
Abstract
A novel series of 1,2,3-triazole/1,2,4-oxadiazole hybrids (7a–o) was developed as dual inhibitors of EGFR/VEGFR-2. Compounds 7a–o were evaluated as antiproliferative agents with Erlotinib as the reference drug. Results demonstrated that most of the tested compounds showed significant antiproliferative action with GI50 values ranging from 28 to 104 nM, compared to Erlotinib (GI50 = 33 nM), and compounds 7i–m were the most potent. Compounds 7h, 7i, 7j, 7k, and 7l were evaluated as dual EGFR/VEGFR-2 inhibitors. These in vitro experiments demonstrated that compounds 7j, 7k, and 7l are potent antiproliferative agents that may operate as dual EGFR/VEGFR-2 inhibitors. Compounds 7j, 7k, and 7l were evaluated for their apoptotic potential activity, where findings indicated that compounds 7j, 7k, and 7l promote apoptosis by activating caspase-3, 8, and Bax and down-regulating the anti-apoptotic Bcl-2. Molecular docking simulations show the binding mode of the most active antiproliferative compounds within EGFR and VEGFR-2 active sites.
Introduction
Despite enormous efforts over the last two decades to find effective therapies, cancer remains a menace to the globe todayCitation1. While conventional cancer treatments cannot distinguish between healthy and malignant cells, targeted therapy kills cancer cells while preserving healthy cells by disrupting critical metabolic pathways or oncoproteins required for tumour cell growth and survivalCitation1,Citation2.
Protein tyrosine kinases are important in signal transduction pathways that govern cellular processes such as proliferation, differentiation, migration, and angiogenesisCitation3,Citation4. The epidermal growth factor receptor (EGFR) is a type of membrane receptor tyrosine kinase that is overexpressed in various tumours. Because EGFR tyrosine kinase signal transduction is closely linked to tumour progression, inhibiting receptor activity can effectively inhibit tumoursCitation5–7. Another significant receptor tyrosine kinase that can induce angiogenesis is vascular endothelial growth factor receptor (VEGFR-2)Citation8. VEGFR-2, a VEGFR family member, is a major mediator in tumour angiogenesis and is essential for solid tumour formation. Inhibiting VEGFR-2 has been proposed as an effective method for preventing angiogenesisCitation9,Citation10.
VEGFR-2 and EGFR have been identified as promising therapeutic targets for cancer treatment. They are essential in the signalling pathways that regulate tumour cell proliferation, differentiation, migration, and angiogenesisCitation11–14. EGFR and VEGFR-2 typically share common downstream signalling pathways as a complex signal network of interconnected circuits. EGFR inhibition can reduce VEGF expression and attenuate angiogenesis while increasing VEGFR-2 expression, eventually leading to EGFR inhibitor resistanceCitation15,Citation16. As a result, inhibiting both EGFR and VEGFR-2 simultaneously has become an effective cancer therapeutic strategy with a synergistic impactCitation17–19.
The 1,2,4-oxadiazole heterocycle has been widely studied over the last four decades, yielding many analogs with diverse biological effects. Due to the potential for particular interactions (such as hydrogen bonding), the 1,2,4-oxadiazole achieves bioisosteric equivalence with amide and ester moietiesCitation20,Citation21. The 1,2,4-oxadiazole nucleus is the basic core of a number of drugs on the market today, including Oxolamine, Butalamine, Prenoxdiazine, Fasiplon, and ProxazoleCitation22–24. A new series of 1,2,4-oxadiazole-based compounds was developed and tested as anticancer agents that target the EGFR inhibitory pathway. The optimal derivative was compound I (), demonstrating equipotent antiproliferative action against a panel of five NSCLC cell lines (IC50 = 0.2–0.6 µM). Cell cycle investigations demonstrated that I's antiproliferative activity is linked to its ability to cause G2/M arrest and, to a lesser extent, apoptosisCitation25.
Figure 1. Structure of anticancer 1,2,4-oxadiazoles I–III.
Saritha et al.Citation26 reported on the design, synthesis, and antiproliferative efficacy of a new series of molecular hybrids containing benzimidazole, thiazolidine-2,4-dione, and 1,2,4-oxadiazole scaffolds against three human cancer cell lines, MCF-7, A-549, and HepG2. Compound (II, ) demonstrated greater activity against all cell lines, with the highest activity against MCF-7 (IC50 = 1.32 ± 0.05 µM) as compared to the reference Erlotinib (IC50 = 4.15 ± 0.12 µM). Similarly, new isatin-1,2,4-oxadiazole hybrids were produced and tested for cytotoxicity against four human cancer cell lines. Their potential VEGFR-2 inhibition assay was also investigated, with compound (III, ) having nearly double the inhibitory potency as Sorafenib, with IC50 = 35.64 ± 1.56 nM against VEGFR-2 enzyme assay.
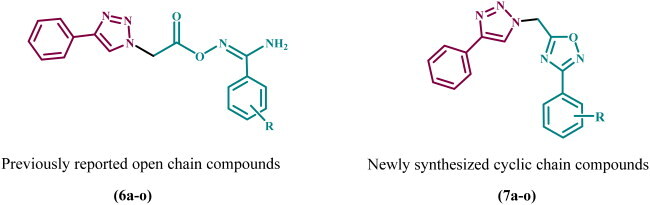
In a recent publicationCitation27, we describe the discovery of a new series of 1,2,3-triazole-based antiproliferative agents, compounds 6a–o (). The newly synthesised compounds were tested for antiproliferative activity against a panel of four cancer cell lines, with compound 6k (R = 3,4-di-OMe) being the most potent derivative with a GI50 value of 31 nM against the tested four cancer cell lines, outperforming the reference Erlotinib (GI50 = 33 nM). The EGFR and VEGFR-2 inhibitory assay results showed that compound 6k (R = 3,4-di-OMe), the most potent antiproliferative agent, was also the most potent anti-EGFR and anti-VEGFR-2 agent, with IC50 values of 83 ± 05 and 1.80 ± 0.05 nM, respectively, compared to Erlotinib (IC50 = 80 ± 4 nM) and Sorafenib (IC50 = 0.17 ± 0.01 nM).
Figure 2. Structures of previously reported compounds 6a–o and newly synthesised 7a–o.
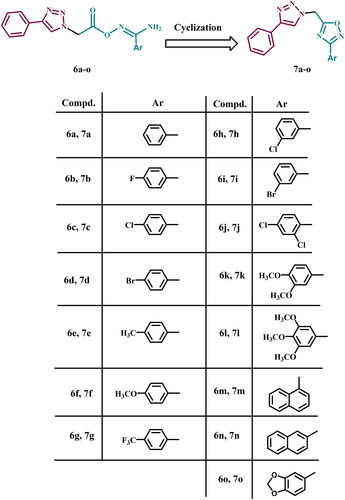
Motivated by previous findings and in pursuit of a new antiproliferative agent with dual or multi-targeted inhibitory actionCitation27–30, we present here the design, synthesis, and antiproliferative activity of a novel series of 1,2,3-triazole/1,2,4-oxadiazole hybrids (7a–o, ) as dual EGFR/VEGFR-2 inhibitors. The newly designed 1,2,4-oxadiazoles 7a–o were considered to be sterically fixed counterparts of the previously discovered linear carboximidamide derivatives 6a–o (). Such a rigid conformation may result in tight fitting within the EGFR and VEGFR-2 active sites, increasing the efficacy of newly synthesised compounds. The antiproliferative effect of compounds 7a–o against four cancer cell lines was investigated using Erlotinib as the reference drug. Next, the most effective compounds were evaluated for inhibitory action against EGFR and VEGFR-2, which were identified as possible targets for their action. In addition, the apoptotic potential of the most active compounds was assessed. Finally, docking analysis and ADME studies were performed for the most potent derivatives.
Results and discussion
Chemistry
Scheme 1 outlines the synthetic steps for the target compounds 7a–o. Compound 1 was hydrolysed in aqueous NaOH (20%) and refluxed for two hours to produce compound 2Citation31. On the other hand, compounds 4a–o were prepared according to a reported procedure in THF via an iodine-catalyzed transformation of the formyl group in 3a–o to a cyano group utilising ammonia as a nitrogen sourceCitation32. Amidoximes 5a–o were prepared in THF by nucleophilic attack of hydroxylamine at the cyanide carbon of 4a–oCitation33. Aryl carboximidamides 6a–o were prepared by reacting carboxylic acid 2 with the appropriate amidoximes 5a–o in dry acetonitrile using the activating agent N,N'-carbonyl diimidazole (CDI)Citation27.
