
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Staphylococcus aureus shikimate dehydrogenase (SaSDH) plays a crucial role in the growth of Staphylococcus aureus (S. aureus), but absent in mammals and therefore a potential target for antibacterial drugs to treat drug-resistant S. aureus infection. In this study, a 3D model of SaSDH was constructed by homology modelling and inhibitors of SaSDH were screened through virtual screening. (-)-Gallocatechin gallate and rhodiosin were identified as inhibitors with Kis of 2.47 μM and 73.38 μM, respectively. Molecular docking and isothermal titration calorimetry showed that both inhibitors interact with SaSDH with a KD of 44.65 μM for (-)-gallocatechin gallate and 16.45 μM for rhodiosin. Both inhibitors had antibacterial activity, showing MICs of 50 μg/mL for (-)-gallocatechin gallate and 250 μg/mL for rhodiosin against S. aureus. The current findings have the potential for identification of drugs to treat S. aureus infections by targeting SaSDH.
Introduction
Antimicrobial resistance (AMR) threatens public health and methicillin-resistant Staphylococcus aureus (MRSA) is on the WHO list of antibiotic-resistant "priority pathogens". The average MRSA infection rate among 76 countries was about 35% in 2021, an increase of 15% from 2017. Resistance to the first-line drug, vancomycin, and to the new-generation drugs, linezolid and daptomycin has been reportedCitation1–3. There is an urgent need to develop anti-MRSA drugs using innovative approaches to overcome resistance.
The shikimate pathway is responsible for the synthesis of aromatic amino acids, flavonoids and other aromatic compounds in plants and microorganisms but is absent from mammalsCitation4. The pathway begins with erythrose phosphate and phosphoenolpyruvate, eventually synthesises chorismite through seven enzymatically catalysed steps by 3-deoxyarabinoheptanulose-7-phosphate synthetase, dehydroquinic acid synthetase, 3-dehydroquinic acid dehydratase, shikimate dehydrogenase, shikimate kinase, 5-enolpyruvyl shikimate-3-phosphate synthetase and chorismite synthetase. Knockout of genes encoding these enzymes decreases bacterial proliferation and virulenceCitation5,Citation6. Enzymes in shikimate pathway are potential new antimicrobial targets. Shikimate dehydrogenase (SDH) converts 3-dehydroshikimate to shikimate (SHK) in the 4th step. The SDH protein structure includes an α/ß fold which is highly conserved among plants and microbes with subtle variations at the active siteCitation7. SDH is vital for bacterial survival and SDH inhibitorsCitation8–11 are attractive antimicrobial targets.
Staphylococcus aureus shikimate dehydrogenase (SaSDH) is a 29 kDa monomer for which antibacterial inhibitors have yet to be identified. SaSDH was expressed and purified during the current work and high-throughput virtual screening against a structurally diverse, commercially available compound library performed to identify SaSDH inhibitors. Antimicrobial potency was determined using an in vitro susceptibility test and binding mode investigated.
Materials and methods
SaSDH expression and purification
The SaSDH gene was amplified from the genomic DNA of Staphylococcus aureus ATCC49775 strain. Primers were F:5′-CTCGGATCCATGAAATTTGCAGTTA-3′ and R:5′-CCGCTCGAGTTATTCTCCTTTTAA-3′ with underlining representing the restriction sites for XHoI and BamHI. The PCR product was cloned into the pET-28a(+) vector (Youbio, China) and the recombinant plasmid transfected into E. coli BL21(DE3)pLysS cells (TianGen, China). Recombinant bacteria were cultured in 500 ml LB medium containing 50 μg/mL kanamycin at 37 °C until the absorbance at 600 nm reached 0.6–0.8 when protein expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 25 °C for 8 h. Cells were harvested by centrifugation at 8,000 g for 5 min at 4 °C, resuspended in 20 mM Tris-HCI, pH 8.0 containing 300 mM NaCI (Buffer A) and lysed by 800 bar high pressure. Cell debris was removed by centrifugation at 10,000 g for 30 min and the supernatant filtered through a 0.45 μm membrane. SaSDH was eluted over a 10–300 mM imidizole gradient in Buffer A through a Ni-NTA affinity column (Cytiva, America) using AKTA pure system (GE, America).
