.83, 130.28, 128.46, 127.94, 127.53, 125.72, 122.0, 117.26, 116.27, 115.86, 114.10, 103.97, 69.73, 25.50, 21.42, 17.65; ESI-HRMS (m/z): calcd. for C27H26O4Na+ [M + Na] + 437.1723, found 437.1725.
The procedure for the preparation of 6
To a solution of IBC (298 mg, 0.92 mmol) in CHCl3 (8 ml) was added 25% NaOH solution (4 ml) at 30 °C for 9 h. The reaction was quenched with water, and adjusted with 10% HCl to acidity. Then the mixture was extracted three times with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure. Finally, the crude residue was purified by silica gel column chromatography using CH2Cl2/MeOH = 30:1 to afford pure compound 6.
(E)-2,4-Dihydroxy-5-(3-(4-hydroxyphenyl)acryloyl)-3-(3-methylbut-2-en-1-yl)benzylaldehyde (6)
Yellow solid, yield 4.1%; 1H-NMR (300 MHz, DMSO-d6) δ 14.61 (1H, s, -OH), 11.93 (1H, s, -OH), 10.30 (1H, s, H-7), 9.89 (2H, s, -OH), 8.85 (1H, s, H-6), 7.87 (2H, d, J = 16.6 Hz, H-α, H-β), 7.82 (2H, d, J = 8.3 Hz, H-2′, H-6′), 6.89 (2H, d, J = 8.3 Hz, H-3′, H-5′), 5.16 (m, H-2″), 3.28 (2H, d, J = 7.2 Hz, H-1″), 1.74 (3H, s, H-5″), 1.63 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 195.76, 192.37, 167.53, 163.84, 160.87, 146.17, 137.95, 131.74, 131.66, 125.50, 121.01, 116.46, 115.98, 115.47, 114.57, 113.73, 25.45, 20.57, 17.72; ESI-HRMS (m/z): calcd. for C21H21O5 +[M + H] + 353.1311, found 353.1383.
The procedure for the preparation of 7
To a solution of 6 (70 mg, 0.2 mmol) with aminoguanidine carbonate (27 mg, 0.2 mmol) in EtOH (3 ml) was added glacial acetic acid (0.5 ml), and the mixture was stirred at 50 °C for 5 h. The reaction mixture was extracted with ethyl acetate, washed with aqueous NaHCO3 and dried over anhydrous Na2SO4 and concentrated under reduced pressure. Finally, the crude residue was purified by silica gel column chromatography using CH2Cl2/MeOH = 20:1 to afford pure compound 7.
2-((E)-2,4-Dihydroxy-5-((E)-3-(4-hydroxyphenyl)acryloyl)-3-(3-methylbut-2-en-1-yl) benzylidene)hydrazine-1-carboximidamide (7)
Yellow solid, yield 7.9%; 1H-NMR (300 MHz, DMSO-d6) δ 14.27 (1H, s, -OH), 10.39 (1H, s, -NH), 10.30 (1H, s, -OH), 8.51 (1H, s, -CH = N-), 8.37 (1H, s, H-6), 7.95 (1H, m, H-α), 7.89 (1H, m, H-β), 7.86 (3H, s, -NH2, =NH), 7.80 (2H, d, J = 8.6 Hz, H-2′, H-6′), 6.89 (2H, d, J = 8.5 Hz, H-3′, H-5′), 5.17 (m, H-2″), 3.29 (2H, d, J = 6.3 Hz, H-1″), 1.75 (3H, s, H-5″), 1.63 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 192.25, 164.48, 160.70, 160.31, 154.81, 148.54, 145.55, 131.91, 131.59, 131.34, 125.58, 121.67, 116.91, 115.94, 115.82, 113.56, 111.50, 25.48, 21.21, 17.81; ESI-HRMS (m/z): calcd. for C22H25N4O4 + [M + H] + 409.18758, found 409.18719.
