
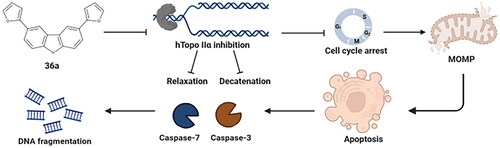
Abstract
Human DNA topoisomerases are essential for crucial cellular processes, including DNA replication, transcription, chromatin condensation, and maintenance of its structure. One of the significant strategies employed in cancer treatment involves the inhibition of a specific type of topoisomerase, known as topoisomerase II (Topo II). Carbazole derivatives, recognised for their varied biological activities, have recently become a significant focus in oncological research. This study assesses the efficacy of three symmetrically substituted carbazole derivatives: 2,7-Di(2-furyl)-9H-carbazole (27a), 3,6-Di(2-furyl)-9H-carbazole (36a), and 3,6-Di(2-thienyl)-9H-carbazole (36b) – as anticancer agents. Among investigated carbazole derivatives, compound 3,6-di(2-furyl)-9H-carbazole bearing two furan moieties emerged as a novel catalytic inhibitor of Topo II. Notably, 3,6-di(2-furyl)-9H-carbazole effectively selectively inhibited the relaxation and decatenation activities of Topo IIα, with minimal effects on the IIβ isoform. These findings underscore the potential of compound 3,6-Di(2-furyl)-9H-carbazole as a promising lead candidate warranting further investigation in the realm of anticancer drug development.
Graphical Abstract
Keywords:
Introduction
Cancer, a complex disease that progresses through multiple stages, is influenced by global population growth, ageing, and advances in economic development. According to the World Health Organization, cancer stands as the second leading cause of mortality in developing countries, following cardiovascular diseases. It is also believed that some types of cancers are modern civilisation diseases, which depend on lifestyle. The contemporary treatment of cancer involves a variety of therapeutic methods, including surgery, radiotherapy, immunotherapy, and commonly used chemotherapyCitation1–3. Despite the rising rates of cancer and the growing problem of drug resistance, chemotherapy with a use of small molecules continues to be the primary strategy for addressing this diseaseCitation4. To address these challenges in combating cancer, we present research on the concept of symmetrically substituted carbazole derivatives as potential agents in cancer treatment.
The carbazole scaffold is already known to play a crucial role as a fundamental structural element in numerous biologically active compounds. These compounds include both natural and synthetic anticancer agentsCitation5. The anticancer mechanism of action of some unsymmetrical carbazole derivatives comprises cell cycle arrestCitation6, intercalation into DNACitation7, inhibition of human topoisomerasesCitation8,Citation9, mitochondria disruptionCitation10, and apoptosis inductionCitation11. It is noteworthy that some modified carbazole-skeleton segments of anticancer agents, such as alectinib and midostaurin, have already received approval from the US Food and Drug Administration (FDA) for use in chemotherapyCitation12. These successes underscore the potential of carbazole scaffolds in the pursuit of new and effective anticancer drug candidates.
Some compounds possessing the carbazole scaffold act as topoisomerase II (Topo II) inhibitors. Elliptinium (also known as celiptium), introduced in 1983, has demonstrated effectiveness as an efficient antineoplastic drug for the treatment of metastatic breast cancer. It exerts its action by poisoning Topo II and intercalating into DNA ()Citation13. Depending on the structural modifications of carbazole, new derivatives are continuously being reported. They can act as either poisons ()Citation14 or catalytic inhibitors ()Citation9 of Topo II. Consequently, our investigations into symmetrically substituted carbazoles, a new idea of the scaffold, were focused on confirming whether these compounds can interact with topoisomerases.
Figure 1. Chemical structures of Topo II inhibitors based on the carbazole scaffold (a–c), and symmetrically substituted carbazoles containing furan and thiophene (d).

In this study, we have designed three symmetrically substituted carbazoles with a focus on their chemical synthetic availability and the incorporation of substituents (furan and thiophene). Previous research suggests that these modifications, when integrated into the core structure (), could potentially confer biological activity. It is known that furan and thiophene motifs act as pharmacologically active pharmacophores, and compounds containing these rings have garnered increasing attention due to their promising anticancer activityCitation15,Citation16. Notably, such compounds can be found as FDA-approved anticancer medications, including lapatinibCitation17 and raltitrexedCitation18. Our research can be seen as an initial idea to test the hypothesis regarding the potential of symmetrically substituted carbazoles as anticancer agents. Consequently, we have designed and synthesised only three compounds: 2,7-di(2-furyl)-9H-carbazole (27a), 3,6-di(2-furyl)-9H-carbazole (36a), and 3,6-di(2-thienyl)-9H-carbazole (36b) to gain preliminary insights into their activities.
The human genome encodes for six topoisomerases (Topo 1, Topo1mt, Topo IIα, Topo IIβ, Topo IIIα, and Topo IIIβ), which play essential roles in catalysing topological changes in both nuclear and mitochondrial DNACitation19. These enzymes function by alleviating unwanted tensions that arise during DNA replication or transcription, involving the creation of transitional single-stranded breaks by type I topoisomerases or double-stranded breaks in the DNA double helix by type II topoisomerasesCitation20. In both cases, the process relies on the formation of a transient enzyme-DNA adduct known as the Topo cleavage complex (Topo cc). Given the uncontrolled proliferation of cancer cells, topoisomerases have emerged as important targets for numerous anticancer drugsCitation21.
Topo II exists in two isoforms, Topo IIα and Topo IIβ, both of which require the presence of Mg2+ ions and ATP hydrolysis to carry out their functions. Drugs targeting Topo II can be classified into two broad groups based on their mechanism of action. The first group consists of compounds known as Topo II poisons, which stabilise the covalent complex of Topo II with DNA, leading to increased levels of DNA damage in cells and ultimately triggering apoptosisCitation22. These poisons can further be categorised based on their ability to bind to DNA: those that do not intercalate with DNA, such as etoposide (ETP) and teniposide, and those that do intercalate, including doxorubicin (DOXO), amsacrine (m-AMSA), and mitoxantroneCitation23.
The second category of agents includes catalytic inhibitors that target the critical enzymatic activity of Topo II. These inhibitors induce cytotoxic effects without significantly increasing DNA damage, achieved by stabilising the Topo II/DNA complex. Catalytic inhibitors exert their effects through various mechanisms, such as preventing ATP hydrolysis (e.g. ICRF-187, ICRF-193), competing for the ATP binding site (e.g. novobiocin), or preventing DNA cleavage (e.g. merbarone)Citation20,Citation24. Collectively, targeting the functions of Topo II represents a systematic approach in oncology therapy, providing potential avenues for developing effective treatments for cancerCitation25.
In our study, we conduct a biological evaluation of symmetrically substituted carbazole derivatives containing furan or thiophene (). The novelty of our approach involves the symmetrical substitution and investigation of these compounds’ potential anticancer properties. Our assessment is based on existing literature reports that highlight carbazole’s activity towards Topo II and the potential impact of furan or thiophene presence on increased anticancer activity. The primary focus of the evaluation lies in the molecular pharmacology of three derivatives, specifically examining their inhibitory activity against human Topo II. Additionally, we explore their potential as anticancer agents across various cancer cell lines, encompassing analysis of their cytotoxic, antiproliferative, and proapoptotic properties. To the best of our knowledge, such study of symmetrical carbazole derivatives has never been reported elsewhere. Moreover, our results presented in this work might be crucial for other scientists and shed light on cancer treatment based on this scaffold.
Synthesis of compounds
The three compounds under investigation are as follows: 2,7-di(2-furyl)-9H-carbazole (27a), 3,6-di(2-furyl)-9H-carbazole (36a), and 3,6-di(2-thienyl)-9H-carbazole (36b) (). These compounds were synthesised, purified, and subjected to physiochemical characterisation before being tested in topoisomerase assays.
Figure 2. The synthetic routes of the three carbazole derivatives (27a, 36a, 36b) (a). The structures of compounds 27a, 36a, and 36b, including atom numbering, are displayed to facilitate the interpretation of NMR spectra (b).