Scheme 1. Synthetic pathway of compounds 7a–o
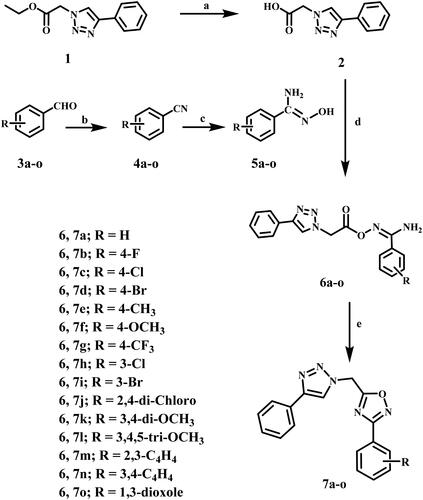
Reagents and reaction conditions: (a) 20% aq. NaOH, reflux, 2 h, 80%; (b) 33% aq. NH3, THF, I2, r.t 3–5 h, 40–85%; (c) NH2OH·HCl, Na2CO3, methanol, reflux, 6–8 h; (d) CDI, dry CH3CN, r.t 10 h; (e) CH3CN, reflux, overnight, 46–90%.
The target compounds 7a–o were synthesised by refluxing the appropriate carboximidamides 6a–o in dry acetonitrile. After the reaction was completed (as monitored by TLC), the excess solvent evaporated under reduced pressure. The precipitates were washed with cold water and filtered, crystalised from acetonitrile to yield 1,2,4-oxadiazoles 7a–o in good yields.
The structures of new compounds 7a–o were elucidated using NMR, IR, and elemental microanalysis. FTIR spectra of target compounds 7a–o confirmed the disappearance of (NH2) and (C = O) peaks of the precursor aryl carboximidamides 6a–o. The presence of (CH) peaks at 3144, 3001, 2965 cm−1, (C = N and C = C) peaks at 1602, 1579, 1468 cm−1, and (Ar–CH bending) at 856 cm−1 in the FTIR spectrum of compound 7g as a representative example verified the prior observations.
Also,1H NMR spectra proved the cyclisation reaction by revealing the absence of amino (NH2) wide signals in compounds 6a–e and the appearance of (N–CH2) signals at (δ = 6.37–6.14 ppm) compared with compounds’ 6a–o (N–CH2) signals that appeared in the range of (δ = 6.66–5.61 ppm). 1H NMR spectrum of compound 7g confirmed the elucidated structure by showing a singlet triazole C–H signal at δ = 8.82 ppm, and nine aromatic protons appeared as five distinct signals in the aromatic region. Moreover, 13C NMR spectra confirmed the cyclisation reaction by the disappearance of the C = O signal of their precursors 6a–o at δ = 166 ppm and the appearance of (N–CH2) signal at (δ = 45.6–45 ppm). 13C NMR spectrum of compound 7g confirmed the elucidated structure by the appearance of 12 aromatic signals at δ = 175.1–122.9, CF3 quartette signal at δ = 124 with J = 272.7 Hz, and N–CH2 signal as δ = 45.3 ppm.
Biology
Cell viability assay
A cell viability experiment was performed on the MCF-10A (normal human mammary gland epithelial) cell line to assess the effect of compounds 7a–o on normal cell linesCitation34,Citation35. Before testing cell viability, 50 µM of the examined compound is used in this investigation for four days. Compounds 7a–o have no cytotoxic impact and are greater than 87% cell viability, as shown in .
Table 1. Antiproliferative activity of compounds 7a–o and Erlotinib.
Antiproliferative assay
The antiproliferative activity of 1,2,4-oxadiazoles 7a–o was evaluated against four different human cancer cell lines, namely, colon cancer (HT-29) cell line, lung cancer (A-549) cell line, pancreatic cancer (Panc-1) cell line, and breast cancer (MCF-7) cell line using Erlotinib as the reference drugCitation36,Citation37. The median inhibitory concentration (IC50) is shown in . The results showed that the tested compounds 7a–o had promising antiproliferative action with GI50 values ranging from 28 to 104 nM. The six most active derivatives were 7h–m, with GI50 values ranging from 28 to 45 nM, compared to Erlotinib’s 33 nM.
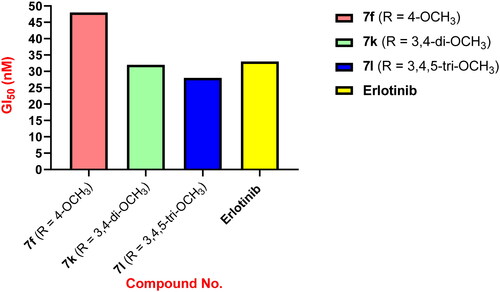
Compound 7l (R = 3,4,5-tri-OMe) was the most potent derivative, with a GI50 value of 28 nM against the four cancer cell lines examined, outperforming the reference Erlotinib (GI50 = 33 nM). Compound 7l was shown to be more potent than Erlotinib against all four human cancer cell lines examined. Compound 7k (R = 3,4-dimethoxy) scored second in activity against the four cancer cell lines with a GI50 value of 32 nM, equipping to the reference Erlotinib (GI50 = 33 nM). Compound 7k was more potent than Erlotinib against the breast cancer (MCF-7) cell line, having an IC50 value of 35 ± 3 nM versus 40 ± 3 nM for Erlotinib. According to the results of compounds 7f (R = 4-OMe) and 7k (R = 3,4-di-OMe), the number of methoxy groups substantially impacts the activity of these compounds. Compounds 7f (R = 4-OMe) and 7k (R = 3,4-di-OMe) were less potent than compound 7l (R = 3,4,5-tri-OMe), indicating that the trimethoxy groups were best suited for antiproliferative action, .
Figure 3. GI50 of compounds 7f, 7k, and 7l compared to Erlotinib.
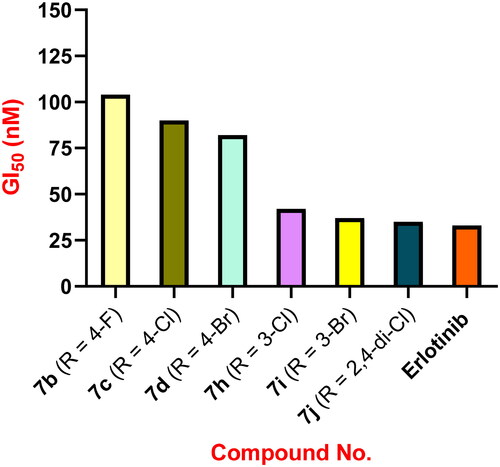
Compound 7j (R = 2,4-di-Cl) was the third most active against the four cancer cell lines, with a GI50 value of 35 nM. Compound 7j was likewise more potent than Erlotinib against the breast cancer (MCF-7) cell line, with an IC50 value of 36 ± 3 nM versus Erlotinib’s IC50 of 40 ± 3 nM. Another key point drawn from the findings in is the effect of halogen atom type, position, and number on antiproliferative activity. Compounds 7b (R = 4-F), 7c (R = 4-Cl), 7d (R = 4-Br), 7i (R = 3-Br), and 7h (R = 3-Cl) demonstrated good antiproliferative action with GI50 values ranging from 37 to 104 nM, being less potent than compound 7j (R = 2,4-di-Cl) indicating that the number of halogen atoms had a significant impact on the antiproliferative action of these compounds and that dihalo derivatives are more potent than mono halo substituted derivatives.
Compounds 7i (R = 3-Br) and 7h (R = 3-Cl) demonstrated significant antiproliferative activity, with GI50 values of 37 and 42 nM, respectively, demonstrating that the bromine atom is better tolerated for antiproliferative action than the chlorine atom. Furthermore, compounds 7c (R = 4-Cl) and 7d (R = 4-Br) had weak antiproliferative activity, with GI50 values of 90 nM and 82 nM, respectively, approximately twice as weak as the 3-halosubstituted derivatives 7i and 7h. Finally, with a GI50 value of 104 nM, the 4-fluoro derivative 7b (R = 4-F) was the least potent of all synthesised derivatives, 2.5-fold less active than the 3-halo-substituted derivatives 7i and 7h. These findings indicated that the type, position, and number of halogen atoms had a significant effect on antiproliferative activity, with activity increasing in the order 2,4-di-Cl > 3-Br > 3-Cl > 4-Br > 4-Cl > 4-F, .
Figure 4. GI50 of compounds 7b, 7c, 7d, 7h, 7i, 7j and Erlotinib.