The general procedure for the preparation of 8–17
To a solution of IBC (149 mg, 0.46 mmol) in MeCN (4 ml) was added aldehydes (4-pyridine-carboxaldehyde, 0.69 mmol; benzaldehyde, 0.69 mmol; 4-bromobenz-aldehyde, 0.69 mmol; 4-chlorobenzaldehyde, 0.69 mmol; 4-fluorobenzyl chloride, 0.69 mmol; 4-nitrobenzaldehyde, 0.69 mmol; 4-carboxy-benzaldehyde, 0.69 mmol; 4-methoxy-bezaldehyde, 0.69 mmol; 3,4,5-trimethoxybenzaldehyde, 0.69 mmol; 4-trifluoro-methylbenzaldehyde, 0.69 mmol) and morpholine (0.86 mmol). After stirred at 80 °C for 12 h, the reaction mixture was quenched with water and extracted three times with ethyl acetate. The combined extracts were dried over anhydrous Na2SO4 and concentrated under reduced pressure to provide the crude products. Finally, the crude residues were purified by silica gel column chromatography using petroleum/ethyl acetate = 3:1 to afford pure compounds 8–17, respectively.
(E)-1-(2,4-Dihydroxy-3-(3-methylbut-2-en-1-yl)-5-(morpholino(pyridin-4-yl)methyl) phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (8)
Yellow solid, yield 66.2%; 1H-NMR (300 MHz, DMSO-d6) δ 13.93 (1H, s, -OH), 12.25 (1H, s, -OH), 10.25 (1H, s, -OH), 8.73 (2H, d, J = 5.6 Hz, H-4‴, H-6‴), 8.12 (1H, s, H-6), 7.87 (2H, d, J = 6.1 Hz, H-2′, H-6′), 7.80 (1H, d, J = 15.3 Hz, H-β), 7.77 (2H, d, J = 6.8 Hz, H-3‴, H-7‴), 7.72 (1H, d, J = 15.4 Hz, H-α), 6.90 (2H, d, J = 8.6 Hz, H-3′, H-5′), 5.15 (m, H-2″), 4.93 (1H, s, H-1‴), 3.75 (2H, m, H-9‴, H-10‴), 3.27 (2H, d, J = 6.7 Hz, H-1″), 2.54 (2H, t, J = 8.2 Hz, H-8‴, H-11‴), 1.71 (3H, s, H-5″), 1.61 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.67, 163.08, 160.57, 160.52, 147.13, 147.08, 144.77, 131.29, 131.03, 129.31, 125.06, 124.02, 121.95, 116.98, 115.91, 115.77, 112.99, 99.80, 65.63, 59.75, 51.79, 25.47, 21.37, 17.73; ESI-HRMS (m/z): calcd. for C30H32N2O5Na+ [M + Na] + 523.22034, found 523.22064.
(E)-1-(2,4-Dihydroxy-3-(3-methylbut-2-en-1-yl)-5-(morpholino(phenyl)methyl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (9)
Yellow solid, yield 43.5%; 1H-NMR (300 MHz, DMSO-d6) δ 13.93 (1H, s, -OH), 10.20 (1H, s, -OH), 8.00 (1H, s, H-6), 7.78 (1H, d, J = 13.7 Hz, H-β), 7.76 (2H, d, J = 8.9 Hz, H-3‴, H-7‴), 7.71 (1H, d, J = 15.5 Hz, H-β), 7.54 (1H, d, J = 7.4 Hz, H-2′, H-6′), 7.36 (m, H-4‴, H-5‴, H-6‴), 6.89 (2H, d, J = 8.5 Hz, H-3′, H-5′), 5.21 (m, H-2″), 4.80 (1H, s, H-1‴), 3.70 (m, H-9‴, H-10‴), 3.30 (2H, d, J = 6.8 Hz, H-1″), 2.57 (2H, t, J = 11.0 Hz, H-8‴, H-11‴), 1.74 (3H, s, H-5″), 1.63 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.55, 162.68, 161.20, 160.38, 144.44, 131.26, 130.85, 129.03, 128.95, 128.12, 127.96, 125.66, 122.16, 117.33, 117.08, 115.86, 115.42, 112.60, 65.72, 59.75, 51.84, 25.50, 21.36, 17.75; ESI-HRMS (m/z): calcd. for C31H33NO5Na+ [M + Na] + 522.22509, found 522.22534.
