
Abstract
Novel coumarin derivatives were synthesised and tested for their cytotoxicity against human cancer cells (PC-3 and MDA-MB-231). Compounds 5, 4b, and 4a possessed potent cytotoxic activity against PC-3 cells with IC50 3.56, 8.99, and 10.22 µM, respectively. Compound 4c displayed cytotoxicity more than erlotinib in the MDA-MB-231 cells with IC50 8.5 µM. Moreover, compound 5 exhibited potent inhibitory activity on EFGR with IC50 0.1812 µM, as well as PI3Kβ inhibitory activity that was twofold higher than LY294002, suggesting that this compound has a dual EGFR and PI3Kβ inhibiting activity. Docking aligns with the in vitro results and sheds light on the molecular mechanisms underlying dual targeting. Furthermore, compound 5 decreased AKT and m-TOR expression in PC-3 cells, showing that it specifically targets these cells via the EGFR/PI3K/Akt/m-TOR signalling pathway. Simultaneously, compound 5 caused cell cycle arrest at S phase and induced activation of both intrinsic and extrinsic apoptotic pathways.
Introduction
Prostate cancer is the second largest cause of cancer death in males, as in 2020, 1 414 259 new cases and 375 304 deaths were documented globally Citation1,Citation2. The most prevalent cause of death from prostate cancer is incurable metastatic illnesses Citation3. Many people with early-stage prostate cancer have a good outlook after having a range of treatments, such as a prostatectomy, radiation, hormone therapy, and even active surveillance; however, the prognosis of late-stage and metastatic prostate cancer patients remains challenging Citation4. Even though the FDA has approved several drugs for the treatment of prostate cancer due to the low tumour response, researchers never stop looking for novel therapeutics to provide more options for castration-resistant prostate cancer Citation5–7.
Different forms of cancer are influenced by the phosphoinositide 3-kinase (PI3K). PI3Ks are lipid kinases that play important roles in the control of cell growth, proliferation, and angiogenesis Citation8. PI3K enzymes can be categorised into three classes based on their structure and function; class I has undergone the most research, and its subclasses include PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ Citation9. On the other hand, a proto-oncogenic serine/threonine kinase called AKT participates in apoptosis, cell migration, glucose metabolism, and proliferation of cells Citation10. AKT can inhibit the BAX and caspase families of proteins to demonstrate its antiapoptotic effects Citation11. Additionally, a typical serine/threonine kinase called mTOR controls how cells develop and metabolise food and energy in response to external stimuli Citation12,Citation13. The PI3K/AKT/mTOR axis connects various signalling pathways within cells in prostate cancer. This axis has direct and indirect influence on various biological processes, including cell cycle progression, cellular proliferation, autophagy, and angiogenesis Citation14–16. Consequently, the selective inhibition of the PI3K enzyme offers a promising approach for developing effective anticancer medications.
A considerable increase in the levels of epidermal growth factor receptor (EGFR), a protein kinase, in 20–40% of prostate cancer patients is recorded. This increase is linked to higher-grade tumours, more aggressive phases, a worse prognosis, and an increased risk of establishing a castration-resistant phenotype and cancer spreading Citation17,Citation18. As a result, medicines that may inhibit both the PI3K/AKT/mTOR and the EGFR signalling pathways at the same time may have therapeutic potential in advanced prostate cancer. The diverse biological activities of coumarins (benzopyrones) and their derivatives are caused by the noncovalent interactions with various active sites. As a result, the development of anti-cancer medicines has frequently exploited benzopyrone as a structural subunit Citation19. Numerous naturally occurring coumarin derivatives have cytotoxic properties. For example, isofraxidin (IF) (), a naturally occurring coumarin derivative, suppresses Akt phosphorylation in human colorectal cancer cells and shows anti-cancer effects in human hepatoma cells Citation20. While 4-methylumbelliferone (4-MU) is a natural powerful chemotherapeutic and chemo-preventive drug for breast, prostate, ovarian, renal cell, and pancreatic cancers () Citation21,Citation22. Esculetin, a natural coumarin derivative, significantly slows the growth of prostate cancer by enhancing cell cycle arrest, modifying the PTEN/AKT signalling pathway, and inducing apoptosis via the EGFR/PI3K/Akt signalling pathway Citation23,Citation24. Several coumarin derivatives have been described in the literature as cytotoxic medicines for solid tumours as compounds I–IIIa,b that function as PI3K inhibitors () Citation19,Citation22,Citation25. On the other hand, compound IV has potent cytotoxicity on breast cancer due to binding interactions with the hydrophobic core of EGFR kinase Citation26. Coumarin derivative V showed high EGFR inhibition impact, demonstrating 97.67% erlotinib’s potency along with the high cytotoxic activity over MDA-MB-231 Citation27. In addition, compound VI possessed high EGFR inhibitory activity and high cytotoxic activity against cervical cancer Citation28. By optimising coumarin as the primary scaffold (), a novel series of coumarin derivatives was discovered in the current study. These novel agents were then tested as potential antineoplastic agents against PC-3 prostate cancer cells and MDA-MB-231 breast cancer cells. The anticancer characteristics of the coumarin-acetohydrazide hybrids IIb and IIIb produced in our earlier research study Citation25 served as the inspiration for series A (Scheme 1) that formed through chain elongation with either a hydrazone (4a–5) or with thiosemicarbazide (6a–c). In series B (Scheme 2), the coumarin core’s cyclisation produced an amino triazole derivative 7 and it was then alkylated to form compound 8. Finally in series C, alkylation of coumarin core afforded compounds 9a–e (Scheme 3).
Scheme 1. Synthesis of the target compounds 4a–e, 5, and 6a–c.
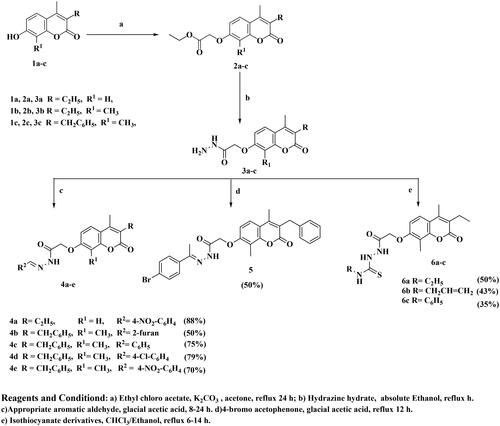
Scheme 2. Synthesis of the target compounds 7 and 8.
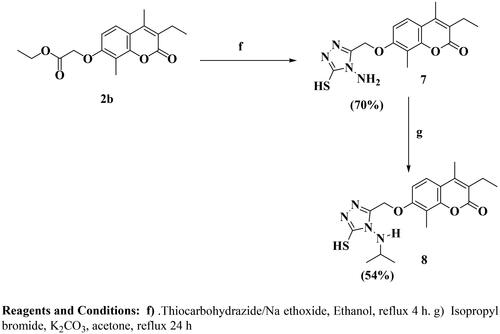
Scheme 3. Synthesis of the target compounds 9a–e.
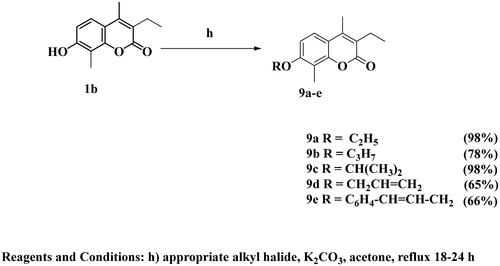
Figure 1. Some natural and synthetic coumarin derivatives with cytotoxic activity.
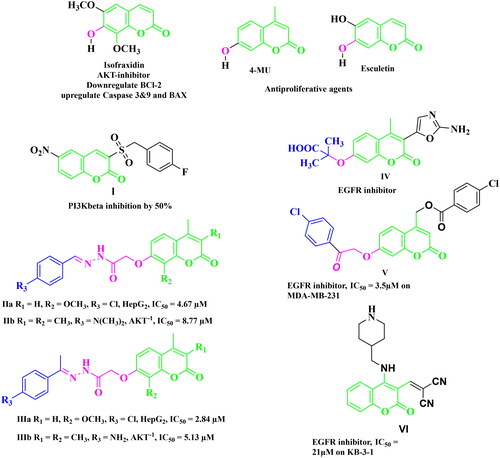
Figure 2. Design strategy of the desired compounds.
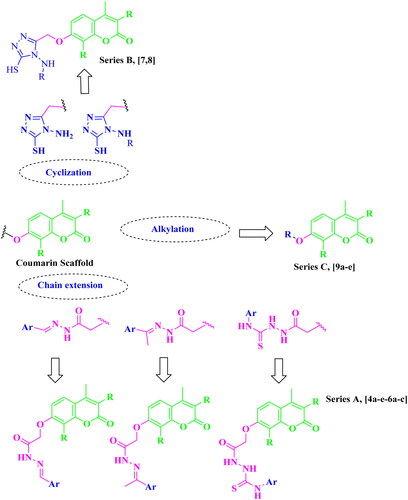
The most effective compound over PC-3 cells was examined for its suppression of the EGFR/PI3K/AKT/mTOR pathway by gene and protein expression levels, cell cycle analysis, and apoptosis studies (), and then docking studies were performed to confirm the mechanism of action.
Figure 3. A diagrammatic sketch illustrating the signalling pathway of the EGFR/PI3K/AKT/mTOR and coumarin targeting retrieved by Biorender.com templates Citation29.