The antiproliferative impact of the 1-naphthyl derivative 7m (R = 2,3-C4H4) was promising, with a GI50 value of 45 nM, which was 1.4-fold less potent than the reference Erlotinib (GI50 = 33 nM). On the other hand, the 2-naphthyl derivative 7n (R = 3,4-C4H4) demonstrated weak antiproliferative activity with a GI50 value of 77 nM, being 2.4-fold and 1.7-fold less potent than Erlotinib and the 1-naphthyl derivative 7m, respectively.
The unsubstituted derivative, compound 7a (R = H), and the 4-methyl derivative 7e (R = 4-CH3) were 3-fold less potent than the reference Erlotinib, with GI50 values of 96 and 101 nM, respectively, demonstrating the effect of the substitution pattern on the phenyl ring of the 1,2,4-oxadiazole moiety.
EGFR inhibitory assay
The inhibitory effects of the most potent antiproliferative derivatives 7h–l on EGFR, as a possible target for their antiproliferative action, were investigated using Erlotinib as the reference drugCitation28,Citation38. shows the results as IC50 values.
Table 2. IC50 values of compounds 7h, 7i, 7j, 7k, 7l, Erlotinib and Sorafenib against EGFR and VEGFR-2.
Generally, compounds 7h–l displayed potential anti-EGFR action, with IC50 values ranging from 76 to 105 nM. The results of this cell-based experiment are consistent with those of the antiproliferative assay, in which the most potent antiproliferative agent, 7l (R = 3,4,5-tri-OMe), was also the most potent anti-EGFR, with an IC50 value of 76 ± 06 nM, exceeding the reference Erlotinib, which had an IC50 value of 80 ± 05 nM. Compounds 7k (R = 3,4-dimethoxy) and 7j (R = 2,4-di-Cl) were placed second and third in EGFR inhibitory activity, with IC50 values of 82 and 89 nM, respectively, and were comparable to the reference Erlotinib.
These findings highlighted the significance of the methoxy group and the number of chlorine atoms in antiproliferative and anti-EGFR activities. Compounds 7h (R = 3-Cl) and 7i (R = 3-Br) displayed moderate anti-EGFR activity, with IC50 values of 105 ± 10 and 97 ± 9 nM, respectively, and were found to be less potent than the reference Erlotinib, which had an IC50 value of 80 ± 05. These findings indicated that derivatives 7j, 7k, and 7l, which require additional structural modifications to develop more potent derivatives, may be capable of inhibiting cancer cell proliferation by targeting the EGFR.
VEGFR inhibitory assay
The inhibitory efficacy of compounds 7h–l against VEGFR-2 was determined by applying kinase-glo-luminescent kinase assays with Sorafenib as the control medicationCitation39. displays the results as IC50 values.
The results showed that the investigated compounds inhibited VEGFR-2 significantly, with IC50 values ranging from 2.40 to 6.90 nM, compared to Sorafenib, which had an IC50 value of 0.17 nM. The most potent derivatives were 7j, 7k, and 7l, with IC50 values of 2.40, 3.80, and 4.70 nM, respectively. Again, compound 7l was the most potent derivative as a VEGFR-2 inhibitor, with an IC50 value of 2.40 ± 0.02 nM. These findings demonstrate that compounds 7j, 7k, and 7l are potent antiproliferative agents that may act as dual EGFR/VEGFR-2 inhibitors.
Apoptosis assay
Cancer can be treated by regulating or stopping the uncontrolled multiplication of cancer cells. Using the cell’s natural dying process is a highly effective method. Apoptosis evasion is a trait of cancer and is not specific to the aetiology or kind of cancer; hence, targeting apoptosis is useful for many types of cancer. Many anticancer drugs target various stages of both the intrinsic and extrinsic pathways Citation40–42. Compounds 7j, 7k, and 7 l, the most potent derivatives in all in vitro studies, were tested for their capacity to initiate the apoptosis cascade and reveal their proapoptotic potential.
Caspase-3 activation assay
Caspases are vital for the induction and maintenance of apoptosis. Caspase-3 is an essential caspase that cleaves many cell proteins, causing apoptosis Citation43,Citation44. Compounds 7j, 7k, and 7 l were investigated as caspase-3 activators against the human epithelial (A-594) cancer cell line Citation45, and the findings are shown in .
Table 3. Caspase-3, caspase-8, Bax, and Bcl-2 levels of compounds 7j, 7k, and 7l.
Compounds 7k and 7l showed a promising increase in caspase-3 protein levels of up to 530 ± 5 and 587 ± 5 pg/mL, respectively. They increased caspase 3 protein levels in the A-594 cancer cell line by 8- and 9-fold compared to untreated control cells. Compounds 7k and 7l were more active than the Staurosporine control, which exhibited 465 ± 4 pg/mL caspase-3 overexpression. Compound 7j was the least active derivative, with a caspase-3 overexpression of 460 ± 4 pg/mL and was comparable to the reference Staurosporine as a caspase-3 level inducer. According to these results, the examined compounds 7j, 7k, and 7l may have apoptotic potential activity, which may account for their antiproliferative effects.
Caspase-8, Bax, and Bcl-2 levels assays
Using Staurosporine as a control, the effects of compounds 7j, 7k, and 7l on the levels of caspase-8, Bax, and the anti-apoptotic protein Bcl-2 against the A-594 cancel cell line were further examined. presents the results.
demonstrated that compound 7l (2.55 ng/mL) had the highest levels of caspase-8 overexpression, followed by compound 7k (2.30 ng/mL) and finally compound 7j (2.00 ng/mL) when compared to the reference Staurosporine (1.85 ng/mL). The investigated compounds 7j, 7k, and 7l elevated caspase-8 levels by 22, 25, and 28 times, respectively, compared to the untreated control cell.
Furthermore, compounds 7k and 7l boosted Bax induction 34- and 40-fold (310 and 362 pg/mL, respectively) over untreated A-594 cancer cells, outperforming Staurosporine (288 pg/mL, a 32-fold induction), while compound 7j, on the other hand, induces Bax at a level comparable to Staurosporine. They were finally compared to Staurosporine, derivatives 7j, 7k, and 7l significantly reduced anti-apoptotic Bcl-2 protein levels in the A-594 cell line (5-, 6-, and 8-folds, respectively). These data suggest that compounds 7j, 7k, and 7l promote apoptosis by activating caspase-3, 8, and Bax and down-regulating the anti-apoptotic Bcl-2.
Docking study
EGFR active site
In silico docking simulations were performed for the most potent antiproliferative compounds, 7h, 7i, 7j, 7k, and 7l, to study their molecular interaction with the epidermal growth factor receptor tyrosine kinase EGFR. Molecular operating environment (MOE) softwareCitation46 was used, as well as the crystal structure of the EGFR in complex with Erlotinib (PDB: 1M17)Citation7.
MOE minimizations were performed with the force field (OPLS-AA) using Born solvation. Before simulations, the protein–ligand complex was protonated and corrected. The simulation results of the compounds were compared with Erlotinib, and the data are shown in . The docking protocol was validated by redocking the co-crystallised ligand into the EGFR binding site, where the docking S score obtained for the docked ligand was −10.70 kcal/mol with an RMSD value of 1.48 Å.
Table 4. Ligand–protein complex interactions of the tested compounds 7h, 7i, 7j, 7k, and 7l within the active site of EGFR.
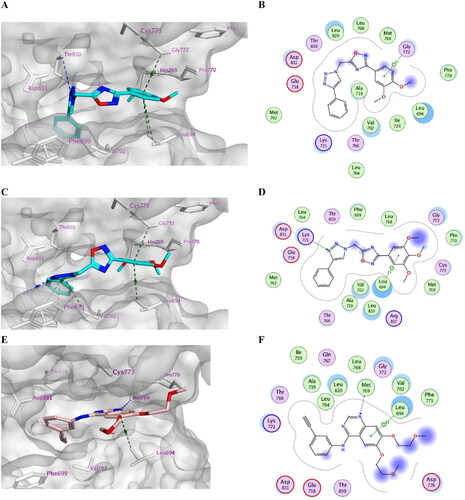
Concerning docking score analysis, compounds 7k and 7l exhibited the highest negative values (−9.52 and −9.57 kcal/mol, respectively), compatible with their in vitro EGFR inhibition effects (). Inspection of the ligand–protein complexes exposed that the ligand–protein interactions were mainly hydrophobic. The new 1,2,4-oxadiazole compounds are sterically fixed analogs of the open carboximidamide derivatives of previous workCitation27. Such a rigid conformation resulted in tight fitting within the EGFR active site with derivatives (R = H, 4-OMe, 3-Cl, 3-Br, 2,4-di-Cl, and 3,4,5-tri-OMe) as the cyclized structure better fits within the bioactive conformation. Compounds 7j, 7k, and 7l adopted an orientation within the large binding site where the phenyl triazole scaffold was inserted deeply into the hydrophobic pocket, aligned with Erlotinib phenyl acetylene moiety. The latter scaffold formed stacking with Phe699 and pi–H interaction with Val702. At the same time, the ligand 2,4-dichloro, 3,4-di-methoxy, or 3,4,5-trimethoxyphenyl moiety directed past the Erlotinib ether linkages at the gate of the binding site. This substituted phenyl moiety formed stacking with Pro770 and pi–H interaction with Leu694 and other hydrophobic interactions with nearby residues Lys721, Asp831, Val702, Leu694, and Gly772.