(E)-1-(5-((4-Bromophenyl)(morpholino)methyl)-2,4-dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (10)
Yellow solid, yield 16.0% 1H-NMR (300 MHz, DMSO-d6) δ 13.91 (1H, s, -OH), 10.20 (1H, s, -OH), 7.96 (1H, s, H-6), 7.72 (m, H-α, H-β, H-4‴, H-6‴), 7.64 (1H, s, -OH), 7.59 (1H, d, J = 6.4 Hz, H-2′, H-6′), 7.47 (2H, d, J = 6.7 Hz, H-3‴, H-7‴), 6.87 (2H, d, J = 6.7 Hz, H-3′, H-5′), 5.20 (m, H-2″), 4.70 (1H, s, H-1‴), 3.67 (2H, t, J = 16.1 Hz, H-9‴, H-10‴), 3.27 (2H, d, J = 7.0 Hz, H-1″), 2.45 (2H, t, J = 11.5 Hz, H-8‴, H-11‴), 1.73 (3H, s, H-5″), 1.62 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.55, 162.70, 161.20, 160.39, 144.43, 139.14, 131.89, 131.25, 130.90, 130.22, 130.19, 129.11, 125.69, 122.16, 121.07, 117.10, 115.89, 115.50, 112.66, 73.46, 65.91, 48.63, 25.52, 21.37, 17.77; ESI-HRMS (m/z): calcd. for C31H33BrNO5 +[M + H] + 578.15366, found 578.15399.
(E)-1-(5-((4-Chlorophenyl)(morpholino)methyl)-2,4-dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (11)
Yellow solid, yield 21.7%; 1H-NMR (300 MHz, DMSO-d6) δ 13.90 (1H, s, -OH), 13.08 (1H, s, -OH), 10.14 (1H, s, -OH), 7.95 (1H, s, H-6), 7.76 (1H, d, J = 14.6 Hz, H-α), 7.72 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.68 (1H, d, J = 15.5 Hz, H-β), 7.54 (2H, d, J = 8.5 Hz, H-4‴, H-6‴), 7.45 (2H, d, J = 8.4 Hz, H-3‴, H-7‴), 7.36 (m, H-5‴), 6.88 (2H, d, J = 8.5 Hz, H-3′, H-5′), 5.20 (m, H-2″), 4,73 (1H, s, H-1‴), 3.68 (2H, t, J = 15.1 Hz, H-9‴, H-10‴), 3.28 (2H, d, J = 6.4 Hz, H-1″), 2.45 (2H, t, J = 12.5 Hz, H-8‴, H-11‴), 1.73 (3H, s, H-5″), 1.62 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.53, 162.68, 161.14, 160.37, 144.41, 138.71, 132.47, 131.22, 130.85, 129.84, 128.93, 125.66, 123.84, 122.14, 117.11, 115.85, 115.46, 112.64, 73.40, 65.89, 51.83, 21.33, 17.74; ESI-HRMS (m/z): calcd. for C31H32ClNO5Na+ [M + Na] + 556.18612, found 556.18616.
(E)-1-(5-((4-Fluorophenyl)(morpholino)methyl)-2,4-dihydroxy-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (12)
Yellow solid, yield 18.21%; 1H-NMR (300 MHz, DMSO-d6) δ 13.91 (1H, s, -OH), 13.19 (1H, s, -OH), 10.16 (1H, s, -OH), 7.96 (1H, d, J = 16.1 Hz, H-β), 7.75 (3H, d, J = 8.7 Hz, H-2′, H-4‴, H-6′), 7.71 (1H, s, H-6), 7.69 (1H, d, J = 15.5 Hz, H-α), 7.54 (m, H-3‴, H-7‴), 7.21 (m, H-6‴), 6.88 (m, H-3′, H-5′), 5.21 (m, H-2″), 4.73 (1H, s, H-1‴), 3.67 (2H, t, J = 11.0 Hz, H-9‴, H-10‴), 3.28 (2H, d, J = 7.1 Hz, H-1″), 2.44 (2H, t, J = 11.8 Hz, H-8‴, H-11‴), 1.73 (3H, s, H-5″), 1.63 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.53, 162.64, 161.22, 160.37, 156.66, 144.39, 139.43, 131.23, 130.84, 130.01, 129.08, 125.66, 122.16, 117.37, 115.86, 115.60, 115.44, 114.77, 112.60, 73.34, 65.89, 63.30, 25.50, 21.35, 17.74; ESI-HRMS (m/z): calcd. for C31H32FNO5Na+ [M + Na] + 540.2157, found 540.2159.
