
Abstract
Colorectal cancer (CRC) is one of the most common cancers worldwide. Nowadays, owing to the complex mechanism of tumorigenesis, simultaneous inhibition of multiple targets is an important anticancer strategy. Recent studies have demonstrated receptor tyrosine kinase AXL (AXL) and histone deacetylase 2 (HDAC2) are closely associated with colorectal cancer. Herein, we identified five hit compounds concurrently targeting AXL and HDAC2 using virtual screening. Inhibitory experiments revealed these hit compounds potently inhibited AXL and HDAC2 in the nanomolar range. Among them, Hit-3 showed the strongest inhibitory effects which were better than that of the positive control groups. Additionally, MD assays showed that Hit-3 could bind stably to the AXL and HDAC2 active pockets. Further MTT assays demonstrated that Hit-3 showed potent anti-proliferative activity. Most importantly, Hit-3 exhibited significant in vivo antitumor efficacy in xenograft models. Collectively, this study is the first discovery of dual-targeting AXL/HDAC2 inhibitors for colorectal cancer treatment.
Introduction
Cancer is a multifaceted and complex disease that is the main cause of mortality worldwide and one of the primary public health challenges harming the health of people globallyCitation1,Citation2. Colorectal cancer (CRC) is the third leading cause of cancer-related death, with an estimated 153,020 persons diagnosed and 52,550 dying from the disease by 2023Citation2,Citation3. The incidence of CRC has grown in recent years, and around 20% of CRC patients have metastasis at diagnosis, with up to 50% of individuals with initial localised disease developing metastasisCitation4. Radiotherapy and chemotherapy are still the main strategies for cancer treatmentCitation5,Citation6. Many marketed drugs now only act on a single target, which may result in drug resistance throughout the treatment process due to mutation and plasticity-driven drug targets that do not achieve good therapeutic resultsCitation7–9. It has been reported that more than 90% of mortality in cancer patients is attributed to resistance to chemotherapy drugs, making drug resistance a major obstacle to cancer treatmentCitation10,Citation11. Combining various medications may improve efficacy while also reducing the emergence of drug resistanceCitation12,Citation13. However, side effects and limits of combination therapy are common, including drug-drug interactions (DDI), unpredictable pharmacokinetics, and enhanced restrictive toxicityCitation8,Citation14. As an alternative strategy to combination therapy, dual-targeting drugs can effectively reduce drug interactions and toxicities while also having better pharmacokinetic profiles, which have been effective in an increasing number of clinical studiesCitation15–17. Therefore, dual-targeting drugs have been widely used in more cancer treatments in recent years, providing more therapeutic alternatives for cancer treatment.
In 1991, the receptor tyrosine kinase AXL (AXL) transformed gene was identified from primary myeloid leukaemia cells from two patients with chronic myeloid leukaemia (CML)Citation18. AXL proteins belong to the TAM receptor family (Tyro 3, AXL, and Mer), which are members of the receptor tyrosine kinase (RTK) familyCitation19. AXL protein overexpression has been associated with a variety of human cancers, such as liver cancer, non-small cell cancer, breast cancer, cervical cancer and so on, which are associated with poor prognosis and treatment resistanceCitation20–23. Meanwhile, aberrant activation of AXL signalling induces an epithelial-mesenchymal transition (EMT) in tumour cells, leading to increased migration and invasivenessCitation24,Citation25. Furthermore, the vitamin K-dependent protein growth arrest-specific 6 (Gas 6), a soluble glycoprotein, is a common ligand for the TAM receptor, with the highest affinity for AXLCitation26,Citation27. Gas 6 promotes AXL activation, thereby facilitating cell survival and proliferationCitation28. In addition, the Gas 6/AXL signalling pathway is essential for cell invasion, cell migration and angiogenesis, highlighting AXLCitation28,Citation29. Aside from the Gas 6/AXL signalling pathway, research has revealed that genetic and pharmacological AXL inhibition can affect downstream signalling pathways such as PI3K-AKT, AK-STAT, etcCitation30, which are crucial for the survival, migration, invasion, and angiogenesis of tumour cellsCitation28. Combining AXL inhibitors and PI3K/AKT pathway inhibitors can promote the inhibition of PI3K/AKT pathway inhibitors, which increases the rate of apoptosis and slows down the growth and proliferation of tumour cellsCitation31–33. Through downstream signal transduction of the JAK/STAT pathway, AXL also drives cancer development and metastasis cascadesCitation34. Therefore, AXL may be a potential target for cancer treatment. The reported small molecule inhibitors of AXL () can be classified according to the inhibitor-kinase binding mode into Type I inhibitors such as TP0903Citation35 and BGB324Citation36, which preferentially occupy the ATP-binding pocket of the kinase in the active “aspartate-phenylalanine-glycine (DFG) – in” conformation, and Type II inhibitors, like BMS-777607Citation37 and NPS-1034Citation38, adopt linear structures to form inactive DFG-out conformations. However, these AXL small molecule inhibitors face challenges of innate or acquired resistance to single-targeting therapies or even radiotherapy in tumour treatment, which leads to severe limitations in the clinical application of AXL small molecule inhibitorsCitation39–41. Therefore, we considered combining AXL with other potential targets to develop novel, potent, dual-targeting inhibitors to overcome these problems.
Figure 1. The reported AXL inhibitors and HDAC inhibitors.
Histone deacetylases (HDACs) are a crucial class of epigenetic enzymes that regulate gene expression by removing lysine residues from histonesCitation42,Citation43. HDACs are key regulators of several vital biological processes, including cell growth, cell differentiation, and cell apoptosisCitation44,Citation45. In addition, HDACs are involved in the regulation of protein expression, including tumour suppressors (p53, p21, etc.) and transcription factors (TFIIE, TCF, SF1, etc.)Citation46. Abnormalities in HDACs are closely associated with cancer development, which is mainly manifested by high levels of HDAC expression and HDAC mutationsCitation47. Among the HDACs, HDAC2 is a significant target in tumour development, as its abnormal expression promotes cancer progression primarily by silencing pro-apoptotic proteins such as NOXA and APAF1 (caspase 9-activating protein) and inactivating the tumour suppressor p53Citation48. In recent years, several HDAC inhibitors have been approved by the U.S. Food and Drug Administration (FDA) including Vorinostat (SAHA), Belinostat and so on ()Citation49,Citation50. Unfortunately, most of the HDAC inhibitors that have been reported with very limited efficacy against human solid tumours, which limits their partial application in oncological indicationsCitation5. Furthermore, these small molecule inhibitors are associated with adverse gastrointestinal, hematological, and cardiac side effectsCitation17,Citation51.
