
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
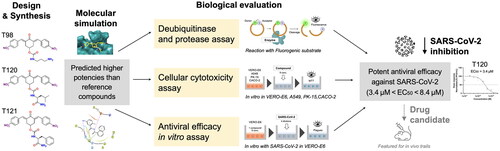
The viral genome of the SARS-CoV-2 coronavirus, the aetiologic agent of COVID-19, encodes structural, non-structural, and accessory proteins. Most of these components undergo rapid genetic variations, though to a lesser extent the essential viral proteases. Consequently, the protease and/or deubiquitinase activities of the cysteine proteases Mpro and PLpro became attractive targets for the design of antiviral agents. Here, we develop and evaluate new bis(benzylidene)cyclohexanones (BBC) and identify potential antiviral compounds. Three compounds were found to be effective in reducing the SARS-CoV-2 load, with EC50 values in the low micromolar concentration range. However, these compounds also exhibited inhibitory activity IC50 against PLpro at approximately 10-fold higher micromolar concentrations. Although originally developed as PLpro inhibitors, the comparison between IC50 and EC50 of BBC indicates that the mechanism of their in vitro antiviral activity is probably not directly related to inhibition of viral cysteine proteases. In conclusion, our study has identified new potential noncytotoxic antiviral compounds suitable for in vivo testing and further improvement.
GRAPHICAL ABSTRACT
Introduction
Coronavirus outbreaks
Several members of the Coronaviridae family constantly circulate in the human population and usually cause mild respiratory diseaseCitation1. In contrast, the severe acute respiratory syndrome coronavirus (SARS-CoV) and the Middle East respiratory syndrome coronavirus (MERS-CoV) are transmitted from animals to humans and cause acute respiratory diseases in infected individualsCitation2. The coronavirus disease COVID-19 started to spread among humans in December 2019. Since its first outbreak in China, it quickly became a global pandemic. The outbreak of COVID-19 showed us how inadequately prepared we are for diseases caused by emerging (e.g., zoonotic) viruses. Vaccines represent the best strategy to suppress the COVID-19 pandemic. However, their waning protection and a large pool of unvaccinated individuals provide the ground for the spread of the virus and the generation of novel variants, which represent a constant threat for vulnerable people with inadequate immune responses. For those who become infected, more efficient treatment will need to be developed.
Viral cycles are highly dependent on cellular factors and cellular metabolic and signalling pathways; therefore, the number of possible antiviral drug targets is limited. However, almost all viruses encode unique proteins and enzymes that can serve as specific targets for antiviral therapy. One of the main goals of modern drug development efforts is to design compounds that specifically inhibit viral targets or cellular targets essential for virus replication. To complement the weaponry available to combat the SARS-CoV-2 virus, here we focus on the development and evaluation of new small-molecule inhibitors targeted against viral enzymes.
Variants, vaccines, and antivirals interconnected with genome stability
Genetic diversification of SARS-CoV-2 was initially considered slow when the virus spread in 2019 and early 2020. The first official variant, a single spike D614G mutation found in early European lineages, was linked to more efficient transmissionCitation3 and rapidly spread to become the dominant viral strain worldwide. Later in 2020, multiple variants emerged that launched regional epidemics. According to the WHO (World Health Organisation), five main variants have been identified (Alpha, Beta, Gamma, Delta, and Omicron). All have characteristic mutations closely described at: www.ecdc.europa.eu/en/covid-19/variants-concern. The spike glycoprotein appears especially prone to accumulate mutationsCitation4 and all circulating variants have some mutations that favour evasion from the host immune responseCitation5. However, sera studies and emerging real-world evidence indicate that Omicron escapes adaptive immunity initiated by previous infection or vaccinationCitation6. Therefore, it is important to develop antiviral agents acting directly on SARS-CoV-2 as an alternative tool in the fight against the virus. Several antiviral therapeutics have been approved or are advancing in clinical developmentCitation7. As an example of the direct-acting small-molecule SARS-CoV-2 antivirals that have received approval or emergency use authorisation do not bind to the variable spike protein, but target conserved viral RNA-dependent RNA polymerase or conserved viral cysteine proteases (Mpro or PLpro). Remdesivir, a mono-phosphoramidate prodrug of the nucleoside GS-441524, originally developed to treat Ebola virus infections, inhibits the RNA polymerase of SARS-CoV-2. It was the first antiviral approved or authorised for emergency use to treat COVID-19 in several countries. Additionally, molnupiravir (MK-4482 or EIDD-2801), a small molecule ribonucleoside prodrug of N-hydroxycytidine, originally developed against different RNA viruses such as influenzaCitation8, has recently received an emergency use authorisation for the treatment of mild-to-moderate COVID-19. Another approved small-molecule antiviral chemotherapeutic drug, paxlovid (nirmatrelvir/ritonavir), irreversibly inhibits viral protease Mpro and stops viral replication of SARS-CoV-2Citation9.
Proteases play an essential role in the replication of SARS-CoV-2 and are genetically stable
The SARS-CoV-2 viral genome encodes several structural proteins (e.g., capsid spike glycoprotein), non-structural proteins (e.g., 3-chymotrypsin-like or main protease abbreviated as 3CLpro or Mpro, papain-like protease PLpro, helicase, and RNA-dependent RNA polymerase) and accessory proteins. Some of these components undergo genetic variationsCitation10, while mutations within cysteine proteases Mpro and PLpro were also found and recently also documentedCitation11. The viral Mpro cleaves the two polyproteins (pp1a and pp1ab) of SARS-CoV-2 at multiple locations, resulting in various non-structural proteins, which are key for viral replicationCitation12. PLpro is similarly involved in viral replication, but it also plays a role in altering the host antiviral immune responseCitation13. Both enzymes were validated as potential antiviral drug targetsCitation14,Citation15 and their higher genomic stability in all variants of SARS-CoV-2 makes them attractive in this respect. For example, nirmatrelvir (PF-07321332), is an oral-available Mpro inhibitor. When treatment is started during the first days after the appearance of symptoms, it results in approximately 90% protection against severe COVID-19 and hospitalisationCitation9. Unfortunately, nirmatrelvir – the active component of paxlovid – is rapidly metabolised in the liver. Therefore, ritonavir is co-administered with nirmatrelvir to dampen metabolic conversion by cytochrome CYP3A, which can cause adverse effects in polymorbid patientsCitation16,Citation17. Moreover, the construction and analysis of several recombinant SARS-CoV-2 clones showed that the main protease mutations mediated only low-level resistance to nirmatrelvir, whereas greater resistance required accumulation of additional mutationsCitation18. Although, these findings indicate that SARS-CoV-2 resistance to nirmatrelvir does readily arise via multiple pathways in vitro, the specific mutations form a foundation to study the mechanism of resistance in more detailCitation18. Although the Mpro and PLpro genes can be affected by evolutionary mutationsCitation18,Citation19, the viability of the SARS-CoV-2 does not appear to be compromisedCitation20,Citation21.
Deubiquitination, an important function of the SARS-CoV-2 machinery
In addition to polyprotein processing activity, SARS-CoV-2 PLpro possesses a characteristic deubiquitinating activity (DUB) (including the deconjugation of other ubiquitin-like modifiers)Citation21–23, as was already observed for analogous SARS-CoV PLproCitation24,Citation25 and for an adenoviral proteaseCitation24,Citation26. This specific activity interferes with host cell processes and contributes to the ability of the SARS-CoV-2 virus to evade cell defence mechanismsCitation13. The deubiquitination function would greatly impact the value of PLpro as a therapeutic target and provide a framework for the development of antivirals to treat SARS-CoV-2Citation27–29. Strategies for the design of PLpro inhibitors must also consider the potentially overlapping specificity of this protease with those of cellular deubiquitinating enzymes.
Bis(benzylidene)cyclohexanones (BBC) are potent deubiquitinase inhibitors
Dienones are synthetic analogues of curcumin known for their antineoplastic, anti-inflammatory, antiviral, and antiparasitic propertiesCitation30. These varied biological activities are due to the ability of the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore to covalently target biological thiols, including the catalytic cysteines present in many enzymes ()Citation31.
Figure 1. Left. Reaction of the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore with thiol group at the catalytic site (showing Cys111 and His272) of SARS-CoV-2 PLpro. Initially, a non-covalent enzyme-inhibitor association is formed (E:I, 1) leading to a covalent adduct. Right. Structure of BBC DU-UC15 (2c) with the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore in red.
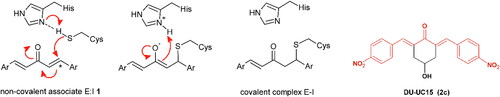
Recently, the Trieste University group has described the development of new antineoplastic agents in which the 1,5-diaryl-3-oxo-1,4-pentadienyl pharmacophore is embedded in the 4-hydroxycyclohexanone scaffoldCitation32. BBC DU-UC15 (formerly: 2c) is a potent inhibitor of several cysteine-dependent deubiquitinases, preventing hydrolytic cleavage of the bond between ubiquitin and target proteins. This, in turn, affects the degradation of damaged proteins by the ubiquitin proteasome system, causing proteotoxic stress and apoptosis in tumour cells. Accordingly, the DU-UC15 inhibitor shows antiproliferative activity in several models, while modified inhibitors, optimised for delivery, show anticancer activity in vivoCitation32–35.
As both proteases encoded by SARS-CoV-2 are cysteine dependent, we foresee the possibility that the compound DU-UC15 and its congeners might covalently inhibit these enzymes with the same mechanism shown in . Substitutions in compound DU-UC15 were introduced to furnish longer chains ending with functional groups capable of reaching the catalytic centres of protease or deubiquitinase of PLpro through relatively narrow substrate channels. Such chains contain amino acids and short peptides or cationic end groups to enhance the interactions with the deubiquitinase site. In addition, inhibition of the specific deubiquitinase activity of PLpro would strengthen cellular defences against infection and provide a new approach to SARS-CoV-2 therapy.