(E)-1-(2,4-Dihydroxy-3-(3-methylbut-2-en-1-yl)-5-(morpholino(4-nitrophenyl)methyl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (13)
Yellow solid, yield 20.38%; 1H-NMR (300 MHz, DMSO-d6) δ 13.91 (1H, s, -OH), 10.19 (1H, s, -OH), 8.26 (2H, d, J = 8.7 Hz, H-4‴, H-6‴), 8.06 (1H, s, H-6), 7.84 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.78 (1H, d, J = 15.3 Hz, H-β), 7.76 (2H, d, J = 8.4 Hz, H-3‴, H-7‴), 7.71 (1H, d, J = 15.3 Hz, H-α), 6.89 (2H, d, J = 8.5 Hz, H-3′, H-5′), 5.17 (m, H-2″), 4.96 (1H, s, H-1‴), 3.74 (m, H-9‴, H-10‴), 3.28 (2H, d, J = 6.6 Hz, H-1″), 2.61 (m, H-8‴, H-11‴), 1.72 (3H, s, H-5″), 1.61 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.62, 162.91, 160.71, 160.46, 146.99, 144.62, 144.60, 131.26, 130.97, 129.30, 129.20, 125.63, 124.18, 122.02, 117.03, 116.22, 115.89, 115.71, 112.86, 72.88, 65.65, 51.86, 25.47, 21.35, 17.74; ESI-HRMS (m/z): calcd. for C31H32N2O7Na+ [M + Na] + 567.21017, found 567.21033.
(E)-4-((2,4-Dihydroxy-5-(3-(4-hydroxyphenyl)acryloyl)-3-(3-methylbut-2-en-1-yl)phenyl)(morpholino)methyl)benzoic acid (14)
Yellow solid, yield 24.52%; 1H-NMR (300 MHz, DMSO-d6) δ 14.01 (1H, s, -COOH), 13.92 (1H, s, -OH), 7.99 (1H, s, H-6), 7.94 (2H, d, J = 8.1 Hz, H-4‴, H-6‴), 7.72 (m, H-2′, H-6′, H-α, H-β), 7.64 (2H, d, J = 8.1 Hz, H-3‴, H-7‴), 6.88 (2H, d, J = 8.3 Hz, H-3′, H-5′), 5.21 (m, H-2″), 4.77 (1H, s, H-1‴), 3.69 (m, H-9‴, H-10‴), 3.27 (2H, d, J = 8.0 Hz, H-1″), 2.42 (m, H-8‴, H-11‴), 1.73 (3H, s, H-5″), 1.62 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.55, 167.74, 162.73, 161.20, 160.47, 144.46, 144.26, 131.28, 130.89, 129.98, 129.93, 129.20, 128.05, 125.66, 122.16, 117.10, 117.02, 115.89, 115.50, 112.68, 73.93, 65.92, 51.85, 25.51, 21.37; ESI-HRMS (m/z): calcd. for C31H32N2O7Na+ [M + Na] + 566.2149, found 566.2151.
(E)-1-(2,4-Dihydroxy-5-((4-methoxyphenyl)(morpholino)methyl)-3-(3-methylbut-2-en-1-yl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (15)
Yellow solid, yield 26.23%; 1H-NMR (300 MHz, DMSO-d6) δ 13.91 (1H, s, -OH), 13.42 (1H, s, -OH), 10.15 (1H, s, -OH), 7.91 (1H, s, H-6), 7.75 (2H, d, J = 8.7 Hz, H-2′, H-6′), 7.72 (m, H-α, H-β), 7.40 (2H, d, J = 8.6 Hz, H-3‴, H-7‴), 6.94 (2H, d, J = 8.6 Hz, H-4‴, H-6‴), 6.87 (2H, d, J = 8.5 Hz, H-3′, H-5′), 5.22 (m, H-2″), 4.65 (1H, s, H-1‴), 3.71 (1H, s, H-12‴), 3.66 (m, H-9‴, H-10‴), 3.28 (2H, d, J = 6.5 Hz, H-1″), 2.43 (m, H-8‴, H-11‴), 1.74 (3H, s, H-5″), 1.63 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.49, 162.55, 161.55, 160.34, 158.80, 144.29, 131.49, 131.21, 130.77, 129.35, 129.04, 125.70, 122.26, 117.77, 117.13, 115.84, 115.30, 114.25, 112.49, 73.71, 65.95, 55.08, 51.78, 25.51, 21.36, 17.74; ESI-HRMS (m/z): calcd. for C32H35NO6Na+ [M + Na] + 529.2464, found 530.2537.