Enzyme activity and kinetic parameters
SaSDH activity was measured as previously reportedCitation12. Briefly, the reaction medium contained 2 mM NADP, 8 mM SHK and 1.54 ng purified recombinant SaSDH in 100 mM Tris-HCI, pH 9.0, at 37 °C and was initiated by SaSDH addition and absorbance monitored at 340 nm every minute for 60 min using SpectraMax Paradigm (Molecular Devices, America). Vmax and Km for NADP and SKH were determined by varying the concentration of one substrate, 0.4, 0.5, 1, 2, 4 mM NADP and 8, 4, 2, 1, 0.5, 0.25 mM SKH, while the other substrate was kept at constant saturation. Kinetic data were analysed by double reciprocal plots.
Homology modelling and virtual screening
Homology modelling was performed using SWISS-MODEL referring to the crystal structure of Staphylococcus epidermidis shikimate dehydrogenase (SeSDH: PDB ID:3DON) which has 70% sequence homology with SaSDHCitation13. The SaSDH model was optimised by the Protein Preparation Wizard module in Schrödinger Maestro 11.4. The amino acid residues, SER13/SER15/THR60/LYS64/ASN85/ASP100/PHE236, form the binding site of SaSDHCitation14 and inhibitors were identified by molecular docking in Glide module in Schrödinger Maestro 11.4 using compounds from the Life Chemicals HTS Compound Collection, Specs HTS Compounds Library and MCE Bioactive Compound Library PlusA. Three-dimensional low-energy conformations of compounds and atomic partial charges were obtained using the LigPrep Module and compounds were selected according to docking scores.
Enzyme inhibition
Enzyme inhibition potency of inhibitors was measured, as above, in a medium containing 1 mM NADP, 1 mM SHK, 1.54 ng SaSDH and inhibitor of different concentration in 100 mM Tris-HCI, pH 9.0, at 37 °C and absorbance at 340 nm measured at t = 0 and t = 30 min. Negative controls lacking inhibitor compounds were included. Inhibition was calculated according to the following equation:
(1)
(1)
(2)
(2)
The 50% inhibition concentration(IC50) was calculated using GraphPad prism. Inhibition constants (Ki) were measured with 4, 2, 1, 0.5 and 0.25 mM SHK and 3 inhibitors concentrations with absorbance at 340 nm measured at 1 min intervals for 60 min. Kis were calculated from Lineweaver-Burke plots using GraphPad prism.
Isothermal titration calorimetry (ITC)
ITC was performed in 100 mM Tris-HCI, containing inhibitors, pH 9.0, at 25 °C with constant stirring at 200 rpm on a MicroCal iTC200 system (MicroCal, England), as previously reportedCitation15. The cell contained 100 μM compound and the syringe 1000 μM SaSDH. 5 μL SaSDH was injected into the cell after equilibration to trigger binding. Data analysis, including baseline correction and evaluation, was carried out with Nano ITC and all SaSDH injections fitted, except the first, to calculate maximum heat and the equilibrium dissociation constant (KD).
S. aureus susceptibility
The minimum inhibitory concentration (MIC) for S. aureus was determined by micro-broth two-fold serial dilution. Compounds were dissolved in DMSO and diluted with MH broth medium in 96-well microtiter plates before the addition of 100 μL bacterial suspension (1 × 106 CFU/ml) and incubation at 37 °C for 16 h. The negative control was compound-free and the positive control included ampicillin.
Growth curves were measured by incubating 1 × 107CFU/ml bacterial suspension at 37 °C and adding the MIC of inhibitor. Absorbance at 600 nm was read for 200 μL sample at 2 h intervals and time-kill curves plotted.
Scanning electron microscopy (SEM)
Overnight S. aureus culture was diluted to 107 CFU/ml and incubated with MIC and 1/2 MIC drug concentrations for 1.5 h before 10 μL samples were settled on a cationic adsorption membrane for 30 min and the medium aspirated. 10 μL 2.5% glutaraldehyde was added and fixed for 30 min, the upper fixative discarded and the membrane immersed in the fixative for 12 h. The membrane was washed twice with 10xPBS, dehydrated by ethanol gradient, dried, sprayed with gold and bacterial morphology observed by SEM (FEI Quanta x50 FEG, America).