Results and discussion
Chemistry
The target compounds were synthesised in accordance with Schemes 1–3. Six compounds 4a–e and 5 were synthesised based on the nucleophilic attack of acid hydrazides 3a–c on the appropriate aromatic aldehyde or acetophenone derivative.1H NMR, 13C NMR, and FT-IR were used for the elucidation of the synthesised compounds. FT-IR showed peaks referring to NH and N═C groups at 3205–3468 and 1519–1606 cm−1, respectively. 1H NMR spectra showed a singlet signal resonating at δ 7.98–8.44 ppm pointing to N═CH proton in addition to peaks at the aromatic region δ 6.91–8.31 ppm referring to the added aromatic rings in compounds 4a–e. On the other hand, compound 5 showed singlet signal referring to CH3 group at δ 2.26 ppm along with the aromatic proton of the added phenyl group at the expected chemical shift. 13C NMR spectra showed a peak referring to N═CH at δ 124.13–146.85 ppm in compounds 4a–e and N═C–CH3 at δ 2.26 ppm in compound 5. Carbothioamide derivatives 6a–c were prepared via a reaction of acid hydrazide 3b with the appropriate isothiocyanate derivative. 1H NMR spectra revealed that all compounds had three NH groups resonating at δ 7.97–10.23 ppm. 13C NMR spectra showed a peak at δ 181.51–182.37 ppm pointing to C═S, as shown in Scheme 1. Upon heating the ester derivative 2 with thiocarbohydrazide in sodium methoxide under reflux, amino triazole derivative 7 was prepared. The structure of compound 7 was confirmed by 1H NMR through the disappearance of peaks corresponding to the ester group while the appearance of two singlet signals at δ 3.68 and 5.66 ppm confirmed the presence of SH and NH2 groups substituting the 1,2,4-tiazole ring as shown in Scheme 2. When compound 7 was alkylated using isopropyl chloride in acetone in the presence of anhydrous sodium carbonate, the result was the N-alkyl derivative 8, which was confirmed by 1H NMR spectrum through the disappearance of the singlet exchangeable peak of NH2 and the appearance of singlet peak of NH group resonating at δ 5.76 ppm in addition to two doublet peaks at δ 1.32 and 1.36 ppm and multiplet peak resonating at δ 3.73–3.80 ppm referring to two CH3 group and CH proton of isopropyl moiety, respectively. Due to the existence of a peak at 165.68 ppm referring to the C5 of the 1,2,4-tiazole moiety connected to the SH group in the 13C NMR spectrum, it was determined that the final molecule is N-alkyl rather than S-alkyl. Finally, Scheme 3 depicts the formation of O-alkyl derivatives 9a–e by reacting 7-hydroxy coumarin derivative 1b with the appropriate alkyl halide in acetone in the presence of anhydrous sodium carbonate. The final compounds’ structures were validated by 1H NMR spectra. For example, compound 9a revealed triplet and quartette peaks resonating at δ 1.03 and δ 2.52–2.57 ppm, indicating CH3 and CH2 protons, respectively, with no peak indicating OH proton. In addition, 13C NMR spectrum revealed two peaks of the added ethyl group at δ 14.76 and 64.84 ppm.
Biological studies
In vitro cytotoxicity studies
The novel synthesised compounds were screened for their cytotoxic activity against breast cancer cell line (MDA-MB-231) and prostate cancer cell line (PC-3) using erlotinib (EGFR inhibitor) as a reference drug. The IC50 values of the synthesised molecules were determined and are shown in . Some of the synthesised compounds had high to moderate activity against the tested human cancer cell lines, while others had minimal activity. Among the synthesised compounds, compounds 5, 4b, and 4a demonstrated the most effective cytotoxic activity against PC-3 cell line as proven by their IC50 values of 3.56, 8.99, and 10.22 µM, respectively, which represent 3.45-, 1.36-, and 1.2-fold, respectively, superior to IC50 of the erlotinib. In accordance with MDA-MB-231 cell line, compound 4c displayed more powerful and effective cytotoxic action than erlotinib with IC50 value of 8.5 µM. Additionally, compound 4b showed high cytotoxic activity comparable to erlotinib with IC50 value of 11.78 µM. On the other hand, compounds 4c and 4e showed moderate cytotoxic activity over PC-3 cell line with IC50 values of 37.58 and 12.13 µM respectively while, compounds 4a and 4e possessed moderate cytotoxic activity over MDA-MB-231 cell line with IC50 value 23.60 and 24.28 µM, respectively. The rest of the compounds had a weak cytotoxic activity on both cell lines. In order to evaluate the in vitro results, structure–activity relationship (SAR) found that, the most active compounds are those bearing hydrazone moiety on C7 of the coumarin ring. High efficacy on both cell lines was obtained when hydrazone is bearing heteroaromatic -furan- ring as in compound 4b. Also, the cytotoxicity increased when the hydrazone is substituted with a strong deactivating electron withdrawing group (4-nitrobenzylidene moiety) as in compounds 4a and 4e than compound 4d bearing weak deactivating electron withdrawing group (4-chlorobenzylidene moiety). Moreover, the best activity on MDA-MB-231 cell line was obtained when the hydrazone was bearing unsubstituted benzylidene moiety as in compound 4c. It was clear that the best cytotoxic activity on PC-3 cell line was obtained when the hydrazone is substituted with weak deactivating electron withdrawing group (4-bromobenzylidene moiety) as in compound 5. Substitution of the hydrazide with either carbothioamide as in compounds 6a–c or cyclisation with amino triazole moiety as in compounds 7 and 8 results in weak cytotoxicity. Finally, alkylation of coumarin at C8 showed weak cytotoxic activity.
Table 1. Cytotoxicity of the tested compounds and erlotinib over MDA-MB-231 and PC-3 (IC50, µM).
Further investigation of the cytotoxic activity of compound 5 was performed on colorectal carcinoma (HCT-116) and hepatocellular carcinoma (HEPG-2) using erlotinib as reference control as indicated in . Compound 5 showed cytotoxic activity 1.5 times higher than erlotinib on HEPG-2 cell line, but comparable to erlotinib on HCT-116 cell line (IC50 values of 19.80 and 17.81, respectively). Moreover, compound 5 was tested on normal human primary prostate epithelial cells (HPrEC) to determine the cytotoxic effects in comparison to erlotinib. The results revealed that the tested compound suppressed the growth of HPrEC cells with IC50 value (95.54 µM) less than erlotinib (IC50 63.47 µM), suggesting that the synthesised compound has a safe profile and is less cytotoxic to normal cells than erlotinib as illustrated in .
Table 2. Cytotoxicity of compound 5 and erlotinib over HCT-116, HEPG-2, and HPrEC (IC50, µM).
In vitro EGFR and PI3Kβ inhibition assay
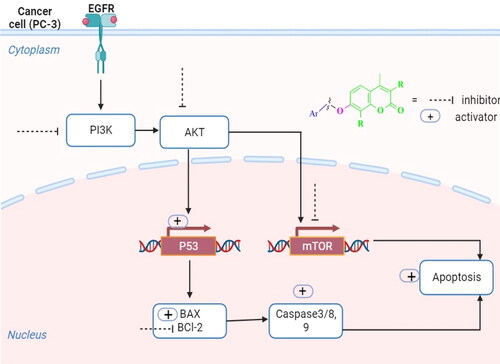
EGFR inhibition assay was performed for the most active compounds over PC-3 cell line. All the tested compounds showed potent inhibitory activity with IC50 values ranging from 0.1812 to 0.5587 µM when compared to erlotinib as a reference EGFR inhibitor (). Coumarin benzylidene derivative 5 with the highest cytotoxic activity on PC-3 cells also, showed the highest inhibitory activity on EFGR (IC50 = 0.1812 µM), which is comparable to erlotinib (IC50 = 0.1344 µM). On the other hand, PI3Kβ inhibitory activity was performed with the same compounds possessing the highest cytotoxic activity on PC-3 cells. The results of the enzyme assay showed that the tested compounds possessed good inhibitory activity with IC50 values ranging from 0.2612 to 0.9923 µM. Compound 5 possessed the highest inhibitory activity twofold more than LY294002 as reference PI3Kβ inhibitor. Compounds 4b and 4c had inhibitory activity comparable to LY294002 while compounds 4a and 4e possessed a good inhibitory activity (0.7918 and 0.9923 µM), respectively ().
Figure 4. (A) Graphical representation for comparison of IC50 EGFR (µM) of compounds 4a–c, 4e, 5, and erlotinib, (B) graphical representation for comparison of IC50 PI3Kβ (µM) of compounds 4a–c, 4e, 5 LY294002. Both enzymes were performed using four-dose protocol. The data are expressed as mean ± SD (n = 3 experiments), statistical analysis was performed by two-way ANOVA followed by Tukey’s post hoc test with *significant at p > 0.05, **significant at p > 0.01, ***significant at p > 0.001, and ****significant at p > 0.0001 and nsno significance.
The most active compound, 5 was tested for PI3K inhibition using class IA (α and γ) and IB (δ) isoforms. demonstrates that the tested compound had strong inhibitory activity on several isoforms (α, γ, and δ) with IC50 values (2.89, 5.04, and 5.49 µM, respectively) which is comparable to the IC50 values of the reference drug LY294002 (2.6, 6.76, and 7.42 µM, respectively), suggesting that compound 5 is a class IA and IB inhibitor.