The synthesis of the two carbazole derivatives, 27a and 36a, was carried out according to the protocol outlined by Oğuztürk et al.Citation26, while the synthesis of compound 36b was described by Damit et al.Citation27. Compounds 36a and 36b, which contain the 3,6-locants in the carbazole skeleton, differ by the heteroatoms in the substituent rings. In 36a, the oxygen atoms in the heterocyclic substituents (furyl) were replaced by sulphur atoms (thienyl) to create the 36b carbazole derivative. Interestingly, compounds 27a and 36a are constitutional isomers, with substituents located in the 2,7 and 3,6 positions of the carbazole skeleton, respectively.
Biological studies
Carbazole derivatives inhibit human Topo II
To investigate the potential of carbazole derivatives as Topo II inhibitors, we conducted a pBR322 DNA relaxation assay in the presence of Topo IIα/IIβ. As illustrated in , the tested compounds displayed concentration-dependent activity against both Topo II isoforms. Notably, compound 36a fully inhibited DNA relaxation in the presence of Topo IIα at the highest concentrations (20–100 µM), yet did not impact Topo IIβ activity. These results suggest that compound 36a selectively inhibits the relaxation activity of Topo IIα. Moreover, compounds 36b and 27a moderately affected the functionality of both Topo IIα and Topo IIβ, inhibiting the relaxation of supercoiled DNA at the highest tested concentrations. In contrast, ETP and ICRF-187 completely inhibited relaxation activity towards Topo IIα, but exerted relatively lower inhibitory effects against Topo IIβ compared to the negative control ().
Figure 3. Inhibition of Topo IIα (a) and IIβ (b) mediated pBR322 relaxation. The experiment was carried out either with Topo IIα/IIβ in the presence of solvent (DMSO IIα/IIβ (+)) or with various concentrations of carbazole derivatives. ETP (100 µM), and ICRF-187 (100 µM) were used as references. (c) Unwinding assay. Determination of the ability of carbazole derivatives to intercalate into DNA in the presence of topoisomerase I (Top I). ETP and DOXO were used as negative and positive controls, respectively. The displayed gels have been cropped for clarity; full-length gels can be found in Supplementary Figures S1–S3.

To explore the intercalating properties of the tested compounds, we performed a DNA unwinding assay using Topo I and relaxed pBR322 DNA as substrates. ETP, a non-intercalating agent, and DOXO, an intercalating agent, served as controls. The results, shown in , indicate that the carbazole derivatives could not convert relaxed plasmid into supercoiled DNA form in the presence of Topo I. These findings strongly suggest that compounds 36a, 36b, and 27a act as non-intercalating topoisomerase inhibitors, pointing to a distinct mechanism of action from intercalating agents such as DOXO.
Carbazole derivatives are not Topo IIα poisons
Topo II plays a crucial role in decatenation, a process required to separate catenated DNA duplexes at the end of replicationCitation28. To evaluate the capacity of carbazole derivatives to inhibit the catalytic decatenation activity of Topo II, we performed electrophoretic separation experiments using highly knotted circular kDNA and the respective Topo II isoforms ().
Figure 4. Inhibition of Topo IIα/IIβ mediated kDNA decatenation by carbazole derivatives (a, b). DNA cleavage assay in the presence of 36a, 36b, and 27a, respectively (c, d). ETP (100 µM) and ICRF-187 (100 µM) were used as references. The displayed gels have been cropped for clarity; full-length gels can be found in Supplementary Figures S4–S7.

In the absence of Topo II, the high molecular weight kDNA remained stationary in the well of the negative control. Compound 36a demonstrated effective and concentration-dependent inhibition of Topo IIα-mediated decatenation across the concentration range of 4–100 µM (). In contrast, compounds 36b and 27a either completely inhibited decatenation only at the highest concentration tested, or partially inhibited the decatenation of higher-order catenates containing two, three, four, or more minicircles (). These catenates migrated slower through the gel compared to the decatenated kDNA.
To thoroughly assess the inhibitory effects of the compounds on both Topo II isoforms, we conducted a decatenation assay using Topo IIβ. Consistent with the results obtained from the relaxation test (), 36a displayed significantly lower inhibitory activity against Topo IIβ (), partially inhibiting decatenation only at the highest concentration tested. In comparison, compound 36b showed similar levels of activity against both Topo IIα and Topo IIβ. Interestingly, 27a completely inhibited decatenation in the presence of Topo IIβ at a concentration of 100 µM (). It is noteworthy that ICRF-187, known for reducing the catalytic activity of Topo II, showed a more significant effect than ETP, which stabilises the cleavage complex of DNA/Topo II.
To determine if carbazole derivatives could be classified as Topo II poisons, we performed a DNA cleavage assay using Topo IIα and pBR322 plasmid. The results, as shown in , demonstrate that only ETP, a well-known Topo II poison, induced a significant amount of linear plasmid visible on the gel. This suggests that compounds 36a, 36b, and 27a do not stabilise the covalent cleavage complex formed between Topo II and DNA, unlike the effects of ETP. To further confirm this, we repeated the cleavage assay in the presence of ETP to establish whether 36a could inhibit Topo IIα similarly to ICRF-187, by stabilising the non-covalent Topo II/DNA complex after the Topo II/DNA cleavage complex had been induced by ETP treatment (). Co-treatment with 36a reduced the level of linear plasmid compared to ETP and ICRF-187, indicating that 36a prevented the formation of the ETP-induced DNA cleavage in reaction with Topo IIα. Further investigation is necessary to identify the specific step or steps of the Topo II catalytic cycle that 36a impacts.
Carbazoles display a potent cytotoxic effect and inhibit the capability to form colonies
The antiproliferative activity of the carbazole derivatives under study was examined. These compounds were tested at different concentrations over a 72-h period using five distinct human cancer cell lines, specifically A549 (non-small lung cancer), HCT-116 (colon cancer), MCF-7 (breast cancer), U-2 OS (bone cancer), U-87 MG (brain cancer), and a non-malignant human embryonic kidney cell line, HEK293. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was employed to determine the IC50 concentration for each compound in comparison to cells treated with 1% v/v dimethyl sulphoxide (DMSO). The carbazole derivatives demonstrated high antiproliferative efficacy against all tested cancer cell lines (). 27a exhibited the highest activity, with an IC50 below 1 µM for all investigated cell lines, including HEK293. 36b displayed similar growth inhibition activity to most of the studied cell lines as 27a, except for U-87 MG cells where the IC50 was 1.40 ± 0.24 µM. Compound 36a exhibited the highest and lowest cytotoxicity against HCT-116 and U87-MG cell lines, with IC50 values of 0.48 ± 0.06 µM and 2.19 ± 0.30 µM, respectively. The effect of 36a on HEK293 cells was comparable to the IC50 value of ETP, used as a reference.
Table 1. In vitro growth inhibitory activity (IC50 ± SD, µM) of 27a, 36a, and 36bTable Footnotea.
To further investigate the impact of carbazole derivatives on cancer cell growth, A549 and HCT-116 cells were subjected to a clonogenic assay. The results demonstrated that treatment with carbazole derivatives significantly reduced the number of colony-forming cells compared to control cells treated with DMSO (). In particular, treatment with compounds 27a and 36a reduced the colony number in both tested cell lines in a concentration-dependent manner, while 36b demonstrated lower inhibitory activity against the A549 cell line (p > 0.01).
Figure 5. Colony-forming ability of HCT-116 and A549 cells after treatment with 27a, 36a, and 36b. Representative images of the clonogenic assay for HCT-116 and A549 cell lines (a, b) and its quantification (c, d). Data represent the mean ± SD of three independent experiments.