Cell membrane integrity
Overnight S. aureus culture was diluted to 107 CFU/ml and incubated until the logarithmic phase was reached. Drugs were added to a final concentration of MIC with incubation at 37 °C for 3 h before washing 3x with PBS, the addition of Calcein AM/PI and incubation for 15 min in the dark. Samples were observed under the fluorescence microscope (Leica DMi8, Germany).
Results
SaSDH expression and purification
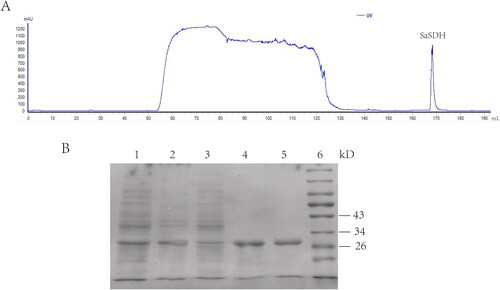
Recombinant SaSDH with a 6His-tag at its N-terminus was expressed in E. coli BL21 (DE3) pLysS induced by 0.5 mM IPTG. The medium was purified by His TrapTM FF column using AKTA pure system and SaSDH eluted with imidazole. The SaSDH elution peak was seen at 100 mM imidazole (). A protein of molecular weight 29 kD was seen after SDS-PAGE (), consistent with SaSDH predicted weight, indicating the presence of recombinant SaSDH with high purity.
Figure 1. SaSDH purification. (A) Chromatogram from AKTA pure. (B) SDS-PAGE: lane 1: bacterial medium; lane 2: bacterial precipitate; lane 3: sample flow through column; lane 4 and lane 5: SaSDH eluted with 100 mM imidazole; lane 6: marker.
Enzyme activity and kinetic parameters
SaSDH activity was assayed in the reverse catalytic direction producing NADP+-dependent oxidation of SHK to form NADPH and 3-dehydroshikimate, accompanied by increased absorbance at 340 nm until a plateau at 60 min. No absorbance change was seen in the control assay lacking the enzyme (). One unit (U) of SaSDH activity was defined as the amount of enzyme required to catalyse the production of 1 μM NADPH per minute in a 100 μL mixture at 37 °C. Purified SaSDH had an activity of 913 KU/mg and a hyperbolic plot was produced, indicating that Michaelis-Menten kinetics were obeyed (). The Km for SHK was 0.04614 mM and Vmax 3.362 mM/min/μg. The Km for NADP was 0.09816 mM and Vmax 3.785 mM/min/μg ().
Figure 2. (A) SaSDH activity. Activity was determined by continuous monitoring of the increase of NADPH concentration at 340 nm. (B) Reciprocal plots of initial velocities under conditions where either NADP or SHK was varied at different fixed concentrations of the co-substrate.
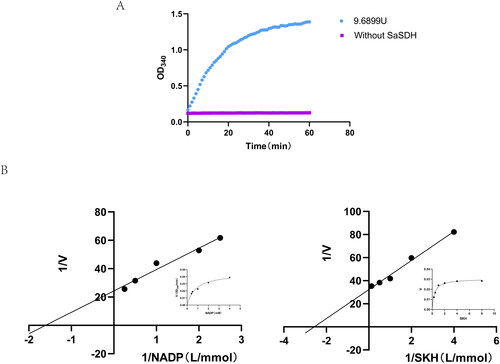
Table 1. Apparent kinetics parameters for SaSDH.
Homology modelling and virtual screening
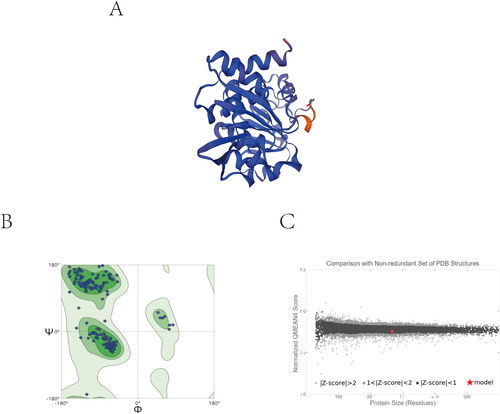
A 3D SaSDH model was generated using SeSDH as template by homology modelling for virtual screening (). The stereochemical quality of the model was revisited and validated by two different programs. Only 0.4% of the residues were in the disallowed regions in Ramachandran plot which is used to reflect whether the protein is conformationally sound or not, 3.8% were in the allowed region and 95.8% in the favoured region of the plot (). Q-MEAN reported a normalised QMEAN4 score of 0.84 (close to 1 is the ideal) and a Z-score of −0.85 (close to zero is the ideal) (). Virtual screening was performed using a library of 720,000 small molecule compounds from three different commercial compound libraries. Three rounds of screening, involving high-throughput screening pattern, standard screening pattern and high-precision screening pattern, enabled the identification of the 200 compounds with the highest docking scores.