Table 3. IC50 values of PI3K isoforms of compound 5 and (IC50, µM).
Measurement of gene expression AKT/m-TOR in PC-3 cells treated with compound 5 with IC50 3.56 µM
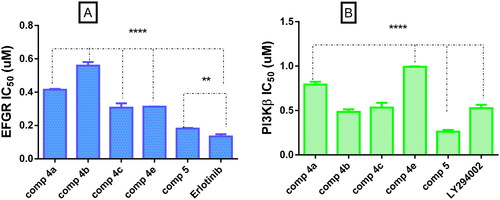
To investigate the effective molecular targets of compound 5 as a potential compound displaying the highest cytotoxic activity over PC-3 cells and the highest inhibitory activity against EGFR and PI3Kβ, the AKT/mTOR pathway was explored. It is known that EGFR/PI3K/AKT/mTOR signalling cascade is crucial for a variety of cellular functions, such as apoptosis, metabolism, cell growth, and proliferation, which aid in the development of tumours Citation30. As shown in , overexpression of AKT and m-TOR genes was observed in untreated PC-3 cells, while treatment of these cells with coumarin derivative 5 resulted in downregulation of AKT and m-TOR by 2.5- and 2.6-fold, respectively, comparing to the untreated cells. These findings demonstrated that compound 5 was successful in producing a promising inhibitor for the EGFR/PI3K/Akt/m-TOR axis to overcome the resistance of chemotherapeutic agents.
Figure 5. A graph represents the effect of compound 5 on the gene expression of AKT and m-TOR compared with negative control and erlotinib. The data are expressed as mean ± SD (n = 3 experiments), statistical analysis was performed by two-way ANOVA followed by Tukey’s post hoc test with *significant at p > 0.05, **significant at p > 0.01, ***significant at p > 0.001, ****significant at p > 0.0001, and nsno significance.
Cell cycle analysis (DNA flow cytometry)
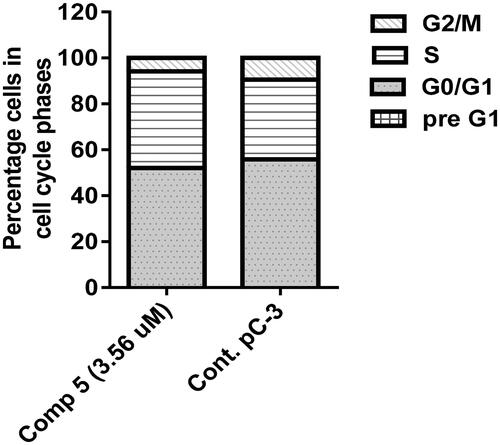
Cell cycle analysis described the percentage of cell accumulation in each phase during the cell cycle Citation31. As described in and , the percentage of G1, S, and G2/M phases when PC-3 cells were incubated in a vehicle were 55.85%, 34.71%, and 9.44%, respectively. In contrast, these percentages varied in PC-3 cells treated with compound 5, particularly in the S phase because the percentage of cells in G1, S, and G2/M phases seemed to be 52.09, 42.11, and 5.8, respectively. The result of this study showed that the coumarin derivative 5 caused cell cycle arrest at the S phase throughout the accumulation of the cells (42.11%) more than the untreated cells (34.71%).
Figure 6. Cell cycle analysis of compound 5 at its IC50 on PC-3 cancer cells.
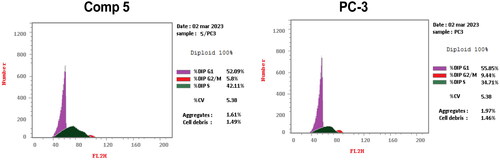
Figure 7. Graphical representation of cell cycle analysis of compound 5 at its IC50 on PC-3 cells.
Annexin V/propidium iodide staining for apoptosis analysis
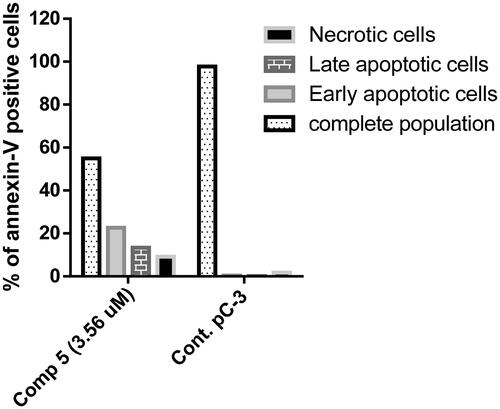
Annexin V/propidium iodide staining was performed to examine whether the cytotoxicity of compound 5 was associated with apoptosis by comparing PC-3 cells treated with compound 5 to untreated cells. The proportion of apoptotic cells was dramatically increased in cells treated with compound 5 ( and ), the proportion of early and late apoptotic cells was 22.62% and 13.33%, respectively. Contrarily, untreated cells displayed higher total population percentages (97.72%) and lower percentages of early and late apoptotic cells (0.39% and 0.15%, respectively). Thus, compound 5 seemed to be an effective apoptotic agent.
Figure 8. Annexin V/propidium iodide staining for apoptosis analysis of compound 5 at its IC50 on PC-3 cells.
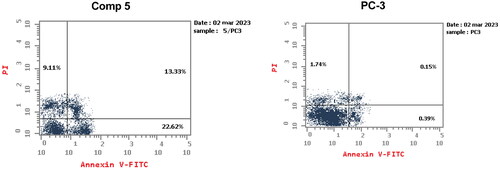
Figure 9. A graphical representation of Annexin V/propidium iodide staining for apoptosis analysis of compound 5 at its IC50 on PC-3 cells.
Real-time PCR for determination of proapoptotic, apoptotic, and anti-apoptotic mediators throughout intrinsic and extrinsic pathway
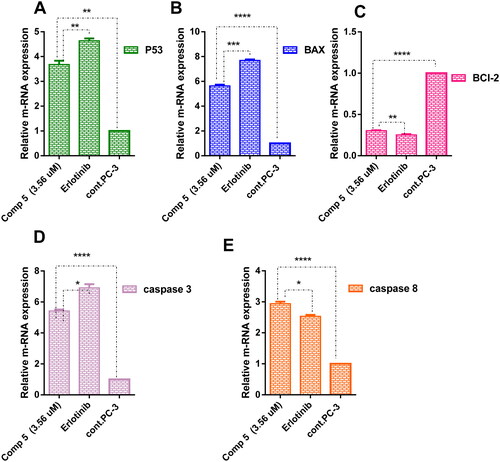
To maintain apoptosis, the balance between cell division and cell death must be regulated. New targeted therapeutics that either induce cancer cell death or make cancer cells more susceptible to recognised cytotoxic agents have been developed as a result of our growing understanding of the mechanisms of intrinsic and extrinsic apoptotic signalling Citation32. The intrinsic mechanism, which primarily comprises a cascade driven by mitochondria includes activation of P53, pro-apoptotic genes (BAX), caspase 3 and suppression of the anti-apoptotic Bcl-2 family but, the extrinsic pathway contains triggers cascades of caspase 8 Citation33,Citation34. PC-3 cells were treated with compound 5 at its IC50 to analyse and compare the gene expression of P53, BAX, BCl-2, caspases 3, and caspases 8 with the untreated PC-3 cells. As observed in , in comparison to PC-3 cells, compound 5 significantly induced high expression of tumour suppression gene P53 by 3.83-fold. BAX as pro-apoptotic gene was highly expressed in PC-3 treated with compound 5 by 5.6-fold than in untreated cells. Also, the upregulation of the apoptotic genes caspases 3 and caspases 8 was recorded by 5.40- and 2.96-fold, respectively. Moreover, compound 5 downregulated the anti-apoptotic gene BCl-2 by 0.30-fold. Therefore, compound 5 suggested induction of intrinsic and extrinsic apoptotic pathways in the treated PC-3 cells by upregulating BAX, P53, caspases-3 and -8 and downregulating BCl-2 gene in comparison to the control.
Figure 10. A graph represents the relative gene expression of (A) P53, (B) BAX, (C) BCl-2, (D) caspase 3, and (E) caspase 8 of compound 5 at its IC50 versus erlotinib and untreated PC-3. The data are expressed as mean ± SD (n = 3 experiment); statistical analysis was performed by two-way ANOVA followed by Tukey’s post hoc test with *significant at p > 0.05, **significant at p > 0.01, ***significant at p > 0.001, ****significant at p > 0.0001 and nsno significance.
Molecular docking studies
A molecular docking study using Molecular Operating Environment (MOE) Software 2020.09 Citation35 was conducted to provide a preliminary SAR based on the binding mode and docking scores of series A, B, and C compounds with EGFR tyrosine kinase (PDB ID: 1M17) Citation36 and PI3Kβ kinase (PDB ID 4JPS) Citation37 (). The docking scores and binding modes for the tested compounds were compared against lead compounds erlotinib and LY294002, both of which served as reference compounds in the in vitro EGFR and PI3Kβ inhibitory assay.
Table 4. Docking score of tested compounds representing series A, B, and C.