Carbazole derivatives show potent antiproliferative activity
The in vitro antiproliferative effect of carbazole derivatives was further demonstrated by evaluating the bromodeoxyuridine (BrdU) incorporation assay on selected cancer cells. Treatment with equitoxic concentrations of compounds led to a time-dependent reduction in DNA synthesis across almost all investigated cell lines (). The U-2 OS cell line exhibited the least antiproliferative effect (), whereas HCT-116 cells showed the most substantial effect (). The BrdU incorporation assay revealed that all tested carbazoles inhibited BrdU incorporation in the A549, HCT-116, and MCF-7 cell lines, with 36b being the most potent. This compound displayed its most significant antiproliferative properties on A549 and HCT-116 lines, leading to an approximately 4-fold and 3.5-fold decrease in BrdU-positive cells, respectively (). Collectively, these findings suggest that carbazole derivatives have the potential to inhibit cancer cell proliferation in vitro.
Figure 6. Cytometric analysis of BrdU incorporation. A549, HCT-116, MCF-7, and U-2 OS cells were treated with 27a, 36a, and 36b for either 24 or 48 h. Representative histograms and statistical analyses after DNA staining (a). The results of the quantification analysis are presented in bar graphs. Error bars represent the mean ± SD of data obtained from three independent experiments (b).

Carbazole derivatives disrupt cell cycle progression
The effects of carbazole derivatives on cell cycle progression were evaluated by monitoring the phases of A549, HCT-116, MCF-7, and U-2 OS cells after exposure to equitoxic concentrations of compounds for 24 and 48 h (). Topoisomerases, essential for DNA replication and crucial in mitotic chromosome condensation and separation, can cause cell cycle arrest and apoptosis upon inhibitionCitation29.
Figure 7. Cell cycle profiles of A549, HCT-116, MCF-7, and U-2 OS cells after treatment with 36a, 36b, and 27a. Representative histograms from PI staining and their corresponding quantification are presented in the bar graphs. Error bars represent the mean ± SD of data obtained in three independent experiments.

The cell-cycle profiles depicted in indicate that all carbazole derivatives caused a significant G0/G1 arrest (p < 0.0001) in A549 cells, associated with a reduced G2/M phase. Among all tested compounds, only 36a displayed a time-dependent increase in the number of cells in the G0/G1 phase in HCT-116 cells (). In MCF-7 cells, all carbazole derivatives resulted in a significant, time-dependent increase in the G0/G1 phase, with a concomitant reduction of the S phase compared to DMSO-treated cells. The most potent cell cycle blockade was observed after 48 h exposure to 36b, with the G0/G1 phase showing a substantial increase (59.85 ± 2.15%; p < 0.00001) (). Time-dependent treatment of U-2 OS cells resulted in a ∼1.7-fold increase in the number of cells in the G2/M phase compared to DMSO-treated cells, which was associated with a reduction in the G0/G1 phase (). The effect of 36a on cell-cycle accumulation in the G1 phase correlated well with its cytotoxic and Topo IIα catalytic inhibitory activitiesCitation30.
These findings suggest that carbazole derivatives can potentially cause G0/G1 arrest in A549 and MCF-7 cells, as well as increase in the number of cells in the G2/M phase in U-2 OS cells. The varied responses of these cells to carbazole treatment could be attributed to their unique genetic profiles and variability in sensitivity to these compounds. Alternatively, the carbazole derivatives may target different biological processes in the two types of cell lines. Further investigation is needed to elucidate the molecular mechanisms underlying the observed cell cycle effects and their relationship to the cytotoxic and Topo II inhibitory activities of the carbazole derivatives.
Carbazoles induce apoptotic cell death via a mitochondrial pathway
Apoptosis evasion is one of the hallmarks of tumour transformationCitation31. Commonly used chemotherapeutics aim to promote cancer cell death through apoptosis activationCitation32,Citation33 and especially Topo inhibitors among others are the most efficient inducers of apoptosisCitation34. The confocal imaging with Annexin V-FITC and Hoechst33342 staining was performed to observe changes in nuclei morphology of A549 and HCT-116 after 24 h treatment with compounds (Figure S8). As depicted in , 27a, 36a, and 36b showed typical features of cells undergoing apoptosis as evidenced by multiple shrunken cells, fragmented nuclei, and apoptotic bodies. The apoptotic potential of the carbazoles was measured by flow cytometry using double staining with Annexin V-FITC and 7-AAD, which allows for determining viable (Annexin V-FITC(–)/7-AAD(–)), early apoptotic (EA) (Annexin V-FITC(+)/7-AAD(–)), late apoptotic (LA) (Annexin V-FITC(+)/7-AAD(+)), and necrotic (Annexin V-FITC(–)/7-AAD(+)) cells. As shown in , after 24 h of A549 cells treatment, tested compounds increased the proportion of apoptotic cells (EA + LA) to above 40%, compared to the control group (8.7 ± 2.3%). Later exposure (48 h) to carbazoles demonstrated a potent increase in the LA phase, from approximately 30–60%, depending on the compound (). Like A549, the treatment of HCT-116 cells with investigated compounds leads to a time-dependent increase of apoptotic cells, whereas the apoptotic potential of carbazoles was similar in both cell lines (). The most substantial pro-apoptotic properties were exhibited by compound 27a, which on both tested cancer lines caused an eightfold increase in the fraction of apoptotic cells compared to the control. Moreover, in both tested cell lines, carbazoles also slightly activated necrosis, and this effect was only statistically significant for 27a and 36b against HCT-116 (**p < 0.001) ( and Figure S9). Importantly, all tested compounds induced apoptosis approximately 2.5 times more than the reference compound ETP, a well-known Topo II inhibitor used in chemotherapies for treating numerous tumours, such as lung cancerCitation35.
Figure 8. Analyses of proapoptotic activities of carbazole derivatives. Confocal imaging of A549 (a) and HCT-116 (b) cells after 24 h of treatment with 27a, 36a, and 36b. The cells were stained with Hoechst33342, scale bar = 10 µm. The arrows indicate cell shrinkage (white), fragmented nuclei (red), and apoptotic bodies (yellow). Quantification of flow cytometry analysis of A549 (c) and HCT-116 (d) cell lines (24 and 48 h) using Annexin V/7-AAD. Modulation of caspase 3/7 in A549 (e) and HCT-116 (f) cells after 24 and 48 h of treatment with 27a, 36a, and 36b. Analysis of changes in mitochondrial potential using JC-1 staining. Bar charts (g) with statistical quantification and confocal images of A549 and HCT-116 cells (h) acquired after 24 h of treatment with the compounds. Error bars represent the mean ± SD of data obtained from three independent experiments. ETP or FCCP served as references.

One of the characteristics of cells undergoing apoptosis is the activation of cysteine proteases known as caspases, Executioner caspases 3 and 7 are known as central players during apoptotic cell deathCitation36. To determine, whether carbazoles triggered caspase-dependent cell death, A549 and HCT-116 cells were treated with equitoxic concentrations of compounds and analysed by flow cytometry. As depicted in , after 24 h of treatment of A549 cells, carbazoles activate caspase-3/7, as revealed by a 3.4-fold, 5.7-fold, and 2.5-fold increase in apoptosis for 27a, 36a, and 36b respectively, as compared to control. Later treatment of A549 cells (48 h) showed a slight increase in apoptotic cells. In comparison, exposure to HCT-116 cells led to a comparable increase in subpopulation cells with activated caspase 3/7 for all carbazole derivatives, as revealed by approximately five times higher than in control (DMSO) (). After 48 h of treatment with the compounds, caspase-3/7 activity in HCT-116 cells was enhanced 1.5- to 2-fold compared to the vehicle (). Moreover, ETP also caused a significant increase in caspase-3/7 activity in both cell lines; however, this effect was more pronounced than that of the carbazoles (, and Figure S10).
Mitochondria play a significant role in cell death, and loss of mitochondrial outer membrane potential (MOMP) is essential to initiate mitochondrial apoptosisCitation37. The change in JC-1 fluorescence demonstrated a loss of ΔΨm from red (aggregates) to green (monomers). As shown in , treatment of A549 and HCT-116 cells with carbazoles for 24 h led to a significant rise in the percentage of cells with dissipated MOMP, as revealed by ∼5-fold increase in JC1-monomers by all carbazoles in comparison to a vehicle (DMSO). On the other hand, the reference compound carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) led to an 11-fold and 6.3-fold increase in JC-1-monomers for A549 and HCT-116 cells, respectively, compared to the control ( and Figures S11–S13).