Figure 3. Homology modelling. (A) 3D model of SaSDH. (B) Ramachandran plot obtained from model protein geometry evaluation (dark green, light green, and green correspond to the favoured, allowed and outlier regions respectively). (C) Normalised QMEAN4 score graphic showing the position (red cross) of the model in the set of PDB structures used for the evaluation and the Z-score value.
Twelve compounds were selected from the MCE Bioactive Compound Library PlusA for inhibition assays. The docking scores and IC50s of these compounds against SaSDH were showed in . Pentagalloylglucose, (-) - gallocatechin gallate, troxerutin, specneuzhenide, rhodiosin and hesperidin all had IC50 less than 100 μM and (-)-gallocatechin gallate showed strongest inhibitory potency against SaSDH with IC50 was 0.85 μM.
Table 2. Inhibition of SaSDH and S. aureus MICs for 12 compounds.
MICs for S. aureus were determined for these compounds (). (-)-Gallocatechin gallate had a MIC of 50 μg/mL, pentagalloylglucose of 100 μg/mL and rhodiosin, 250 μg/mL. As the structure of pentagalloylglucose is similar to that of (-)-gallocatechin gallate, (-)-gallocatechin gallate and rhodiosin were selected for further kinetic and mechanistic studies.
Kinetic study and docking of the SaSDH-inhibitor complex
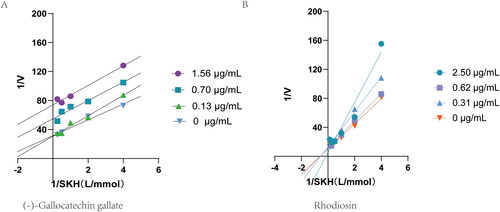
Kis were calculated from Lineweaver-Burke curves to assess the nature of the inhibitory effect of (-)-gallocatechin gallate and rhodiosin (). Rhodiosin had characteristics of competitive inhibition with SHK giving a Ki of 73.38 μM and (-)-gallocatechin gallate of uncompetitive inhibition with a Ki of 2.47 μM.
Figure 4. Double-reciprocal plot of initial velocities versus substrate concentration in the presence of different concentrations of (-)-Gallocatechin gallate and Rhodiosin.
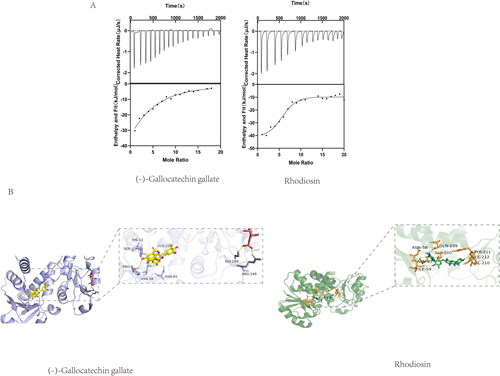
Molecular docking was performed for (-)-gallocatechin gallate and rhodiosin separately to the binding pocket of SaSDH using AutoDock Vina. As rhodiosin was competitive inhibitor and (-)-gallocatechin gallate was uncompetitive inhibitor, (-)-gallocatechin gallate was docked to enzyme-substrate complex, while rhodiosin was docked to free enzyme. (-)-Gallocatechin gallate formed hydrogen bonds with Pro 9, Ser 13, Ser15, Asn 58, Asn 85 and Glu 239. Rhodiosin formed hydrogen bonds with Asn 58, Ile 59, Asn 85, Asp 100 and Glu 239 (). ITC showed that both (-) -gallocatechin gallate and rhodiosin interacted with SaSDH with moderate affinity, giving a KD for (-) -gallocatechin gallate of 44.65 μM and for rhodiosin of 16.45 μM (). We conclude that both (-)-gallocatechin gallate and rhodiosin achieved inhibition of SaSDH activity by binding to the enzyme.