Protease inhibition and antiviral effect – Searching for new scaffolds
The viral proteases Mpro and PLpro play an essential role in coronavirus replication by digesting viral polyproteins at many sites; thus, they appear as high-profile targets for antiviral drug discoveryCitation36–40. The discovery of S-217622 (also called the Shionogi’s ensitrelvir), the first oral non-covalent, non-peptidic SARS-CoV-2 Mpro inhibitor as a clinical candidate provided new opportunity in SARS-CoV-2 antiviral developmentCitation41, while covalent inhibitorsCitation42,Citation43 also show great promise for the development of new therapeutics for SARS-CoV-2 infection.
Here, we investigate the inhibitory activity of compound DU-UC15 and several other BBCs against both viral proteases. Some known inhibitors of cysteine proteases and proteasome-associated deubiquitinating enzymes have also been included in the study. PLpro inhibition of both protease (PA) and deubiquitinase (DUA) activities was studied using kits from BPS Bioscience Inc. (San Diego, CA, USA). The in vitro antiviral activity of the studied compounds and reference inhibitors was also evaluated in SARS-CoV-2 infected cell cultures.
Overall, our study provides new compounds with potential antiviral activity against SARS-CoV-2 with expected mechanism of action related to inhibition of Mpro and PLpro protease activity and PLpro deubiquitinase activity. Some of the compounds identified here represent efficient SARS-CoV-2 inhibitors that can impede SARS-CoV-2 replication in vitro.
Materials and methods
Cell lines
The cell line VERO-E6 (ECACC 85020206, ATCC-CRL-1586) isolated from the kidney of an African Green Monkey, human colon adenocarcinoma cells Caco-2 (ATCC-HTB-37), porcine kidney cells PK-15 (DSMZ-ACC640), and human lung adenocarcinoma cells A549 (DSMZ-ACC107) are mammalian epithelial cell lines used in a variety of biomedical research, nowadays are also the most widely used to replicate and isolate the SARS-CoV-2 virus. The selected cell models are also commonly used in drug tests and were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, Braunschweig, Heidelberg, Germany) or from the American Type Culture Collection (ATCC, LGC Promochem GmbH, Wesel, Germany). VERO-E6, PK-15, and A549 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 5% heat-inactivated foetal bovine serum (FBS) and L-glutamine (2 mM). Caco-2 cells were cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% heat-inactivated foetal bovine serum (FBS) and L-glutamine (2 mM). The cells were kept in a humidified incubator at 37° C and 5% CO2. Cells were subcultured regularly 2–3 times a week, keeping them in the exponential growth phase. All cell lines are regularly tested by PCR for common species of mycoplasma by using primers: 5′-ACACCATGGGAGYTGGTAAT-3′ (forward), 5′-CTTWTCGACTTYCAGACCCAAGGCAT-3′ (reverse); and nested PCR primers: 5′-GTGSGGMTGGATCACCTCCT-3′ (forward), 5′-GCATCCACCAWAWACYCTT-3′ (reverse)Citation44,Citation45.
Preparation of inhibitors
Commercial reagents and solvents were purchased from Sigma-Aldrich. Column flash chromatography was performed on silica gel 60 (230,400 mesh). Reactions were monitored by thin layer chromatography on silica gel plates using UV light as visualising and KMnO4 as developing agent. NMR spectra were recorded on a Varian 500 MHz spectrometer at 500 MHz (1H) and 125 MHz (13C). Chemical shifts are given in ppm (δ). The coupling constants J are given in Hertz. NMR 1H and 13C resonances were assigned using a combination of DEPT, COSY, and HSQC spectra. Electrospray mass spectra were obtained on a Bruker Daltonics Esquire 4000 spectrometer. Yields refer to spectroscopically homogeneous materials (1H NMR).
The structures of the inhibitors related to DU-UC15 (T97), which are discussed in this study, are reported in .
Table 1. Bis(benzylidene)cyclohexanones studied in this work.
The general approach to the synthesis of BBC starts with aldol condensation of 4-hydroxycyclohexanone with the appropriate aromatic aldehyde, after which the 4-hydroxycyclohexanone scaffold is modified as indicated (Scheme 1).
Scheme 1. Synthesis of bis(benzylidene)cyclohexanone inhibitors.
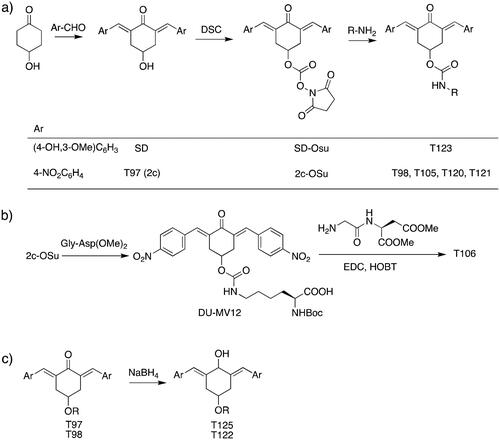
Double condensation of 4-hydroxycyclohexanone with 4-nitrobenzaldehyde gives compound T97. Compounds T98, T105, T120 and T121 were synthesised from the carbonate 2c-OSu Citation32 by reaction with the appropriate primary amine as shown in Scheme 1a. Similarly, the reaction of 2c-OSu with N-Boc-Lys gave compound DU-MV12 from which T106 was obtained by condensation with the dipeptide Gly-Asp(OMe)2 (Scheme 1b). Compound T123 was obtained by aldol condensation of 4-hydroxycyclohexanone with vanillin under acidic conditions, followed by carbonylation of the resulting alcohol with N,N’-disuccinimidyldicarbonate (DSC) and reaction with 1,3-diaminopropane. T125 and T122 were obtained by borohydride reduction of T97 and T98, respectively (Scheme 1c).
The synthesis of compounds T97 (DU-UC15, 2c), T98 and T124 has previously been reported (T97Citation32; T98, T124Citation34). Full details of the syntheses of T120 and T121 will be reported elsewhere.
T105. A solution of 2c-OSu (0.639 g, 1.22 mmol) and εN-Boc-Lys (0.351 g, 1.47 mmol) in dichloromethane was adjusted to pH 8–9 with triethylamine and stirred for 12 h until complete conversion was observed (TLC). The reaction mixture was extracted with 20% aqueous citric acid and the organic layer was dried over Na2SO4 and evaporated to obtain the desired product. Yield 83%.1H NMR (CDCl3, 500 MHz) δ (ppm): 8.27–8.24 (m, 4H), 7.92–7.84 (m, 2H), 7.59–7.54 (m, 4H), 5.43 (m, 1H), 5.25–5.07 (m, 1H), 4.79–4.55 (m, 1H), 4.34–3.89 (m, 1H), 3.39–2.85 (m, 6H), 1.86–1.54 (m, 2H), 1.51–1.20 (m, 4H), 1.40 (s, 9H).13C NMR (CDCl3, 125 MHz) δ (ppm): 187.6, 175.6, 156.5, 155.2, 147.6, 141.7, 137.6, 134.4, 130.9, 123.9, 79.7, 67.8, 53.7, 40.0, 33.3, 31.6, 29.8, 28.5, 22.3. ESI-MS: m/z 675.2 [MNa]+, 691.2 [MK]+.
T106. 2c-OSu (0.856 g, 1.64 mmol) and αN-Boc-Lys (0.458 g, 1.86 mmol), as described above for T105, gave the carbamate DU-MV12 (86%). 1H NMR (CDCl3, 500 MHz) δ (ppm): 8.30–8.26 (m, 4H), 7.93–7.87 (m, 2H), 7.62–7.57 (m, 4H), 5.24–5.12 (m, 1H), 5.07 (d, 1H, J = 7.3 Hz), 4.73 (t, 1H, J = 5.9 Hz), 4.32–4.04 (m, 1H), 3.31–2.93 (m, 6H), 1.86–1.57 (m, 2H), 1.53–1.24 (m, 4H), 1.42 (s, 9H). 13C NMR (CDCl3, 125 MHz) δ (ppm): 187.9, 175.9, 155.9, 155.5, 147.7, 141.7, 137.4, 134.6, 130.9, 124.0, 80.5, 67.2, 53.2, 40.6, 33.4, 32.0, 29.3, 28.5, 22.8. ESI-MS: m/z 675.2 [MNa]+, 691.2 [MK]+. This compound (0.484 g, 0.742 mmol) was stirred with EDC.HCl (0.213 g, 1.113 mmol) and HOBt (0.150 g, 1.113 mmol) in dichloromethane at 0 °C for 30 min. After 10 min, Gly-Asp dimethyl ester trifluoroacetate (0.256 g, 0.771 mmol) in dichloromethane was slowly added to the mixture together with the minimum volume of triethylamine to reach an apparent pH 8–9. The resulting solution was stirred at 25 °C for 18 h till complete conversion of DU-MV12 to the product (TLC) and the mixture was extracted with 20% aqueous citric acid (3 x) and sat. aqueous NaHCO3 (3 x). The organic layer was dried over Na2SO4 and evaporated in vacuo to obtain the crude product, which was purified by flash chromatography (eluent: CHCl3/MeOH from 100:0 to 97:3). Yield = 39% from DU-MV12; yellow solid. 1H NMR (CDCl3, 500 MHz) δ (ppm): 8.30–8.23 (m, 4H), 7.95–7.84 (m, 2H), 7.65–7.54 (m, 4H), 7.06 (d, J = 8.1 Hz, 1H), 6.61 (t, J = 5.1 Hz, 1H), 5.30–5.09 (m, 2H), 4.94 (t, J = 5.7 Hz), 4.82 (dt, J = 8.1, 4.7 Hz), 4.15–3.89 (m, 3H), 3.72 (s, 3H), 3.66 (s, 3H each), 3.33–2.91 (m, 7H) 2.83 (dd, J = 17.2, 4.7 Hz, 1H), 1.93–1.70 (m, 2H), 1.69–1.21 (m, 4H), 1.40 (s, 9H). 13C NMR (CDCl3, 125 MHz) δ (ppm) 187.7, 172.6, 171.4, 170.9, 168.6, 155.9, 155.4, 147.7, 141.7, 137.3, 134.6, 130.9, 124.0, 80.4, 67.1, 54.4, 53.1, 52.3, 48.7, 42.9, 40.5, 36.0, 33.4, 32.0, 29.3, 28.4, 22.6. ESI-MS: m/z 875.4 [MNa]+, 981.3 [MK]+.