The docking scores, presented in , provide insights into the binding affinities of the tested compounds with both EGFR tyrosine kinase (PDB ID: 1M17) and PI3Kβ kinase (PDB ID: 4JPS). Notably, compound 5 exhibited the highest docking scores for both targets, indicating a strong binding affinity with EGFR and PI3Kβ. Compounds 4b, 4c, and 4d also demonstrated favourable docking scores for both kinases. These results are coherent with findings in vitro EGFR and PI3Kβ inhibitory assay. Further investigation of binding modes and 2D/3D interaction diagram was conducted to rationalise the inactivity of compounds 6a–c and series B and C in the cytotoxic assay.
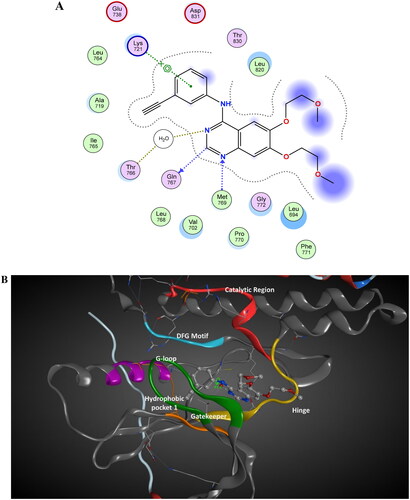
The binding mode of docking score of tested compounds in series A, B, and C with EGFR tyrosine kinase was evaluated against the bioactive conformation of erlotinib (). The analysis of 2D/3D interactions of erlotinib showed that erlotinib’s carbon atom (C19) forms a hydrogen bond with GLN 767 (3.15 Å). The nitrogen atom (N2) engages in a hydrogen bond with MET 769, serving as a hydrogen acceptor (2.70 Å). The N3 atom of erlotinib acts as a hydrogen acceptor forming a hydrogen bond with a water molecule (HOH 10, 2.78 Å). This water acts as a bridge quinazoline nitrogen atom (N3) of erlotinib and the side chain of Thr766. At the same time, the aniline ring of erlotinib participates in a pi-cation interaction with the NZ atom of Lys 721 (4.53 Å). The 3D geometry of erlotinib revealed interplanar angle of aromatic ring systems between the phenyl ring and quinazoline is 41.6° Citation36 ((B)). This 3D arrangement allows the phenyl ring to be held in the proper direction to interact with Lys 721 in pi-cation while orienting the quinoline ring in the proper position to interact with Met 769 and Gln 768 Citation36.
Figure 11. The 2D/3D interaction diagram of bioactive confirmation of erlotinib within the binding site of EGFR tyrosine kinase domain (PDB ID: 1M17). (A) The 2D interaction diagram, amino acids in contact with erlotinib are displayed as spheres. Violet spheres represent polar amino acids while green sphere represents hydrophobic amino acids. Solvent-exposed regions of erlotinib are highlighted in blue. The dotted lines represent non-bonding interactions. (B) The 3D interaction of erlotinib reveals a 41.6° dihedral angle between the phenyl ring and quinazoline, facilitating optimal interactions with Lys 721 in a pi-cation configuration, along with interactions with Met 769 and Gln 768.
Docking of series, A, B, and C compounds within the binding site of EGFR kinase and comparing the binding mode and these compounds with the bioactive conformation of erlotinib was conducted. The series A active compounds (4a–5) showed binding mode demonstrated in . This binding mode was similar to the reported binding mode of erlotinib Citation36. The coumarin scaffold was equivalent to the quinazoline ring in erlotinib having hydrophobic pi–pi hydrogen interaction with Val 702 in G-Loop. The 2-oxo of the coumarin ring is equivalent to the N3 atom of erlotinib acting as a hydrogen acceptor forming a hydrogen bond with a water molecule (HOH 10). This water acts as a bridge between the quinazoline nitrogen atom (N3) of erlotinib and series A active compounds (4a, 4b) and the side chain of Thr766. The three alkyl/aryl group interactions with hydrophobic pocket 1 amino acids including Lys 721 and Ile 720. This interaction is equivalent to aniline ring interaction in erlotinib. The 2-acetohydrazide moiety with various linkers protruded in the solvent-exposed regions. These pharmacophoric features of compounds 4a–e and 5, provided optimum 3D geometry forcing the coumarin ring to be non-planer with alky/aryl group in interacting with hydrophobic pocket 1. The interplanar angle of active compounds 4a–e and 5 was around 60°, comparable to the coplanar angle or aromatic system of erlotinib. The inactive compounds in series A, B, and C showed different interaction patterns than erlotinib. For example, compounds 6a–c, having thiosemicarbazone group binding with a flipped pattern in the binding site, coumarin ring did not perform the crucial interaction described in . Compounds 7, 8, and 9 were smaller in size and did provide the proper 3D geometric arrangement for selective and specific binding with EGFR kinase domain ().
Figure 12. Schematic diagram of the interaction of series A active compounds (4a–5).
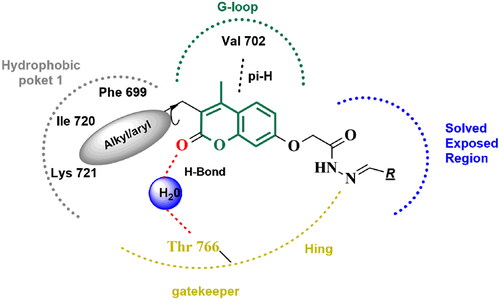
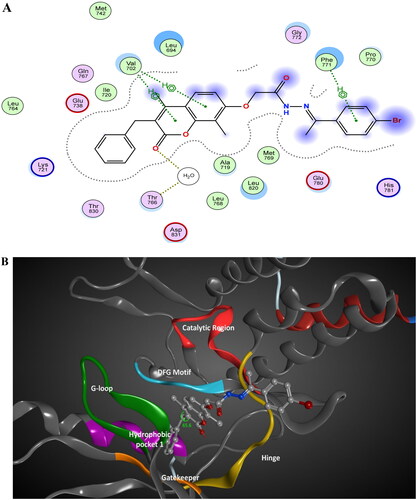
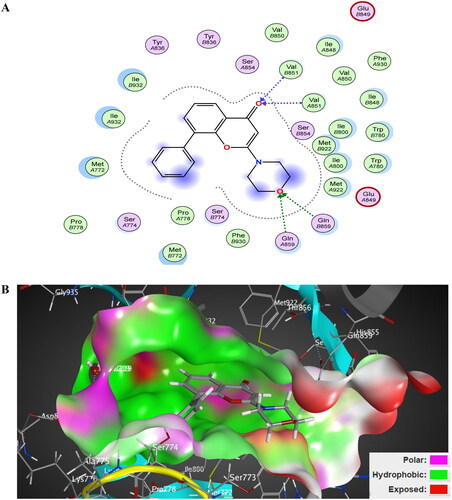
Figure 13. The 2D/3D interaction diagram of compound 5 within the binding site of EGFR tyrosine kinase domain (PDB ID: 1M17). (A) Showing the 2D interaction diagram, amino acids in contact with compound 5 are displayed as spheres. Violet spheres represent polar amino acids while green sphere represents hydrophobic amino acids. Solvent-exposed regions of compound 5 are highlighted in blue. The dotted lines represent non-bonding interactions. (B) The 3D interaction diagram of compound 5 reveals a 65.6° dihedral angle between the phenyl ring and coumarin scaffold allowing optimum 3D geometry for the coumarin ring to interact with Val 702 in G-loop and side chain of Thr766 via water bridge.
Docking of series, A, B, and C within the binding site of binding site of PI3K kinase comparing the binding mode and these compounds with the docked pose of the in vitro assay Lead LY294002 was conducted. The docking scores of designed compounds ranged from −6.2472 to −8.5675 kcal/mol compared to LY294002 docking score of −6.175 kcal/mol. Compound 5 showed the highest docking score equals −8.5675 kcal/mol. The docking score aligned with the in vitro enzyme inhibitory assay. The 2D/3D interaction diagram of compound 5 and LY294002 within the binding site of PI3K kinase (PDB ID 4JPS) is described in and . The coumarin rings in LY29400 and design compounds are deeply buried with a hydrophobic pocket, while morpholine group in LY294002 or equivalent in 2-acetohydrazide moiety with various linkers protruded in the solvent-exposed regions. The 2-acetohydrazide moiety is involved in hydrogen bond interactions anchoring the structure within the binding site. The presence of the three alkyl, aryl group in designed compounds contributes to the increased binding affinity of tested compounds compared to LY29400 due to hydrophobic interaction with activation loop hydrophobic amino acids such as Ile 932.
Figure 14. The 2D/3D interaction diagram of compound 5 within the binding site of PI3K kinase (PDB ID 4JPS). (A) The 2D interaction diagram, amino acids in contact with compound 5 are displayed as spheres. Violet spheres represent polar amino acids while green spheres represent hydrophobic amino acids. Solvent-exposed regions of compound 5 are highlighted in blue. The dotted lines represent non-bonding interactions. (B) The 3D interaction diagram of compound 5 with in binding site of PI3K represented as a coloured surface. The polar binding site regions are represented in magenta, hydrophobic regions in green, and solvent-exposed regions are coloured in red.