The trimethoxy derivative 7l showed the best binding mode and hydrophobic interactions with the surrounding amino acid residues owing to its increased van der Waals volume within the active site relative to the other derivatives (). However, the ligand loses H-bond interaction with the key amino acid residue Met769 compared with the H-bond formed by the quinazoline nitrogen of Erlotinib (3.56 Å) (). Alternatively, the ligand stabilises its complex by accepting a strong H-bond from Lys721 in the case of compound 7l with (3.50 Å) and a weak one from Thr830 in the case of compound 7k at the hydrophobic hinge ().
Figure 5. Docking representation models of compound 7k, 7l, and Erlotinib within the binding site of EGFR (H-bond: blue dashed lines, Pi–H; green dashed lines). (A) 3D-docked model of compound 7k (cyan) showing the protein surface (grey); (B) 2D-docked model of compound 7k; (C) 3D-docked model of compound 7l (cyan) showing the protein surface (grey); (D) 2D-docked model of compound 7l; (E) 3D-docked model of compound Erlotinib (pink) showing the protein surface (grey); (F) 2D-docked model of compound Erlotinib.
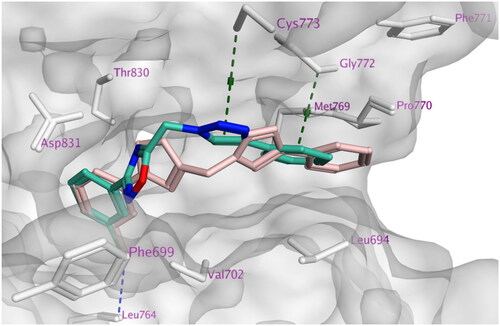
On the other hand, compounds 7h and 7i exhibited another orientation where m-chloro and m-bromophenyl moiety were better accommodated within the active site than 4-chloro and 4-bromo substituents. The m-halophenyl moiety stacked with Phe699 deeply inside the hydrophobic cleft of the enzyme and formed a halogen bond interaction with Leu764 in the case of compound 7i with (R = m-Br). However, the phenyl triazole scaffold formed stacking with Pro770 and pi–H interactions with Cys773 and Gly772 at the gate of the binding site. This indicated that the active site tolerated the m-halophenyl rather than the p-halophenyl moiety with a bulkier halogen atom ().
Figure 6. Docking representation model of compound 7h (cyan) aligned with compound 7i (pink) showing the protein surface (grey) of the site of EGFR (H-bond or halogen bond: blue dashed lines, Pi–H; green dashed lines).
VEGFR active site
Moreover, the most potent VEGFR-2 inhibitors, 7h, 7i, 7j, 7k, and 7l, were docked against vascular endothelial growth factor VEGFR-2. The crystal structure of VEGFR-2 in complex with Sorafenib (PDB: 4ASD)Citation47 was used in the present study. The docking protocol was validated by redocking the co-crystallised ligand with an S score of −10.73 kcal/mol with an RMSD value of 0.46 Å. Again, the trimethoxy compound 7l showed the highest negative score (−9.10 kcal/mol) among the tested compounds (). Inspection of the docked complexes revealed that the compounds bind tightly within the binding pocket. Compounds 7l and 7k exhibited good fitting (–C), where the phenyl triazole scaffold bi-stacked between Phe918 and Phe92 forming pi–H interaction with Leu840. However, there were missing H-bond interactions with Cys919 at the gate of the binding site. This indicated the significance of introducing an H-bond forming substituent at the phenyl ring to enhance the fitting. On the opposite end of the active site, the dimethoxy or trimethoxy phenyl moiety formed stacking with His1026 and pi–H interaction with Glu885. Interestingly, the oxadiazole moiety accepted a H-bond from Asp1046 and formed pi–H interactions with Phe1047 (7k) or Lys868 (7l).
Figure 7. Docking representation model of compounds 7k and 7l within the binding site of VEGFR-2 (H-bond: blue dashed lines, Pi–H; green dashed lines). (A) 3D-docked model of compound 7k (yellow) aligned with compound 7l (cyan) showing the lipophilicity surface of active site (purple; hydrophilic, white; neutral; green; lipophilic); (B) 2D-docked model of compound 7k; (C) 2D-docked model of compound 7l.
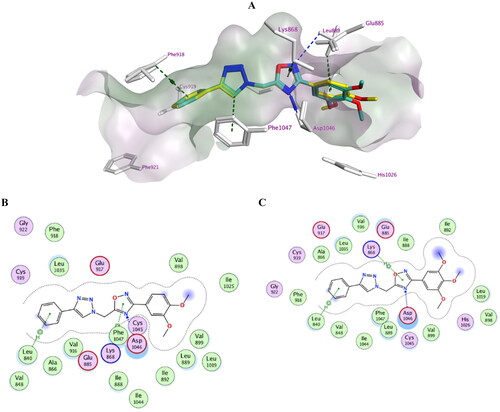
Table 5. Ligand–protein complex interactions of the tested compounds 7h, 7i, 7j, 7k, and 7l within the active site of VEGFR-2.
Other derivatives showed comparable binding modes within the active site; however, compound 7l showed the best fitting due to its enhanced van der Waals volume. Results of the docking simulations attributed to investigate the impact of introducing fixed geometry to the open carboximidamide derivatives on the binding modes within active sites and confirm dual EGFR/VEGFR-2 kinase inhibitory effects of compounds 7h, 7i, 7j, 7k, and 7l.
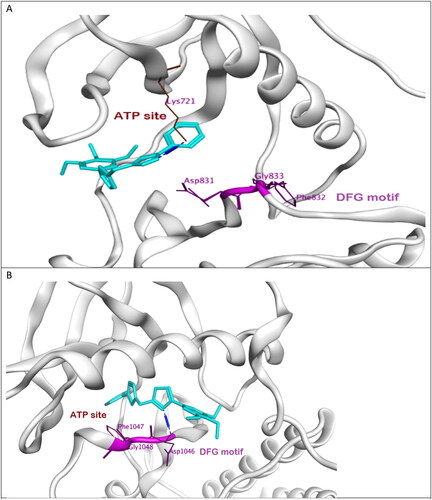
Moreover, the most potent compound 7l adopts DFG-in conformation of active form EGFR as erlotinib forming H-bond with Lys721 at the ATP binding site. However, the compound missed interaction with Asp831 at the DFG sequence. On the other hand, the same compound probes DFG-out conformation of inactive VEGFR-2 forming H-bond with Asp1046 that is equivalent to Asp831 at EGFR. We assumed that this new ligand could act as dual type I/II kinase inhibitor as stabilising DFG-in conformation at EGFR as well as DFG-out conformation at VEGFR-2 and thus could also inhibit phosphorylation, .
Figure 8. (A) Binding mode of compound 7l (cyan) to active site of EGFR showing DFG motif in purple. (B) Binding mode of compound 7l (cyan) to active site of VEGFR-2 showing DFG motif in purple.
In silico ADME/pharmacokinetics studies
The most potent antiproliferative 1,2,4-oxadiazole hybrids 7h, 7i, 7j, 7k, and 7l were examined for their ADME/Pharmacokinetics properties using the web tool Swiss ADMECitation48 by entering a list of the compounds’ SMILES (Simplified Molecule Input Line Entry Specification) provided by ChemDraw software.