T123 (DU-SD2). Following the procedure described for T105, compound DU-SD2 was synthesised from 2c-OSu (0,752 g, 1.43 mmol) and 1,3-diaminopropane (0,128 g, 0,145 ml, 1,72 mmol). Yield 39%. 1H NMR (400 MHz, D6-DMSO) δ (ppm): 9.58 (s, 2H), 7.65 (s, 2H), 7.58 (m, 3H) 7.16 (br, 1H), 7.09 (s, 2H), 7.01 (apparent d, 2H), 6.85 (apparent d, 2H), 5.02 (br, 1H), 3.80 (s, 6H), 3.16 − 3.05 (m, 4H), 2.91 (m, 2H), 2.49 (m, 2H), 1.56 (m, 2H) ppm. 13C NMR (100 MHz D6-DMSO) δ (ppm): 187.55, 159.00, 148.48, 147.93, 138.62, 129.80, 126.99, 124.51, 116.05, 115.32, 56.14, 46.16, 37.65, 37.14, 33.22, 27.92.
T125. To a solution of T97 (2c) (0.152 g, 0.4 mmol) in 25 ml 1:10 MeOH/THF, 0.017 g of NaBH4 (0.44 mmol) were added. The resulting solution was stirred at 25° C for 1.5 h until complete conversion to product (TLC). After adding 25 ml of brine, the mixture was extracted with diethyl ether (3 x). The organic layer was dried over Na2SO4 and evaporated in vacuo to obtain the product as a mixture of cis/trans diastereoisomers. Yield = 93%., 1H NMR (400 MHz, D6-DMSO) δ (ppm): 8.19, 8.16 (2d, 4H, J = 8.5 Hz), 7.57, 7.53 (2d, 4H, J = 8.5 Hz), 6.76, 6.74 (2s, 2H), 5.81, 5.73 (2d, 2H, J = 4.6 Hz), 5.04, 4.98 (2d, 2H, J = 4.6 Hz), 4.74, 4.66 (2 m, 2H), 3.86, 3.65 (2 m, 2H), 2.95, 2.75 (2dd, 2H, J = 4.0 Hz, J = 13.5 Hz), 2.56, 2.31 (2dd, 2H, J = 3.0 Hz, J = 13.5 Hz). 13C NMR (100 MHz, D6-DMSO) δ (ppm): 188.27, 148.22, 147.87, 137.73, 131.37, 127.36, 124.44, 115.97, 115.22, 64.11, 56.11, 36.58, 188.27, 148.22, 147.87, 137.73, 131.37, 127.36, 124.44, 115.97, 115.22, 64.11, 56.11, 36.58
T122. Reduction of T98 (120 mg, 0.25 mmol), as described for T125, gave the product as a mixture of cis/trans diastereoisomers. Yield 67%. 1H NMR (500 MHz, D6-DMSO) δ (ppm): 8.18, 8.16, (2d, 4H, J = 8.7 Hz), 7.53, 7.51 (2d, 4H, J = 8.7 Hz), 7.11 (bt, 1H), 6.82, 6.79 (2s, 2H), 5.91, 5.86 (2 d, J = 4 Hz each, 1H), 5.83 (d, J = 4.0 Hz, 1H), 5.29 (2t, J = 2.1 Hz, 1H), 3.30 − 3.10 (m, 4H), 2.85 (m, 2H), 2.61 (bm, 2H), 1.53 (quint, 2H); ESI-MS: m/z 483.2 [MH]+, 505.1 [MNa]+.
Determination of IC50 by fluorogenic enzyme assay kits
Half-maximal inhibitory concentrations IC50 of the compounds tested for SARS-CoV-2 Mpro and PLpro were measured using commercially available kits from BPS Bioscience Inc. (San Diego, CA, USA), specifically SARS-CoV-2 Mpro untagged assay kit (catalog #78042–1), SARS-CoV-2 PLpro assay kit, protease activity (catalog #79995–1) and SARS-CoV-2 PLpro assay kit, deubiquitinase activity (catalog # 79996), all in 96-well format. The assay kits come with purified recombinant enzymes (Mpro and PLpro), assay buffers and control inhibitors (GC376 for Mpro and GRL0617 for PLpro), and fluorogenic substrates. According to information from the manufacturer’s website (bpsbioscience.com), all substrates were the following quenched compounds. For SARS-CoV-2 Mpro assay it was 14-mer fluorogenic peptide (DABCYL-KTSAVLQSGFRKME-EDANS), and upon proteolysis by Mpro the peptide substrate is cleaved between glutamine and serine generating the highly fluorescent peptide fragment (SGFRKME-EDANS). For SARS-CoV-2 PLpro protease activity assay it was 5-mer fluorogenic peptide (Z-RLRGG-AMC), and upon proteolysis by PLpro fluorescent AMC dye (7-amino-4-methylcoumarin) is generated. For SARS-CoV-2 PLpro deubiquitinase activity assay it was ubiquitin conjugated by C-terminal Gly with AMC (Ub-AMC), and upon proteolysis by PLpro fluorescent AMC dye is generated. The manufacturer has recommended excitation/emission wavelengths for the determination of enzyme activity for all kits at 360/460 nm. The assay procedures were performed according to the manufacturer’s recommendations, shortly as follows. The inhibitors were screened against recombinant SARS-CoV-2 Mpro and PLpro enzymes in the assay buffers delivered with the kits containing 1 mM DTT. The final concentration of DMSO in the assays did not exceed 1%. The steady-state measurements were started by adding the substrate solution to the wells and the incubation with slow shaking lasted for 4 h at room temperature (Mpro assay), or for 1 h at 37 °C (PLpro assays). The fluorescence intensity was measured by a microtiter plate-reading fluorimeter BioTek Synergy HT (BioTek, Winooski, VT, USA). Enzyme activity was defined as the amount of enzyme that converts 1 µmol of substrate per minute. Data were fitted and IC50 values and the corresponding Ki values were calculated from the Dixon plot using GraphPad Prism 9.5.0 (GraphPad Software, LLC).
In addition to the BBC studied, we have included a set of known cysteine protease inhibitors and proteasome-associated deubiquitinating enzyme inhibitors as reference compounds: RA-9Citation30, PR-619 (C4)Citation46,Citation47, RA-190Citation30, GRL0617 (C9)Citation48, GC376 (C10)Citation49,Citation50 (). The table also includes an additional reference inhibitor remdesivir (C8)Citation51–54 with different mechanism of action, thus was added later for the cellular assay as a control for SARS-CoV-2 inhibition.
Table 2. Chemical structures of the reference inhibitors of Mpro and PLpro of SARS-CoV-2 or proteasome-associated deubiquitinating enzyme inhibitors included in this study.
Compounds stocks
Stock solutions of individual compounds in solid form (10 mg/ml, at approx. 10–50 mM concentrations depending on the individual Mw) were prepared by dissolving in sterile dimethyl sulfoxide (DMSO) and filtered through a 0.2 µm filter (4 mm syringe filter) for fluorogenic enzyme assays. For cell lines, the stock solutions containing DMSO were diluted to the 1 mM concentration in PBS buffer. All stock solutions were kept under dark conditions and kept at −20 °C. For the cytotoxicity and plague reduction assay, stocks were further diluted to appropriate concentrations ranging from 0.1 µM up to 30 µM in cell condition medium.
Virus
The original SARS-CoV-2 strain was isolated from a clinical sample of a patient with COVID-19 in Slovakia. The isolate was deposited in the European virus archive (EVA) GLOBAL. The Slovak strain called Slovakia/SK-BMC5/2020 (available at https://www.european-virus-archive.com/virus/sars-cov-2-strain-slovakiask-bmc52020) represents the strain circulating in Europe in spring 2020 and carries the Spike D614G mutation (lineage B0.1). The identity of the virus was confirmed by sequencing. The complete genome sequence was deposited in the GISAID.org database under the accession ID EPI_ISL_417879. The virus was isolated from a clinical sample, passaged and propagated in VERO-E6 cells, and the presence of mycoplasma was tested with a negative result before its use in this study.
Establishment of a workflow for in vitro biological evaluation
To establish the effective workflow for the evaluation of therapeutic potential, we sought to optimise the testing strategy in several steps. As the first step in biological evaluation, we selected model cell lines based on their known enzymatic characteristics and the level of available data on the RNA expression of relevant genes (e.g., angiotensin converting enzyme-2 (ACE2) entry receptor, transmembrane protease serine 2 (TMPRSS2), and others). Several different types of host cells have been used in SARS-CoV-2 infection assays, as well as for determination of viability and cytotoxicity; therefore, the following models were considered in our studies: VERO-E6Citation55, Caco-2Citation56, PK-15Citation57, and A549Citation58. The most common cell type recently used in coronavirus-related studies are primate-derived epithelial kidney VERO-E6 cells due to their ability to propagate SARS-CoV-2 very effectivelyCitation55. Short-time SARS-CoV-2 infection was reported to produce relevant cytopathic effects in the VERO-E6 cell line. However, since VERO-E6 cells are not an accurate mimic of human airway and lung epithelial cells, the primary site of SARS-CoV-2 infection, we also conducted the experiment with other parallel cell lines of human origin. Two of these model systems are Caco-2 and A549, of which the first has the more sufficient expression levels of ACE2 to allow efficient infection with SARS-CoV-2. Caco-2 is an immortalised human colorectal adenocarcinoma cell line that is a primary model of the human intestinal epithelial barrier and has higher expression of the RNA serine protease TMPRSS2, which is known to mediate pathways of coronavirus infection; for example, priming of SARS-CoV-2 S proteinCitation7. A549 cells are more relevant to the primary infection site of coronavirus since they represent a human pulmonary epithelial cell model used to study drug metabolismCitation58. However, these cells do not maintain sufficient levels of ACE2. The last cell line used in our studies is the mammalian-origin PK-15 model, which is the kidney epithelial cell line and serves as a good model for the multiplication of various mammalian viruses while studying host immune responsesCitation57. Due to the individual specificities of the cell models, we sought to evaluate the cytotoxicity of the compounds in all of them, while for the evaluation of antiviral efficacy we report the data collected in VERO-E6 cells only, while including some gathered remarks from other models.