Despite the limitations of AXL and HDAC single-targeting inhibitors in different aspects, it has been shown that combining AXL inhibitors with HDAC inhibitors is effective in the treatment of tumoursCitation52,Citation53. In addition, HDAC inhibitors with a high affinity for HDAC2 have been demonstrated to induce apoptosis and cell cycle arrest while suppressing mitomycin C (MMC) induced expression of AXL and the expression of related moleculesCitation54. The multi-targeting HDAC, EGFR, and HER2 inhibitor CUDC-101 has been reported to promote the reduction of AXL levels to overcome treatment resistance caused by AXL overexpressionCitation55. Although these studies have demonstrated the relevance of AXL and HDAC in tumour therapy, no dual-targeting AXL/HDAC2 inhibitors for tumour treatment have been reported so far.
Based on the correlation between AXL and HDAC2, we used structure-based virtual screening technology to screen and obtain the dual-targeting AXL/HDAC2 inhibitorCitation56,Citation57, relying on its low cost, high efficiency, and ability to replace traditional screening methods, which avoid the drawbacks that exist in mono-targeting drugs and drug combination therapy. Here, we combined structure-based pharmacophore modelling, virtual screening, and molecular docking to obtain the hit compounds. Molecular dynamics simulations were performed to determine whether the best-scored compounds have good stability, and further in vitro and in vivo evaluations were performed to determine whether they have potential antitumor activity. The results showed that we successfully discovered a novel and potent dual-targeting AXL/HDAC2 inhibitor, providing a new candidate for tumour therapy.
Materials and methods
Materials
Cells and reagents. All cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, New York, NY, USA) (A549, MCF7, HepG2, HCT116 (KRAS(mut)/p53(wt)) and Hela) or Roswell Park Memorial Institute (RPMI) −1640 medium (Gibco, Grand Island, NY, USA) (H1975 and Anoikis-resistant H460) with 10% FBS, 1% penicillin/streptomycin comprising 5% CO2 controlling the temperature at 37 °C. All compounds were purchased from WuXi AppTec (Shanghai, China). Proteins were obtained from Abcam (Cambridge, UK). AXL and HDACs [HDAC1-11] proteins were obtained from Abcam (Cambridge, MA, USA). The 6-week-old BALB/c nude mice were bought from Changzhou Cavens Experimental Animal Limited Company (Changzhou, China).
Anoikis-resistant H460 cells. Anoikis-resistant H460 cells were established according to previously described methodsCitation58. First, attached H460 cells were trypsinized with 0.05% trypsin/0.02% EDTA. The cell suspensions were then mixed with serum-free RPMI-1640 medium at a density of 2 × 105 and seeded in 6-well plates (Corning, New York, NY, USA) for 48 h. After collection of the suspended cells, the cells were treated with 1 mM EDTA. After finally washing the cells with RPMI medium, cell viability was tested and viable cells were used for further experiments.
Establishment of AXL pharmacophore model
The X-ray 3D structures of AXL (PDB ID: 5U6B) and HDAC2 (PDB ID: 4LXZ) were obtained from the Protein Data Bank. All the structures were prepared in the Molecular Operating Environment (MOE) Program, which included partial charge calculation, protonation and energy minimisation of the two proteins. Subsequently, the structures of proteins were analysed, and the interactions between ligands and proteins were examined by using the ligand interaction tool. Based on the key interactions between AXL and small molecule inhibitors, the pharmacophore model of AXL was established with the pharmacophore query editor of MOE. All the established pharmacophores correspond to key amino acid residues of AXL.
Docking-based virtual screening
First, the In-house database of 43,000 compounds was established for virtual screening. These compounds were made up of 35,000 compounds reported in previous studies and 8000 compounds from the commercially available SPECS database (http://www.specs.net)Citation59. The MOE energy optimisation algorithm changed all of the compounds from two-dimensional structures to three-dimensional structures. The AXL pharmacophore model was selected as a constraint condition and the selected hit compounds interacted with AXL and HDAC2 active sites respectively. The native ligand and docking ligand were used for further docking verification, and the overlaps were assessed using the RMSD value calculation. MOE docking scores were used to evaluate the binding affinity and the docking scores of positive controls were set as the screening threshold value. Finally, five hits that met both scoring thresholds were chosen for further analysis.
Enzymatic assays for AXL and HDAC2
The in vitro HDACs enzymatic inhibitory experiments were performed as previously reportedCitation60. 10 μL of HDAC2 enzyme solution and 50 μL different concentrations of compounds were added to the 96-well plate. Following 5 min incubation at 37 °C, 40 μL of the fluorescent substrate Boc-Lys (acetyl) – AMC was added to the plates. The 100 L developer containing trypsin and TSA were then added to the mixture after the plates had been incubated at 37 °C for 30 min. Finally, after 20 min, the fluorescence intensity at 390 nm and 460 nm were measured by a microplate reader Perkin Elemer.
The in vitro AXL enzymatic assay was performed using the method previously reportedCitation61. The reaction was performed in the buffer containing 100 mM Hepes, pH 7.5, 10 mM MgCl2, 1 × Halt™ Protease and Phosphatase Inhibitor Cocktail, EDTA-Free, 1 mM DTT (Dithiothreitol), 0.003% of Brij35 and 0.004% of Tween-20. The total volume of the reaction was 26 μL including 1 nM AXL, 63 µM ATP (KmATP) and 1.5 μM compound. The plate was incubated at RT for 1.5 h and then stopped by adding 45 μL termination buffer containing 10 mM EDTA.
All the experiments were performed in triplicate. GraphPad Prism 6.0 software (GraphPad Software Inc, San Diego, CA) was used to analyse the competition binding data, and nonlinear curve fitting was used to get the inhibition constants (IC50).
Molecular dynamics simulation
The GROMACS software (version 2021.5) was used to simulate the stability of Hit-3 over time in the AMBER99SB-ILDN field. The molecular dynamics simulation was performed under periodic boundary conditionsCitation62,Citation63. First, the Hit-3 was solvated with a single point charge (SPC) water molecule in a cube box that was 1.0 nm distant from the complex, and Na+ and Cl− were used to neutralise the energy of the system instead of water molecules. The steepest descent algorithm was then used to set the energy of the system minimised with 5000 steps. A 100 ps NVT simulation with a V-rescale thermostat kept the system temperature at 300 K. This was followed by 100 ps NPT simulations using the Parinello-Rahman constant pressure device, which maintained the system pressure at 1 bar. Finally, the 50 ns MD simulation of the system was performed, and the RMSD and RMSF analysis of the system were recorded. Track data was saved every 10 ps.