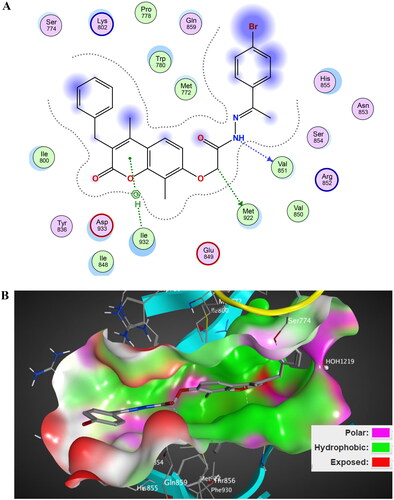
Figure 15. The 2D/3D interaction diagram of LY294002 within the binding site of PI3K kinase (PDB ID 4JPS). (A) The 2D interaction diagram, amino acids in contact with compound 5 are displayed as spheres. Violet spheres represent polar amino acids while green spheres represent hydrophobic amino acids. Solvent-exposed regions of compound 5 are highlighted in blue. The dotted lines represent non-bonding interactions. (B) The 3D interaction diagram of LY294002 within the binding site of PI3K is represented as a coloured surface. The polar binding site regions are represented in magenta, hydrophobic regions in green, and solvent-exposed regions are coloured in red.
In conclusion, the molecular docking studies of designed compounds within EGFR and PI3K kinase binding sites demonstrated promising interactions, suggesting their potential as dual-targeted agents in cancer therapy. The compounds exhibited favourable affinities and key interactions, providing a foundation for further exploration.
Conclusions
Novel coumarin analogues were synthesised and evaluated for their cytotoxic activity against two human cancer cells namely, PC-3 and MDA-MB-231. Some compounds showed comparable or better antiproliferative activity than erlotinib, among which benzylidene derivatives 5, 4b, and 4a with IC50 values 3.56, 8.99, and 10.22 µM, respectively, against PC-3 cells and compound 4c displayed cytotoxic activity more potent than erlotinib with IC50 value 8.5 µM on the MDA-MB-231 cell line. The effect of compound 5 on EGFR/PI3Kβ/AKT/mTOR signalling pathway on PC-3 cells was explored and the results showed down-regulation of AKT and mTOR expression in addition to the inhibitory activity on PI3Kβ and EGFR enzymes with IC50 values 0.2612 and 0.1812 µM, respectively. Moreover, compound 5 inhibited other PI3K isoforms (α, γ, and δ) with IC50 comparable to LY294002. Compound 5 caused cell cycle arrest at the S phase. Also, compound 5 elevated the proportion of early and late apoptotic cells. Confirmation of both extrinsic and intrinsic pathways was conducted by upreulating caspases (3 and 8) P53 and BAX while downregulating the antiapoptotic gene BCl-2 by compound 5. Docking studies ensured that compound 5 showed a high binding affinity to both enzymes which proves compound 5 as PI3Kβ and EGFR dual inhibitor.
Experimental
General
Aldrich (St. Louis, MO) supplied the solvents and reagents, which were used exactly as received unless otherwise noted. The open capillary tube approach, which uses the Stuart SMP10 melting point apparatus, was used to determine the melting points. Wave number υmax (cm−1) was used to express the infra-red spectra, which were captured as potassium bromide discs using a Bruker FT-IR spectrophotometer (Billerica, MA) at Misr University for Science and Technology. The Regional Centre for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt performed the elemental analysis using a Thermo Scientific TM FLASH 2000 CHNS/O analyser (Waltham, MA). The National Research Centre in Cairo, Egypt and Ain Shams University (Cairo, Egypt) conducted 1H NMR and 13C NMR spectra on Jeol ECA (Tokyo, Japan) 500 MHz and Bruker 400 MHz spectrophotometers (Billerica, MA), respectively, for proton and with Jeol ECA 125 MHz and Bruker (Billerica, MA) 100 MHz for carbon using TMS as internal standard, chemical shifts (δ) were recorded in ppm on δ scale. At the Regional Centre for Mycology & Biotechnology, Al-Azhar University, Cairo, Egypt, mass spectra were conducted on a Hewlett Packard 5988 spectrometer (Palo Alto, CA) or a Shimadzu QP-2010 plus (Kyoto, Japan). Using precoated aluminium sheets and silica gel (Merck 60F 254, Rahway, NJ) as the eluting solution, the TLC method was used to track the reactions’ progress.
The starting materials 1a–c, 2a–c, and 3a–c were synthesised according to adopted procedures in literatures Citation38–43.
General procedure for synthesis of 2-(3-substituted-4,8-disubstituted-2-oxo-2H-chromen-7-yl)oxy-N′-substituted acetohydrazides (4a–e)
An equimolar amount of the appropriate acid hydrazide 3a and 3c (0.001 mol) in glacial acetic acid (20 mL), and appropriate aromatic aldehyde derivative (0.001 mol) was heated under reflux for 8–24 h. The solvent was removed under vacuum after completion of the reaction and the formed precipitate was recrystallised from ethanol to afford compounds 4a–e.
2-((3-Ethyl-4-methyl-2-oxo-2H-chromen-7-yl)oxy)-N'-(4-nitrobenzylidene) acetohydrazide (4a)
White powder, yield 88%. mp 240–242 °C. IR υmax/cm−1: 3468 (NH), 3112, 3080 (CH aromatic), 2972 (CH aliphatic), 1681, 1608 (2 C═O), 1519 (C═N).1H NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (t, 3H, J = 7.2 Hz, CH2CH3), 2.40 (s, 3H, CH3), 2.50–2.59 (q, 2H, J = 7.2 Hz, CH2CH3), 4.85 (s, 1H, OCH2), 5.34 (s, 1H, OCH2), 6.97–7.05 (m, 2H, H-Ar), 7.70 (d, 1H, J = 9.2 Hz, H-Ar), 8.01 (d, 2H, J = 10.8 Hz, H-Ar), 8.28 (d, 2H, J = 7.6 Hz, H-Ar), 8.44 (s, 1H, CH=N), 11.93 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz): 13.40, 14.81, 20.68, 66.67, 101.83, 112.90 (2C), 114.73, 124.55, 126.79 (2C), 130.65, 131.75, 132.20, 146.85, 153.33, 160.13, 161.16, 166.50, 166.70, and 168.57. MS m/z (%): 409.88 (M+, 21.66%), 97.02 (100%). Anal. Calcd. for C21H19N3O6 (409.40): C, 61.61; H, 4.68; N 10.26. Found: C, 61.98; H, 4.85; N, 10.44.
2-((3-Benzyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)-N'-(furan-2-ylmethylene)acetohydrazide (4b)
White powder, yield 50%. mp 184–186 °C. IR υmax/cm−1: 3384 (NH), 3057 (CH aromatic), 2976, 2925 (CH aliphatic), 1699, 1679 (2 C═O), 1605 (C═N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 2.26 (s, 3H, CH3), 2.39 (s, 3H, CH3), 3.95 (s, 2H, CH2), 4.91 (s, 1H, OCH2), 5.24 (s, 1H, OCH2), 6.62 (s, 1H, H-Ar), 6.91–6.98 (m, 2H, H-Ar), 7.15–7.28 (m, 5H, H-Ar), 7.57 (d, 1H, J = 8.8 Hz, H-Ar), 7.83–8.20 (m, 2H (H-Ar + CH═N), 11.54 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz): 8.55, 15.92, 32.93, 65.40, 109.21, 112.83, 114.17, 115.24, 121.19, 124.20, 126.60, 127.88 (2C), 128.93 (2C), 134.28, 139.93, 145.66, 148.67, 149.65, 151.31, 158.67, 162.38, 165.00, 168.38. MS m/z (%): 430.36 (M+, 36.57%), 77.11 (91.22%), 52.26 (100%). Anal. Calcd. for C25H22N2O5 (430.46): C, 69.76; H, 5.15; N 6.51. Found: C, 69.97; H, 5.37; N, 6.80.
2-((3-Benzyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)-N'-benzylideneacetohydrazide (4c)
White powder, yield 75%. mp 219–220 °C. IR υmax/cm−1: 3341 (NH), 3065 (CH aromatic), 2942 (CH aliphatic), 1721, 1683 (2 C═O), 1606 (C═N). 1H NMR (500 MHz, DMSO-d6) δ ppm: 2.26 (s, 3H, CH3), 2.38 (s, 3H, CH3), 3.92 (s, 2H, CH2), 4.80 (s, 1H, OCH2), 5.30 (s, 1H, OCH2), 6.94 (d, 1H, J = 8.6 Hz, H-Ar), 7.14–7.23 (m, 5H, H-Ar), 7.39–7.44 (m, 3H, H-Ar), 7.57–7.67 (m, 3H, H-Ar), 7.98 (s, 1H, CH═N), 11.59 (s, 1H, NH, D2O exchangeable). 13C NMR (125 MHz): 9.84, 16.53, 33.48, 66.99, 109.90, 115.25, 122.29, 124.92, 127.38, 128.29 (2C), 128.49 (2C), 129.34 (4C), 130.11 (2C), 131.32, 135.29, 140.70, 145.33, 149.75, 152.12, 162.62, 170.03. MS m/z (%): 439.96 (M+, 63.14), 312.19 (90.66), 65.25 (100). Anal. Calcd. for C27H24N2O4 (440.50): C, 73.62; H, 5.49; N 6.36. Found: C, 73.43; H, 5.72; N, 6.62.