(E)-1-(2,4-Dihydroxy-3-(3-methylbut-2-en-1-yl)-5-(morpholino(3,4,5-trimethoxyphe-nyl)methyl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (16)
Yellow solid, yield 12.03%; 1H-NMR (300 MHz, DMSO-d6) δ 13.94 (1H, s, -OH), 13.48 (1H, s, -OH), 10.16 (1H, s, -OH), 7.92 (1H, s, H-6), 7.77 (1H, d, J = 15.3 Hz, H-β), 7.74 (2H, d, J = 7.7 Hz, H-2′, H-6′), 7.69 (1H, d, J = 15.2 Hz, H-α), 6.86 (2H, d, J = 8.6 Hz, H-3‴, H-7‴), 6.84 (2H, d, J = 7.1 Hz, H-3′, H-5′), 5.24 (m, H-2″), 4.59 (1H, s, H-1‴), 4.11 (3H, s, H-12‴, H-13‴, H-14‴), 3.75 (m, H-9‴, H-10‴), 3.61 (m, H-1″), 3.33 (m, H-8‴, H-11‴), 1.74 (3H, s, H-5″), 1.60 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.49, 162.43, 161.73, 160.35, 152.95, 144.32, 136.95, 135.27, 131.19, 130.73, 129.09, 125.71, 122.17, 117.46, 117.15, 115.85, 115.48, 112.51, 74.63, 65.95, 59.91, 59.76, 55.73, 25.47, 21.32, 17.73; ESI-HRMS (m/z): calcd. for C34H39NO8Na+ [M + Na] + 612.25679, found 612.25696.
(E)-1-(2,4-Dihydroxy-3-(3-methylbut-2-en-1-yl)-5-(morpholino(4-(trifluoromethyl) phenyl)methyl)phenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one (17)
Yellow solid, yield 12.38%; 1H-NMR (300 MHz, DMSO-d6) δ 13.91 (1H, s, -OH), 10.18 (1H, s, -OH), 7.99 (1H, s, H-6), 7.75 (m, H-2′, H-6′, H-α, H-β, H-3‴, H-4‴, H-6‴, H-7‴), 6.88 (2H, d, J = 8.5 Hz, H-3′, H-5′), 5.20 (m, H-2″), 4.82 (1H, s, H-1‴), 4.10 (m, H-9‴, H-10‴), 3.69 (m, H-8‴, H-11‴), 3.28 (2H, d, J = 6.9 Hz, H-1″), 1.72 (3H, s, H-5″), 1.62 (3H, s, H-4″); 13C-NMR (75 MHz, DMSO-d6) δ 191.54, 162.77, 161.07, 160.40, 144.44, 144.40, 131.22, 130.86, 129.19, 129.16, 129.14, 128.79, 125.93, 125.65, 122.10, 117.05, 116.77, 115.86, 115.56, 112.70, 73.53, 65.87, 51.84, 25.48, 21.35, 17.73; ESI-HRMS (m/z): calcd. for C32H32F3NO5Na+ [M + Na] +590.21248, found 590.21252.
Cell culture
Four cell lines (H1975, A549, PC9, and Aml-12) were cultured in RPMI 1640 medium containing 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin in an atmosphere with 5% CO2 and 95% relative humidity at 37 °C.
MTT assay
Cell viability was examined using the MTT assay. The cells grown in the logarithmic phase were seeded in 96-well plates at a density of 5 × 103 per well. The cells were treated with various concentrations (0, 5, 10, 20, 40, and 80 μM) of the compound at different times, respectively. At the end of time point, MTT solution (5 mg/ml) was added to each well and incubated at 37 °C for 4 h. Next, the media was removed, and formazan was dissolved in DMSO. Finally, absorbance was measured with a microplate spectrophotometer at 490 nm (Synergy HT, BioTek, VT, USA).
Colony-formation assay
Cells were seeded at a density of 1.5 × 103 cells/well into 6-well plates. When colonies formed, the media was replaced with fresh media at various concentrations of compound 16 (0, 1, 2, and 4 μM) and incubated for 10 days. At the end of the incubation time, the cells were washed three times with PBS, fixed with 4% paraformaldehyde for 15 min, stained with crystal violet solution for 10 min, and photographed.
Flow cytometry with Annexin V/PI dual staining
Cells were seeded (3 × 105 per well) in 6-well plates and incubated overnight. After adherence, the cells were treated with various concentrations (0, 10, 20, 40, and 80 μM) of compound 16 for 24 h. Furthermore, the cells were harvested, washed three times with PBS, and stained with Annexin V (FITC Annexin V) and propidium iodide (PI) staining solution for 30 min in the dark. The percentage of cell death was measured by flow cytometry (Becton-Dickinson, Franklin Lakes, NJ, USA).