DNA fragmentation is the hallmark of the last step of apoptosis, achieved by the activation of endonucleases, which cleave genomic DNA in dying cells into internucleosomal DNA fragmentsCitation38. To confirm the proapoptotic properties of the investigated carbazoles, terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) was assessed after treating A549 and HCT-116 cells for 24 and 48 h. As depicted in Figure S14, all compounds led to a time-dependent, significant increase in the rates of TUNEL-positive cells on both cancer cell lines; however, observed DNA fragmentation was more pronounced on HCT-116 than on A549 cells, where the most massive effect induced compound 27a which is consistent with results obtained from Annexin V-FITC/7-AAD assay ().
Discussion
The global burden of cancer has been growing steadily, leading to increasing incidence and mortality rates. According to Globocan 2020 report, there were 19.3 million newly diagnosed cancer cases and 10 million deaths worldwideCitation39. Notably, lung cancer accounted for 18% of cases, marking it as one of the most lethal forms of the disease. Similarly, colon cancer accounted for 9.4% of cases, posing a significant health threat. These two types of cancer were particularly prominent among men, whereas, among women, the leading types were breast, colon, and lung cancersCitation39. The incidence of lung and colorectal cancer is projected to rise significantly in the future, driven by factors such as genetic mutations, an ageing population, and growing resistance to conventional pharmacological treatmentsCitation40,Citation41. Given this alarming trend, it is crucial to prioritise the search for novel and effective treatments, especially for lung, colon, and breast cancers.
Several drugs have been identified to inhibit the catalytic activity of Topo II, and recent research suggests that these catalytic inhibitors act through diverse mechanisms. The specific cellular effects of these drugs can vary significantly depending on the step or steps of the Topo II catalytic cycle they targetCitation42. In our study, we found that the tested carbazole compounds did not stabilise the DNA/Topo II complex involved in the cleavage process. Compound 36a exhibited promising inhibitory properties against the catalytic activity of Topo IIα. It demonstrated high selectivity for the Topo IIα isoform, contrasting with most human Topo II inhibitors that act on both α and β isoformsCitation43. This isoform selectivity is particularly noteworthy given recent research emphasising the need for isoform-selective poisons and catalytic inhibitors to mitigate side effects associated with Topo II poisons, such as cardiomyopathy and secondary malignanciesCitation44. Moreover, targeting Topo IIβ with chemotherapeutics has been linked to adverse effects, underscoring the increasing interest in the search for compounds that preferentially inhibit one isoformCitation45–47.
Further, we discovered that 36a might exert its mechanism of action by inhibiting a specific step in the catalytic cycle of the enzyme before the formation of the cleavable complex between Topo II and DNA. We also explored the possibility that the tested carbazole derivatives could disrupt the interaction between Topo II and DNA by distorting the DNA's helical structure. Previous studies have suggested that the carbazole moiety has the potential to intercalate with DNACitation48–50. However, our findings indicate that none of the compounds investigated in this study bound to DNA, which suggests that their inhibitory effect on Topo II was direct and not mediated through DNA binding.
To assess their antiproliferative activity, we evaluated carbazole derivatives in various cancer cell lines, including bone, breast, colon, and lung. Among the tested cancer cell lines, 36a, 36b, and 27a showed significant nanomolar IC50 values, effectively suppressing cancer cell proliferation and inhibiting colony formation. In most of the tested cancer cell lines, these compounds demonstrated potent activity. Interestingly, when considering cytotoxicity, 36a showed a relatively weaker impact on the HEK293 cell line compared to the other cancer cell lines investigated in this study. However, despite this observation, its effectiveness was on par with that of the reference compound, ETP. The studies carried out by Ortega’s groupCitation51 revealed that after 72 h, merbarone, acting as a catalytic inhibitor, exhibited significantly reduced cytotoxic activity towards A549 and MCF-7 cell lines, with IC50 values of 40 ± 2.7 µM and 83.9 ± 3.0 µM, respectively, in comparison to 36a. These findings underscore the potent inhibitory effect of 36a on Topo II catalytic activity, as well as its ability to suppress the proliferation of lung and colon cancer cells at nanomolar levels.
Cell cycle regulation plays a critical role in the development of malignancy and the emergence of chemotherapy resistanceCitation52. Notably, various catalytic inhibitors of Topo IIα have been found to operate through distinct mechanisms of cell cycle suppression compared to Topo II poisonsCitation53. Among all the compounds, 36a demonstrated significant reduction in cell proliferation and induced G1-phase cell cycle arrest in the majority of evaluated cancer cell lines. However, in U-2-OS cells, we observed a time-dependent block in the G2/M phase. These findings align with the research conducted by Perdih’s group, who discovered a novel chemical class of Topo IIα catalytic inhibitors that caused G1-phase cell cycle arrest in MCF-7 and Hep-G2 cellsCitation30. Similarly, Kang et al. reported a distinct effect of a catalytic inhibitor of Topo IIα, which arrested the cell cycle in the S phase and suppressed the viability of ovarian cancer cellsCitation54. Conversely, Chen et al. described a G2/M delay in fibrosarcoma cells treated with ICRF-193Citation55. These studies provide further evidence that the cellular response to treatment with catalytic inhibitors of Topo II differs from that of Topo II poisons, particularly regarding their effects on cell cycle progression.
Current pharmacological therapies for cancer often exert their anticancer effects by inducing a decrease in mitochondrial membrane potential (ΔΨm) and permeabilisation of the mitochondrial membrane, leading to the release of apoptotic factors. Consequently, this triggers the activation of caspases, which initiate cell degradation through the limited proteolysis of various cellular proteinsCitation56. Topoisomerases, being important biological targets, play a role in multiple signalling pathways, including apoptosis, when their function is inhibited. In our study, we investigated the effects of carbazole derivatives on Topo II activity and its impact on apoptosis. Our research also revealed that these derivatives effectively inhibit the function of Topo II, triggering the mitochondria-dependent apoptosis pathway. This was evidenced by the activation of effector caspases 3/7 and DNA fragmentation in lung and colon cancer cells. Notably, many well-known Topo II poisons, such as ETP, DOXO, and mitoxantrone, are recognised as apoptosis inducersCitation57,Citation58. Similarly, previous reports have demonstrated that Topo II catalytic inhibitors, such as ICRF-187 and ICRF-193, induce apoptosis by activating caspases and causing internucleosomal DNA fragmentationCitation59,Citation60.
Conclusions
This study provides compelling in vitro evidence of the anticancer efficacy of symmetric carbazole derivatives (27a, 36a, and 36b). In particular, compound 36a, which contains a furyl moiety, effectively inhibits proliferation and induces G1-phase arrest in cancer cells. 36a represents a novel and promising class of non-intercalating catalytic inhibitors targeting Topo II. It exhibits significant selectivity in inhibiting the relaxation and decatenation activity of the Topo IIα isoform, and its mode of action involves the induction of apoptosis through the intrinsic pathway, accompanied by DNA fragmentation. This compound is a promising example of symmetrically substituted carbazoles with potent anticancer properties demonstrated in vitro. However, to fully understand the mechanism by which compound 36a inhibits the catalytic activity of Topo IIα, further comprehensive studies are warranted. Such future investigations will elucidate the intricate molecular interactions and signalling pathways contributing to the anticancer effects of this compound.
Furthermore, the flattening of the structure (27a) or the replacement of the O-heteroatom in the pyrrole ring (36a) with an S-heteroatom in the thiophene substituent (36b) significantly alters the properties of the compounds. These alterations are clearly evident in the results of biological tests conducted on Topo II. This information could have a substantial impact on the design of novel carbazole derivative structures. Compound 36a is particularly appealing because it features two highly electronegative oxygen atoms that are configurationally compatible with the active site of the enzyme under investigation. On the contrary, 36b, which contains sulphur atoms (larger and less electronegative than oxygen in 36a), is unable to form such strong interactions with the isoforms of Topo II. In the case of 27a, the linear structure may be the reason for its incompatibility with the size of the active centre pocket of the Topo II isoforms considered in this study.