MTT cell viability assay
According to the previously described methodCitation64, cell suspensions were prepared at the concentration of 5 × 104/ml and then cell lines were inoculated overnight in 96-well plates. Cells were exposed to different concentrations of inhibitors and incubated at 37 °C for 72 h. The incubation was maintained for an additional 4 h after adding 0.5 mg/ml MTT solution to each well. Insoluble crystals were dissolved with dimethyl sulfoxide and absorbance was measured at 570 nm according to the microplate reader. The data were analysed with the GraphPad Prism 6.0 software and half-maximal inhibitory concentration (IC50) values and cell viability was calculated from dose-response curves displayed by nonlinear regression (curve fitting).
In vivo anti-tumour activity
We injected tumour cells (200 µL, 1 × 107 cells) into the right subcutaneous space of 6-week-old BALB/c nude mice according to the previous in vivo antitumor activity assay methodCitation56. The Animal Ethics Committee of China Pharmaceutical University gave its approval to all the experimental protocols.[药代动方法]
The BALB/c nude mice were divided into four groups at random, with five mice in each group, while the tumours of BALB/c nude mice reached 80–120 mm3. The BALB/c nude mice were then injected intraperitoneally with vehicle (PBS buffer pH 7.4), Hit-3 (1 mg/kg), Hit-3 (10 mg/kg), and Hit-3 (20 mg/kg), respectively. The injection volume capacity of each group was 0.2 ml and the Hit-3 was all in PBS vehicle. Each group was injected every 3 days for a total of five times. Tumour volume was measured and computed as (c × c × d)/2 (c, minimum diameter; d, maximum diameter).
After that, the BALB/c nude mice were injected intraperitoneally with vehicle (PBS buffer pH 7.4), TP0903 (10 mg/kg), SAHA (10 mg/kg), and Hit-3 (10 mg/kg), respectively. The injection volume capacity of each group was 0.2 ml and each group was injected every 3 d for a total of five times. The body weights of the BALB/c nude mice were recorded, and the tumour volumes were measured and calculated according to the above method.
Results
Structure-based virtual screening of dual-targeting AXL/HDAC2 inhibitors
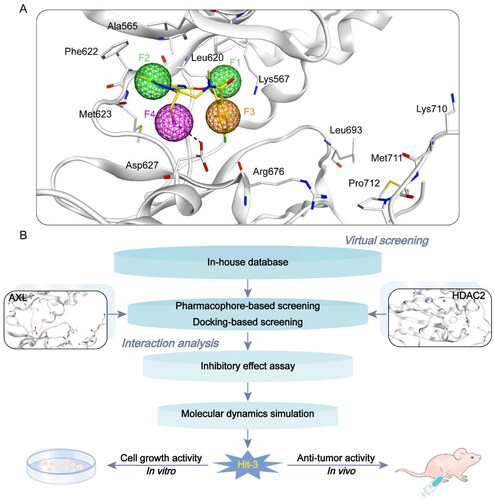
In this study, we identified novel and potent dual-targeting AXL/HDAC2 inhibitors from the In-house database (), and the identification steps were shown in . Firstly, we used a structure-based technique to construct the AXL pharmacophore model depending on the AXL structure of the crystal (PDB ID: 5U6B). As shown in , we generated four typical pharmacophore features (called F1-F4), two hydrophobic features (F1 and F2), one aromatic feature (F3), and one hydrogen bond donor feature (F4). Hydrophobic characteristics F1 and F2 corresponded to amino acid residues around the ligand, Leu620, Phe622, and Met623. The Arg676 residue was related to the aromatic character F3. The hydrogen bond interaction established by the amino acid residue Asp627 with the ligand corresponded to characteristic F4. According to the four pharmacophore characteristics of F1–F4, the pharmacophore model was established to prepare for the docking virtual screening.
Figure 2. The chemical structures of Hits 1–5.
Figure 3. Identification of dual-targeting AXL/HDAC2 inhibitors. (A) Pharmacophore model based on AXL (F1 and F2 are hydrophobic characteristics, F3 is aromatic characteristics, and F4 is hydrogen bond donor characteristics). Pharmacophore characteristics are represented by spheres. Hydrogen bonds are represented by the black dotted line. (B) The identification process of dual–targeting AXL/HDAC2 inhibitors.
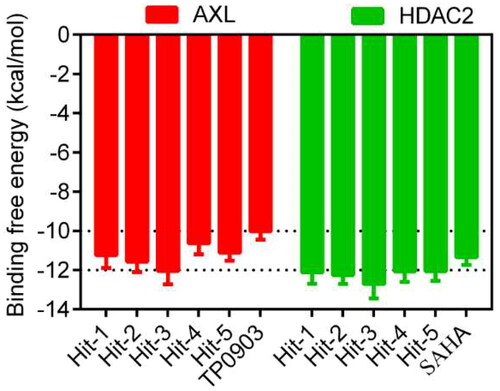
First, the small molecule database of 43,000 compounds was established for virtual screening and the 2D structure of each compound was converted into the 3D structure by energy minimisation. Following that, 379 hit compounds meeting all pharmacophore features were obtained by using the AXL pharmacophore model as a screening limitation. Before the docking-based virtual screening, the validations of native ligand and docking ligand with co-crystal ligand and protein complex were performed respectively. The validation results were shown in , showing the high overlaps between the native and docking ligands. The RMSD values were calculated with 0.009 nm of AXL and 0.012 nm of HDAC2. These 379 compounds were then screened and docked to AXL active sites and HDAC2 active sites. The binding free energy was utilised to forecast the binding affinity of hit compounds to the active sites, usually, the lower binding free energy indicated the stronger binding affinity. The active site of the compound connected to AXL was screened, and the docking score of each hit was calculated. The molecular docking score of TP0903 was −10.02 kcal/mol. Therefore, the reasonable docking score of −10.02 kcal/mol was set as the AXL threshold. A total of 49 compounds were identified. Then, 379 compounds were docked to the crystal structure of HDAC2 (PDB ID: 4LXZ), and 30 compounds were screened with docking scores of less than −11.35 kcal/mol (The molecular docking score of SAHA was −11.35 kcal/mol). Based on docking scores of less than −10.02 kcal/mol and less than −11.35 kcal/mol, we identified five potential dual-targeting AXL/HDAC2 candidate compounds (called Hits 1–5) that simultaneously met both scoring thresholds. The docking scores of Hits 1–5 were shown in . The interactions between Hits 1–5 and AXL/HDAC2 were further analysed.