The second important step was to estimate the exposure interval to cover the full effect of the compounds studied. As the compounds tested are expected to act as inhibitors of SARS-CoV-2 viral proteases, we set the exposure interval of the compounds at least 72 h after 3 h of incubation with SARS-CoV-2 alone. The last parameter to set was the multiplicity of infection (MOI). The cells were infected at low MOI to achieve more replication cycles. Together, the fully optimised strategy for this biological evaluation shown here (Figure S1) allowed us to thoroughly test and analyse the inhibitory potency of new BBC.
Virus titre determination
Classical plaque assays, according to De Madrid et al.Citation59, were performed in VERO-E6 cells grown at 37 °C and 5% CO2 in DMEM supplemented with 5% FBS and 2 mM L-glutamine. In summary, cells were seeded 24 h prior to virus titration to receive around 80% confluency. The next day, serial 10-fold dilutions of virus were added to the cells. After 1 h, the suspension was washed with PBS and overlaid with 1.5% (w/v) CMC in DMEM. Following a 4-day incubation at 37 °C and 5% CO2, cells were fixed for 20 – 30 min with formalin and plaques were visualised by staining with 0.5% crystal violet (1.01408.0025, Merck) at room temperature for 10 – 20 min. After washing the wells with water, the number of plaques was counted. The virus titre was expressed as plaque-forming units (PFU) per ml. The virus stocks were kept in the dark at −80 °C.
Measurement of cytotoxicity and cell viability
For the cell cytotoxicity assay, A549, VERO-E6, Caco-2, and PK-15 cells were seeded into 96-well plates (10 000 cells of VERO-E6, PK-15, and A549 and 40 000 cells of Caco-2). Cells were cultured overnight at 37 °C in a humidified atmosphere. Cells were grown 72 h after the addition of individual compounds in appropriate concentrations (0.5, 1.0, 5.0, 10, 15, and 30 µM) and followed by microscopy inspection. Untreated cells were considered negative control, and DMSO treated cells were considered vehicle control. The classical MTT assayCitation60 was used to measure cellular metabolic activity as an indicator of cell viability, proliferation, and cytotoxicity. Briefly, this colorimetric assay is based on the reduction of a yellow tetrazolium salt (dimethylthiazol-diphenyltetrazolium bromide, MTT) to purple formazan crystals by metabolically active cells. The viable cells contain oxidoreductase enzymes that reduce MTT to formazan thus resulting in a coloured solution that is quantified by measuring absorbance at 570 nm using a spectrophotometer. The darker the solution, the greater the number of viable, metabolically active cells. The results were plotted in a graph of cell viability (represented by a fold change normalised to an untreated control) against compound concentrations. All experimental and reference compounds were measured using at least 2 replicates for each.
Quantitative SARS-CoV-2 plaque reduction assay
The plaque reduction assay with the SARS-CoV-2 virus (performed in a Biosafety level 3 containment laboratory BSL3 at the Biomedical Research Centre of the Slovak Academy of Sciences) was used to determine the inhibition capacity of several new and commercially available reference compounds. The SARS-CoV-2 virus at multiplicity of infection (MOI) = 1.0 was added to the monolayer of VERO-E6 cells (at a concentration of 2.5 x 105 cells per well seeded overnight) in 12-well plates and incubated at 37 °C for 3 h. Serial dilutions (0.1, 0.25, 0.5, 1, 2.5, 5, 10, 15, and 30 µM) of compounds in DMSO-PBS-DMEM solution were then added to SARS-CoV-2 infected VERO-E6 cells and incubated at 37 °C for another 72 h. Cells with supernatant were collected, stored at −80 °C and virus titre was determined by plaque assay as described above. The results were plotted in a plaque reduction represented by the normalised response of PFU/ml to the untreated control against compound concentrations. The inhibition activity (represented by EC50) was determined from the normalised response plotted against logarithmic compound concentrations as the reciprocal of the highest dilution resulting in a 50% infection reduction. Data were processed by regression analysis and fitted to a logistic 4-parameter sigmoidal dose response curve using GraphPad Prism 9.5.0 (GraphPad Software, LLC). The goodness of fit is represented by the R2 parameter (for most experimental compounds, the data fit the model very well with R2 ∼ 0.85–0.91).
Molecular modelling and in silico screening
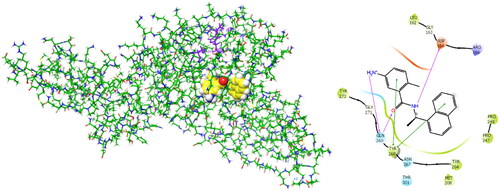
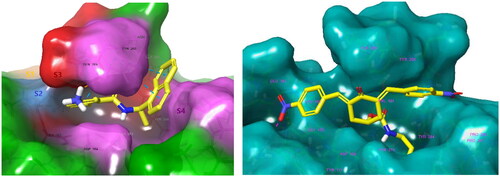
BBC with observed deubiquitinating activity towards the analogous SARS-CoVCitation22–25 were also studied by molecular modelling. We have modelled inhibitor-enzyme interactions at the ligand binding site using the crystal structure of the PLpro-GRL0617 complex available from the Protein Data BankCitation61 (PDB entry 7CMDCitation62) shown in . GRL0617 is a potent noncovalent inhibitor of SARS-CoV-2 PLpro (IC50 = 2.1 µM)Citation48 that shows the highest structural similarity to the studied BBC among known PLpro inhibitors with the published crystal structure of the enzyme-inhibitor complex. Previously, potent PLpro inhibitors were derived from GRL0617, which occupies the BL2 groove of the PLpro binding site (i.e., S4 - S3 pockets) and prevents substrate binding. These covalent inhibitors enhance the methyl group of the benzamide ring using a Gly-Gly-mimetic linker that fills the narrow S2 - S1 substrate channel and an electrophilic warhead that extends to the catalytic Cys111Citation63. We have modelled the formation of a noncovalent enzyme-inhibitor complex (E:I 1, ) that precedes the eventual formation of a covalent adduct to the catalytic cysteineCitation64. BBC were docked to the GRL0617 binding site using extra precision algorithm of GlideCitation65 (Schrödinger, LLC, New York, NY, 2021, release 2021–2). The ligand and water molecules were removed from the binding site, which was defined as the space that covers the S4 and S3 binding pockets of the PLpro located near the catalytic centre. PLpro cleaves the non-structural viral proteins nsp1-nsp4, as well as human ubiquitin and ISG15Footnote1 with strong specificity for the sequences L(R/K)GG↓(A/K), respectively, indicating that the S2 and S1 pockets of the active site are narrow and consequently cannot accommodate bulkier inhibitors aloneCitation13. The zinc ion was retained in the crystal structure of PLpro. We have validated the docking protocol by performing redocking of GRL0617, which resulted in a pose of the ligand very similar to that of the crystal structure. The PLpro-inhibitor complexes generated were subsequently energy optimised by molecular mechanics using the OPLS4 force fieldCitation66 considering the effect of solventCitation67 in Maestro (Schrödinger, LLC, New York, NY, 2021, rel. 2021–2). The predicted enzyme-inhibitor () binding energies of the associate:
calculated with help of molecular mechanics (MM) total energies (
), were compared with the determined IC50 values of the enzyme assays on PLpro inhibition. Several laboratories adopted a similar approach to the in silico identification of SARS-CoV-2 PLpro inhibitors among natural compounds and drug repurposingCitation27,Citation28. To assess the ability of the BBC studied to undergo a nucleophilic attack of the thiol group of catalytic cysteine, we have calculated the Mulliken net atomic chargesCitation68 of the carbon C* of the benzylidene double bond as well as LUMOFootnote2 energies of the optimised geometries of studied compounds () using density functional theory (DTF-M06-2X/6–31++G** method)Citation69 in Jaguar (Schrödinger, LLC, New York, NY, 2021, release 2021–2).
Figure 2. Left: 3D structure of the SARS-CoV-2 PLpro-GRL0617 complex (PDB entry 7CMDCitation62). The inhibitor GRL0617 that occupies the S4 - S3 pockets of the substrate binding cleft is shown in the CPK representation. Catalytic triad of PLpro: Cys111 - His272 - Asp286 coloured purple shows the location of the catalytic site. Right: 2D scheme of inhibitor - residue interactions. Binding of GRL0617 induces closure of the flexible blocking loop BL2 (Gly266 - Gly271) and narrows the substrate binding cleft. The residues Tyr268 and Gln269 in the BL2 loop shift towards the bound GRL0617.
Statistical analysis
Statistical methods used for the evaluation of the data are described in the figure legends. The statistical significance of means ± STDEV was evaluated using the two-tailed Student’s t-test. For all statistical analyses, p values < 0.05 were considered significant. All evaluations were performed using GraphPad Prism 9.5.0 (GraphPad Software, LLC), if other software was not explicitly mentioned.