Figure 5. (A) Molecular docking of (-)-Gallocatechin gallate and Rhodiosin with SaSDH. (B) Binding isotherm of SaSDH titrated with (-)-Gallocatechin gallate and Rhodiosin.
Antibacterial action of (-)-gallocatechin gallate and rhodiosin
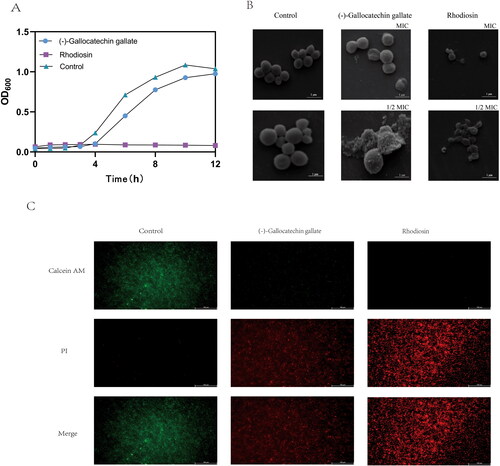
Time-kill curves showed that (-)-gallocatechin gallate inhibited S.aureus growth at 50 μg/mL but did not kill the bacteria. Rhodiosin completely inhibited S.aureus growth at 250 μg/mL (). SEM observations of bacterial morphology after treatment with MIC and 1/2 MIC of (-)-gallocatechin gallate and rhodiosin for 1.5 h showed cell disintegration, cell surface fracturing and wrinkling compared with the smooth, intact, spherical appearance of water-treated cells (). Fluorescent imaging showed the appearance of PI-labeled bacteria, indicating cell membrane destruction, after treatment with MICs of (-)-gallocatechin gallate or rhodiosin compared with red fluorescence of intact cell membranes in control groups (). Both (-)-gallocatechin gallate and rhodiosin affected bacterial morphology by disrupting the integrity of the cell membrane.
Figure 6. Antibacterial action of (-)-Gallocatechin gallate and Rhodiosin.(A) Time-Kill curves for (-)-Gallocatechin gallate and Rhodiosin against S.aureus.(B) SEM of S.aureus treated with (-)-Gallocatechin gallate and Rhodiosin.(C) Calcein AM/PI staining of S.aureus treated with (-)-Gallocatechin gallate and Rhodiosin.
Discussion
Staphylococcus aureus is common and causes skin and soft tissue infection, infective endocarditis, osteomyelitis, bacteraemia and fatal pneumoniaCitation16. β-Lactam drugs are usually used for treatment but most were marketed half a century ago and drug resistance is rife. There is an urgent need to find new anti-Staphylococcus aureus targets to develop new drugs. Many scientists have already demonstrated that SDH is attractive target for new antibacterial drugs but no SaSDH inhibitors with antibacterial activity have been found so far. The current work explored the potential of SDH as a new drug target for anti-Staphylococcus aureus drugs. High-throughput virtual screening based on molecular docking and susceptibility were used to identify SaSDH inhibitors with antibacterial activity against Staphylococcus aureus.
494,000 compounds from Life Chemicals HTS Compound Collection, 208,000 from Specs HTS Compounds Library and 18,000 from MCE Bioactive Compound Library PlusA were screened and clogP, rotating Bonds, TPSA, HB Acceptor and HB Donor identified for the 200 most promising candidates (Supplement 2). Binding to SaSDH depended largely on hydrogen bonds from hydroxyl groups on the compounds, consistent with previous observationsCitation17–19. All compounds in the Life Chemicals HTS Compound Collection and Specs HTS Compounds Library had absolute docking score values of less than 10 but 63 compounds from MCE Bioactive Compound Library PlusA had values greater than 10. Therefore, compounds from MCE Bioactive Compound Library PlusA were selected. We finally chose 12 compounds after multiple considerations, of which 6 had IC50 less than 100 μM and 3 had antibacterial activities: pentagalloylglucose, (-)-gallocatechin gallate and rhodiosin.