Figure 4. The docking validation using the co-crystal ligand and protein complex before setting screening threshold values and docking-based virtual screening. (A) The docking validation of AXL (PDB ID: 5U6B). (B) The docking validation of HDAC2 (PDB ID: 4LXZ). (yellow for native ligand, green for docking ligand).
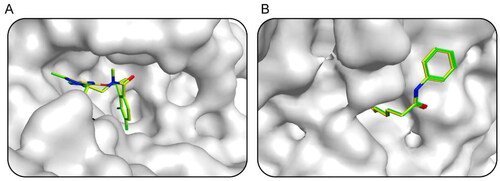
Figure 5. The docking scores of Hits 1–5, TP0903 and SAHA. aBinding free energy between the compounds and the targets (lower binding free energies show stronger binding affinities).
Interaction analysis
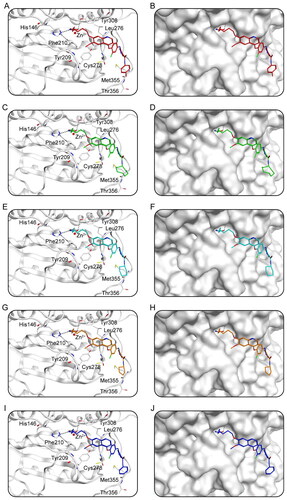
Docking studies predicted the binding conditions of Hits 1–5 to AXL and HDAC2. showed the interaction of Hits 1–5 with AXL. showed that a total of 11 amino acid residues (Ala565, Lys567, Leu620, Phe622, Met623, Asp627, Arg676, Leu693, Lys710, Met711, Pro712) were crucial for the binding of each compound to AXL. Notably, all of the hits generated hydrogen bonds with residues Asp627 and Arg676, which improved the stability of the compound binding to the AXL active site, resulting in a good binding between the compound and the active binding pocket. In addition, each hit formed hydrophobic interactions with Leu620, Phe622, and Met623, making the binding more stable. showed the binding surface maps of Hits 1–5 to AXL, and the compounds obtained from the screening were well accommodated by the AXL active site pocket. The docking binding pattern of Hits 1–5 to HDAC2 was shown in . As shown in , Hits 1–5 formed interactions with 8 amino acid residues (His146, Tyr209, Phe210, Leu276, Cys278, Tyr308, Met355, Thr356). The hydroxamic acid of the compound produced ionic bonds with zinc ions and hydrogen bonds with residues Phe210 and Tyr308, which could increase the stability of the compound. Besides, the surface maps in showed that the hydroxamic acid groups of Hits 1–5 were inserted into the HDAC2 active pocket and Hits 1–5 could fit comfortably in the active pocket of HDAC2. Therefore, these docking poses suggest that Hits 1–5 can interact with key residues of both AXL and HDAC2, indicating that Hits 1–5 may have the potential effect of inhibiting target proteins.
Figure 6. The X-ray crystal structure and the predicted docking poses of Hits 1–5 at the AXL active site: (A,B) Hit-1; (C,D) Hit-2; (E,F) Hit-3; (G,H) Hit-4; (I,J) Hit-5. Compounds are indicated as sticks with different atom colours (red for Hit-1, green for Hit-2, cyan for Hit-3, orange for Hit-4, dark blue for Hit-5). AXL is colour-coded by gray. The surfaces of AXL are colour-coded by gray. Hydrogen bonds are indicated by dashed black lines.
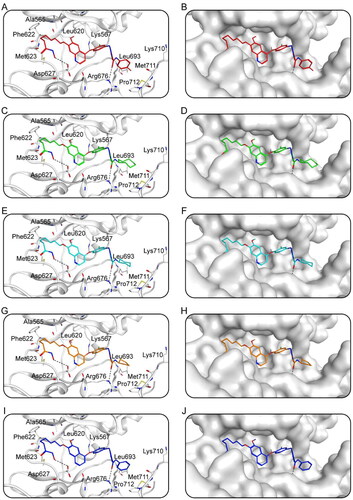
Figure 7. The X-ray crystal structure and the predicted docking poses of Hits 1–5 at the HDAC2 active site: (A,B) Hit-1; (C,D) Hit-2; (E,F) Hit-3; (G,H) Hit-4; (I,J) Hit-5. Compounds are indicated as sticks with different atom colours (red for Hit-1, green for Hit-2, cyan for Hit-3, orange for Hit-4, dark blue for Hit-5). HDAC2 is colour-coded by gray. The surfaces of HDAC2 are colour-coded in gray. Hydrogen bonds are indicated by dashed black lines.
Enzymatic inhibition assays of AXL and HDAC2 by Hits 1–5
To further evaluate the inhibitory effects of the selected Hits 1–5 on the dual targets, in vitro inhibitory effect assays were performed on Hits 1–5. The IC50 values for the inhibitory effects of Hits 1–5 against AXL were 19.52, 14.33, 9.41, 24.38 and 21.25 nM, respectively, all of which were lower than the positive control of 26.23 nM (). The IC50 values for HDAC2 inhibitory effects were 7.49, 6.65, 4.06, 8.37 and 8.89 nM, respectively, all with lower IC50 values than the positive control of 9.02 nM. Among the positive controls, TP0903 inhibited AXL, but not HDAC2. SAHA inhibited HDAC2 but did not affect AXL activity. Notably, among Hits 1–5, Hit-3 showed the best inhibitory activity against AXL and HDAC2, and the inhibitory effects were stronger than that of the positive controls, with IC50 values about 2.8 times that of TP0903 and 2.2 times that of SAHA. In addition, we selected Hit-3 to evaluate the IC50 values of other HDAC enzymes. The results show that the inhibitory effect of Hit-3 on other HDAC enzymes was not as good as that of HDAC2 (). Therefore, we further performed molecular dynamics simulations of Hit-3.
Table 1. The enzymatic inhibitory effects of Hits 1–5, TP0903 and SAHA in cell-free assays.
Table 2. The inhibitory effects of Hit-3 on other HDAC enzymes.