Results and discussion
Validation of methods for determination of inhibitory potencies and effectivities
The half-maximal inhibitory concentrations IC50 of the selected reference inhibitors () towards Mpro and PLpro of SARS-CoV-2 were determined by enzyme assays. shows the half-maximal IC50 and EC50 determined for selected reference inhibitors. The half-maximal effective in vitro antiviral concentrations (EC50) measured here by the in vitro plaque reduction assay in VERO-E6 cells (e.g., GC376 (C10) showed EC50 of 1.0 µM) correspond well to the values reported in the literature (EC50 of 3.37 µM)Citation50 obtained by performing a similar assay. Likewise, inhibitory concentrations IC50 measured here by fluorogenic enzyme assays (e.g., for GRL0617, the IC50 of 3.2 µM) are comparable to IC50 obtained from the literature (e.g., GRL0617 IC50 of 1.37 − 2.10 µM) for inhibition of protease activityCitation48,Citation62. Furthermore, the measured inhibitory concentrations IC50 for reference inhibitors that showed inhibitory potency in enzymatic assays (PR-619, GRL0617, and GC376) are lower, but comparable to their measured antiviral effective concentrations EC50 (). In the case of the inhibitor PR-619 (C4), we observed the inhibitory effect on both Mpro and PLpro (both protease and deubiquitinase activities), and here we find that inhibitory concentrations IC50 are lower than those obtained from the literatureCitation46. However, such differences are common when comparing results of different enzymatic assays using different substrates and different sources of enzymes. Bis-benzylidene piperidone RA-190 binds directly and covalently to Cys88 of the RPN13 Pru ubiquitin receptor domain in the regulatory particle 19S and inhibits proteasome function, triggering rapid accumulation of polyubiquitinated proteinsCitation70. RA-9 is a non-specific inhibitor that irreversibly inhibits DUBs by exposing its carbonyl group to a nucleophilic attack from the cysteine -SH groupCitation71. Furthermore, the inclusion of remdesivir (C8) in our study was not a random choice. Today, most in vitro efficacy studies have reported positive results on remdesivir (C8) anticoronaviral activityCitation51–54. Therefore, this commercially available compound was included as a positive control for the validation of the method in our studies on infected human cells. Remdesivir did not show any inhibitory effect on Mpro and PLpro of SARS-CoV-2 () since its mechanism of antiviral activity is related to the viral RNA-dependent RNA polymerase. Strangely enough, some authors in the recent literatureCitation72 came to a wrong conclusion based on molecular simulations claiming that remdesivir is an efficient inhibitor of Mpro. In any case, the effective antiviral concentration of remdesivir measured here in cells infected with SARS-CoV-2 (EC50 of 1.8 µM) corresponds well to the data (EC50 of 0.77 µM) reported inCitation54 (). Thus, all these findings collectively validate the methods used in this work to determine the antiviral activities of the BBC.
Table 3. Determined IC50 and EC50 and cytotoxicity of selected reference inhibitors of cysteine proteases.
Determination of cytotoxic effect of studied compounds
To determine the actual cytotoxicity in vitro of the biologically active compounds listed in , we tested cellular viability using MTT assay in multiple cell lines. Additionally, a series of commercially available reference inhibitors listed in were tested in parallel to validate the method. The four model cell lines VERO-E6, Caco-2, PK-15, and A549 were used as described above. Cells were incubated with each compound at concentrations ranging from 0.5 to 30 µM and any possible cytotoxic effect was monitored for 72 h. Novel compounds were dissolved in dimethyl sulfoxide (DMSO), and compounds showing any signs of insolubility were excluded from the analysis. The cytotoxic effect (CTE) is represented here as the concentration that causes a reduction of more than 40% in number of viable cells (Figure S2). Cell viability results were averaged for all screened cell lines and are shown in - and Figure S2. Interestingly, all experimental compounds were relatively well tolerated in selected cell lines, except T97. This was considered cytotoxic even at lower concentrations (starting at approximately 5 µM) in the VERO-E6 cell line. Our findings for T97 (DU-UC15) are truly in line with its previously reported antiproliferative properties in several models, while an analogue of T97 modified for elevated solubility and bioavailability shows anticancer activity in vivoCitation32–35. However, considering also the small CTE of DMSO itself (shown at the end of graphs B-E, Figure S2), compound T97 was excluded from further biological evaluations with live virus. The most pronounced cytotoxicity was observed in the case of the commercial inhibitors PR-619 and RA-190 that exhibited higher CTE at concentrations of approximately 5–10 µM over all cell lines.
Table 4. Determined IC50 and EC50 values of bis(benzylidene)cyclohexanones T97 - T125 and reference inhibitors of proteasome-associated deubiquitinating enzymes RA-9 and RA-190.
These findings correspond very well to previously reported in vitro experiments that showed that PR-619 could decrease cell viability, mainly by inducing caspase3/7-dependent cell apoptosisCitation73. Furthermore, Anchoori reported that compound RA-190 is selectively toxic to cellsCitation70. Although PR-619 and RA-190 appear to be unsuitable as biological probes due to their widespread reactivityCitation30, such compounds could be profoundly cytotoxic to apoptosis-resistant tumour cells. On the other hand, compounds T98, T120, and T121 were better tolerated in the human adenocarcinoma cell line Caco-2 and A549 (Figure S2-E) compared to epithelial kidney VERO-6 and PK-15 cells. In this case, T98 exhibited very mild CTE at concentrations >15 µM and similarly T120 and T121 showed CTE at concentration >10–15 µM.
In conclusion, all compounds that exhibited cytotoxicity only at higher concentrations (CTE >15 µM) or had no observed cytotoxic effect within the screened concentration range (e.g., T105 and T125), were further subjected to antiviral efficacy testing and estimation of EC50 in the presence of live SARS-CoV-2 virus. On the other hand, selectivity index (SI = CTE/EC50) of the most active compounds is low.
Antiviral efficacy of new compounds against live SARS-CoV-2 isolate
To investigate the therapeutic potential of our newly synthesised inhibitors T97, T98, T105, T106, T120-T125, all compounds that passed the cytotoxicity requirements (CTE >15 µM in the VERO-E6 cell line) were tested for inhibition of SARS-CoV-2. EC50 inhibitory concentrations were measured by the in vitro plaque reduction assay in VERO-E6 cells. Known cysteine protease and deubiquitinase inhibitors were included in this study for comparison or as a reference: RA-9Citation30; PR-619 (C4)Citation46,Citation47; RA-190Citation30; remdesivir (C8)Citation51–54; GRL0617 (C9)Citation48; GC376 (C10)Citation49,Citation50.
Typical outcomes measured in such in vitro studies include the amount of virus (e.g. copies of certain ORF, or viral RNA); the number of virus-infected cells in the culture (e.g., number of plaque-forming units, PFU) or changes in viral replication rates. Therefore, in our biological experiment, the reduction in viral titre (in PFU/ml) was monitored and used to evaluate the inhibitory effect (EC50) against the live SAR-CoV-2 virus. For the BBC studied, the following inhibitory concentrations were determined: T98 EC50 = 6.7 µM, T120 EC50 = 3.4 µM, T121 EC50 = 8.4 µM, T122 EC50 = 17.0 µM, and T124 EC50 = 15.9 µM, respectively (, ). Three new compounds T98, T120 and T121 show an inhibitory effect in vitro similar or higher than the reference compounds with similar chemical structures RA-9 and RA-190, as well as GRL0617 (C9) (, Figure S3). The compound T120 showed the lowest EC50 value among the experimental compounds tested and represents bis(benzylidene)cyclohexanone with the highest inhibitory effect on the SARS-CoV-2 virus in vitro, while its CTE remains mild. In comparison, T98 exhibited inhibition of SARS-CoV-2 in the same plaque reduction assay with the EC50 of 6.7 µM, which is approximately 2-fold weaker than T120; however, T98 was better tolerated by model epithelial cells (e.g., in VERO-E6 was CTE >15 µM vs. >10 µM, respectively). Therefore, T98 is also considered in this study a promising compound with the highest inhibitory effect against the SARS-CoV-2 virus in vitro. On the other hand, for the experimental compounds T105, T106, T123 and T125, no antiviral effect was observed within the concentration range (0.1–30.0 µM) tested. Furthermore, the antiviral effect of known reference cysteine protease inhibitors determined in this work corresponds well to the EC50 available in the literature ()Citation49,Citation50,Citation54,Citation74. Although antiviral effect was observed for the six reference inhibitors (), EC50 was successfully determined for four of them: C4, C8, C9, and C10. The last two inhibitors RA-9 and GRL0617 (C9) give EC50 values outside the acceptable R2 range (Figure S3). This was perhaps due to a minimal number of points analysed and/or the concentration scale being out of range.
When comparing the observed EC50 of our three most promising experimental compounds, T98, T120, T121, with the most potent reference inhibitors included in this study, GC376 (C10) and remdesivir (C8) (), the experimental compounds showed in vitro efficiency EC50 against SARS-CoV-2 somewhat inferior, that is, approximately 3-4 times weaker (EC50 of 3.4 − 8.4 µM vs. 1.0 − 1.8 µM, respectively). However, both GC376 (C10)Citation49,Citation50 and remdesivir (C8)Citation51–54 represent a different mechanism of inhibition of the virus than the inhibition of PLpro proposed for the novel compounds. Although remdesivir was used as a positive reference for virus depletion, the GC376 inhibitor was chosen as a reference for viral protease activity.
Finally, the experimental plaque reduction assay correlated with evidence collected from the MTT viability assay, identified 3 (out of 10) new compounds with potent antiviral activity against SARS-CoV-2 in the concentration range as low as EC50 = 3.4 µM.
Determination of inhibitory potencies of bis(benzylidene)cyclohexanones in enzymatic assays
We did not find any reported information on inhibitory concentrations IC50 for reference proteasome-associated deubiquitinating enzyme inhibitors RA-9 and RA-190 measured using enzyme assays towards Mpro or PLpro of SARS-CoV-2, and we did not observe any inhibitory effect for these two inhibitors (IC50 >100 µM, ). The IC50 values of BBC were determined using the same enzymatic assays against Mpro and PLpro of SARS-CoV-2 as were used for the reference inhibitors (). Of the 12 compounds tested, we observed some inhibitory effect (IC50 < 100 µM) in 50% of them (T98, T106, T120, T122, T123, and T125), and . In all cases, the compounds inhibited the protease and deubiquitinase activities of PLpro. Only compound T106 also showed inhibitory potency towards the protease activity of Mpro. All the IC50 values for the compounds tested fall in the range of 35-90 µM, so they were relatively high. The best inhibitory efficiency showed the compound T106 (). However, T106 did not show any inhibitory efficiency (similar to compounds T123 and T125) in in vitro experiments on model epithelial cell lines infected with SARS-CoV-2. Possible reasons for these findings, which may include, for example, different cellular permeabilities of the compounds or a different mechanism of action, are discussed in the following.