The in silico pharmacokinetic data () showed that all 1,2,4-oxadiazole hybrids are orally active as they obey Lipinski’s rules of five with zero violation. Also, those hybrids exhibit high intestinal absorbance and are non-substrate for P-gp. None of those hybrids are likely to cross BBB except 7h, 7i, and 7j. Based on Lipinski’s rules, log P should be ≤5, 1,2,4-oxadiazole tested hybrids exhibited good permeability as indicated by log P values in the range of 2.77–3.85. Amongst compounds, the most potent antiproliferative compound, 7l, exhibited the lowest log P values and the highest TPSA, indicating the relevance of the latter physicochemical parameters to its highest activity. All tested compounds will likely be metabolised by CYP1A2, CYP2C19, and CYP2C9 but will be CYP2D3 inhibitors. However, only compounds 7k and 7l are considered substrates for CYP3A4. The predicted ADME properties are shown in ().
Table 6. Physicochemical and pharmacokinetic properties (Lipinski parameters) of compounds 7h, 7i, 7j, 7k, and 7l.
Table 7. ADME properties of compounds 7h, 7i, 7j, 7k, and 7l.
Structure activity relationship (SAR) analysis
In general, cyclisation of amidoximes 6a–o to 1,2,4-oxadiazole derivatives 7a–o increased the activity of the newly synthesised compounds. The new 1,2,4-oxadiazole compounds are sterically fixed analogues of the open carboximidamide derivatives 6a–o. Such a rigid conformation may result in tight fitting within the receptor active site.
The substitution on the phenyl group of the 1,2,4-oxadiazole moiety significantly increased the activity (except for methyl group) of 7a–o, and substitution with methoxy groups increased the activity more than halogen atoms. Similar reasoning applies to the amidoxime derivatives 6a–o, where substitution of the amidoxime moiety’s phenyl group with electron donating or electron withdrawing groups’ increases activity.
The number of methoxy groups substantially impacts the activity of 7a–o compounds where the trimethoxy derivative > dimethoxy > mono-methoxy one. In 6a–o, the dimethoxy derivative was the most tolerated for activity.
The type, position, and number of halogen atoms had a significant effect on antiproliferative activity of 7a–o, with activity increasing in the order 2,4-di-Cl > 3-Br > 3-Cl > 4-Br > 4-Cl > 4-F. The dihalo derivative was also more active than the mono-halo derivative in 6a–o, but the 4-position is better tolerated than the 3-position.
When the phenyl group in 6a–o and 7a–o is replaced with a naphthalene moiety, the activity increases, and 1-naphthyl is tolerated better than 2-naphthyl.
Conclusion
Fifteen novel 1,2,3-triazole/1,2,4-oxadiazole hybrids (7a–o) were synthesised and tested as antiproliferative agents with dual EGFR/VEGFR-2 inhibitory activity. The newly synthesised compounds showed promising antiproliferative activity, with the hybrid 7h–l being the most potent class. In vitro investigations revealed that compounds 7j, 7k, and 7l were effective antiproliferative agents that could operate as dual EGFR/VEGFR-2 inhibitors. Moreover, apoptotic-inducing activity experiments indicate that compounds 7j, 7k, and 7l enhance apoptosis by activating caspase-3, 8, and Bax and down-regulating the anti-apoptotic Bcl-2. Docking simulations highlighted the output of including 1,2,4-oxadiazole scaffold to improve the binding of the compounds within the active sites of EGFR and VEGFR-2. The cyclized structures fit well within the bioactive conformations. In addition, the phenyl triazole scaffold bound considerably within the hydrophobic pocket of binding sites.
Moreover, there is a future concern about introducing an H-bond forming substituent at the phenyl ring to tolerate binding with Cys919 within the VEGFR-2 active site. Also, docking results revealed that 7l and 7k exhibited the best binding mode within both active sites. In silico ADME and pharmacokinetic study, the compounds were predicted to have acceptable bioavailability and pharmacokinetic profiles.
Materials and methods
Chemistry
General details: see Appendix A
Compounds 1, 2, 5a–o, and 6a–o were prepared according to previously reported literatureCitation27,Citation31–33.
General procedures for the synthesis of compounds 7a–o
A stirred solution of the appropriate N'-(((4-phenyl-1H-1,2,3-triazol-1-yl)acetyl)oxy)benzene-carboximidamide (6a–o) (0.78 mmole) in dry acetonitrile (20 mL) was refluxed overnight. After the reaction was completed (monitored with TLC), the excess solvent evaporated under reduced pressure. The obtained precipitate was washed with cold water, filtered, and crystallised from acetonitrile.
3-Phenyl-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7a)Citation49
Yield: 0.19 g (78%), white solid, mp: 146–148 °C, Rf: 0.72 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3126, 3100, 2964 (CH), 1596, 1580, 1467 (C = N and C = C), 766 and 689 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.81 (s, 1H, triazole-CH), 7.99 (d, J = 7.9 Hz, 2H, Ar–H), 7.90 (d, J = 8.5 Hz, 2H, Ar–H), 7.62–7.56 (m, 3H, Ar–H), 7.48 (t, J = 7.6 Hz, 2H, Ar–H), 7.37 (t, J = 7.4 Hz, 1H, Ar–H), 6.29 (s, 2H, N–CH2). Anal. Calc. (%) for C17H13N5O: C, 67.32; H, 4.32; N, 23.09. Found: C, 67.49; H, 4.60; N, 23.37.
3-(4-Fluorophenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7b)
Yield: 0.12 g (48%), white solid, mp: 137–139 °C, Rf: 0.74 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3125, 3099, 2979, 2952 (CH), 1608, 1578, 1469 (C = N and C = C), 848, 770, 700 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.81 (s, 1H, triazole-CH), 8.05 (q, J = 4.8 Hz, 2H, Ar–H), 7.90 (d, J = 7.4 Hz, 2H, Ar–H), 7.48 (t, J = 7.4 Hz, 2H, Ar–H), 7.39 (q, J = 8.6 Hz, 3H, Ar–H), 6.30 (s, 2H, N–CH2). 13C NMR (100 MHz, δ ppm DMSO-d6): 175.1, 167.7, 166.2, 162.9, 147.3, 130.7, 130.2 (d, J = 9.2 Hz), 129.5, 128.6, 125.7, 123.1, 117.0 (d, J = 22.4 Hz), 45.5. Anal. Calc. (%) for C17H12FN5O: C, 63.55; H, 3.76; N, 21.80. Found: C, 63.38; H, 3.91; N, 22.03.
3-(4-Chlorophenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7c)
Yield: 0.16 g (61%), white solid, mp: 150–152 °C, Rf: 0.71 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3137, 3086, 2977, 2957 (CH), 1598, 1569, 1467 (C = N and C = C), 844, 766, 697 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 8.00 (d, J = 8.5 Hz, 2H, Ar–H), 7.89 (d, J = 7.4 Hz, 2H, Ar–H), 7.63 (d, J = 8.5 Hz, 2H, Ar–H), 7.47 (t, J = 7.6 Hz, 2H, Ar–H), 7.36 (t, J = 7.3 Hz, 1H, Ar–H), 6.29 (s, 2H, N–CH2). Anal. Calc. (%) for C17H12ClN5O: C, 60.45; H, 3.58; N, 20.73. Found: C, 60.71; H, 3.69; N, 20.95.
3-(4-Bromophenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7d)
Yield: 0.22 g (74%), white solid, mp: 158–160 °C, Rf: 0.73 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3122, 3093 (CH), 1592, 1564, 1459 (C = N and C = C), 838, 766, 697 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 7.92 (d, J = 8.6 Hz, 2H, Ar–H), 7.90 (d, J = 7.1 Hz, 2H, Ar–H), 7.78 (d, J = 8.6 Hz, 2H, Ar–H), 7.48 (t, J = 7.7 Hz, 2H, Ar–H), 7.37 (t, J = 7.5 Hz, 1H, Ar–H), 6.29 (s, 2H, N–CH2). Anal. Calc. (%) for C17H12BrN5O: C, 53.42; H, 3.16; N, 18.32. Found: C, 53.66; H, 3.40; N, 18.59.
3-(4-Methylphenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7e)
Yield: 0.20 g (83%), white solid, mp: 140–142 °C, Rf: 0.69 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3121, 3089, 2966, 2935 (CH), 1604, 1574, 1466 (C = N and C = C), 834, 769, 693 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.81 (s, 1H, triazole-CH), 7.92–7.87 (m, 4H, Ar–H), 7.48 (d, J = 7.5 Hz, 2H, Ar–H), 7.40–7.35 (m, 3H, Ar–H), 6.28 (s, 2H, N–CH2), 2.38 (s, 3H, Ar–CH3). 13C NMR (100 MHz, δ ppm DMSO-d6): 174.7, 168.4, 147.3, 142.4, 130.7, 130.4, 129.5, 128.7, 127.5, 125.8, 123.3, 123.1, 45.6, 21.6. Anal. Calc. (%) for C18H15N5O: C, 68.13; H, 4.76; N, 22.07. Found: C, 68.35; H, 4.88; N, 21.89.