Molecular dynamics simulations of Hit-3
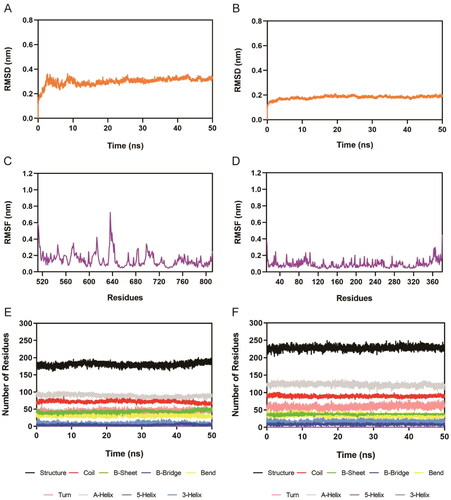
The binding stability of the system over time was analysed using 50 ns molecular dynamics simulations of the Hit-3-AXL complex and Hit-3-HDAC2 complex. The RMSD of Hit-3 in AXL and HDAC2 complexes were depicted in . RMSD was one of the most essential factors in describing the stability. Better binding stability is indicated by lower RMSD values. The final average value of the Hit-3-AXL complex was less than 0.4 nm, and the final average value of the Hit-3-HDAC2 complex was less than 0.2 nm. These show that Hit-3 can tightly bind both AXL and HDAC2 and the Hit-3-AXL and Hit-3-HDAC2 complexes are stable in the molecular dynamics simulation. The RMSF of the C atom of the amino acid residues was then measured. The RMSF values were related to the interactions between AXL/HDAC2 and Hit-3. Generally, low RMSF indicates minimal residual motion and a stable system. As shown in , there were large fluctuations in the RMSF of both complexes at the C-/N-terminus, which might be connected with less interaction. Moreover, the fluctuations of key amino acid residues related to Hit-3-AXL and Hit-3-HDAC2 complexes were mostly below 0.4 nm. showed the secondary structures of the complexes including A-Helix, Coil, Turn, B-Sheet, Bend, 3-Helix, B-Bridge, and 5-Helix in Hit-3-AXL and Hit-3-HDAC2. Within 50 ns, these secondary structures were fluctuating stably, indicating that the proteins were in a stable state. Therefore, the molecular dynamics simulations show that Hit-3 can stably bind to AXL and HDAC2 and Hit-3 may be a novel and potent dual-targeting inhibitor of AXL/HDAC2.
Figure 8. Molecular dynamics simulation of the Hit-3 with AXL and HDAC2. (A,B) RMSD of Hit-3 in complex with AXL and HDAC2, respectively. (C,D) RMSF of amino acid residues of Hit-3 in complex with AXL and HDAC2, respectively. (E,F) The secondary structures analysis of Hit-3 in complex with AXL and HDAC2, respectively. (black for Structure, red for Coil, green for B-Sheet, dark blue for B-Bridge, yellow for Bend, pink for Turn, grey for A-Helix, purple for 5-Helix and blue for 3-Helix).
Cell inhibition rate and cell viability analysis
To further analyse the anti-proliferative effect of the Hit-3, we also evaluated the cell inhibition rate of 5 hits on the HCT116 cells at 0.7 μM concentration. The experimental results are shown in . The cell inhibition rate of Hits 1–5 on the HCT116 cells was more than 70%, with the best inhibitory effect of Hit-3 reaching more than 90%. In addition, the IC50 value of Hit-3 was determined by the MTT method. In general, the lower the IC50 values indicated the stronger ability of the compound to induce apoptosis. As shown in , Hit-3 had growth inhibitory activity on HCT116 cells with an IC50 value of 0.07 μM. At the concentration of 0.01 μM, the cell viability rate was about 80%, which decreased with increasing concentration. Therefore, these indicate that Hit-3 has good anti-tumour ability and maintains good cell viability at IC50 concentration. We also evaluated the IC50 values of A549, MCF-7, HepG2 and HeLa cells. The results show that the inhibitory effect of Hit-3 on HCT116 cells was stronger than that of other cells. In the evaluation of the inhibition effect of EGFRi-resistant H1975 and AXLi-resistant H460, Hit-3 shows an improvement in drug resistance of them ().
Figure 9. Inhibitory effects of Hits 1–5, TP0903 and SAHA on HCT116 cells.
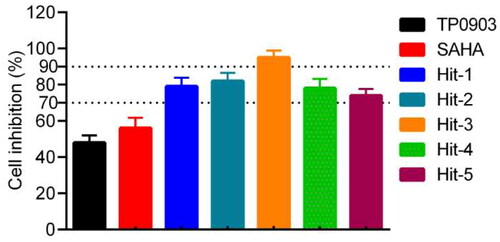
Figure 10. Inhibitory effects of Hit-3 on HCT116 cells detected by using MTT assay.
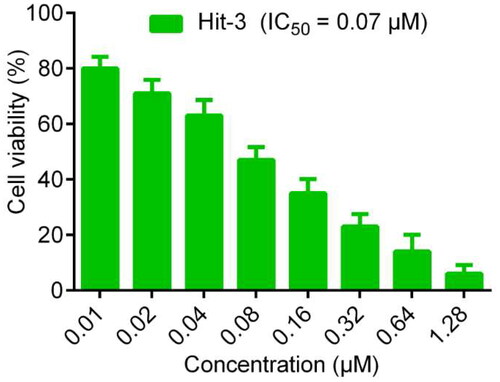
Table 3. The inhibitory activity of Hit-3 on other cancer cells and drug-resistant cancer cells.
In vivo anti-tumour activity
The metabolic stability and pharmacokinetics profiles of Hit-3 were evaluated before in vivo antitumor activity. The results in show that the T1/2 of Hit-3 is 8.41 h and the Cmax of Hit-3 is 1307 ng ml−1 at 10 mg/kg dose. To further analyse the in vivo anti-tumour activity of Hit-3, HCT116 cells were injected into BALB/c nude mice to establish the tumour models, and the mice were divided into four groups: vehicle, 1 mg/kg Hit-3, 10 mg/kg Hit-3, and 20 mg/kg Hit-3. As shown in , compared with the vehicle, the tumour volumes of mice given injections of Hit-3 at doses of 1 mg/kg, 10 mg/kg, and 20 mg/kg were substantially reduced. Therefore, these indicate that Hit-3 has a strong in vivo anti-tumour activity. Furthermore, compared to mice given injections of 1 mg/kg Hit-3 and 10 mg/kg Hit-3, the mice treated with 20 mg/kg Hit-3 showed a better notable reduction in tumour volume. These suggest that Hit-3 inhibits tumour growth in a dose-dependent manner.
Figure 11. Changes of tumour volume with injecting HCT116 (KRAS(mut)/p53(wt)) cells in BALB/c nude mice over time. The data are shown as mean ± SD, n = 5. ***p < .001 means a significant difference versus the vehicle group.
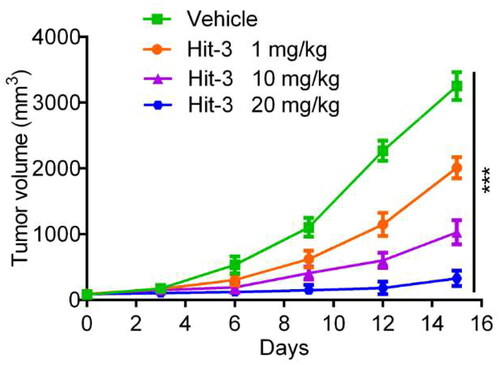
Table 4. In vivo pharmacokinetic parameters of Hit-3 after intraperitoneal administration in micea.