Fluorescence staining
Cells were seeded at a density of 5 × 103 cells/well in 12-well plates and treated with different concentrations (0, 10, 20, 40, and 80 μM) of 16. After incubated for 24 h, the cells were stained with Calcein/PI solutions and subsequent incubated at 37 °C for 30 min. The fluorescent intensity of the cells was observed using a fluorescence microscope (Olympus, Japan).
Western blotting
Cells were seeded in 100 mm dishes at a density of 3 × 106 cells/well and treated with different concentrations (0, 10, 20, 40, and 80 μM) of compound 16. After incubated for 24 h, the cells were harvested, and washed three times with PBS solution. Next, the cells were lysed with RIPA lysis buffer for 30 min on ice. Protein quantification was determined using the BCA protein assay kit. Equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membrane. The membrane was blocked with 5% non-fat milk and incubated with primary antibodies (Bax, Bcl-2, Akt, Cyt C, Caspase-9, Caspase-3, RIP1, RIP3, p-RIP3, MLKL, p-MLKL, and β-actin) for 2 h at room temperature, followed by the secondary antibodies for 2 h at room temperature. Finally, the blots were detected by a chemiluminescent gel imaging system (Bio-Rad, Hercules, CA, USA).
Electron microscopy
Cells were seeded in a 6-well plate (1.5 × 103 cells/well) and treated with compound 16 (40 μM) and control for 6 h. At the end of time point, the cells were washed with PBS, fixed with 2.5% glutaraldehyde, and stored at 4 °C. Post-fixed samples were sent to Servicebio (Wuhan, China) for subsequent experiments using transmission electron microscopy (TEM).
Knockdown of RIP3 using siRNA
RIP3 siRNAs were purchased from Gene-Pharma (Shanghai, China). The siRNAs were transiently transfected into H1975 cells using a lipofectamine 2000 reagent kit (Invitrogen, CA, USA) according to the manufacturer’s protocol. After 48 h of transfection, the cells were collected for western blot analysis. The sequences of siRNA used for experiments with human RIP3 were as follows: positive control sense, 5′-UGA CCU CAA CUA CAU GGU UTT-3′ and antisense, 5′-AAC CAU GUA GUU GAG GUC ATT-3′; negative control sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′ and antisense, 5′-ACG UGA CAC GUU CGG AGA ATT-3′; RIP3 homo-1687 sense, 5′-GCC ACA GGG UUG GUA UAA UTT-3′ and antisense, 5′-AUU AUA CCA ACC CUG UGG CTT-3′; RIP3 homo-305 sense, 5′-GCG GUC AAG AUC GUA AAC UTT-3′ and antisense, 5′-AGU UUA CGA UCU UGA CCG CTT-3′; RIP3 homo-1503 sense, 5′-GAC CGC CUC GUU AAC AUA ATT-3′ and antisense, 5′-UAU AUG UUA ACG AGC GGU CTT-3′; and RIP3 homo-1171 sense, 5′-CCA GCAC CUC UCG UAA UGA ATT-3′ and antisense, 5′-AUC AUU ACG AGA GUG CUG GTT-3′.
Detection of mitochondrial membrane potential with JC-1
Cells were seeded in 6-well plates at a density of 3 × 105 cells/well and treated with various concentrations (0, 10, 20, 40, and 80 μM) of compound 16. After incubated for 24 h, the cells were harvested, stained with JC-1 solution, and incubated for 30 min at 37 °C. Finally, the cells were detected and analysed using a flow cytometry (Becton-Dickinson, Franklin Lakes, NJ, USA).
ATP detection
Cells were seeded in 6-well plates at a density of 3 × 105 cells/well and incubated with different concentrations (0, 10, 20, 40, and 80 μM) of compound 16 for 6 h. Afterwards, the cells were washed three times with PBS, lysed with RIPA buffer, and the lysates were centrifuged at 12 000 rpm for 5 min. The intracellular ATP levels were measured using a luminoskan luminometer (Thermo Scientific, Atlanta, GA, USA), according to the ATP assay kit’s protocol.
Detection of ROS
Cells were seeded in 6-well plates at a density of 3 × 105 cells/well and incubated with different concentrations (0, 10, 20, 40, and 80 μM) of compound 16 or pre-treated with NAC (20 μM) incubated for 6 h, the cells were co-incubated with DCFH-DA for 20 min at 37° C in the dark. Finally, the intracellular ROS levels were measured using flow cytometry (Becton-Dickinson, Franklin Lakes, NJ, USA).