3-(4-Methoxyphenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7f)
Yield: 0.19 g (75%), white solid, mp: 148–150 °C, Rf: 0.70 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3133, 2977, 2843 (CH), 1617, 1576, 1469 (C = N and C = C), 843, 769, 698 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 7.93 (d, J = 8.9 Hz, 2H, Ar–H), 7.90 (d, J = 7 Hz, 2H, Ar–H), 7.48 (t, J = 7.5 Hz, 2H, Ar–H), 7.37 (t, J = 7.4 Hz, 1H, Ar–H), 7.11 (d, J = 8.9 Hz, 2H, Ar–H), 6.26 (s, 2H, N–CH2), 3.83 (s, 3H, Ar–OCH3). Anal. Calc. (%) for C18H15N5O2: C, 64.86; H, 4.54; N, 21.01. Found: C, 65.08; H, 4.67; N, 21.28.
3-(4-Trifluoromethylphenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7g)
Yield: 0.13 g (46%), white solid, mp: 166–168 °C, Rf: 0.7 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3144, 3001, 2965 (CH), 1602, 1579, 1468 (C = N and C = C), 856, 767, 694 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.82 (s, 1H, triazole-CH), 8.21 (d, J = 8.1 Hz, 2H, Ar–H), 7.94 (d, J = 8.4 Hz, 2H, Ar–H), 7.90 (d, J = 7.5 Hz, 2H, Ar–H), 7.48 (t, J = 7.5 Hz, 2H, Ar–H), 7.37 (t, J = 7.3 Hz, 1H, Ar–H), 6.33 (s, 2H, N–CH2). 13C NMR (100 MHz, δ ppm DMSO-d6): 175.1, 167.2, 147.1, 131.8 (q, J = 32.2 Hz), 130.3, 129.6, 129.2, 128.4, 128.2, 126.5 (q, J = 3.5 Hz), 125.5, 124.0 (q, J = 272.7 Hz), 122.9, 45.3. Anal. Calc. (%) for C18H12F3N5O: C, 58.22; H, 3.26; N, 18.86. Found: C, 58.43; H, 3.39; N, 19.07.
3-(3-Chlorophenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7h)
Yield: 0.17 g (67%), white solid, mp: 128–130 °C, Rf: 0.71 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3121, 3095, 2966 (CH), 1606, 1574, 1465 (C = N and C = C), 892, 796, 767, 736, 693 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 7.97–7.94 (m, 2H, Ar–H), 7.89 (d, J = 7.3 Hz, 2H, Ar–H), 7.69 (d, J = 8.7 Hz, 1H, Ar–H), 7.60 (t, J = 8.1 Hz, 1H, Ar–H), 7.47 (t, J = 7.6 Hz, 2H, Ar–H), 7.37 (t, J = 7.3 Hz, 1H, Ar–H), 6.30 (s, 2H, N–CH2). 13C NMR (100 MHz, δ ppm DMSO-d6): 174.8, 167.0, 146.9, 134.0, 131.8, 131.5, 130.2, 129.0, 128.2, 127.5, 126.5, 125.8, 125.3, 122.7, 45.1. Anal. Calc. (%) for C17H12ClN5O: C, 60.45; H, 3.58; N, 20.73. Found: C, 60.72; H, 3.79; N, 21.01.
3-(3-Bromophenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7i)
Yield: 0.23 g (77%), white solid, mp: 139–141 °C, Rf: 0.72 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3143, 3067, 3002 (CH), 1595, 1564, 1471 (C = N and C = C), 903, 800, 767, 737, 694 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 8.10 (t, J = 1.7 Hz, 1H, Ar–H), 7.99 (two t, J = 8, 2.5 Hz, 1H, Ar–H), 7.89 (d, J = 7.1 Hz, 2H, Ar–H), 7.84–7.80 (m, 1H, Ar–H), 7.54 (t, J = 7.9 Hz, 1H, Ar–H), 7.48 (t, J = 7.5 Hz, 2H, Ar–H), 7.37 (t, J = 7.4 Hz, 1H, Ar–H), 6.30 (s, 2H, N–CH2). Anal. Calc. (%) C17H12BrN5O: C, 53.42; H, 3.16; N, 18.32. Found: C, 53.68; H, 3.29; N, 18.57.
3-(2,4-Dichlorophenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7j)
Yield: 0.16 g (54%), white solid, mp: 137–139 °C, Rf: 0.73 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3146, 3080, 2992 (CH), 1602, 1588, 1469 (C = N and C = C), 881, 762, 690 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 7.93–7.88 (m, 4H, Ar–H), 7.64 (two d, J = 8.4, 2.1 Hz, 1H, Ar–H), 7.48 (t, J = 8.4 Hz, 2H, Ar–H), 7.37 (t, J = 7.4 Hz, 1H, Ar–H), 6.32 (s, 2H, N–CH2). 13C NMR (100 MHz, δ ppm DMSO-d6): 174.1, 166.0, 146.9, 136.8, 133.2, 133.0, 130.6, 130.2, 129.0, 128.2, 128.1, 125.3, 123.8, 122.7, 45.0 Anal. Calc. (%) C17H11Cl2N5O: C, 54.86; H, 2.98; N, 18.82. Found: C, 54.98; H, 3.12; N, 19.09.
3-(3,4-Dimethoxyphenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7k)
Yield: 0.25 g (89%), white solid, mp: 174–176 °C, Rf: 0.68 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3125, 3096, 2943, 2838 (CH), 1601, 1583, 1499 (C = N and C = C), 864, 765, 687 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 7.89 (d, J = 7.3 Hz, 2H, Ar–H), 7.57 (two d, J = 8.4, 1.8 Hz, 1H, Ar–H), 7.49–7.45 (m, 3H, Ar–H), 7.36 (t, J = 7.3 Hz, 1H, Ar–H), 7.11 (d, J = 8.5 Hz, 1H, Ar–H), 6.26 (s, 2H, N–CH2), 3.82 (s, 6H, Ar–(OCH3)2). 13C NMR (100 MHz, δ ppm DMSO-d6): 174.0, 167.8, 151.7, 149.1, 146.9, 130.3, 129.0, 128.2, 125.3, 122.7, 120.7, 117.8, 112.0, 109.5, 55.7, 55.6, 45.1. Anal. Calc. (%) C19H17N5O3: C, 62.80; H, 4.72; N, 19.27. Found: C, 62.67; H, 4.89; N, 19.51.
3-(3,4,5-Trimethoxyphenyl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7l)
Yield: 0.21 g (69%), white solid, mp: 154–156 °C, Rf: 0.74 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3143, 3000, 2947, 2847 (CH), 1604, 1581, 1467 (C = N and C = C), 762, 689 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.81 (s, 1H, triazole-CH), 7.89 (d, J = 7 Hz, 2H, Ar–H), 7.48 (t, J = 7.5 Hz, 2H, Ar–H), 7.37 (t, J = 7.4 Hz, 1H, Ar–H), 7.26 (s, 2H, Ar–H), 6.27 (s, 2H, N–CH2), 3.85 (s, 6H, Ar–(OCH3)2), 3.74 (s, 3H, Ar–OCH3). Anal. Calc. (%) C20H19N5O4: C, 61.06; H, 4.87; N, 17.80. Found: C, 61.28; H, 4.95; N, 18.04.
3-(Naphthalen-1-yl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7m)
Yield: 0.21 g (78%), white solid, mp: 144–146 °C, Rf: 0.69 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3120, 3088, 2975, 2943 (CH), 1607, 1592, 1464 (C = N and C = C), 767, 747, 694 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.86 (s, 1H, triazole-CH), 8.76 (d, J = 8.3 Hz, 1H, Ar–H), 8.19 (d, J = 7.7 Hz, 2H, Ar–H), 8.08 (d, J = 7.9 Hz, 1H, Ar–H), 7.91 (d, J = 7.2 Hz, 2H, Ar–H), 7.72–7.63 (m, 3H, Ar–H), 7.48 (t, J = 7.5 Hz, 2H, Ar–H), 7.37 (t, J = 7.3 Hz, 1H, Ar–H), 6.37 (s, 2H, N–CH2). 13C NMR (100 MHz, δ ppm DMSO-d6): 173.6, 168.3, 146.9, 133.5, 132.3, 130.3, 129.7, 129.5, 129.0, 128.9, 128.2, 127.9, 126.7, 125.5, 125.4, 125.3, 122.7, 122.5, 45.2. Anal. Calc. (%) C21H15N5O: C, 71.38; H, 4.28; N, 19.82. Found: C, 71.09; H, 4.41; N, 20.04.