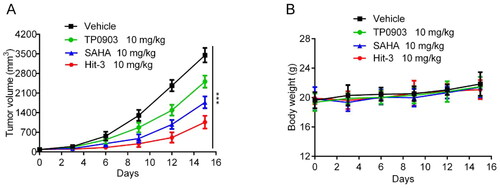
As shown in , BALB/c nude mice were divided into four groups, one of which was used as the vehicle group, and the other three groups were injected with TP0903, SAHA and Hit-3 at a dose of 10 mg/kg respectively to evaluate the in vivo anti-tumour effect. showed that the tumour volume of mice injected with positive control drugs and the Hit-3 group was smaller than that of the vehicle group, and the tumour volume of Hit-3 was the smallest. In , the body weight changes of the four groups showed a stable trend. Therefore, in vivo experimental results suggest that Hit-3 is effective in inhibiting tumour growth with good safety.
Figure 12. In vivo anticancer activity. (A) Tumour volume evaluations at the dose of 10 mg/kg for TP0903, SAHA, Hit-3 and vehicle. (B) Body weight evaluations after injecting vehicle, TP0903, SAHA and Hit-3 at the dose of 10 mg/kg. The data are shown as mean ± SD, n = 5. ***p < .001 means a significant difference versus the vehicle group.
Conclusion
Single-targeting medications and combination therapy have shown limitations in various parts of cancer therapy, thus the development of novel dual-targeting drugs is essential. AXL and HDAC2 are highly correlated with targeting cancer therapy, and the dual-targeting AXL/HDAC2 inhibitor may provide new options for cancer treatment. However, no dual-targeting AXL/HDAC2 inhibitors have been reported so far. In this study, five hit compounds targeting both AXL and HDAC2 were rationally identified through pharmacophore modelling, virtual screening, and molecular dynamics simulations. Interaction analysis showed that Hits 1–5 formed key interactions with residues Ala565, Lys567, Leu620, Phe622, Met623, Asp627, Arg676, Leu693, Lys710, Met711, and Pro712 of AXL. All hit compounds formed hydrogen bonds with residues Asp627 and Arg676 and hydrophobic interactions with Leu620, Phe622, and Met623, making the compounds occupy the AXL pocket completely. Meanwhile, Hits 1–5 formed interactions with HDAC2 residues His146, Tyr209, Phe210, Leu276, Cys278, Tyr308, Met355, and Thr356, which ensured that the compounds were stably bound to the active site. Further in vitro inhibitory experiments showed that the affinity of Hits 1–5 for both AXL and HDAC2 were within the micromolar range and the cellular inhibition rate was greater than 70%. The most effective inhibitor Hit-3, was selected for molecular dynamics simulations to analyse the binding stability of Hit-3 to AXL and HDAC2. The results showed that Hit-3 was able to bind stably to AXL and HDAC2. In the MTT assay, Hit-3 had a significant inhibitory effect on HCT116 cells and ensured cell viability well. According to in vivo data, Hit-3 suppressed tumour growth in a dose-dependent manner in the HCT116 cells xenograft tumour mouse model.
However, it is worth noting that although the in vivo activity evaluation of Hit-3 indicates that it has good anti-tumour activity, the anti-tumour mechanism of Hit-3 remains to be further explained. Previous research has revealed that increased HDAC2 expression promotes the positive feedback mechanism, which causes phosphoinositol molecules to activate AKT phosphorylationCitation65. Previously reported HDAC2 inhibitors have been shown to exert anti-tumour activities by significantly inhibiting the PI3K/AKT/mTOR pathwayCitation66,Citation67. Furthermore, studies have shown that AXL-HER2 dimerisation can also affect the PI3K/AKT pathway, hence affecting treatment efficacyCitation68. As mentioned above, we conclude that Hit-3 may have activation or inhibitory effects on other pathways, implying that we can further research the anti-tumour mechanism of Hit-3 in the future. Based on existing experimental studies, we reported and identified the dual-targeting AXL/HDAC2 inhibitor for the first time, and evaluated both the HDACs enzyme activity and the anti-tumour activity of Hit-3, which may provide a new therapeutic strategy for future cancer treatment.
Authors contributions
YZ, HH and XQ contributed to the conception of the study. XW and MMN performed experiments and acquired data. XQ, SC, XW and MMN performed statistical analysis. YZ, HH, XQ, XW and MMN contributed significantly to manuscript preparation. All authors had contributed and approved the manuscript.
Ethics statement
All experimental protocols were reviewed and approved by the Animal Ethics Committee of China Pharmaceutical University.
Acknowledgements
We gratefully acknowledge the support from the Taizhou People’s Hospital.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Khan N, Dhritlahre RK, Saneja A. Ruchika Recent advances in dual-ligand targeted nanocarriers for cancer therapy. Drug Discov Today. 2022;27(8):2288–2299.
- Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48.
- Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. 2023;73(3):233–254.
- Ciardiello F, Ciardiello D, Martini G, Napolitano S, Tabernero J, Cervantes A. Clinical management of metastatic colorectal cancer in the era of precision medicine. CA Cancer J Clin. 2022;72(4):372–401.
- Ren Y, Li S, Zhu R, Wan C, Song D, Zhu J, Cai G, Long S, Kong L, Yu W. Discovery of STAT3 and Histone Deacetylase (HDAC) dual-pathway inhibitors for the treatment of solid cancer. J Med Chem. 2021;64(11):7468–7482.
- Navya PN, Kaphle A, Srinivas SP, Bhargava SK, Rotello VM, Daima HK. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019;6(1):23.
- Assaraf YG, Brozovic A, Gonçalves AC, Jurkovicova D, Linē A, Machuqueiro M, Saponara S, Sarmento-Ribeiro AB, Xavier CPR, Vasconcelos MH. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist Updat. 2019;46:100645.
- Sun D, Zhao Y, Zhang S, Zhang L, Liu B, Ouyang L. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Eur J Med Chem. 2020;188:112025.
- Qin S, Jiang J, Lu Y, Nice EC, Huang C, Zhang J, He W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct Target Ther. 2020;5(1):228.
- Bukowski K, Kciuk M, Kontek R. Mechanisms of multidrug resistance in cancer chemotherapy. Int J Mol Sci. 2020;21(9):3233.
- Ding S, Chen X, Shen K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020;40(8):329–344.