Molecular docking
The compound 16 bound to RIP3 (PDB code: 6oko) was used for molecular docking experiment using MOE software. PyMOL software was used for further visualisation, figure preparation, and conformational analyses.
Statistical analysis
All data were analysed using GraphPad Prism software version 8.0. Student’s t-tests were performed to analyse the differences between the two groups. The results are presented as the mean ± SD from three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 were considered statistically significant.
Supplemental Material
Download PDF (3.8 MB)Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
- Travis WD, Brambilla E, Burke AP, Marx A, Nicholson AG. Introduction to the 2015 World Health Organization classification of tumors of the lung, pleura, thymus, and heart. J Thorac Oncol. 2015;10(9):1240–1242.
- Kocher F, Hilbe W, Seeber A, Pircher A, Schmid T, Greil R, Auberger J, Nevinny-Stickel M, Sterlacci W, Tzankov A, et al. Longitudinal analysis of 2293 NSCLC patients: a comprehensive study from the TYROL registry. Lung Cancer. 2015;87(2):193–200.
- Check JH, Poretta T, Check D, Srivastava M. Lung cancer–standard therapy and the use of a novel, highly effective, well tolerated, treatment with progesterone receptor molulators. Anticancer Res. 2023;43(3):951–965.
- Spiro SG, Rudd RM, Souhami RL, Brown J, Fairlamb DJ, Gower NH, Maslove L, Milroy R, Napp V, Parmar MK, et al. Chemotherapy versus supportive care in advanced non-small cell lung cancer: improved survival without detriment to quality of life. Thorax. 2004;59(10):828–836.
- Clegg A, Scott DA, Hewitson P, Sidhu M, Waugh N. Clinical and cost effectiveness of paclitaxel, docetaxel, gemcitabine, and vinorelbine in non-small cell lung cancer: a systematic review. Thorax. 2002;57(1):20–28.
- Lu X, Yu L, Zhang Z, Ren X, Smaill JB, Ding K. Targeting EGFRL858R/T790M and EGFRL858R/T790M/C797S resistance mutations in NSCLC: current developments in medicinal chemistry. Med Res Rev. 2018;38(5):1550–1581.
- Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, Chen Y, Han B. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. 2022;7(1):286.
- Gibellini L, Moro L. Programmed cell death in health and disease. Cells. 2021;10(7):1765.
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528–542.
- Wu Y, Dong G, Sheng C. Targeting necroptosis in anticancer therapy: mechanisms and modulators. Acta Pharm Sin B. 2020;10(9):1601–1618.
- Yin L, Liu P, Jin Y, Ning Z, Yang Y, Gao H. Ferroptosis-related small-molecule compounds in cancer therapy: strategies and applications. Eur J Med Chem. 2022;244:114861.
- Morana O, Wood W, Gregory CD. The apoptosis paradox in cancer. Int J Mol Sci. 2022;23(3):1328.
- Nyiramana MM, Cho SB, Kim EJ, Kim MJ, Ryu JH, Nam HJ, Kim NG, Park SH, Choi YJ, Kang SS, et al. Sea hare hydrolysate-induced reduction of human non-small cell lung cancer cell growth through regulation of macrophage polarization and non-apoptotic regulated cell death pathways. Cancer. 2020;12(3):726.
- Wu CZ, Gao MJ, Chen J, Sun XL, Zhang KY, Dai YQ, Ma T, Li HM, Zhang YX. Isobavachalcone induces multiple cell death in human triple-negative breast cancer MDA-MB-231 cells. Molecules. 2022;27(20):6787.
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz M, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119.
- Lu B, Gong X, Wang ZQ, Ding Y, Wang C, Luo TF, Piao MH, Meng FK, Chi GF, Luo YN, et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 ncrosome formation. Acta Pharmacol Sin. 2017;38(11):1543–1553.
- Xu B, Xu M, Tian Y, Yu Q, Zhao Y, Chen X, Mi P, Cao H, Zhang B, Song G, et al. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Discov. 2017;3(1):16096.
- Ma YM, Peng YM, Zhu QH, Gao AH, Chao B, He QJ, Li J, Hu YH, Zhou YB. Nover CHOP activator LGH00168 induces necroptosis in A5149 human lung cancer cells via ROS-mediated ER stress and NF-κB inhibition. Acta Pharmacol Sin. 2016;37(10):1381–1390.