(-)-Gallocatechin gallate is a naturally-occurring polyphenolic compound derived from green teaCitation20 for which antitumorCitation21, antiviralCitation22,Citation23, anti-depressiveCitation24, antioxidantCitation25 and cardioprotective propertiesCitation26 have been reported. It has also been reported to have antibacterial activity with an unknown mechanismCitation27–29. Rhodiosin is also naturally-occurring and is a flavonoid derived from Rhodiola roseaCitation30 for which antitumorCitation31, antiviralCitation32, antioxidant and anti-inflammatoryCitation33, but not antibacterial activities have been identified. (-)-Gallocatechin gallate was found to have a Ki for SaSDH activity of 2.47 μM and rhodiosin of 73.38 μM using Lineweaver-Burk double-reciprocal plots during the current work. Different modes of inhibition were found for the two compounds with rhodiosin being competitive with respect to SKH while (-)-gallocatechin gallate was uncompetitive, perhaps due to structural differences. Both (-)-gallocatechin gallate and rhodiosin were found to form hydrogen bonds with SaSDH by molecular docking and ITC giving respective KDs of 44.65 μM and 16.45 μM, indicating moderate affinity. Previously, Claudia et alCitation14 firstly reported three inhibitors of SaSDH through virtual screening, two of which were competitive inhibitors with Ki were 9.53 μM and 19.76 μM respectively, another was mixed competitive having a Ki of 58.3 μM. Afterwards Claudia et alCitation9 adopted a structure similarity search strategy based on these three inhibitors to find new SaSDH inhibitors, and successfully found four more inhibitors with Ki of 106.1 μM, 75.3 μM, 83.1 μM and 8.3 μM. Each of these four inhibitors had a very different behaviour in binding to SaSDH. All these inhibitors were synthetic compound with low molecular weight, not natural product. Almost all of these compounds seemed to have stronger enzymatic inhibitory activity than rhodiosin found in our work, but the antibacterial activities of these inhibitors previously reported were not determined. (-)-Gallocatechin gallate and rhodiosin we found offer new structures of SaSDH inhibitors reported actually.
Antibacterial activity MICs of 50 μg/mL for (-)-gallocatechin gallate and 250 μg/mL for rhodiosin were found, indicating fairly weak antibacterial actions, similar to most antibacterial natural products. (-)-Gallocatechin gallate was found to slow bacterial growth, unlike rhodiosin which showed complete growth inhibition, by time-kill curves. The chemical instability of (-)-gallocatechin gallate may be responsible for its more limited performance which limits its utility as an antibacterial agent. Both compounds were found to disrupt the bacterial cell wall but we believe they are unlikely to be single-target compounds and further study is necessary to determine the full extent of their antibacterial effects.
In summary, recombinant SaSDH protein was firstly expressed and purified, and then inhibitors were identified from three commercial databases through virtual screening. (-)-Gallocatechin gallate and rhodiosin were found to inhibit SaSDH catalytic activity. Both (-)-gallocatechin gallate and rhodiosin had antibacterial activity against S.aureus in vitro, disrupting the integrity of the bacterial cell membrane. The two novel SaSDH inhibitors with antimicrobial activity have potential for the development of drugs for the treatment of S.aureus infection.
Authors’ contributions
ZMF and DQ conceived and designed the experiments. ZMF and QJF prepared the materials and performed the experiments. ZMF, QJF and DQ analysed the computational data and results. The manuscript was organised and written by ZMF and DQ.
Supplemental Material
Download PDF (1.1 MB)Acknowledgements
The authors would like to express their gratitude to EditSprings (https://www.editsprings.cn) for the expert linguistic services provided.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Guo Y, Song G, Sun M, Wang J, Wang Y. Prevalence and therapies of antibiotic-resistance in Staphylococcus aureus. Front Cell Infect Microbiol. 2020;10:107.
- Nichols CN, Wardlow LC, Coe KE, Sobhanie MME. Clinical outcomes with definitive treatment of methicillin-resistant Staphylococcus aureus bacteremia with retained daptomycin and ceftaroline combination therapy vs de-escalation to monotherapy with vancomycin, daptomycin, or ceftaroline. Open Forum Infect Dis. 2021;8(7):ofab327.
- Silva V, Almeida F, Silva A, Correia S, Carvalho JA, Castro AP, Ferreira E, Manageiro V, Caniça M, Igrejas G, et al. First report of linezolid-resistant cfr-positive methicillin-resistant Staphylococcus aureus in humans in Portugal. J Glob Antimicrob Resist. 2019;17:323–325.