Experimental
Apparatus and measurement methods
The MALDI-TOF mass spectra of the studied compounds 27a, 36a, and 36b were recorded on an autoflex TOF/TOF maX instrument (Brüker Daltonics, Billerica, MA) with DHB matrix in the range of m/z 100–1000. Additionally, the 1H (500 Hz) and 13C NMR (125 Hz) spectra of the studied compounds 27a, 36a, and 36b in d6-DMSO solutions were obtained with a Brüker AVANCE III 700 MHz spectrometer (Billerica, MA). UV absorption spectra of 27a, 36a, and 36b acetonitrile solutions were recorded on an Evolution 300 (Thermo Scientific, Waltham, MA) spectrophotometer with a data interval of 1.0 nm, a slit width of 1.0 nm and a scan speed of 240 nm/min. The fluorescence emission spectra of all compounds studied in acetonitrile (λexc = 300 nm for 36a and λexc = 340 nm for 27a and 36b) were recorded using FL 6500 Fluorescence spectrophotometer (Perkin Elmer, Waltham, MA) in the wavelength area between 350 and 550 nm. The results obtained for all studied samples, 27a, 36a, and 36b by the fluorometric method are in excellent agreement with those reported previouslyCitation27.
General procedure for the Stille cross-coupling reaction
Pathways and the consecutive steps of the three syntheses are shown in .
First synthesis step: 3,6-Dibromo-9H-carbazole or 2,7-dibromo-9H-carbazole (substrate) (1 equiv.) and Pd[(C6H5)3P]4 (0.025 equiv.) as catalyst was dissolved in 7 ml of dry toluene. The toluene solution was vigorously stirred under argon (Ar) atmosphere at room temperature for 20 min. Then, 2-(tributylstannyl)thiophene (precursor of 36b; CAS no.: 54663-78-4) or 2-(tributylstannyl)furan (precursor of 27a or 36a; CAS no.: 118486-94-5) (2.5 equiv.) was added and the reaction mixture was heated at 90 °C for 24 h. The exact weight and yield of the reagents are given in .
Table 2. Molecular formula, mass, and yield of the reagents and crude products 27a, 36a, and 36b.
Second synthesis step: After the reactions were completed (confirmed by TLC), the toluene solutions were cooled to room temperature. The solvent was removed under reduced pressure and crude products were extracted with ethyl acetate (EtOAc; 3 × 20 ml), washed twice with water (first fraction), saturated KF (second fraction), and dichloromethane (DCM; third fraction) solvents and all fractions dried over Na2SO4. Initial TLC and UV analysis confirmed that only the DCM fraction contained the expected product.
Third synthesis step: After evaporation, the crude products, oily for 27a and powdery for 36a and 36b, were purified by column chromatography. In the case of 27a derivative 1:10 (v/v) of the ethyl acetate:petroleum, ether mixture was used as an eluent, and in the case of 36a and 36b derivatives, the different, a specific mixture of toluene and petroleum ether (v/v = 1:1) was used. The structures of the objects studied and their numbering style are introduced in , and the equipment used as well as TLC monitoring made during syntheses are presented in Figure S15.
Structures confirmation
2,7-Di(2-furyl)-9H-carbazole (27a)
MALDI-TOF spectrum registered for 27a, DHB matrix (m/z signal; found (calc.)): [M] 299.101 (299.05) (Figure S16). ATR signals (cm−1): 3398 ν(N–H); 2956–2854 ν(C–H); 1599 ν(C═C); 1454, 1422, and 1377 ν(C–N); 1326 (C–C)interring; 1240 ν(C–N); 1155, 1075, and 1012 ν(C–C)interring (Figure S17). Electronic absorption and fluorescence spectra obtained for 27a were included in the SI file (Figure S18) and agreed with the data reported in Refs.Citation26,Citation27. The images of the colourful carbazole derivatives solutions in d6-DMSO are included in Figure S19.
1H NMR chemical shifts (500 Hz; d6-DMSO); δ (ppm): 11.46 (s, 1H, H-9); 8.18–8.09 (d, 2H, J4,3 = J5,6 = 6.95, H-5, H-4); 7.79 (s, 1H, H-8), 7.72 (s, 1H, H-1), 7.64–7.61 (d, 2H, H-3 and H-6); 7.34 (dd, 2H, J5′,4′ = 3.4, J4′,3′ = 1.79 both H-4′); 7.02 (d, 2H, both H-5′), 6.63 (d, 2H, both H-3′) (Figure S20).
13C NMR chemical shifts (125 Hz, d6-DMSO); δ (ppm): 154.1 (both C-2′); 143.4 (both C-5′); 141.3 (C-8a, C-9a); 131.9 (C-2, C-7); 129.4 (C-4, C-5); 122.7 (C-3, C-6); 117.2 (C-1, C-8); 114.3 (both C-4′); 112.8 (C-4a, C-4b); 106.1 (both C-3′) (Figure S21). Additionally, the 2D COSY (Figure S22) and HSQC (Figure S23) of 27a were registered. Based on the last analysis, the assignment positions of carbons were established and the complementary pairs C–H were reported for 27a.
3,6-Di(2-furyl)-9H-carbazole (36a)
MALDI-TOF spectrum registered for 36a, DHB matrix (m/z signal; found (calc.)): [M + 2H+] 301.045 (301.0) (Figure S24). ATR signals (cm−1): 3420 ν(N–H); 3252 ν(C–H) in furan ring; 2957–2854 ν(C–H); 1635 and 1609 ν(C═C); 1467 and 1379 ν(C–N); 1325 (C–C)interring; 1290 ν(C–N); 1237 and 1153 ν(C–N); 1073 νsym(–C═C–); 1011 ν(C–C)ring. (Figure S25). Electronic absorption and fluorescence spectra obtained for 36a were included in the SI file (Figure S26) and agreed with the data presented in Refs.Citation26,Citation27.
1H NMR chemical shifts (500 Hz; d6-DMSO); δ (ppm): 11.53 (s, 1H, H-9); 8.53 (s, 1H, H-4); 8.44 (s, 1H, H-5); 7.72 (d, 2H, both H-5′), 7.52–7.47 (m, 4H, J1,2 = J7,8 = 8.67 Hz, H-1, H-2, H-7, H-8); 6.87 (d, 2H, J3′,4′ = 3.16, both H-3′); 6.61 (t, 2H, J4′,5′ = 2.94 Hz, J3′,4′ = 1.17 Hz, both H-4′) (Figure S27).
13C NMR chemical shifts (125 Hz, d6-DMSO); δ (ppm): 154.7 (both C-2′); 142.4 (both C-5′); 139.9 (C-8a); 139.3 (C-9a); 128.7 (C-2); 124.9 (C-7); 123.5 (C-3); 123.1 (C-6); 122.5 (C-4); 122.2 (C-5); 116.3 (C-4a, C-4b); 113.6 (C-1); 112.5 (C-8); 112.1 and 111.3 (both C-4′); 104.3 (C-3′) (Figure S28).
The 2D COSY (Figure S29) and HSQC (Figure S30) of 36a were registered. Based on the last analysis, the assignment positions of carbons were established and the complementary pairs C–H were reported for 36a.
3,6-Di(2-thienyl)-9H-carbazole (36b)
MALDI-TOF spectrum registered for 36b, DHB matrix (m/z signal; found (calc.)): [M] 331.03 (331.05) (Figure S31). ATR signals (cm−1): 3411 ν(N–H); 3259–3100 ν(C–H) in thiophene ring; 2955–2844 ν(C–H); 1629; 1602 ν(C═C); 1468; 1378 ν(C–N); 1288, 1234, 1176, and 1158 ν(C–N); 1070 and 1050 νsym(–C═C–); 1017 ν(C–C)ring (Figure S32). Electronic absorption and fluorescence spectra obtained for 36b were included in the SI file (Figure S33) and correlated with the data presented elsewhereCitation61.