- Sun W, Yu J, Gao H, Wu X, Wang S, Hou Y, Lu JJ, Chen X. Inhibition of lung cancer by 2-methoxy-6-acetyl-7-methyljuglone through induction of necroptosis by targeting receptor-interacting protein 1. Antioxid Redox Signal. 2019;31(2):93–108.
- Lan W, Santofimia-Castaño P, Xia Y, Zhou Z, Huang C, Fraunhoffer N, Barea D, Cervello M, Giannitrapani L, Montalto G, et al. Targeting NUPR1 with the small compound ZZW-115 is an efficient strategy to treat hepatocellular carcinoma. Cancer Lett. 2020;486:8–17.
- Bhalla VX, Nayak UR, Dev S. Some new flavonoids from Psoralea corylifolia. Tetrahedron Lett. 1968;9(20):2401–2406.
- Ouyang Y, Li J, Chen X, Fu X, Sun S, Wu Q. Chalcone derivatives: role in anticancer therapy. Biomolecules. 2021;11(6):894.
- Zhao S, Cui J, Cao L, Han K, Ma X, Chen H, Yin S, Zhao C, Ma C, Hu H. Xanthohumol inhibits non-small cell lung cancer via directly targeting T-lymphokine-activated killer cell-originated protein kinase. Phytother Res. 2023;37(7):3057–3068.
- Cheah SC, Lai SL, Lee ST, Hadi AH, Mustafa MR. Panduratin A, a possible inhibitor in metastasized A549 cells through inhibition of NG-kappa B translocation and chemoinvasion. Molecules. 2013;18(8):8764–8778.
- Deng N, Qiao M, Li Y, Liang F, Li J, Liu Y. Anticancer effects of licochalcones: a review of the mechanisms. Front Pharmacol. 2023;14:1074506.
- Kris MG, Natale RB, Herbst RS, Lynch TJ, Prager D, Belani CP, Schiller JH, Kelly K, Spiridonidis H, Sandler A, et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. J Am Med Assoc. 2003;290(16):2149–2158.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, et al. Erlotinib in previously treated non-small cell lung cancer. N Engl J Med. 2005;353(2):123–132.
- Reck M, Remon J, Hellmann MD. First-line immunotherapy for non-small-cell lung cancer. J Clin Oncol. 2022;40(6):586–597.
- Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–454.
- Liu X, Zhang H, Cao J, Zhuo Y, Jin J, Gao Q, Yuan X, Yang L, Li D, Wang Y. Isobavachalcone activates antitumor immunity on orthotopic pancreatic cancer model: a screening and validation. Front Pharmacol. 2022;13:919035.
- He H, Wang C, Liu G, Ma H, Jiang M, Li P, Lu Q, Li L, Qi H. Isobavachalcone inhibits acute myeloid leukemia: potential role for ROS-dependent mitochondrial apoptosis and differentiation. Phytother Res. 2021;35(6):3337–3350.
- Shi J, Chen Y, Chen W, Tang C, Zhang H, Chen Y, Yang X, Xu Z, Wei J, Chen J. Isobavachalcone sensitizes cells to E2-induced paclitaxel resistance by down-regulating CD44 expression in ER+ breast cancer cells. J Cell Mol Med. 2018;22(11):5220–5230.
- Carneiro BA, Ei-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417.
- Gao W, Xiao F, Wang X, Chen T. Artemisinin induces A549 cell apoptosis dominantly via a reactive oxygen species-mediated amplification activation loop among caspase-9, -8, and -3. Apoptosis. 2013;18(10):1201–1213.
- Jiao C, Chen W, Tan X, Liang H, Li J, Yun H, He C, Chen J, Ma X, Xie Y, et al. Ganoderma lucidum spore oil induces apoptosis of breast cancer cells in vitro and in vivo by activating caspase-3 and caspase-9. J Ethnopharmacol. 2020;247:112256.
- Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, Cheng H, Jin K, Ni Q, Yu X, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18(1):100.
- Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16(7):689–697.
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336.
- Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76(1):129–150.
- Yao Z, Zhang P, Guo H, Shi J, Liu S, Liu Y, Zheng D. RIP1 modulates death receptor mediated apoptosis and autophagy in macrophages. Mol Oncol. 2015;9(4):806–817.
- Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–736.
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159.
- Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J, Bazhin AV. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016;231(12):2570–2581.
- Moloney JN, Cotter TG. ROS signaling in the biology of cancer. Semin Cell Dev Biol. 2018;80:50–64.