- Mir R, Jallu S, Singh TP. The shikimate pathway: Review of amino acid sequence, function and three-dimensional structures of the enzymes. Crit Rev Microbiol. 2015;41(2):172–189.
- Parish T, Stoker NG. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology (Reading)). 2002;148(Pt 10):3069–3077.
- Duque-Villegas MA, Lopes Abbadi B, Romero PR, Matter LB, Galina L, Dalberto PF, da Silva Rodrigues-Junior V, Ducati RG, Roth CD, Scheibler Rambo R, et al. EPSP synthase-depleted cells are aromatic amino acid auxotrophs in Mycobacterium smegmatis. Microbiol Spectr. 2021;9(3):e0000921.
- Peek J, Lee J, Hu S, Senisterra G, Christendat D. Structural and mechanistic analysis of a novel class of shikimate dehydrogenases: evidence for a conserved catalytic mechanism in the shikimate dehydrogenase family. Biochemistr. 2011; 50(40):8616–8627.
- Díaz-Quiroz DC, Cardona-Félix CS, Luis Viveros-Ceballos J, Reyes-González MA, Bolívar F, Ordoñez M, Escalante A. Synthesis, biological activity and molecular modelling studies of shikimic acid derivatives as inhibitors of the shikimate dehydrogenase enzyme of Escherichia coli. J Enzyme Inhib Med Chem. 2018;33(1):397–404.
- Enríquez-Mendiola D, Téllez-Valencia A, Sierra-Campos E, Campos-Almazán M, Valdez-Solana M, Gómez Palacio-Gastélum M, Avitia-Domínguez C. Kinetic and molecular dynamic studies of inhibitors of shikimate dehydrogenase from methicillin-resistant Staphylococcus aureus. Chem Biol Drug Des. 2019;94(2):1504–1517.
- Isa A, Majumdar S, Haider S. In Silico Identification of potential inhibitors against shikimate dehydrogenase through virtual screening and toxicity studies for the treatment of tuberculosis. Int Microbiol. 2019;22 (1):7–17.
- Zhu N, Wang X, Li D, Lin Y, You X, Jiang J, Xu Y, Jiang W, Si S. IMB-T130 targets 3-dehydroquinate synthase and inhibits Mycobacterium tuberculosis. Sci Rep. 2018; 8 (1):17439.
- Fonseca IO, Magalhães MLB, Oliveira JS, Silva RG, Mendes MA, Palma MS, Santos DS, Basso LA. Functional shikimate dehydrogenase from Mycobacterium tuberculosis H37Rv: purification and characterization. Protein Expr Purif. 2006;46(2):429–437.
- Han C, Hu T, Wu D, Qu S, Zhou J, Ding J, Shen X, Qu D, Jiang H. X-ray crystallographic and enzymatic analyses of shikimate dehydrogenase from Staphylococcus epidermidis. FEBS J. 2009; 276(4):1125–1139.
- Avitia-Domínguez C, Sierra-Campos E, Salas-Pacheco JM, Nájera H, Rojo-Domínguez A, Cisneros-Martínez J, Téllez-Valencia A. Inhibition and biochemical characterization of methicillin-resistant Staphylococcus aureus shikimate dehydrogenase. Molecules. 2014;19(4):4491–4509.
- Wei B, Zhang T, Wang P, Pan Y, Li J, Chen W, Zhang M, Ji Q, Wu W, Lan L, et al. Anti-infective therapy using species-specific activators of Staphylococcus aureus ClpP. Nat Commun. 2022;13(1):6909.
- Chen Y, Sun L, Ba X, Jiang S, Zhuang H, Zhu F, Wang H, Lan P, Shi Q, Wang Z, et al. Epidemiology, evolution and cryptic susceptibility of methicillin-resistant Staphylococcus aureus in China: a whole-genome-based survey. Clin Microbiol Infect. 2022; 28(1):85–92.
- Xu Z, Zhang Q, Shi J, Zhu J W. Underestimated noncovalent interactions in protein data bank. J Chem Inf Model. 2019; 59(8):3389–3399.
- Ray M, Sarkar S, Rath SN. Druggability for COVID-19: in silico discovery of potential drug compounds against nucleocapsid (N) protein of SARS-CoV-2. Genomics Inform. 2020; 18(4):e43.