- Duan YC, Zhang SJ, Shi XJ, Jin LF, Yu T, Song Y, Guan YY. Research progress of dual inhibitors targeting crosstalk between histone epigenetic modulators for cancer therapy. Eur J Med Chem. 2021;222:113588.
- Liang Q, Wang J, Zhao L, Hou J, Hu Y, Shi J. Recent advances of dual FGFR inhibitors as a novel therapy for cancer. Eur J Med Chem. 2021;214:113205.
- Ye J, Wu J, Liu B. Therapeutic strategies of dual-target small molecules to overcome drug resistance in cancer therapy. Biochim Biophys Acta Rev Cancer. 2023;1878(3):188866.
- Zou Y, Li W, Zhou J, Zhang J, Huang Y, Wang Z. ERK inhibitor enhances everolimus efficacy through the attenuation of dNTP pools in renal cell carcinoma. Mol Ther Nucleic Acids. 2019;14:550–561.
- Ince W, Eisen T. Combination therapies in clinical trials for renal cell carcinoma: how could they impact future treatments? Expert Opin Investig Drugs. 2021;30(12):1221–1229.
- Thakur A, Tawa GJ, Henderson MJ, Danchik C, Liu S, Shah P, Wang AQ, Dunn G, Kabir M, Padilha EC, et al. Design, synthesis, and biological evaluation of quinazolin-4-one-based hydroxamic acids as dual PI3K/HDAC inhibitors. J Med Chem. 2020;63(8):4256–4292.
- O'Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, Espinosa R, 3rd, Le Beau MM, Earp HS, Liu ET. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11(10):5016–5031.
- Antony J, Huang RY. AXL-driven EMT state as a targetable conduit in cancer. Cancer Res. 2017;77(14):3725–3732.
- Myers SH, Brunton VG, Unciti-Broceta A. AXL inhibitors in cancer: a medicinal chemistry perspective. J Med Chem. 2016;59(8):3593–3608.
- Tanaka M, Siemann DW. Therapeutic targeting of the Gas6/Axl signaling pathway in cancer. Int J Mol Sci. 2021;22(18):9953.
- Wium M, Ajayi-Smith AF, Paccez JD, Zerbini LF. The role of the receptor tyrosine kinase Axl in carcinogenesis and development of therapeutic resistance: an overview of molecular mechanisms and future applications. Cancers. 2021; 13(7):1521.
- Hsu CH, Huang YH, Lin SM, Hsu C. AXL and MET in hepatocellular carcinoma: a systematic literature review. Liver Cancer. 2022;11(2):94–112.
- Cichoń MA, Szentpetery Z, Caley MP, Papadakis ES, Mackenzie IC, Brennan CH, O'Toole EA. The receptor tyrosine kinase Axl regulates cell-cell adhesion and stemness in cutaneous squamous cell carcinoma. Oncogene. 2014;33(32):4185–4192.
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–290.
- Di Stasi R, De Rosa L, D'Andrea LD. Therapeutic aspects of the Axl/Gas6 molecular system. Drug Discov Today. 2020;25(12):2130–2148.
- Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang S, Li T, Chen F, Yang Y. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. 2018;17(1):20.
- Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18(1):153.
- Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769–785.
- Tanaka M, Siemann DW. Gas6/Axl signaling pathway in the tumor immune microenvironment. Cancers. 2020;12(7):1850.
- Gao X, Xue D, Cheng J, Zhang X, Cai X. Inhibition of Axl promotes the therapeutic effect of targeted inhibition of the PI3K/Akt pathway in NRAS mutant melanoma cells. J Oncol. 2022;2022:2946929–2946929.
- Hafizi S, Dahlbäck B. Signalling and functional diversity within the Axl subfamily of receptor tyrosine kinases. Cytokine Growth Factor Rev. 2006;17(4):295–304.
- Lauter M, Weber A, Torka R. Targeting of the AXL receptor tyrosine kinase by small molecule inhibitor leads to AXL cell surface accumulation by impairing the ubiquitin-dependent receptor degradation. Cell Commun Signal. 2019;17(1):59.
- Bhalla S, Gerber DE. AXL inhibitors: status of clinical development. Curr Oncol Rep. 2023;25(5):521–529.
- Jeon JY, Buelow DR, Garrison DA, Niu M, Eisenmann ED, Huang KM, Zavorka Thomas ME, Weber RH, Whatcott CJ, Warner SL, et al. TP-0903 is active in models of drug-resistant acute myeloid leukemia. JCI Insight. 2020;5(23):e140169.
- Ben-Batalla I, Erdmann R, Jørgensen H, Mitchell R, Ernst T, von Amsberg G, Schafhausen P, Velthaus JL, Rankin S, Clark RE, et al. Axl blockade by BGB324 inhibits BCR-ABL tyrosine kinase inhibitor-sensitive and -resistant chronic myeloid leukemia. Clin Cancer Res. 2017;23(9):2289–2300.
- Kasikara C, Davra V, Calianese D, Geng K, Spires TE, Quigley M, Wichroski M, Sriram G, Suarez-Lopez L, Yaffe MB, et al. Pan-TAM tyrosine kinase inhibitor BMS-777607 enhances anti-PD-1 mAb efficacy in a murine model of triple-negative breast cancer. Cancer Res. 2019;79(10):2669–2683.
- Rho JK, Choi YJ, Kim SY, Kim TW, Choi EK, Yoon SJ, Park BM, Park E, Bae JH, Choi CM, et al. MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 2014;74(1):253–262.
- Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Coan JP, Pearson HE, Bahrar H, Fowler TL, Bednarz BP, et al. AXL is a logical molecular target in head and neck squamous cell carcinoma. Clin Cancer Res. 2015;21(11):2601–2612.
- Tian Y, Zhang Z, Miao L, Yang Z, Yang J, Wang Y, Qian D, Cai H, Wang Y. Anexelekto (AXL) increases resistance to EGFR-TKI and activation of AKT and ERK1/2 in non-small cell lung cancer cells. Oncol Res. 2016;24(5):295–303.
- Kim KC, Baek SH, Lee C. Curcumin-induced downregulation of Axl receptor tyrosine kinase inhibits cell proliferation and circumvents chemoresistance in non-small lung cancer cells. Int J Oncol. 2015;47(6):2296–2303.
- Sanchez GJ, Richmond PA, Bunker EN, Karman SS, Azofeifa J, Garnett AT, Xu Q, Wheeler GE, Toomey CM, Zhang Q, et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 2018;46(4):1756–1776.