1H NMR chemical shifts (500 Hz; d6-DMSO); δ (ppm): 11.54 (s, 1H, H-9); 8.52 (s, 1H, H-4); 8.52 (s, 1H, H-4); 8.48 (s, 1H, H-5); 8.44 (d, 2H, both H-5′); 7.53–7.48 (m, 4H, J1,2 = J7,8 = 5.72 Hz H-1, H-2, H-7, H-8); 7.74 (t, 2H, J4′,5′ = 4.32 Hz, J3′,4′ = 3.71 Hz, both H-3′); 7.15 (d, 2H, both H-4′) (Figure S34).
13C NMR chemical shifts (125 Hz, d6-DMSO); δ (ppm): 145.3 (both C-2′); 139.2 (C-8a, C-9a); 129.2 (C-3, C-6); 128.8 (both C-5′); 124.8 (C-4, C-5); 123.8 (C-2, C-7); 122.8 (C-1, C-8); 118.1 (C-4a; C-4b); 113.7 (both C-4′); 112.2 (both C-3′) (Figures S35).
Moreover, the 2D COSY (Figure S36) and HSQC (Figure S37) of 36b were registered. Based on the last analysis, the assignment positions of carbons were established and the complementary pairs C–H were reported for 36b.
The structural characterisation of the three carbazole derivatives studied based on data obtained is in good agreement with those reported in Refs.Citation26,Citation27,Citation61.
Cell culture
A549 (CCL-185), HCT-116 (CVCL-427), MCF-7 (HB-8065), U-2 OS (HTB-96), U-87 MG (HTB-14), and HEK-293 (CRL-1573) cells used in this study were acquired from ATCC (Manassas, VA). The non-small cell lung cancer cell line A-549 and breast cancer cell line MCF-7 were cultured in RPMI-1640 medium, while the bone cancer cell line U-2 OS and colon cancer cell line HCT-116 were cultured in McCoy’s 5A medium. The brain cancer cell line U-87 MG was cultured in MEM medium, while the non-malignant embryonic kidney cell line HEK293 was cultured in DMEM. All culture media were supplemented with 10% foetal bovine serum, 2 mM l-glutamine, and antibiotics (penicillin 62.6 µg/ml and streptomycin 40 µg/ml). The cells were cultured in a humidified atmosphere containing 5% CO2 at 37 °C and were routinely screened for Mycoplasma contamination. All reagents used in this study were purchased from Corning (Corning, NY) unless otherwise stated.
Drug sensitivity assay
Cell viability was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide) assay. The cells were seeded into 96-well plates and treated with the compounds under investigation at concentrations ranging from 0 to 50 µM for 72 h. ETP (Sigma-Aldrich, St. Louis, MO) and m-AMSA (Cayman Chemical, Arbor, MI) were used as reference compounds. After treatment, the cells were incubated with MTT solution (0.4 mg/ml in PBS) for three hours at 37 °C. The medium was removed, and the formazan crystals were dissolved in 100 μl of DMSO. The absorbance was measured at 540 nm using an ASYS UVM340 microplate reader (Biochrom Ltd., Cambridge, UK). The experiment was conducted independently in triplicate.
Clonogenic assay
At a density of 400 cells/well, A549 and HCT-116 cells were seeded into six-well plates. The cells were treated with various concentrations of investigated carbazole derivatives for 24 h followed by a wash and cultured for eight days. The methanol was used to fix the cells and then followed by 0.5% crystal violet staining. The visible colonies were counted using ImageJ software (Bethesda, MD), and viability was then calculated relative to the control.
Flow cytometry
For each flow cytometry experiment, A549, HCT-116, MCF-7, and U-2 OS cells were seeded onto tissue culture plates and allowed to attach for 24 h. Cells were incubated with the compounds under investigation at their respective IC50 concentrations for the designated time for each experiment. Ten thousand events were analysed using Guava easyCyte 8 cell sorter (Merck Millipore, Burlington, MA) and FlowJo v10 software. Each experiment was repeated independently three times.
Cell cycle analysis
A549, HCT-116, MCF-7, and U-2 OS cells were treated with carbazole derivatives for either 24 or 48 h. After that, the cells were harvested, fixed in ice-cold 75% ethanol, and stored overnight at –20 °C. In the final step, after centrifugation, the cells were rinsed with PBS and stained with 20 μg/μl PI (Sigma-Aldrich, St. Louis, MO) and 100 μg/μl RNaseA (Thermo Fisher Scientific, Waltham, MA) in PBS for 20 min at RT.
BrdU incorporation
To detect DNA synthesis, A549, HCT-116, MCF-7, and U-2 OS cells were incubated with 20 μM BrdU (5-bromo-2′-deoxyuridine) (Sigma-Aldrich, St. Louis, MO) for 1 h before the end of treatment. Next, samples were harvested with trypsin solution and fixed in 75% ethanol (overnight or longer, −20 °C). Following 10 min rehydration with PBS, samples were denatured with 2 M HCl (45 min, RT), and the suspension was neutralised with 0.1 M sodium tetraborate, pH 8.5 (10 min, RT). 1% w/v bovine serum albumin (BSA) in PBS was used for blocking (30 min, RT). Following this, samples were incubated with rat anti-BrdU antibody (for 1 h at 37 °C at a 1:100 dilution; Abcam, #ab6326, Cambridge, UK), and then with goat anti-rat conjugated antibody (for 30 min at 37 °C at a 1:200 dilution; Abcam, #ab150157, Cambridge, UK). Twenty micrograms per microlitres PI and 100 μg/μl RNaseA in PBS (20 min, RT) were used to stain the DNA.
JC-1 staining
After the treatment period ended, the culture medium of the A549 and HCT-116 cells was replaced with fresh medium containing 5 µg/ml JC-1 dye (Sigma-Aldrich, St. Louis, MO) and incubated in the dark for an additional 20 min at 37 °C. Then, the cells were washed two times with PBS and measured. As a reference, 50 µM FCCP (Sigma-Aldrich, St. Louis, MO) was added 15 min before the end of drug treatment incubation.
Apoptosis and caspase 3,7 activation
Briefly, after incubation with tested compounds, ETP, A549, and HCT-116 cells were harvested by trypsinisation, rinsed twice with PBS, and stained with Annexin V FITC conjugate (Thermo Fisher Scientific, #A13199, Waltham, MA) for apoptosis assay and with CellEvent™ Caspase-3/7 Green Flow Cytometry Assay Kit (Thermo Fisher Scientific, #C10427, Waltham, MA) for caspase-3/7 activation according to the manufacturer’s protocols. ETP was used as a reference.
DNA fragmentation analysis
A549 and HCT-116 cells were tested using TUNEL Assay Kit – FITC (Abcam, #ab66108, Cambridge, UK). After drug treatment, samples were harvested using trypsin solution, washed with PBS, and fixed with 1% formaldehyde. The experiment was performed according to the manufacturer’s protocol. ETP was used as a reference.
Relaxation/decatenation of human Topo IIα/IIβ
Determination of the inhibitory activity of investigated compounds was performed according to the manufacturer’s protocol (Inspiralis; #HT205, Norwich, UK). In brief, a mixture containing 250 ng supercoiled pBR322 (Thermo Fisher; #SD0041, Waltham, MA), tested compounds, and a reaction buffer was prepared. The reaction was then initiated by adding diluted human Topo IIα or IIβ in assay buffer, and the samples were incubated for 30 min at 37 °C. The reaction was terminated by adding a loading buffer (New England BioLabs; #B7024S, Ipswich, MA). Samples were loaded onto 1% (w/v) agarose gel and subjected to electrophoresis in 1xTBE at 20 V for 18 h. The gel was stained with ethidium bromide, destained in H2O, and photographed by ChemiDoc Imaging System (Bio-Rad, Hercules, CA). Two hundred and fifty nanograms of kDNA (Inspiralis, #K1002, Norwich, UK) was used for the decatenation assay. ETP and ICRF-187 (Cayman Chemical, Ann Arbor, MI) were used as references.