2-((3-Benzyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)-N'-(4-chlorobenzylidene)acetohydrazide (4d)
White powder, yield 79%. mp 232–234 °C. IR υmax/cm−1: 3205 (NH), 3064 (CH aromatic), 2964, 2924 (CH aliphatic), 1696, 1682 (2 C═O), 1603 (C═N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 2.27 (s, 3H, CH3), 2.42 (s, 3H, CH3), 3.97 (s, 2H, CH2), 4.87 (s, 1H, OCH2), 5.33 (s, 1H, OCH2), 6.98 (d, 1H, J = 7.6 Hz, H-Ar), 7.16–7.28 (m, 5H, H-Ar), 7.50 (d, 2H, J = 8.4 Hz, H Ar), 7.61 (d, 1H, J = 7.6 Hz, H-Ar), 7.75 (d, 2H, J = 8.4 Hz, H-Ar), 8.02 (s, 1H, CH═N), 11.72 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz): 8.92, 15.55, 32.64, 66.07, 109.23, 113.20, 114.19, 121.85, 123.59, 126.24, 127.59 (2C), 128.57 (2C), 129.92 (2C), 130.60 (2C), 133.60, 134.97, 142.95, 149.28, 151.68, 157.62, 159.36, 161.03, 169.67. MS m/z (%): 476.77 (M++1, 4.41), 474.80 (M+, 12.71), 91.24 (100). Anal. Calcd. for C27H23ClN2O4 (474.94): C, 68.28; H, 4.88; N 5.90. Found: C, 68.41; H, 4.95; N, 6.14.
2-((3-Benzyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)-N'-(4-nitrobenzylidene)acetohydrazide (4e)
White powder, yield 70%. mp 252–254 °C. IR υmax/cm−1: 3372 (NH), 3059 (CH aromatic), 2924 (CH aliphatic), 1761, 1685 (2 C═O), 1603 (C═N).1H NMR (500 MHz, DMSO-d6) δ ppm: 2.26 (s, 3H, CH3), 2.38 (s, 3H, CH3), 3.92 (s, 2H, CH2), 4.85 (s, 1H, OCH2), 5.35 (s, 1H, OCH2), 6.96 (d, 1H, J = 9.5 Hz, H-Ar), 7.14–7.23 (m, 5H, H-Ar), 7.57 (d, 1H, J = 8.6 Hz H-Ar), 7.95 (d, 2H, J = 8.6 Hz, H-Ar), 8.08 (s, 1H, CH═N), 8.23 (d, 2H, J = 8.6 Hz, H-Ar), 11.94 (s, 1H, NH, D2O exchangeable). 13C NMR (125 MHz): 8.67, 15.71, 32.67, 66.16, 109.10, 112.63, 113.06, 114.46, 124.46 (2C), 126.57 (2C), 128.44 (2C), 128.53, 128.93 (2C), 139.87, 140.76, 142.13, 148.33, 148.91, 151.30, 158.39, 161.78, 164.96, 169.67. MS m/z (%): 485.20 (M+, 24.52), 102.32 (87.48), 51.02 (100). Anal. Calcd. for C27H23N3O6 (485.50): C, 66.80; H, 4.78; N 8.66. Found: C, 67.06; H, 4.89; N, 8.92.
Procedure for synthesis of 2-((3-benzyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)-N'-(1-(4-bromophenyl)ethylidene)acetohydrazide (5)
To a solution of the acid hydrazide 3c (0.352, 0.001 mol) in glacial acetic acid (20 mL), 4-bromo acetophenone (0.199 g, 0.001 mol) was added and the mixture was heated under reflux for 12 h. The solvent was removed under vacuum and the precipitate formed was recrystallised from DMF.
White powder, yield 50%. mp 240–242 °C. IR υmax/cm−1: 3213 (NH), 3071 (CH aromatic), 2923 (CH aliphatic), 1685, 1602 (2 C═O), 1569 (C═N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 2.26 (s, 6H, 2 CH3), 2.39 (s, 3H, CH3), 3.96 (s, 2H, CH2), 4.95 (s, 1H, OCH2), 5.34 (s, 1H, OCH2), 6.94–6.99 (m, 1H, H-Ar), 7.09–7.35 (m, 5H, H-Ar), 7.57–7.66 (m, 3H, H-Ar), 7.70–7.82 (m, 2H, H-Ar), 10.96 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz): 8.61, 13.84, 15.64, 32.59, 66.44, 108.91, 112.84, 114.26, 121.34, 123.10, 123.75, 126.50 (2C), 128.47 (2C), 128.69, 128.87 (2C), 131.72 (2C), 137.57, 139.81, 147.85, 148.85, 151.21, 158.85, 161.73, 170.08. MS m/z (%): 535.04 (M++2, 12.77), 533.22 (M+, 13.03), 43.19 (100). Anal. Calcd. for C28H25BrN2O4 (533.42): C, 63.05; H, 4.72; N 5.25. Found: C, 63.28; H, 4.89; N, 5.49.
General procedure for synthesis of N-substituted-2-(2-((3-ethyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)acetyl)hydrazinecarbothioamide (6a–c)
To a solution of the acid hydrazide 3b (0.29 g, 0.001 mol) in a mixture of ethyl alcohol and chloroform (1:1), the appropriate isothiocyanate derivative (0.001 mol) was added and the mixture was refluxed for 6–14 h. The solvent was removed under vacuum and the formed precipitate was recrystallised from ethanol to give compounds 6a–c.
N-Ethyl-2-(2-((3-ethyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)acetyl)hydrazine-1-carbothioamide (6a)
White powder, yield 50%. mp 214–216 °C. IR υmax/cm−1: 3446, 3377, 3277 (3NH) 3099 (CH aromatic), 2970 (CH aliphatic), 1708, 1687 (2 C═O), 1602 (C═N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.02–1.13 (m, 6H, 2 CH2CH3), 2.26 (s, 3H, CH3), 2.37 (s, 3H, CH3), 2.53–2.58 (q, 2H, J = 7.6 Hz, CH2CH3), 3.43–3.50 (s, 2H, CH2CH3), 4.74 (s, 2H, OCH2), 6.96 (d, 1H, J = 8.8 Hz, H-Ar), 7.57 (d, 1H, J = 8.8 Hz, H-Ar), 7.97 (s, 1H, NH, D2O exchangeable), 9.24 (s, 1H, NH, D2O exchangeable), 9.95 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz): 8.62, 13.41 (2C), 14.79, 14.87, 20.65, 67.01, 108.80, 113.10, 114.77, 123.57, 124.31, 146.98, 150.88, 157.89, 161.29, 167.57, 181.62. MS m/z (%): 377.45 (M+, 11.2%), 329.13 (100), 77.08 (90.75). Anal. Calcd. for C18H23N3O4S (377.46): C, 57.28; H, 6.14; N 11.13. Found: C, 57.47; H, 6.31; N, 11.40.
N-Allyl-2-(2-((3-ethyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)acetyl)hydrazine-1-carbothioamide (6b)
White powder, yield 43%. mp 180–182 °C. IR υmax/cm−1: 3460, 3446, 3398 (3NH), 3035 (CH aromatic), 2966, (CH aliphatic), 1705, 1602 (2 C═O), 1577 (C═N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (t, 3H, J = 6.8 Hz, CH2CH3), 2.26 (s, 3H, CH3), 2.38 (s, 3H, CH3), 2.50–2.58 (m, 2H, CH2CH3), 4.11 (s, 2H, CH2CH═CH2), 4.73 (s, 2H, OCH2), 5.03–5.30 (m, 2H, CH2CH═CH2), 5.78–5.85 (m, 1H, CH2CH═CH2), 6.98 (d, 1H, J = 8.4 Hz, H-Ar), 7.58 (d, 1H, J = 8.4 Hz, H-Ar), 8.10 (s, 1H, NH, D2O exchangeable), 9.36 (s, 1H, NH, D2O exchangeable), 9.56 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz): 8.63, 13.42, 14.82, 20.65, 46.30, 67.09, 108.88, 113.10, 114.79, 115.77, 123.61, 124.32, 135.22, 147.08, 150.90, 157.96, 161.37, 166.12, 182.37. MS m/z (%): 389.24 (M+, 12.21), 388.32 (55.02), 56.19 (100). Anal. Calcd. for C19H23N3O4S (389.47): C, 58.59; H, 5.95; N 10.79. Found: C, 58.78; H, 6.12; N, 11.03.
2-(2-((3-Ethyl-4,8-dimethyl-2-oxo-2H-chromen-7-yl)oxy)acetyl)-N-phenylhydrazine-1-carbothioamide (6c)
White powder, yield 35%. mp 160 °C. IR υmax/cm−1: 3446 (NH), 3034 (CH aromatic), 2966 (CH aliphatic), 1705, 1602 (2 C═O), 1577 (C═N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (t, 3H, J = 7.2 Hz, CH2CH3), 2.28 (s, 3H, CH3), 2.36 (s, 3H, CH3), 2.53–2.58 (q, 2H, J = 7.2 Hz, CH2CH3), 4.80 (s, 2H, OCH2), 7.00 (d, 1H, J = 9.2 Hz, H-Ar), 7.17 (t, 1H, J = 7.2 Hz, H-Ar), 7.34 (t, 2H, J = 7.6 Hz, H-Ar), 7.43 (d, 2H, J = 7.2 Hz, H-Ar), 7.57 (d, 1H, J = 9.2 Hz, H-Ar), 9.67 (s, 1H, NH, D2O exchangeable), 9.76 (s, 1H, NH, D2O exchangeable), 10.23 (s, 1H, NH, D2O exchangeable). 13C NMR (100 MHz): 8.63, 13.40, 14.75, 20.65, 67.11, 108.78, 113.09, 114.76, 123.50, 124.28 (2C), 125.70, 128.60 (2C), 129.29, 139.44, 146.88, 150.86, 157.84, 161.26, 167.77, 181.51. MS m/z (%): 425.39 (M+, 14.96), 377.03 (60.95), 297.25 (100). Anal. Calcd. for C22H23N3O4S (425.50): C, 62.10; H, 5.45; N 9.88. Found: C, 62.37; H, 5.62; N, 10.07.