Figure 3. Evaluation of antiviral efficacy with calculation of the effective inhibitory concentration EC50 for compounds: T98 (A1, B1), T120 (A2, B2), T121 (A3, B3), T122 (A4, B4) and T124 (A5, B5). Monitoring the trend of decreasing the SARS-CoV-2 virus titre (normalised to untreated control, presented as mean with SEM) with increasing compound concentration (A1-A5). Determination of half-maximal effective concentrations EC50 [M] (B1-B5) from the sigmoidal model with the corresponding regression coefficient R2 (the goodness of fit). Data (B1-B5) are presented as mean and error with SEM.
![Figure 3. Evaluation of antiviral efficacy with calculation of the effective inhibitory concentration EC50 for compounds: T98 (A1, B1), T120 (A2, B2), T121 (A3, B3), T122 (A4, B4) and T124 (A5, B5). Monitoring the trend of decreasing the SARS-CoV-2 virus titre (normalised to untreated control, presented as mean with SEM) with increasing compound concentration (A1-A5). Determination of half-maximal effective concentrations EC50 [M] (B1-B5) from the sigmoidal model with the corresponding regression coefficient R2 (the goodness of fit). Data (B1-B5) are presented as mean and error with SEM.](/cms/asset/f1d0a03d-c7fb-4929-aa28-7a4bd4300842/ienz_a_2301772_f0003_b.jpg)
Molecular modelling of PLpro inhibition
Computational predictions of enzyme-inhibitor interaction energies ( in kcal/mol) of the studied bis(benzylidene)cyclohexanone inhibitors and PLpro from SARS-CoV-2 (), which were based on the model crystal structure of the noncovalent complex PLpro-GRL0617, resulted in the following order of calculated
Footnote3 of the compounds to PLpro:
In the PLpro-inhibitor complexes of compounds T97 - T125, the head group occupied similar positions in the S4 - S3 binding pockets, while most of the tail chains (substituents in the 4-position of the cyclohexanone) were directed towards the catalytic site (S2 - S1). As expected, PLpro with proven DUB activity recognises and preferentially binds thin aliphatic chains ending with a protonated amine group that mimics the side chain of lysine (as in T123, T122, and T98). These three compounds are predicted to form more stable associates with PLpro () than the reference inhibitor GRL0617 (IC50 = 1.37 − 2.10 µM)Citation46,Citation48,Citation62, . The calculated are based on a non-covalent attachment of inhibitors to PLpro disregarding possible subsequent nucleophilic addition of the thiol group of the catalytic Cys111. Therefore, it is not surprising that predicted
of the compounds T98 and T122 are similar.
Figure 4. Left: Molecular surface of the binding site of GRL0617 that occupies the S4 - S3 pockets of the substrate binding cleft S4 - S3 - S2 - S1 of PLpro of SARS-CoV-2. The flexible BL2 loop G266-NYQC-G271 stabilises the bound ligand in its position by closing on the inhibitor (PDB entry 7CMD) Citation62. Right: Binding site of the model of noncovalent complex PLpro - T98. The partially transparent molecular surface of the enzyme illustrates the shape of the pockets of the substrate binding cleft. The amine tail is oriented downward and is not fully visible. Atom colouring scheme: H, white; C, yellow; N, blue; O, red. Only polar hydrogens are shown.
Irreversible inhibitors capable of forming a covalent bond with the catalytic cysteine of PLpro will, according to our computational predictions, display inhibitory potencies related to their ability to undergo nucleophilic attack by the thiol group of Cys111 and consequently inactivate the enzyme, i.e., in the order of decreasing energy of the LUMO orbital of irreversible inhibitors. It was shown that within groups of structurally similar Michael acceptors, relative reactivities correlate with the LUMO energiesCitation76. Based on the DFT calculations, we can rank the BBC according to their LUMO energies (in a. u.):
Compounds T122 and T125 were included in this array for comparison, despite not being Michael acceptors. According to LUMO orbital energies, the most reactive compounds T98 and T123 should be as potent or better irreversible inhibitors as the reference compound RA-190, . In general, molecular modelling suggests that compounds T98, and T123 not only form stable initial noncovalent associates with PLpro but should be potentially able to undergo nucleophilic addition of thiol group of cysteine to form a covalent complex with the PLpro, thus eventually lowering the catalytic efficiency of this enzyme.
On the other hand, the observed enzymatic inhibition by most BBC tested is relatively weak (IC50 values from 40 µM to >100 µM, ), which suggests that PLpro denaturation may have occurred rather than active site binding, as it was recently observed in high-throughput screenings of compound libraries for SARS-CoV-2 PLpro inhibitionCitation77,Citation78
Concluding remarks
In summary, we evaluated the in vitro antiviral efficacy of 10 newly synthesised and 6 known inhibitors against SARS-CoV-2 using the cellular plaque reduction assay and explored the possible mechanism of the antiviral effect of the compounds by performing enzyme inhibition assays of the viral cysteine proteases Mpro and PLpro. Although in vivo animal models are preferred experimental systems for evaluating antiviral efficacy, in vitro testing using mammalian cells is a feasible option for assessing antiviral efficacy when animal models are not readily available. For that, cytotoxic activities were evaluated in various epithelial cell models such as VERO-E6, A549, PK-15, and Caco-2. This study was supplemented with evaluation of the enzymatic activities (deubiquitinase and protease activity) of the selected compounds. Gratifyingly, three compounds T98, T120, and T121 showed a potent antiviral efficacy against SARS‐CoV‐2 (3.4 µM < EC50 < 8.4 µM) in vitro using VERO-E6 cells, while being tolerated by these cells (CTE > 10–15 µM). Furthermore, two of these three compounds (T98 and T120) also exhibited a direct inhibitory effect towards the deubiquitinase and protease activities of PLpro measured by enzyme assays (IC50), however, approximately 10 times weaker than the measured EC50 in the viral assay. Two more compounds T122 and T124 showed acceptable inhibitory potency against SARS-CoV-2 (EC50 of 17.0 and 15.9 µM, respectively) in vitro using the same cells, while being very well tolerated by these cells (CTE > 30 µM). One of these two compounds (T122) exhibited a direct inhibitory effect towards both the deubiquitinase and protease activities of PLpro measured by enzyme assays (IC50), although approximately 4 times weaker than the measured EC50 in the viral assay.
The observed inhibitory activities (IC50 and EC50) towards SARS-CoV-2 do not correlate well with molecular modelling predictions of binding energies () and reactivities (qC* and ELUMO) related to the multistep nucleophilic addition of the catalytic cysteine thiol group. The possible reason is, that molecular models assumed the binding mode of the bis(benzylidene)cyclohexanone headgroup to PLpro similar to the noncovalent inhibitor GRL0617Citation62. Based on the molecular models and calculated binding energies
were able to identify analogs T98, T106, T122, and T123, which then showed the highest inhibitory effect towards the protease activity of PLpro (IC50 = 40 – 60 µM) in the group of studied compounds, but
could not reproduce the relative order of observed activity. Thus, we cannot conclude that BBC fully share the mode of action of GRL0617, which is a potent inhibitor of PLpro (IC50 = 3.2 µM)Citation62. Likewise, the order of LUMO orbital energies does not correlate well with the observed IC50. Therefore, nucleophilic addition is most probably not crucial for the mode of inhibition of PLpro by the compounds studied. Obviously, the mechanism of action of BBC appears to be more complex and different from that considered in molecular modelling predictions, since enzyme assays showed only weak inhibitory potencies of T98 – T125 towards viral proteases Mpro and PLpro. Known irreversible deubiquitinase inhibitors RA-9 and RA-190 that share the structure of bis(benzylidene)cyclohexanone with the compounds tested also did not show an inhibitory effect on Mpro and PLpro (IC50 > 100 µM). In a drug repurposing studyCitation46, Cho et al. experimentally screened libraries of deubiquitinase and cysteine protease inhibitors containing BBC on their inhibition of PLpro. They found that only one of them NSC63283 showed poor deubiquitinase activity (IC50 > 100 µM). It is, in fact, well known that Michael acceptors of this class can react with a variety of biological thiolsCitation30. Comparable inhibitory potencies in vitro of compounds T98 (EC50 = 6.7 µM) and T122 (EC50 = 17.0 µM) suggest that the reaction of the 1,5-diaryl-3-oxo-1,4-pentadienyl motif with the catalytic Cys111 thiol group is not essential for the observed antiviral effect in virus-infected VERO-E6 cells, as T122 is not a Michael acceptor. Within the homologous series T97, T98, T105, T106, T120, T121, and T124, which differ only in substituent in the 4-position of cyclohexanone, the antiviral effectiveness ranges from EC50 = 3.4 µM to EC50 > 30 µM, while inhibition of the cysteine protease activity of PLpro and Mpro is weak (IC50 > 100 µM, except for T106). Therefore, we must conclude that, in analogy to RA-9 and RA-190, antiviral activity is mainly due to interactions with other targetsCitation35. The reference inhibitors included in our study PR-619, GRL0617, GC376 showed in our experiment inhibitory potencies IC50 corresponding to the data published in the literatureCitation46,Citation48,Citation50,Citation62,Citation73,Citation75. Therefore, it could be hypothesised that the 4-substituent group plays a specific role in inhibiting viral replication in cells by affecting replication pathways or pharmacological targets other than cysteine proteases. The important role of the 4-substituent is also supported by the similar antiviral activity of T122 vs. T98 and T97 vs. T125, but also by the loss of inhibitory activity (EC50 > 30 µM) when the 4-substituent group becomes too bulky (T105) or too long (T106). Compound T122 has moderate antiviral and inhibitory activities while T125 shows only moderate inhibitory activity and no antiviral activity. This reinforces the conclusion that there is no relationship between in vitro antiviral activity and the PLpro and Mpro activity of these compounds.