Formation of cleavable complexes
The composition of the mixture was the same as for the relaxation of human Topo IIα assay, except for the amount of enzyme (five times more was used). The reaction was initiated by adding the enzyme to the samples and incubating for 10 min at 37 °C. Then, 0.35% SDS and 0.3 mg/ml proteinase K (A&A Biotechnology, Gdańsk, Poland) were added, and the probes were additionally incubated at 56 °C for 1 h, before adding loading buffer (New England BioLabs, #B7024S, Ipswich, MA) and chloroform:isoamyl alcohol (24:1 v/v). ETP and ICRF-187 were used as references. The electrophoresis was run in the presence of EtBr (1 µg/ml) in TBEx1.
Intercalation into DNA
The unwinding assay was performed according to the manufacturer’s instructions (Inspiralis, #DUKSR002, Norwich, UK) to determine the intercalating ability of the tested carbazole derivatives. ETP and DOXO (Sigma-Aldrich, St. Louis, MO) were used as references. In brief, wheat germ topoisomerase I, diluted in assay buffer, was added to a mixture containing assay buffer, the tested compounds, and relaxed pBR322. After incubation (30 min, 37 °C), the reaction was terminated by 50 µl of butanol and 20 µl of H2O. Next, samples were vortexed and centrifuged, and the aqueous layer was mixed with chloroform/isoamyl alcohol (24:1 v/v) and loading buffer (New England BioLabs, #B7024S, Ipswich, MA).
Live-cell imaging
For live-cell imaging of A549 and HCT-116 cells, JC-1 and Annexin V-FITC staining were performed by seeding them into glass-bottom 24-well plates. The drug-treated samples were then stained in the same way as flow cytometry. Hoechst33342 (Sigma-Aldrich, St. Louis, MO) was used to visualise cell nuclei under the microscope. Images were obtained using an LSM 800 inverted laser-scanning confocal microscope (Carl Zeiss, Oberkochen, Germany), equipped with a ×63 1.4-NA Plan Apochromat objective (Carl Zeiss, Oberkochen, Germany) and an airyscan detector for high-resolution confocal scanning. The incubation chamber was maintained at 37 °C with 5% CO2 during the analysis. ETP was used as a reference. The microscopy analysis was performed using equipment funded by the Foster Foundation (Kirkland, WA).
Statistical analyses
Statistical analysis was performed using GraphPad Prism 9 software (La Jolla, CA). Uniform significance levels were used throughout the entire manuscript as follows: ns = p > 0.01; *p < 0.01; **p < 0.001; ***p < 0.0001; ****p < 0.00001. Statistical significance was calculated in comparison to the DMSO-treated control (1% v/v) using one or two-way ANOVA.
Author contributions
Conceptualisation: M.O.; methodology: M.O., N.M., A.C, A.M.D., and M.Bi.; validation: M.O., A.C., and A.M.D.; formal analysis: M.O.; writing – original draft preparation: M.O. and N.M.; graphical conceptualisation: M.O. and N.M.; writing – review and editing: M.O., N.M., A.K., J.M.P., M.M., and M.B.
Supplemental Material
Download MS Word (77 MB)Disclosure statement
The authors report no conflicts of interest.
Data availability statement
The datasets presented in the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Upadhyay A. Cancer: an unknown territory; rethinking before going ahead. Genes Dis. 2021;8(5):655–661.
- Berben L, Floris G, Wildiers H, Hatse S. Cancer and aging: two tightly interconnected biological processes. Cancers. 2021;13(6):1400.
- Cancer; 2023 [accessed 2023 Jan 31]. https://www.who.int/health-topics/cancer#tab=tab_1.
- Alfarouk KO, Stock CM, Taylor S, Walsh M, Muddathir AK, Verduzco D, Bashir AH, Mohammed OY, Elhassan GO, Harguindey S, et al. Resistance to cancer chemotherapy: failure in drug response from ADME to P-gp. Cancer Cell Int. 2015;15(1):71.
- Tsutsumi LS, Gündisch D, Sun D. Carbazole scaffold in medicinal chemistry and natural products: a review from 2010–2015. Curr Top Med Chem. 2016;16(11):1290–1313.
- Lin W, Wang Y, Lin S, Li C, Zhou C, Wang S, Huang H, Liu P, Ye G, Shen X. Induction of cell cycle arrest by the carbazole alkaloid Clauszoline-I from Clausena vestita D. D. Tao via inhibition of the PKCδ phosphorylation. Eur J Med Chem. 2012;47(1):214–220.
- Dias N, Jacquemard U, Baldeyrou B, Tardy C, Lansiaux A, Colson P, Tanious F, Wilson WD, Routier S, Mérour JY, et al. Targeting DNA with novel diphenylcarbazoles. Biochemistry. 2004;43(48):15169–15178.
- Zembower DE, Xie Y, Koohang A, Kuffel MJ, Ames MM, Zhou Y, Mishra R, Mar AA, Flavin MT, Xu ZQ. Methylenedioxy- and ethylenedioxy-fused indolocarbazoles: potent human topoisomerase I inhibitors and antitumor agents. Anticancer Agents Med Chem. 2012;12(9):1117–1131.
- Li PH, Jiang H, Zhang WJ, Li YL, Zhao MC, Zhou W, Zhang LY, Tang YD, Dong CZ, Huang ZS, et al. Synthesis of carbazole derivatives containing chalcone analogs as non-intercalative topoisomerase II catalytic inhibitors and apoptosis inducers. Eur J Med Chem. 2018;145:498–510.
- Witkowska M, Maciejewska N, Ryczkowska M, Olszewski M, Bagiński M, Makowiec S. From tryptophan to novel mitochondria-disruptive agent, synthesis and biological evaluation of 1,2,3,6-tetrasubstituted carbazoles. Eur J Med Chem. 2022;238:114453.
- Utaipan T, Athipornchai A, Suksamrarn A, Jirachotikoon C, Yuan X, Lertcanawanichakul M, Chunglok W. Carbazole alkaloids from Murraya koenigii trigger apoptosis and autophagic flux inhibition in human oral squamous cell carcinoma cells. J Nat Med. 2017;71(1):158–169.
- Issa S, Prandina A, Bedel N, Rongved P, Yous S, Le Borgne M, Bouaziz Z. Carbazole scaffolds in cancer therapy: a review from 2012 to 2018. J Enzyme Inhib Med Chem. 2019;34(1):1321–1346.
- Newman DJ, Cragg GM. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J Nat Prod. 2020;83(3):770–803.
- Yoon S, Kim JH, Lee YJ, Ahn MY, Choi G, Kim WK, Yang Z, Lee HJ, Moon HR, Kim HS. A novel carbazole derivative, MHY407, sensitizes cancer cells to doxorubicin-, etoposide-, and radiation treatment via DNA damage. Eur J Pharmacol. 2012;697(1–3):24–31.
- Bukhari SNA, Ejaz H, Elsherif MA, Junaid K, Zaki I, Masoud RE. Design and synthesis of some new furan-based derivatives and evaluation of in vitro cytotoxic activity. Molecules. 2022;27(8):2606.
- Abedinifar F, Babazadeh Rezaei E, Biglar M, Larijani B, Hamedifar H, Ansari S, Mahdavi M. Recent strategies in the synthesis of thiophene derivatives: highlights from the 2012–2020 literature. Mol Divers. 2021;25(4):2571–2604.
- Opdam FL, Guchelaar HJ, Beijnen JH, Schellens JHM. Lapatinib for advanced or metastatic breast cancer. Oncologist. 2012;17(4):536–542.
- Van Cutsem E, Cunningham D, Maroun J, Cervantes A, Glimelius B. Raltitrexed: current clinical status and future directions. Ann Oncol. 2002;13(4):513–522.
- Pommier Y, Nussenzweig A, Takeda S, Austin C. Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol. 2022;23(6):407–427.
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer. 2009;9(5):338–350.