- Henrique Santana Silveira P, Silva da Rocha Pita S. Druggable sites identification in Streptococcus mutans VicRK system evaluated by catechols. J Biomol Struct Dyn. 2023;41(21):12000–12015.
- Khan N, Mukhtar H. Tea polyphenols in promotion of human health. Nutrients. 2018;11(1):39.
- Zhang X, Li J, Li Y, Liu Z, Lin Y, Huang JA. Anti-melanogenic effects of epigallocatechin-3-gallate (EGCG), epicatechin-3-gallate (ECG) and gallocatechin-3-gallate (GCG) via down-regulation of cAMP/CREB/MITF signaling pathway in B16F10 melanoma cells. Fitoterapia. 2020; 145:104634.
- Bo Z, Zhu J, Guo M, Zhang C, Cao Y, Zhang X, Yantao W. Gallocatechin gallate inhibits the replication of pseudorabies virus via suppressing the entry and release stages in its replication cycle. Vet Sci. 2023; 10(3):18.
- Xiao T, Cui M, Zheng C, Zhang P, Ren S, Bao J, Dandi G, Ronghao S, Ming W, Jianping L, et al. Both baicalein and gallocatechin gallate effectively inhibit SARS-CoV-2 replication by targeting Mpro and sepsis in mice. Inflammation. 2022;45(3):1076–1088.
- Ko S, Jang WS, Jeong JH, Ahn JW, Kim YH, Kim S, Hyeon Kyeong C, Seungsoo C. (-)-Gallocatechin gallate from green tea rescues cognitive impairment through restoring hippocampal silent synapses in post-menopausal depression. Sci Rep. 2021; 11(1):910.
- Park DH, Park JY, Kang KS, Hwang GS. Neuroprotective effect of gallocatechin gallate on glutamate-induced oxidative stress in hippocampal HT22 cells. Molecules. 2021; 26(5):1387.
- Ikeda A, Iso H, Yamagishi K, Iwasaki M, Yamaji T, Miura T, Sawada N, Inoue M, Tsugane S, the JPHC Study Group. Plasma tea catechins and risk of cardiovascular disease in middle-aged Japanese subjects: The JPHC study. Atherosclerosis. 2018; 277:90–97.
- Gabr GA, Hassan HMM, Seshadri VD, Hassan NMM. Comparative study of phenolic profile, antioxidant and antimicrobial activities of aqueous extract of white and green tea. Z Naturforsch C J Biosci. 2020; 77(11-12):483–492.
- Javadkhani A, Shokouhi B, Mosayebzadeh A, Safa S, Fahimi M, Sharifi S, Maleki Dizaj S, Salatin S. Nano-catechin gel as a sustained release antimicrobial agent against clinically isolated Porphyromonas gingivalis for promising treatment of periodontal diseases. J Biomedicines. 2023; 11(7):1932.
- Hui X, Hua SH, Wu QQ, Li H, Gao WY. Antimicrobial mechanism of epigallocatechin gallate and gallocatechin gallate: They target 1-deoxy-d-xylulose 5-phosphate reductoisomerase, the key enzyme of the MEP terpenoid biosynthetic pathway. Arch Biochem Biophys. 2017; 622:1–8.
- Péter Zomborszki Z, Kúsz N, Csupor D, Peschel W. Rhodiosin and herbacetin in Rhodiola rosea preparations: additional markers for quality control? Pharm Biol. 2019; 57(1):295–305.
- Zhang X, Zhu J, Yan J, Xiao Y, Yang R, Huang R, Zhou J, Wang Z, Xiao W, Zheng C, et al. Systems pharmacology unravels the synergic target space and therapeutic potential of Rhodiola rosea L. for non-small cell lung cancer. Phytomedicine. 2020; 79:153326.
- Döring K, Langeder J, Duwe S, Tahir A, Grienke U, Rollinger JM, Schmidtke M. Insights into the direct anti-influenza virus mode of action of Rhodiola rosea. Phytomedicine. 2022; 96:153895.
- Zhou J-T, Li C-Y, Wang C-H, Wang Y-F, Wang X-D, Wang H-T, Zhu Y, Jiang M-M, Gao X-M. Phenolic compounds from the roots of Rhodiola crenulata and their antioxidant and inducing IFN-γ production activities. Molecules. 2015;20(8):13725–13739.