Procedure for synthesis of 7-((4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)methoxy)-3-ethyl-4,8-dimethyl-2H-chromen-2-one (7)
To a freshly prepared sodium methoxide solution (0.8 Na metal in 52 mL methanol), the ester derivative 2b (3.04 g, 0.01 mol) and thiocarbohydrazide (1.06 g, 0.01 mol) were added and the mixture was heated under reflux for 4 h. After completion of the reaction, the excess solvent was removed under vacuum and the formed residue was dissolved in water and acidified with acetic acid till pH = 4. The formed product was filtered, washed with water, dried, and recrystallised from ethanol to give compound 7.
Grey powder, yield 70%. mp 220–222 °C. IR υmax/cm−1: 3423, 3284 (NH2), 3178, 3010 (CH aromatic), 2958 (CH aliphatic), 2065 (HS), 1693, 1637 (2 C═O), 1558 (C═N), 1498, 1489, 1415 (NH, C═C). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.03 (t, 3H, J = 8.8 Hz, CH2CH3), 2.17 (s, 3H, CH3), 2.36 (s, 3H, CH3), 2.50–2.55 (m, 2H, CH2CH3), 3.68 (s, 1H, SH), 5.25 (s, 2H, OCH2), 5.66 (s, 2H, NH2, D2O exchangeable), 7.16 (d, 1H, J = 8.8 Hz, H-Ar), 7.58 (d, 1H, J = 8.8 Hz, H-Ar). MS m/z (%): 346.19 (M+, 28.40), 83.95 (91.30), 48.37 (100). Anal. Calcd. for C16H18N4O3S (346.41): C, 55.48; H, 5.24; N 16.17. Found: C, 55.73; H, 5.61; N, 16.45.
Procedure for synthesis of 7-((amino-5-(isopropylthio)-4H-1,2,4-triazol-3-yl)methoxy)-3-ethyl-4,8-dimethyl-2H-chromen-2-one (8)
A mixture of amino triazole derivative 7 (0.346 g, 0.001 mol), isopropyl chloride (1.56 g, 0.002 mol) and anhydrous potassium carbonate (0.552 g, 0.004 mol) in dry acetone was heated under reflux for 24 h. After the completion of the reaction, water was added, and the formed precipitate was recrystallised from methanol to afford compound 8.
White powder, yield 54%. mp 125–127 °C. IR υmax/cm−1: 3431 (NH2), 3176, 3132 (CH aromatic), 2964 (CH aliphatic), 1697, 1604 (2 C═O), 1577 (C═N). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.03 (t, 3H, J = 7.6 Hz CH2CH3), 1.32 (d, 3H, J = 6.8 Hz, CH(CH3)2), 1.36 (d, 3H, J = 6.8 Hz, CH(CH3)2), 2.17 (s, 3H, CH3), 2.35 (s, 3H, CH3), 2.51–2.56 (q, 2H, J = 7.2 Hz CH2CH3), 3.73–3.80 (m, 1H, CH(CH3)2), 5.27 (s, 1H, OCH2), 5.32 (s, 1H, OCH2), 5.76 (s, 1H, NH, D2O exchangeable), 6.02 (s, 1H, SH, D2O exchangeable), 7.23 (d, 1H, J = 6.4 Hz, H-Ar), 7.55(d, 1H, J = 6.4 Hz, H-Ar). 13C NMR (100 MHz): 8.25, 13.59, 14.26, 20.97, 22.93, 37.61, 50.67, 59.75, 109.51, 113.27, 114.86, 123.57, 124.59, 146.26, 150.33, 152.28, 158.68, 161.01, 165.68. MS m/z (%): 388.88 (M+, 5.06), 268.24 (64.16), 151.09 (100). Anal. Calcd. for C19H24N4O3S (388.49): C, 58.74; H, 6.23; N 14.42. Found: C, 58.95; H, 6.31; N, 14.68.
General procedure for synthesis of 3-ethyl-7-substituted-4,8-dimethyl-2H-chromen-2-one (9a–e)
A mixture of compound 1b (0.218 g, 0.001 mol), the appropriate alkyl halide (0.002 mol) and anhydrous potassium carbonate (0.552 g, 0.004 mol) in dry acetone was heated under reflux for 18–24 h. After the completion of the reaction, water was added, and the formed precipitate was recrystallised from methanol to give compounds 9a–e.
7-Ethoxy-3-ethyl-4,8-dimethyl-2H-chromen-2-one (9a)
White powder, yield 98%. mp 180–182 °C. IR υmax/cm−1: 3028 (CH aromatic), 2968 (CH aliphatic), 1703 (C═O), 1550 (C═C). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.03 (t, 3H, J = 7.2 Hz, CH2CH3), 1.37 (t, 3H, J = 7.2 Hz, CH2CH3), 2.16 (s, 3H, CH3), 2.36 (s, 3H, CH3), 2.52–2.57 (q, 2H, J = 7.6 Hz, CH2CH3), 4.10–4.15 (q, 2H, J = 6.8 Hz CH2CH3), 6.98 (d, 1H, J = 8.8 Hz, H-Ar), 7.56 (d, 1H, J = 8.8 Hz, H-Ar). 13C NMR (100 MHz): 8.38, 13.45, 14.76, 15.09, 20.62, 64.48, 108.53, 112.39, 114.14, 123.72, 123.85, 147.03, 150.95, 158.67, 161.32. MS m/z (%): 246.40 (M+, 9.46), 136.73 (94.46), 105.18 (89.78), 74.50 (100). Anal. Calcd. for C15H18O3 (246.31): C, 73.15; H, 7.37. Found: C, 73.37; H, 7.45.
3-Ethyl-4,8-dimethyl-7-propoxy-2H-chromen-2-one (9b)
White powder, yield 78%. mp >300 °C. IR υmax/cm−1: 3068 (CH aromatic), 2960, (CH aliphatic), 1707 (C═O), 1502 (C═C). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.00 (t, 6H, J = 7.2 Hz, CH2CH3, CH2CH2CH3), 1.72–1.78 (m, 2H, CH2CH2CH3), 2.07 (s, 3H, CH3), 2.30 (s, 3H, CH3), 2.47–2.53 (q, 2H, J = 7.2 Hz, CH2CH3), 3.97 (t, 2H, J = 6.4, CH2CH2CH3), 6.91 (d, 1H, J = 8.8 Hz, H-Ar), 7.48 (d, 1H, J = 8.8 Hz, H-Ar). 13C NMR (100 MHz): 8.22, 10.82, 13.41, 14.76, 20.59, 22.52, 69.97, 108.43, 112.31, 114.01, 123.60, 123.70, 146.95, 150.82, 158.67, 161.30. MS m/z (%): 260.47 (M+, 15.15), 55.06 (65.79), 43.22 (100). Anal. Calcd. for C16H20O3 (260.33): C, 73.82; H, 7.74. Found: C, 73.68; H, 7.86.
3-Ethyl-7-isopropoxy-4,8-dimethyl- -2H-chromen-2-one (9c)
White powder, yield 98%. mp 165–167 °C. IR υmax/cm−1: 3061 (CH aromatic), 2974 (CH aliphatic), 1687 (C═O), 1568 (C═C). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.03 (t, 3H, J = 5.6 Hz, CH2CH3), 1.29–1.31 (m, 6H, CH(CH3)2), 2.14 (s, 3H, CH3), 2.36 (s, 3H, CH3), 2.53–2.56 (m, 2H, CH2CH3), 4.70–4.74 (m, 1H, CH(CH3)2), 7.01 (d, 1H, J = 7.2 Hz, H-Ar), 7.54 (d, 1H, J = 7.2 Hz, H-Ar). 13C NMR (100 MHz): 8.54, 13.43, 14.72, 20.61, 22.36 (2C), 70.96, 110.13, 113.45, 114.01, 123.54, 123.86, 146.96, 151.17, 157.80, 161.29. MS m/z (%): 260.80 (M+, 13.29), 242.91 (51.99), 48.32 (75.95), 44.13 (100). Anal. Calcd. for C16H20O3 (260.33): C, 73.82; H, 7.74. Found: C, 74.09; H, 7.89.
7-(Allyloxy)-3-ethyl-4,8-dimethyl-2H-chromen-2-one (9d)
White powder, yield 65%. mp 160–162 °C. IR υmax/cm−1: 3046 (CH aromatic), 2968 (CH aliphatic), 1693 (C═O), 1577 (C═C). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (t, 3H, J = 7.2 Hz, CH2CH3), 2.21 (s, 3H, CH3), 2.38 (s, 3H, CH3), 2.53–2.59 (q, 2H, J = 7.6 Hz CH2CH3), 4.69 (d, 2H, J = 4.8 Hz, CH2CH═CH2), 5.27–5.30 (dd 1H, J = 9.2 Hz, CH2CH═CHH), 5.41–5.47 (dd, 1H, J = 14 Hz, CH2CH═CHH), 6.09–6.13 (m, 1H, –CH2CH═CH2), 7.02 (d, 1H, J = 9.2 Hz, H-Ar), 7.58 (d, 1H, J = 9.2 Hz, H-Ar). 13C NMR (100 MHz): 8.43, 13.45, 14.79, 20.64, 69.22, 108.93, 112.65, 114.37, 117.79, 123.71, 124.04, 133.84, 147.06, 150.96, 158.25, 161.32. MS m/z (%): 258.21 (M+, 26.31), 182.34 (86.80), 75.18 (100). Anal. Calcd. for C16H18O3 (258.32): C, 74.39; H, 7.02. Found: C, 74.57; H, 7.14.