- Delgado JL, Hsieh CM, Chan NL, Hiasa H. Topoisomerases as anticancer targets. Biochem J. 2018;475(2):373–398.
- Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17(5):421–433.
- Hu W, Huang XS, Wu JF, Yang L, Zheng YT, Shen YM, Li ZY, Li X. Discovery of novel topoisomerase II inhibitors by medicinal chemistry approaches. J Med Chem. 2018;61(20):8947–8980.
- Skok Ž, Zidar N, Kikelj D, Ilaš J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J Med Chem. 2020;63(3):884–904.
- Larsen AK, Escargueil AE, Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther. 2003;99(2):167–181.
- Oğuztürk E, Tirkeş S, Önal AM. Electrochemical synthesis of new conjugated polymers based on carbazole and furan units. J Electroanal Chem. 2015;750:1–8.
- Damit EF, Nordin N, Ariffin A, Sulaiman K. Synthesis of novel derivatives of carbazole–thiophene, their electronic properties, and computational studies. J Chem. 2016;2016:1–14.
- Lucas I, Germe T, Chevrier-Miller M, Hyrien O. Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J. 2001;20(22):6509–6519.
- Larsen AK, Skladanowski A, Bojanowski K. The roles of DNA topoisomerase II during the cell cycle. Prog Cell Cycle Res. 1996;2:229–239.
- Bergant Loboda K, Janežič M, Štampar M, Žegura B, Filipič M, Perdih A. Substituted 4,5′-bithiazoles as catalytic inhibitors of human DNA topoisomerase IIα. J Chem Inf Model. 2020;60(7):3662–3678.
- Fernald K, Kurokawa M. Evading apoptosis in cancer. Trends Cell Biol. 2013;23(12):620–633.
- Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395–417.
- Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11(4):342–357.
- Sordet O, Khan QA, Kohn KW, Pommier Y. Apoptosis induced by topoisomerase inhibitors. Curr Med Chem Anticancer Agents. 2003;3(4):271–290.
- Rezonja R, Knez L, Cufer T, Mrhar A. Oral treatment with etoposide in small cell lung cancer – dilemmas and solutions. Radiol Oncol. 2013;47(1):1–13.
- Mcllwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):1–28.
- Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13(8):1396–1402.
- Zhang JH, Xu M. DNA fragmentation in apoptosis. Cell Res. 2000;10(3):205–211.
- Deo SVS, Sharma J, Kumar S. GLOBOCAN 2020 report on global cancer burden: challenges and opportunities for surgical oncologists. Ann Surg Oncol. 2022;29(11):6497–6500.
- Zappa C, Mousa SA. Non-small cell lung cancer: current treatment and future advances. Transl Lung Cancer Res. 2016;5(3):288–300.
- Van Der Jeught K, Xu HC, Li YJ, Lu XB, Ji G. Drug resistance and new therapies in colorectal cancer. World J Gastroenterol. 2018;24(34):3834–3848.
- Pastor N, Domínguez I, Luís Orta M, Campanella C, Mateos S, Cortés F. The DNA topoisomerase II catalytic inhibitor merbarone is genotoxic and induces endoreduplication. Mutat Res. 2012;738–739:45–51.
- Bau JT, Kang Z, Austin CA, Kurz EU. Salicylate, a catalytic inhibitor of topoisomerase II, inhibits DNA cleavage and is selective for the α isoform. Mol Pharmacol. 2014;85(2):198–207.
- Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JY, Wang JC, Liu LF. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc Natl Acad Sci U S A. 2007;104(26):11014–11019.
- Toyoda E, Kagaya S, Cowell IG, Kurosawa A, Kamoshita K, Nishikawa K, Iiizumi S, Koyama H, Austin CA, Adachi N. NK314, a topoisomerase II inhibitor that specifically targets the alpha isoform. J Biol Chem. 2008;283(35):23711–23720.
- Auzanneau C, Montaudon D, Jacquet R, Puyo S, Pouységu L, Deffieux D, Elkaoukabi-Chaibi A, De Giorgi F, Ichas F, Quideau S, et al. The polyphenolic ellagitannin vescalagin acts as a preferential catalytic inhibitor of the α isoform of human DNA topoisomerase II. Mol Pharmacol. 2012;82(1):134–141.
- Ortega JA, Arencibia JM, Minniti E, Byl JAW, Franco-Ulloa S, Borgogno M, Genna V, Summa M, Bertozzi SM, Bertorelli R, et al. Novel, potent, and druglike tetrahydroquinazoline inhibitor that is highly selective for human topoisomerase II α over β. J Med Chem. 2020;63(21):12873–12886.
- Sajewicz W, Dlugosz A. Cytotoxicity of some potential DNA intercalators (carbazole, acridine and anthracene derivatives) evaluated through neutrophil chemiluminescence. J Appl Toxicol. 2000;20(4):305–312.
- Shaikh MS, Karpoormath R, Thapliyal N, Rane RA, Palkar MB, Faya AM, Patel HM, Alwan WS, Jain K, Hampannavar GA. Current perspective of natural alkaloid carbazole and its derivatives as antitumor agents. Anticancer Agents Med Chem. 2015;15(8):1049–1065.
- Das A, Mohammed TP, Kumar R, Bhunia S, Sankaralingam M. Carbazole appended trans-dicationic pyridinium porphyrin finds supremacy in DNA binding/photocleavage over a non-carbazolyl analogue. Dalton Trans. 2022;51(33):12453–12466.
- Ortega JA, Riccardi L, Minniti E, Borgogno M, Arencibia JM, Greco ML, Minarini A, Sissi C, De Vivo M. Pharmacophore hybridization to discover novel topoisomerase II poisons with promising antiproliferative activity. J Med Chem. 2018;61(3):1375–1379.
- Schwartz GK, Shah MA. Targeting the cell cycle: a new approach to cancer therapy. J Clin Oncol. 2005;23(36):9408–9421.
- Jeon KH, Park S, Jang HJ, Hwang SY, Shrestha A, Lee ES, Kwon Y. Ak-i-190, a new catalytic inhibitor of topoisomerase II with anti-proliferative and pro-apoptotic activity on androgen-negative prostate cancer cells. Int J Mol Sci. 2021;22(20):11246.
- Kang K, Nho CW, Kim ND, Song DG, Park YG, Kim M, Pan CH, Shin D, Oh SH, Oh HS. Daurinol, a catalytic inhibitor of topoisomerase IIα, suppresses SNU-840 ovarian cancer cell proliferation through cell cycle arrest in S phase. Int J Oncol. 2014;45(2):558–566.
- Chen L, Zhu X, Zou Y, Xing J, Gilson E, Lu Y, Ye J. The topoisomerase II catalytic inhibitor ICRF-193 preferentially targets telomeres that are capped by TRF2. Am J Physiol Cell Physiol. 2015;308(5):C372–C377.
- Wong RSY. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30(1):87.
- Kolb RH, Greer PM, Cao PT, Cowan KH, Yan Y. ERK1/2 signaling plays an important role in topoisomerase II poison-induced G2/M checkpoint activation. PLOS One. 2012;7(11):e50281.
- Park SH, Lee J, Kang MA, Jang KY, Kim JR. Mitoxantrone induces apoptosis in osteosarcoma cells through regulation of the Akt/FOXO3 pathway. Oncol Lett. 2018;15(6):9687.
- Khélifa T, Beck WT. Induction of apoptosis by dexrazoxane (ICRF-187) through caspases in the absence of c-Jun expression and c-Jun NH2-terminal kinase 1 (JNK1) activation in VM-26-resistant CEM cells. Biochem Pharmacol. 1999;58(8):1247–1257.
- Iguchi K, Usami Y, Hirano K, Hamatake M, Shibata M, Ishida R. Decreased thymosin beta4 in apoptosis induced by a variety of antitumor drugs. Biochem Pharmacol. 1999;57(10):1105–1111.
- Gao W, Zheng M, Li Y. A novel and facile synthesis of 3-(2-benzofuroyl)- and 3,6-bis(2-benzofuroyl)carbazole derivatives. Beilstein J Org Chem. 2011;7:1533–1540.