Although potent inhibition in an enzyme assay cannot guarantee antiviral activity in cell-based assays (nor in animal studies), we accessed both evaluations to determine antiviral efficiency of the new compounds and explore their mechanism of action. Measured inhibitory potencies (EC50 < IC50) were thus influenced by a complex interplay of multiple factors in the cellular environment, which includes pharmacological target(s) other than PLpro and Mpro, metabolic activation of the tested compounds, target location, structural complexity of cells, transport mechanisms, and others. Therefore, it is important to measure both IC50 and EC50 values to fully understand the pharmacological profile of a novel compound, as they provide complementary information about the compound’s potency and efficacy.
It is noteworthy that all our active compounds exhibited effective in vitro inhibitory concentrations EC50 comparable to those of commercially available inhibitors having similar chemical structures (e.g., PR-619, RA-9, RA-190) as well as to those of approved FDA drugs (e.g., remdesivir C8). In general, our study has identified new chemical entities, antiviral compounds as possible candidates for further in vivo testing and for eventual advancement to preclinical development.
Supplemental Material
Download MS Word (658.9 KB)Acknowledgements
The financial support provided by the Slovak Research and Development Agency (APVV) and the Research Grant Agency of the Ministry of Education and the Slovak Academy of Sciences (VEGA), grants PP-COVID-20–0010 (main source), APVV-19–0376, VEGA-02/0026/22, VEGA 1/0223/20 and APVV-21–0108, are gratefully acknowledged.
For providing access to the SARS-CoV-2 strain Slovakia/SK-BMC5/2020, we thank the EVA GLOBAL consortium (funded by the European Union’s Horizon 2020 research and innovation program under grant agreement No. 871029, https://www.european-virus-archive.com/virus/sars-cov-2-strain-slovakiask-bmc52020-fd).
Notes
1 Interferon-stimulated gene 15 (ISG15) is a secreted protein that covalently links to lysine residues on newly synthesized proteins. The effects of protein ISGylation involve activation and inhibition of antiviral immunity.
2 LUMO - lowest unoccupied molecular orbital.
3 More negative value of means stronger binding of the ligand to PLpro.
References
- Corman VM, Lienau J, Witzenrath M. Coronaviruses as the cause of respiratory infections. Internist (Berl)). 2019;60(11):1136–1145.
- Fehr AR, Channappanavar R, Perlman S. Middle east respiratory syndrome: Emergence of a pathogenic human coronavirus. Annu Rev Med. 2017;68(1):387–399.
- Volz E, Hill V, McCrone JT, Price A, Jorgensen D, O'Toole A, Southgate J, Johnson R, Jackson B, Nascimento FF, et al. Evaluating the effects of sars-cov-2 spike mutation d614g on transmissibility and pathogenicity. Cell. 2021;184(1):64–75 e11.
- Saputri DS, Li S, van Eerden FJ, Rozewicki J, Xu Z, Ismanto HS, Davila A, Teraguchi S, Katoh K, Standley DM. Flexible, functional, and familiar: Characteristics of sars-cov-2 spike protein evolution. Front Microbiol. 2020;11:2112.
- Khateeb J, Li Y, Zhang H. Emerging sars-cov-2 variants of concern and potential intervention approaches. Crit Care. 2021;25(1):244.
- Cohen J. Omicron sparks a vaccine strategy debate. Science. 2021;374(6575):1544–1545.
- Nepali K, Sharma R, Sharma S, Thakur A, Liou JP. Beyond the vaccines: A glance at the small molecule and peptide-based anti-covid19 arsenal. J Biomed Sci. 2022;29(1):65.
- Painter WP, Holman W, Bush JA, Almazedi F, Malik H, Eraut N, Morin MJ, Szewczyk LJ, Painter GR. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad-spectrum oral antiviral agent with activity against sars-cov-2. Antimicrob Agents Chemother. 2021;65(5):e02428–02420.
- Owen DR, Allerton CMN, Anderson AS, Aschenbrenner L, Avery M, Berritt S, Boras B, Cardin RD, Carlo A, Coffman KJ, et al. An oral sars-cov-2 m(pro) inhibitor clinical candidate for the treatment of covid-19. Science. 2021;374(6575):1586–1593.
- Ulrich L, Halwe NJ, Taddeo A, Ebert N, Schön J, Devisme C, Trüeb BS, Hoffmann B, Wider M, Fan X, et al. Enhanced fitness of sars-cov-2 variant of concern alpha but not beta. Nature. 2022;602(7896):307–313.
- Moghadasi SA, Heilmann E, Khalil AM, Nnabuife C, Kearns FL, Ye C, Moraes SN, Costacurta F, Esler MA, Aihara H, et al. Transmissible sars-cov-2 variants with resistance to clinical protease inhibitors. Sci Adv. 2023;9(13):eade8778.
- Vangeel L, Chiu W, De Jonghe S, Maes P, Slechten B, Raymenants J, André E, Leyssen P, Neyts J, Jochmans D. Remdesivir, molnupiravir and nirmatrelvir remain active against sars-cov-2 omicron and other variants of concern. Antiviral Res. 105252;198:105252.
- Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, et al. Papain-like protease regulates sars-cov-2 viral spread and innate immunity. Nature. 2020;587(7835):657–662.
- Li G, De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-ncov). Nat Rev Drug Discov. 2020;19(3):149–150.
- Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, Qi F, Bao L, Du L, Liu S, et al. Inhibition of sars-cov-2 (previously 2019-ncov) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30(4):343–355.
- Marzolini C, Kuritzkes DR, Marra F, Boyle A, Gibbons S, Flexner C, Pozniak A, Boffito M, Waters L, Burger D, et al. Recommendations for the management of drug-drug interactions between the covid-19 antiviral nirmatrelvir/ritonavir (paxlovid) and comedications. Clin Pharmacol Ther. 2022;112(6):1191–1200.
- Lamb YN. Nirmatrelvir plus ritonavir: First approval. Drugs. 2022;82(5):585–591.
- Iketani S, Mohri H, Culbertson B, Hong SJ, Duan Y, Luck MI, Annavajhala MK, Guo Y, Sheng Z, Uhlemann AC, et al. Multiple pathways for sars-cov-2 resistance to nirmatrelvir. Nature. 2023;613(7944):558–564.
- Hu Y, Lewandowski EM, Tan H, Zhang X, Morgan RT, Zhang X, Jacobs LMC, Butler SG, Gongora MV, Choy J, et al. Naturally occurring mutations of sars-cov-2 main protease confer drug resistance to nirmatrelvir. ACS Cent Sci. 2023;9(8):1658–1669.
- Ullrich S, Sasi VM, Mahawaththa MC, Ekanayake KB, Morewood R, George J, Shuttleworth L, Zhang X, Whitefield C, Otting G, et al. Challenges of short substrate analogues as sars-cov-2 main protease inhibitors. Bioorg Med Chem Lett. 2021;50(:128333.
- Klemm T, Ebert G, Calleja DJ, Allison CC, Richardson LW, Bernardini JP, Lu BG, Kuchel NW, Grohmann C, Shibata Y, et al. Mechanism and inhibition of the papain-like protease, plpro, of sars-cov-2. Embo J. 2020;39(18):e106275.
- Rut W, Lv Z, Zmudzinski M, Patchett S, Nayak D, Snipas SJ, El Oualid F, Huang TT, Bekes M, Drag M, et al. Activity profiling and crystal structures of inhibitor-bound sars-cov-2 papain-like protease: A framework for anti-covid-19 drug design. Sci Adv. 2020;6(42):eabd4596.
- Freitas BT, Durie IA, Murray J, Longo JE, Miller HC, Crich D, Hogan RJ, Tripp RA, Pegan SD. Characterization and noncovalent inhibition of the deubiquitinase and deisgylase activity of sars-cov-2 papain-like protease. ACS Infect Dis. 2020;6(8):2099–2109.
- Sulea T, Lindner HA, Purisima EO, Ménard R. Deubiquitination, a new function of the severe acute respiratory syndrome coronavirus papain-like protease? J Virol. 2005;79(7):4550–4551.
- Báez-Santos YM, St John SE, Mesecar AD. The sars-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115(:21–38.
- Balakirev MY, Jaquinod M, Haas AL, Chroboczek J. Deubiquitinating function of adenovirus proteinase. J Virol. 2002;76(12):6323–6331.
- Shen Z, Ratia K, Cooper L, Kong D, Lee H, Kwon Y, Li Y, Alqarni S, Huang F, Dubrovskyi O, et al. Design of sars-cov-2 plpro inhibitors for covid-19 antiviral therapy leveraging binding cooperativity. J Med Chem. 2022;65(4):2940–2955.
- Calleja DJ, Lessene G, Komander D. Inhibitors of sars-cov-2 plpro. Front Chem. 2022;10:876212.
- Tan H, Hu Y, Jadhav P, Tan B, Wang J. Progress and challenges in targeting the sars-cov-2 papain-like protease. J Med Chem. 2022;65(11):7561–7580.
- Bazzaro M, Linder S. Dienone compounds: Targets and pharmacological responses. J Med Chem. 2020;63(24):15075–15093.
- Das U, Sharma RK, Dimmock JR. 1,5-diaryl-3-oxo-1,4-pentadienes: A case for antineoplastics with multiple targets. Curr Med Chem. 2009;16(16):2001–2020.
- Cersosimo U, Sgorbissa A, Foti C, Drioli S, Angelica R, Tomasella A, Picco R, Semrau MS, Storici P, Benedetti F, et al. Synthesis, characterization, and optimization for in vivo delivery of a nonselective isopeptidase inhibitor as new antineoplastic agent. J Med Chem. 2015;58(4):1691–1704.
- Tomasella A, Picco R, Ciotti S, Sgorbissa A, Bianchi E, Manfredini R, Benedetti F, Trimarco V, Frezzato F, Trentin L, et al. The isopeptidase inhibitor 2cpe triggers proteotoxic stress and atm activation in chronic lymphocytic leukemia cells. Oncotarget. 2016;7(29):45429–45443.
- Iuliano L, Drioli S, Pignochino Y, Cafiero CM, Minisini M, D'Este F, Picco R, Dalla E, Giordano G, Grignani G, et al. Enhancing proteotoxic stress in leiomyosarcoma cells triggers mitochondrial dysfunctions, cell death, and antitumor activity in vivo. Mol Cancer Ther. 2021;20(6):1039–1051.