3-(Naphthalen-2-yl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7n)
Yield: 0.23 g (84%), white solid, mp: 180–182 °C, Rf: 0.66 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3117, 3088, 2971, 2939 (CH), 1600, 1462 (C = N and C = C), 764, 756, 689 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.81 (s, 1H, triazole-CH), 8.61 (s, 1H, Ar–H), 8.12 (d, J = 7.8 Hz, 1H, Ar–H), 8.06 (t, J = 7.5 Hz, 2H, Ar–H), 8.00 (d, J = 7.8 Hz, 1H, Ar–H), 7.91 (d, J = 7.9 Hz, 2H, Ar–H), 7.66–7.59 (m, 2H, Ar–H), 7.48 (t, J = 7.6 Hz, 2H, Ar–H), 7.37 (t, J = 7.4 Hz, 1H, Ar–H), 6.31 (s, 2H, N–CH2). Anal. Calc. (%) C21H15N5O: C, 71.38; H, 4.28; N, 19.82. Found: C, 71.21; H, 4.52; N, 20.06.
3-(2H-1,3-Benzodioxol-5-yl)-5-[(4-phenyl-1H-1,2,3-triazol-1-yl)methyl]-1,2,4-oxadiazole (7o)
Yield: 0.18 g (67%), white solid, mp: 149–151 °C, Rf: 0.67 (hexane:ethyl acetate, 2:1, v/v). IR (FTIR): νmax (cm−1) 3141, 2982, 2914 (CH), 1583, 1452 (C = N and C = C), 761, 692 (Ar–CH bending). 1H NMR (400 MHz, δ ppm DMSO-d6): 8.80 (s, 1H, triazole-CH), 7.89 (d, J = 7.1 Hz, 2H, Ar–H), 7.55 (two d, J = 8.13, 1.7 Hz, 1H, Ar–H), 7.48 (d, J = 7.6 Hz, 2H, Ar–H), 7.43 (d, J = 1.6 Hz, 1H, Ar–H), 7.37 (t, J = 7.4 Hz, 1H, Ar–H), 7.08 (d, J = 8.13 Hz, 1H, Ar–H), 6.25 (s, 2H, –O–CH2–O–), 6.14 (s, 2H, N–CH2). 13C NMR (100 MHz, δ ppm DMSO-d6): 174.1, 167.6, 150.3, 148.1, 146.9, 130.3, 129.0, 128.2, 125.3, 122.7, 122.3, 119.2, 109.0, 106.5, 102.0, 45.1. Anal. Calc. (%) C18H13N5O3: C, 62.24; H, 3.77; N, 20.16. Found: C, 62.45; H, 3.85; N, 20.42.
Biology
Assay of cell viability effect
To investigate the effect of compounds 7a–o on normal cell lines, a cell viability experiment was performed on the MCF-10A (human mammary gland epithelial) cell line. This experiment uses 50 µM of the investigated compound for four daysCitation34,Citation35. Refer to Appendix A (Supp. File) for more details.
Assay of antiproliferative effect
The antiproliferative activity of 1,2,4-oxadiazoles 7a–o against four human cancer cell lines was assessed using Erlotinib as the reference medicationCitation36,Citation37. See Appendix A for more details.
Assay of EGFR inhibitory activity
The inhibitory effects of the most potent antiproliferative compounds 7h–l on EGFR, as a potential target for their antiproliferative action, were studied using Erlotinib as a control medicationCitation38. For more information, see Appendix A.
Assay of VEGFR-2 inhibitory activity
Compounds 7h–l’s inhibitory activity against VEGFR-2 was assessed using kinase-glo-luminescent kinase assays with Sorafenib as the control drugCitation39. For more information, see Appendix A.
Apoptosis-inducing activity assay
Compounds 7j, 7k, and 7l, the most potent derivatives in all in vitro studies, were tested for their capacity to initiate the apoptosis cascade and reveal their proapoptotic potentialCitation45. For more information, see Appendix A.
Docking
All molecular modelling calculations and docking simulation studies were performed on a Processor Intel (R) Pentium (R) CPU N3510@ 1.99 GHz and 4 GB memory with Microsoft Windows 8.1 pro (64 Bit) operating system using Molecular Operating Environment (MOE 2019.0102, 2020; Chemical Computing Group, Canada) as the computational software. MOE minimizations were performed until an RMSD gradient of 0.01 kcal/mol/Å with the force field (OPLS-AA) to calculate the partial charges automatically using Born solvation. Before simulations, the protein was corrected, hydrogens were added, and ionisation states were assigned; the system was optimised via protonation, distant water molecules were deleted, and the receptor was minimised using the QuickPrep function. Then, both ligand and pocket were isolated in 3D, and molecular surface was drawn around the binding site to visualise the space available for docked ligands. The compounds’ database was created after the compounds’ structures were prepared, partially charged, and minimised. Initially, self-docking was performed, and ligand conformations were generated with the bond rotation method. These are then placed on the site with the Triangle Matcher method and ranked with the London dG scoring function. The retain option specifies the number of poses (30) to pass to the refinement for energy minimisation in the pocket before restoring with the GBVI/WSA dG scoring function. Docking simulation was performed using a compound database mdb file. A triangle matching with London dG scoring was chosen for initial placement, and then the top 30 poses were refined using force field (OPLS-AA) and GBVI/WSA dG scoring. The output database dock file was created with different poses for each ligand and arranged according to the final score function (S), which is the score of the last stage that was not set to zero.
Supplemental Material
Download PDF (4.8 MB)Acknowledgements
This work was funded by the Researchers Supporting Project Number [RSP2023R273] at King Saud University, Riyadh, Saudi Arabia. The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Disclosure statement
The author reported no potential conflicts of interests.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Akram M, Iqbal M, Daniyal M, Khan AU. Awareness and current knowledge of breast cancer. Biol Res. 2017;50(1):33.
- Youssif BGM, Abdelrahman MH, Abdelazeem AH, Abdelgawad MA, Ibrahim HM, Salem OIA, Mohamed MFA, Treambleau L, Bukhari SNA. Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino[1,2-a]indol-1 (2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur J Med Chem. 2018;146:260–273.
- Guo YJ, Pan WW, Liu SB, Shen ZF, Xu Y, Hu LL. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med. 2020;19(3):1997–2007.
- Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr Relat Cancer. 2001;8(3):161–173.
- Huang L, Fu L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 2015;5(5):390–401.
- Yewale C, Baradia D, Vhora I, Patil S, Misra A. Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials. 2013;34(34):8690–8707.
- Mohamed FA, Gomaa HA, Hendawy O, Ali AT, Farghaly HS, Gouda AM, Abdelazeem AH, Abdelrahman MH, Trembleau L, Youssif BG. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg Chem. 2021;112:104960.
- Modi SJ, Kulkarni VM. Vascular endothelial growth factor receptor (VEGFR-2)/KDR inhibitors: medicinal chemistry perspective. Med Drug Discov. 2019;2:100009.
- Falcon BL, Chintharlapalli S, Uhlik MT, Pytowski B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol Ther. 2016;164:204–225.
- Peng F-W, Liu D-K, Zhang Q-W, Xu Y-G, Shi L. VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012–2016). Expert Opin Ther Pat. 2017;27(9):987–1004.
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134.
- Liao JJ-L. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J Med Chem. 2007;50(3):409–424.
- Wheler JJ, Janku F, Naing A, Li Y, Stephen B, Zinner R, Subbiah V, Fu S, Karp D, Falchook GS, et al. TP53 alterations correlate with response to VEGF/VEGFR inhibitors: implications for targeted therapeutics. Mol Cancer Ther. 2016;15(10):2475–2485.
- Xie C, Wan X, Quan H, Zheng M, Fu L, Li Y, Lou L. Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor‐2 inhibitor. Cancer Sci. 2018;109(4):1207–1219.
- Liu Z-L, Chen H-H, Zheng L-L, Sun L-P, Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. 2023;8(1):198.
- Sun S, Zhang J, Wang N, Kong X, Fu F, Wang H, Yao J. Design and discovery of quinazoline- and thiourea-containing sorafenib analogs as EGFR and VEGFR-2 dual TK inhibitors. Molecules. 2017;23(1):24.