- Ferrante F, Giaimo BD, Bartkuhn M, Zimmermann T, Close V, Mertens D, Nist A, Stiewe T, Meier-Soelch J, Kracht M, et al. HDAC3 functions as a positive regulator in Notch signal transduction. Nucleic Acids Res. 2020;48(7):3496–3512.
- Peng X, Sun Z, Kuang P, Chen J. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment. Eur J Med Chem. 2020;208:112831.
- Jia D, Augert A, Kim DW, Eastwood E, Wu N, Ibrahim AH, Kim KB, Dunn CT, Pillai SPS, Gazdar AF, et al. Crebbp loss drives small cell lung cancer and increases sensitivity to HDAC inhibition. Cancer Discov. 2018;8(11):1422–1437.
- Zhang L, Han Y, Jiang Q, Wang C, Chen X, Li X, Xu F, Jiang Y, Wang Q, Xu W. Trend of histone deacetylase inhibitors in cancer therapy: isoform selectivity or multitargeted strategy. Med Res Rev. 2015;35(1):63–84.
- Hu XT, Xing W, Zhao RS, Tan Y, Wu XF, Ao LQ, Li Z, Yao MW, Yuan M, Guo W, et al. HDAC2 inhibits EMT-mediated cancer metastasis by downregulating the long noncoding RNA H19 in colorectal cancer. J Exp Clin Cancer Res. 2020;39(1):270.
- Shetty MG, Pai P, Deaver RE, Satyamoorthy K, Babitha KS. Histone deacetylase 2 selective inhibitors: a versatile therapeutic strategy as next generation drug target in cancer therapy. Pharmacol Res. 2021;170:105695.
- Wan G, Feng Z, Zhang Q, Li X, Ran K, Feng H, Luo T, Zhou S, Su C, Wei W, et al. Design and synthesis of fibroblast growth factor receptor (FGFR) and histone deacetylase (HDAC) dual inhibitors for the treatment of cancer. J Med Chem. 2022;65(24):16541–16569.
- Lee HZ, Kwitkowski VE, Del Valle PL, Ricci MS, Saber H, Habtemariam BA, Bullock J, Bloomquist E, Li Shen Y, Chen XH, et al. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. 2015;21(12):2666–2670.
- Suraweera A, O'Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92.
- Leszczynska KB, Jayaprakash C, Kaminska B, Mieczkowski J. Emerging advances in combinatorial treatments of epigenetically altered pediatric high-grade H3K27M gliomas. Front Genet. 2021;12:742561.
- Meel MH, de Gooijer MC, Metselaar DS, Sewing ACP, Zwaan K, Waranecki P, Breur M, Buil LCM, Lagerweij T, Wedekind LE, et al. Combined therapy of AXL and HDAC inhibition reverses mesenchymal transition in diffuse intrinsic pontine glioma. Clin Cancer Res. 2020;26(13):3319–3332.
- Wang SC, Yu CY, Wu YC, Chang YC, Chen SL, Sung WW. Chidamide and mitomycin C exert synergistic cytotoxic effects against bladder cancer cells in vitro and suppress tumor growth in a rat bladder cancer model. Cancer Lett. 2022;530:8–15.
- Wang J, Pursell NW, Samson ME, Atoyan R, Ma AW, Selmi A, Xu W, Cai X, Voi M, Savagner P, et al. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol Cancer Ther. 2013;12(6):925–936.
- Zheng L, Ren R, Sun X, Zou Y, Shi Y, Di B, Niu MM. Discovery of a dual tubulin and poly(ADP-Ribose) polymerase-1 inhibitor by structure-based pharmacophore modeling, virtual screening, molecular docking, and biological evaluation. J Med Chem. 2021;64(21):15702–15715.
- Zhou Y, Chen Y, Tan Y, Hu R, Niu MM. An NRP1/MDM2-targeted D-peptide supramolecular nanomedicine for high-efficacy and low-toxic liver cancer therapy. Adv Healthc Mater. 2021;10(9):e2002197.
- Winitthana T, Lawanprasert S, Chanvorachote P. Triclosan potentiates epithelial-to-mesenchymal transition in Anoikis-resistant human lung cancer cells. PLOS One. 2014;9(10):e110851.
- Zhou Y, Tang S, Chen T, Niu MM. Structure-based pharmacophore modeling, virtual screening, molecular docking and biological evaluation for identification of potential poly (ADP-Ribose) polymerase-1 (PARP-1) inhibitors. Molecules. 2019;24(23):4258.
- Li X, Inks ES, Li X, Hou J, Chou CJ, Zhang J, Jiang Y, Zhang Y, Xu W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J Med Chem. 2014;57(8):3324–3341.
- Ng PS, Foo K, Sim S, Wang G, Huang C, Tan LH, Poulsen A, Liu B, Tee DHY, Ahmad NHB, et al. Fragment-based lead discovery of indazole-based compounds as AXL kinase inhibitors. Bioorg Med Chem. 2021;49:116437.
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25.
- Páll S, Zhmurov A, Bauer P, Abraham M, Lundborg M, Gray A, Hess B, Lindahl E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J Chem Phys. 2020;153(13):134110.
- Zhou Y, Zou Y, Yang M, Mei S, Liu X, Han H, Zhang CD, Niu MM. Highly potent, selective, biostable, and cell-permeable cyclic d-peptide for dual-targeting therapy of lung cancer. J Am Chem Soc. 2022;144(16):7117–7128.
- Noh JH, Bae HJ, Eun JW, Shen Q, Park SJ, Kim HS, Nam B, Shin WC, Lee EK, Lee K, et al. HDAC2 provides a critical support to malignant progression of hepatocellular carcinoma through feedback control of mTORC1 and AKT. Cancer Res. 2014;74(6):1728–1738.
- Ma S, Liu T, Xu L, Wang Y, Zhou J, Huang T, Li P, Liu H, Zhang Y, Zhou X, et al. Histone deacetylases inhibitor MS-275 suppresses human esophageal squamous cell carcinoma cell growth and progression via the PI3K/Akt/mTOR pathway. J Cell Physiol. 2019;234(12):22400–22410.
- Sun J, Piao J, Li N, Yang Y, Kim KY, Lin Z. Valproic acid targets HDAC1/2 and HDAC1/PTEN/Akt signalling to inhibit cell proliferation via the induction of autophagy in gastric cancer. Febs J. 2020;287(10):2118–2133.
- Adam-Artigues A, Arenas EJ, Martínez-Sabadell A, Brasó-Maristany F, Cervera R, Tormo E, Hernando C, Martínez MT, Carbonell-Asins J, Simón S, et al. Targeting HER2-AXL heterodimerization to overcome resistance to HER2 blockade in breast cancer. Sci Adv. 2022;8(20):eabk2746.