- Ciotti S, Sgarra R, Sgorbissa A, Penzo C, Tomasella A, Casarsa F, Benedetti F, Berti F, Manfioletti G, Brancolini C. The binding landscape of a partially-selective isopeptidase inhibitor with potent pro-death activity, based on the bis(arylidene)cyclohexanone scaffold. Cell Death Dis. 2018;9(2):184.
- Anand K, Ziebuhr J, Wadhwani P, Mesters JR, Hilgenfeld R. Coronavirus main proteinase (3clpro) structure: Basis for design of anti-sars drugs. Science. 2003;300(5626):1763–1767.
- Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, Xie X, Jin Z, Peng J, Liu F, et al. Structure-based design of antiviral drug candidates targeting the sars-cov-2 main protease. Science. 2020;368(6497):1331–1335.
- Jin W, Zhang W, Mitra D, McCandless MG, Sharma P, Tandon R, Zhang F, Linhardt RJ. The structure-activity relationship of the interactions of sars-cov-2 spike glycoproteins with glucuronomannan and sulfated galactofucan from saccharina japonica. Int J Biol Macromol. 2020;163:1649–1658.
- Li J, Zhou X, Zhang Y, Zhong F, Lin C, McCormick PJ, Jiang F, Luo J, Zhou H, Wang Q, et al. Crystal structure of sars-cov-2 main protease in complex with the natural product inhibitor shikonin illuminates a unique binding mode. Sci Bull (Beijing). 2021;66(7):661–663.
- Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of sars-cov-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368(6489):409–412.
- Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, Maruyama Y, Taoda Y, Kasamatsu K, Suto T, et al. Discovery of s-217622, a noncovalent oral sars-cov-2 3cl protease inhibitor clinical candidate for treating covid-19. J Med Chem. 2022;65(9):6499–6512.
- Sharma A, Kaliya K, Maurya SK. Recent advances in the discovery of potent proteases inhibitors targeting the sars coronaviruses. Curr Top Med Chem. 2021;21(4):307–328.
- Su H, Zhou F, Huang Z, Ma X, Natarajan K, Zhang M, Huang Y, Su H. Molecular insights into small-molecule drug discovery for sars-cov-2. Angew Chem Int Ed Engl. 2021;60(18):9789–9802.
- Sung H, Kang SH, Bae YJ, Hong JT, Chung YB, Lee CK, Song S. Pcr-based detection of mycoplasma species. J Microbiol. 2006;44(1):42–49.
- Ogawa M, Uchiyama T, Satoh M, Ando S. Decontamination of mycoplasma-contaminated orientia tsutsugamushi strains by repeating passages through cell cultures with antibiotics. BMC Microbiol. 2013;13(1):32.
- Cho CC, Li SG, Lalonde TJ, Yang KS, Yu G, Qiao Y, Xu S, Ray Liu W. Drug repurposing for the sars-cov-2 papain-like protease. ChemMedChem. 2022;17(1):e202100455.
- Cowell IG, Ling EM, Swan RL, Brooks MLW, Austin CA. The deubiquitinating enzyme inhibitor pr-619 is a potent DNA topoisomerase ii poison. Mol Pharmacol. 2019;96(5):562–572.
- Fu Z, Huang B, Tang J, Liu S, Liu M, Ye Y, Liu Z, Xiong Y, Zhu W, Cao D, et al. The complex structure of grl0617 and sars-cov-2 plpro reveals a hot spot for antiviral drug discovery. Nat Commun. 2021;12(1):488.
- Hu Y, Ma C, Szeto T, Hurst B, Tarbet B, Wang J. Boceprevir, calpain inhibitors ii and xii, and gc-376 have broad-spectrum antiviral activity against coronaviruses. ACS Infect Dis. 2021;7(3):586–597.
- Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, et al. Boceprevir, gc-376, and calpain inhibitors ii, xii inhibit sars-cov-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692.
- Agostini ML, Andres EL, Sims AC, Graham RL, Sheahan TP, Lu X, Smith EC, Case JB, Feng JY, Jordan R, et al. Coronavirus susceptibility to the antiviral remdesivir (gs-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9(2):e00221–00218.
- Pruijssers AJ, George AS, Schäfer A, Leist SR, Gralinksi LE, Dinnon KH, Yount BL, Agostini ML, Stevens LJ, Chappell JD, et al. Remdesivir inhibits sars-cov-2 in human lung cells and chimeric sars-cov expressing the sars-cov-2 rna polymerase in mice. Cell Rep. 2020;32(3):107940.
- Wang J, Reiss K, Shi Y, Lolis E, Lisi GP, Batista VS. Mechanism of inhibition of the reproduction of sars-cov-2 and ebola viruses by remdesivir. Biochemistry. 2021;60(24):1869–1875.
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-ncov) in vitro. Cell Res. 2020;30(3):269–271.
- Zupin L, Fontana F, Gratton R, Milani M, Clemente L, Pascolo L, Ruscio M, Crovella S. Sars-cov-2 short-time infection produces relevant cytopathic effects in vero e6 cell line. Int J Environ Res Public Health. 2021; 18(17):9020–9027.
- Hidalgo IJ, Raub TJ, Borchardt RT. Characterization of the human colon carcinoma cell line (caco-2) as a model system for intestinal epithelial permeability. Gastroenterology. 1989;96(2):736–749.
- Zheng G, Li LF, Zhang Y, Qu L, Wang W, Li M, Yu S, Zhou M, Luo Y, Sun Y, et al. Mertk is a host factor that promotes classical swine fever virus entry and antagonizes innate immune response in pk-15 cells. Emerg Microbes Infect. 2020;9(1):571–581.
- Foster KA, Oster CG, Mayer MM, Avery ML, Audus KL. Characterization of the a549 cell line as a type ii pulmonary epithelial cell model for drug metabolism. Exp Cell Res. 1998;243(2):359–366.
- De Madrid AT, Porterfield JS. A simple micro-culture method for the study of group b arboviruses. Bull World Health Organ. 1969;40(1):113–121.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1-2):55–63.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242.
- Gao X, Qin B, Chen P, Zhu K, Hou P, Wojdyla JA, Wang M, Cui S. Crystal structure of sars-cov-2 papain-like protease. Acta Pharm Sin B. 2021;11(1):237–245.
- Sanders BC, Pokhrel S, Labbe AD, Mathews II, Cooper CJ, Davidson RB, Phillips G, Weiss KL, Zhang Q, O'Neill H, et al. Potent and selective covalent inhibition of the papain-like protease from sars-cov-2. Nat Commun. 2023;14(1):1733.
- Sapse AM, Schweitzer BS, Dicker AP, Bertino JR, Frecer V. Ab initio studies of aromatic-aromatic and aromatic-polar interactions in the binding of substrate and inhibitor to dihydrofolate reductase. Int J Pept Protein Res. 1992;39(1):18–23.
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196.
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, et al. Opls3: A force field providing broad coverage of drug-like small molecules and proteins. J Chem Theory Comput. 2016;12(1):281–296.
- Frecer V, Májeková M, Miertuš S. Approximate methods for solvent effect calculations on biomolecules. Journal of Molecular Structure (Theochem). 1989;183(3-4):403–419.
- Mulliken DK. The protean hydatid mole; report of a case. Obstet Gynecol. 1955;6(2):219–220.
- Geerlings P, De Proft F, Langenaeker W. Conceptual density functional theory. Chem Rev. 2003;103(5):1793–1873.
- Anchoori RK, Karanam B, Peng S, Wang JW, Jiang R, Tanno T, Orlowski RZ, Matsui W, Zhao M, Rudek MA, et al. A bis-benzylidine piperidone targeting proteasome ubiquitin receptor rpn13/adrm1 as a therapy for cancer. Cancer Cell. 2013;24(6):791–805.
- Issaenko OA, Amerik AY. Chalcone-based small-molecule inhibitors attenuate malignant phenotype via targeting deubiquitinating enzymes. Cell Cycle. 2012;11(9):1804–1817.
- Nguyen HL, Thai NQ, Truong DT, Li MS. Remdesivir strongly binds to both rna-dependent rna polymerase and main protease of sars-cov-2: Evidence from molecular simulations. J Phys Chem B. 2020;124(50):11337–11348.
- Lavaud M, Mullard M, Tesfaye R, Amiaud J, Legrand M, Danieau G, Brion R, Morice S, Regnier L, Dupuy M, et al. Overexpression of the ubiquitin specific proteases usp43, usp41, usp27x and usp6 in osteosarcoma cell lines: Inhibition of osteosarcoma tumor growth and lung metastasis development by the usp antagonist pr619. Cells. 2021;10(9):2268–2283.
- Pizzorno A, Padey B, Dubois J, Julien T, Traversier A, Dulière V, Brun P, Lina B, Rosa-Calatrava M, Terrier O. In vitro evaluation of antiviral activity of single and combined repurposable drugs against sars-cov-2. Antiviral Res. 104878;181:104878.
- Fu L, Ye F, Feng Y, Yu F, Wang Q, Wu Y, Zhao C, Sun H, Huang B, Niu P, et al. Both boceprevir and gc376 efficaciously inhibit sars-cov-2 by targeting its main protease. Nat Commun. 2020;11(1):4417.
- Zhuo L-G, Liao W, Yu Z-X. A frontier molecular orbital theory approach to understanding the mayr equation and to quantifying nucleophilicity and electrophilicity by using homo and lumo energies. Asian J Org Chem. 2012;1(4):336–345.
- Ma CL, Wang J. Validation and invalidation of sars-cov-2 papain-like protease inhibitors. ACS Pharmacol Transl Sci. 2022;5(2):102–109.
- Brewitz L, Kamps J, Lukacik P, Strain-Damerell C, Zhao Y, Tumber A, Malla TR, Orville AM, Walsh MA, Schofield CJ. Mass spectrometric assays reveal discrepancies in inhibition profiles for the sars-cov-2 papain-like protease. ChemMedChem. 2022;17(9):e202200016.