- Liu X-J, Zhao H-C, Hou S-J, Zhang H-J, Cheng L, Yuan S, Zhang L-R, Song J, Zhang S-Y, Chen S-W. Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg Chem. 2023;133:106425.
- Farghaly TA, Al-Hasani WA, Abdulwahab HG. An updated patent review of VEGFR-2 inhibitors (2017–present). Expert Opin Ther Pat. 2021;31(11):989–1007.
- Zhang H-Q, Gong F-H, Li C-G, Zhang C, Wang Y-J, Xu Y-G, Sun L-P. Design and discovery of 4-anilinoquinazoline-acylamino derivatives as EGFR and VEGFR-2 dual TK inhibitors. Eur J Med Chem. 2016;109:371–379.
- Biernacki K, Daśko M, Ciupak O, Kubiński K, Rachon J, Demkowicz S. Novel 1,2,4-oxadiazole derivatives in drug discovery. Pharmaceuticals. 2020;13(6):111.
- Wang J-J, Sun W, Jia W-D, Bian M, Yu L-J. Research progress on the synthesis and pharmacology of 1,3,4-oxadiazole and 1,2,4-oxadiazole derivatives: a mini review. J Enzyme Inhib Med Chem. 2022;37(1):2304–2319.
- Coupar I, Hedges A, Metcalfe HL, Turner P. Effect of aminophylline, butalamine and imolamine on human isolated smooth muscle. J Pharm Pharmacol. 1969;21(7):474–475.
- Rotbart HA, Webster AD, Group PTR, Pleconaril Treatment Registry Group. Treatment of potentially life-threatening enterovirus infections with pleconaril. Clin Infect Dis. 2001;32(2):228–235.
- McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, Heydemann P, Kaminska A, Kirschner J, Muntoni F, Clinical Evaluator Training Group; ACT DMD Study Group, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10101):1489–1498.
- Dokla EM, Fang C-S, Abouzid KA, Chen CS. 1,2,4-Oxadiazole derivatives targeting EGFR and c-Met degradation in TKI resistant NSCLC. Eur J Med Chem. 2019;182:111607.
- Venu K, Saritha B, Sailaja BBV. New molecular hybrids containing benzimidazole, thiazolidine-2,4-dione and 1,2,4-oxadiazole as EGFR directing cytotoxic agents. Tetrahedron. 2022;124:132991.
- Mahmoud MA, Mohammed AF, Salem OI, Rabea SM, Youssif BG. Design, synthesis, and antiproliferative properties of new 1,2,3-triazole-carboximidamide derivatives as dual EGFR/VEGFR-2 inhibitors. J Mol Struct. 2023;1282:135165.
- Al-Wahaibi LH, Mahmoud MA, Mostafa YA, Raslan AE, Youssif BG. Novel piperine-carboximidamide hybrids: design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J Enzyme Inhib Med Chem. 2023;38 (1):376–386.
- Al-Wahaibi LH, Mohammed AF, Abdel Rahman FE-ZS, Abdelrahman MH, Gu X, Trembleau L, Youssif BG. Design, synthesis, apoptotic, and antiproliferative effects of 5-chloro-3-(2-methoxyvinyl)-indole-2-carboxamides and pyrido[3,4-b]indol-1-ones as potent EGFRWT/EGFRT790M inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):2218602.
- El-Sheref EM, Bräse S, Tawfeek HN, Alasmary FA, Youssif BG. Synthesis, antioxidant and antiproliferative actions of 4-(1,2,3-triazol-1-yl)quinolin-2 (1H)-ones as multi-target inhibitors. Int J Mol Sci. 2023;24(17):13300.
- Abdu-Allah HH, Youssif BG, Abdelrahman MH, Abdel-Hamid MK, Reshma RS, Yogeeswari P, Aboul-Fadl T, Sriram D. Synthesis and anti-mycobacterial activity of 4-(4-phenyl-1H-1,2,3-triazol-1-yl)salicylhydrazones: revitalizing an old drug. Arch Pharm Res. 2017;40(2):168–179.
- Talukdar S, Hsu J-L, Chou T-C, Fang J-M. Direct transformation of aldehydes to nitriles using iodine in ammonia water. Tetrahedron Lett . 2001;42(6):1103–1105.
- Frejat FOA, Zhai H, Cao Y, Wang L, Mostafa YA, Gomaa HA, Youssif BG, Wu C. Novel indazole derivatives as potent apoptotic antiproliferative agents by multi-targeted mechanism: synthesis and biological evaluation. Bioorg Chem. 2022;126:105922.
- Gomaa HAM, Shaker ME, Alzarea SI, Hendawy OM, Mohamed FAM, Gouda AM, Ali AT, Morcoss MM, Abdelrahman MH, Trembleau L, et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg Chem. 2022;120:105616.
- Gomaa HA, El-Sherief HA, Hussein S, Gouda AM, Salem OI, Alharbi KS, Hayallah AM, Youssif BG. Novel 1,2,4-triazole derivatives as apoptotic inducers targeting p53: synthesis and antiproliferative activity. Bioorg Chem. 2020;105:104369.
- Youssif BG, Gouda AM, Moustafa AH, Abdelhamid AA, Gomaa HA, Kamal I, Marzouk AA. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of BRAFV600E/p38α with potential antiproliferative activity. J Mol Struct. 2022;1253:132218.
- Mekheimer RA, Allam SMR, Al-Sheikh MA, Moustafa MS, Al-Mousawi SM, Mostafa YA, Youssif BGM, Gomaa HAM, Hayallah AM, Abdelaziz M, et al. Discovery of new pyrimido[5,4-c]quinolines as potential antiproliferative agents with multitarget actions: rapid synthesis, docking, and ADME studies. Bioorg Chem. 2022;121:105693.
- Mahmoud MA, Mohammed AF, Salem OI, Gomaa HA, Youssif BG. New 1,3,4‐oxadiazoles linked with the 1,2,3‐triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase. Arch Pharm (Weinheim). 2022;355(6):e2200009.
- Marzouk AA, Abdel-Aziz SA, Abdelrahman KS, Wanas AS, Gouda AM, Youssif BG, Abdel-Aziz M. Design and synthesis of new 1,6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: molecular docking and antiproliferative evaluation. Bioorg Chem. 2020;102:104090.
- Liu Y, Zhu X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl Neurodegener. 2017;6(1):21.
- Villa-Pulgarín JA, Gajate C, Botet J, Jimenez A, Justies N, Varela-M RE, Cuesta-Marbán Á, Müller I, Modolell M, Revuelta JL, et al. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl Trop Dis. 2017;11(8):e0005805.
- Bao H, Zhang Q, Zhu Z, Xu H, Ding F, Wang M, Du S, Du Y, Yan Z. BHX, a novel pyrazoline derivative, inhibits breast cancer cell invasion by reversing the epithelial–mesenchymal transition and down-regulating Wnt/β-catenin signalling. Sci Rep. 2017;7(1):9153.
- Al-Wahaibi LH, Mostafa YA, Abdelrahman MH, El-Bahrawy AH, Trembleau L, Youssif BG. Synthesis and biological evaluation of indole-2-carboxamides with potent apoptotic antiproliferative activity as EGFR/CDK2 dual inhibitors. Pharmaceuticals. 2022;15(8):1006.
- Choudhary GS, Al-Harbi S, Almasan A. Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Methods Mol Biol. 2015;1219:1–9.
- Youssif BG, Mohamed AM, Osman EEA, Abou-Ghadir OF, Elnaggar DH, Abdelrahman MH, Treamblu L, Gomaa HA. 5-Chlorobenzofuran-2-carboxamides: from allosteric CB1 modulators to potential apoptotic antitumor agents. Eur J Med Chem. 2019;177:1–11.
- Shattuck TW. Colby college molecular mechanics exercises MOE (molecular operating environment) exercises. Montreal, QC: Chemical Computing Group ULC; 2011.
- McTigue M, Murray BW, Chen JH, Deng Y-L, Solowiej J, Kania RS. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc Natl Acad Sci U S A. 2012;109(45):18281–18289.
- Daina A, Michielin O, Zoete VJ. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717.
- Pokhodylo NT, Savka RD, Shyyka OY, Obushak MD. One‐pot CuAAC synthesis of (1H‐1,2,3‐triazol‐1‐yl) methyl‐1,3,4/1,2,4‐oxadiazoles starting from available chloromethyl‐1,3,4/1,2,4‐oxadiazoles. J Heterocycl Chem. 2020;57(7):2969–2976.