7-(Cinnamyloxy)-3-ethyl-4,8-dimethyl-2H-chromen-2-one (9e)
White powder, yield 66%. mp 140–142 °C. IR υmax/cm−1: 3041 (CH aromatic), 2962 (CH aliphatic), 1710 (C═O), 1577 (C═C). 1H NMR (400 MHz, DMSO-d6) δ ppm: 1.04 (t, 3H, J = 7.2 Hz, CH2CH3), 2.23 (s, 3H, CH3), 2.37 (s, 3H, CH3), 2.53–2.59 (q, 2H, J = 7.2, Hz, CH2CH3), 4.85 (d, 2H, J = 5.6 Hz, C6H5-CH═CHCH2), 6.50–6.56 (m, 1H, C6H5-CH═CHCH2), 6.79 (d, 1H, J = 16 Hz, C6H5-CH═CHCH2), 7.09 (d, 1H, J = 9.2 Hz, H-Ar), 7.27 (t, 1H, J = 7.2 Hz, H-Ar), 7.35 (t, 2H, J = 7.2 Hz H-Ar), 7.48 (d, 2H, J = 7.2 Hz, H-Ar), 7.59 (d, 1H, J = 8.8 Hz, H-Ar). 13C NMR (100 MHz): 8.54, 13.46, 14.80, 20.65, 69.28, 109.10, 112.77, 114.41, 123.75, 124.05, 125.10, 126.96 (2C), 128.42 (2C), 129.15, 132.90, 136.53, 147.07, 151.00, 158.38, 161.32. MS m/z (%): 334.70 (M+, 46.97), 296.11 (100), 41.40 (89.53). Anal. Calcd. for C22H22O3 (334.42): C, 79.02; H, 6.63. Found: C, 78.87; H, 6.79.
Biological studies
In vitro cytotoxicity studies (MTT protocol)
A 96-well tissue culture plate was inoculated with 1 × 105 cells/mL (100 µL/well) and cultured with PC-3, MDA-MB-231, HCT-116, HEPG-2, and HPrEC cells at 37 °C for 24 h to form a full monolayer sheet. After forming a confluent sheet of cells, the growth medium was decanted from 96-well microtitre plates, and the cell monolayer was washed twice with wash media. Twofold dilutions of the tested material were produced in RPMI medium containing 2% serum (maintenance medium). 0.1 mL of each dilution was examined in various wells, with three wells serving as controls and receiving only maintenance media. The plate was incubated at 37 °C and checked. Cells were examined for any physical evidence of toxicity, such as partial or total monolayer loss, rounding, shrinkage, or cell granulation. MTT solution (5 mg/mL in PBS) was prepared (Bio Basic Canada Inc., Markham, Canada). Each well received an 20 µL MTT solution. Shake the MTT into the medium for 5 min at 150 rpm on a shaking table. Incubate for 4 h (37 °C, 5% CO2) to allow the MTT to be metabolised. Get rid of the media (if required, dry the plate on paper towels to remove residue). Rehydrate formazan (MTT metabolic product) in 200 µL of DMSO. Shake at 150 rpm for 5 min to properly mix the formazan into the solvent. At 560 nm, read the optical density and subtract the background at 620 nm. The optical density should be proportional to the number of cells Citation44,Citation45. The percentage of viability in relation to vehicle control (100%) was used to express the results. Using the commercial software programmed Prism (GraphPad Software, Inc., La Jolla, CA), dose–response curves were produced and IC50 values (drug concentration resulting in a 50% reduction in cell survival) were determined. At least three separate experiments were conducted with all of the experiments. The values were means ± SD, n = 3.
In vitro EGFR and PI3Kβ inhibition assay
In vitro EGFR assay was conducted using EGFR Kinase Assay Kit using Kinase-Glo MAX (catalogue # 40321; BPS Bioscience, Inc., San Diego, CA) and the PI3Kβ (p110β/p85α) Assay Kit was designed to measure PI3Kβ activity for screening and profiling applications, using ADP-Glo® Kinase Assay as a detection reagent (catalogue #79802; BPS Bioscience, Inc., San Diego, CA), and other isoforms using ELISA Kit (Merck Millipore, Burlington, MA) following the manufacturing procedures. Three independent experiments were conducted for each compound and the values were means ± SD.
RT-qPCR analysis of AKT, m-TOR, BAX, BCl-2, P53, caspase 3, and caspase 8
Trypsinisation, centrifugation at 5000 rpm, and PBS washing were all performed on 1 × 105 Caco-2 control and treated cells. Total RNA was isolated from the pelleted Caco cells in accordance with the reported directions, and cDNA was subsequently produced. The expression of AKT, m-TOR, BAX, BCl-2, P53, caspase 3, and caspase 8 mRNA in control Caco-2 and treated cells was assessed using Applied qPCR Biosystems (Foster City, CA) in line with Livak and Schmittgen Citation46,Citation47. The primer sequences were created via integrated DNA Technologies (IDT) software as stated in .
Figure 16. Primer sequences of the targeted genes.

Cell cycle analysis (DNA flow cytometry)
A six-well plate was used, with 2 × 105 PC-3 cells put into each well. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% foetal bovine serum, and incubated for 24 h at 37 °C in a humid environment with 5% CO2 in the air. The synthesised compound 5 was added to a brand-new incubation medium at its IC50 concentration in DMSO (1% v/v). The cell plates underwent a 24 h incubation. The cells were fixed with 70% ice-cold ethanol after being rinsed twice with cold phosphate-buffered saline (PBS). Cells were centrifuged at 2000 rpm for 5 min after being washed with PBS at 37 °C for 30 min. Propidium iodide and DNA fluorochrome were used to stain the cells. Twenty minutes were spent incubating the plates at room temperature in the dark. Following that, the cells were examined using a FACS Calibre flow cytometer (Becton Dickinson, Heidelberg, Germany) Citation48,Citation49.
Annexin V/propidium iodide staining for apoptosis analysis
The determination of apoptosis was carried out using the Annexin V-FITC/PI Apoptosis Detection Kit. The targeted cells were obtained, washed twice with PBS, and then resuspension in binding buffer following the intended injection. Cell co-staining with Annexin V-FITC and PI was then carried out for 20 min in the dark at 37 °C. A FACS Calibur flow cytometer (Becton-Dickinson, Fullerton, CA) was used instantly for the analysis Citation50,Citation51.
Molecular docking studies
Molecular Operating Environment Software 2020.09, was used in the in silico simulation studies. The X-ray crystal structure EGFR tyrosine kinase co-crystallised with erlotinib was downloaded from the protein databank (rcsb.org, ID: 1M17) Citation36 with a resolution of 2.6 Å. The X-ray crystal structure of PI3Kβ co-crystallised with alpelisib was downloaded from the protein databank (rcsb.org, ID: 4JPS) Citation37 with a resolution of 2.20 Å. The 3D structures of enzymes were prepared for docking using prepared protein protocol default parameters taking into consideration retaining water molecule (H2O 10) in EGGFR kinase since it was reported to be crucial for binding interaction Citation52. The binding mode of erlotinib was analysed in 2D/3D to visualise the binding mode of erlotinib, this includes determining the non-bonding interaction and its 3D alignment in the binding site of EGFR tyrosine kinase binding site.
The 3D structures of the tested compounds were drawn and prepared by Molecular Operating Environment Software 2020.09 Citation35. The docking study was conducted using MOE docking protocol default parameters. A triangle matcher was generated as the placement method followed by rigid receptor refinement and GBVI and London dG scoring functions were used for pose ranking. In refinement, repetition was set to 30 poses, with five best poses to be retained. The resulting docking score and binding mode of the docked compounds were analysed to develop a preliminary SAR.
The docking protocol was validated by re-docking of crystallised lead erlotinib and alpelisib derivatives in the binding site EGFR tyrosine kinase and PI3Kβ Citation53, respectively, followed by the alignment of the 3D structure of the X-ray bioactive conformation with the best-fitted docked pose. Good alignment between the X-ray bioactive conformations of erlotinib and alpelisib and their best-fitted docked poses were obtained (Supplementary Data).
Author contributions
The strategy was designed by Lina Amin, Noha Ryad, and Abdulrahman Salim Alharbi; experimental work was performed by Lina Amin, Noha Ryad, and Abdulrahman Salim Alharbi; biological studies were discussed by Noha Ryad, Lina Amin, Tarek S. Ibrahim, Mohamed Elagawany, and Mohamed S. Abdel-Aziz docking studies were performed by Eman El-labbad. All of the authors discussed the results and revised the manuscript. All authors approved the final version of the manuscript.
Supplemental Material
Download PDF (4.9 MB)Acknowledgements
Authors Aknowledge with thanks The Deanship of Scientific Research (DSR) at King Abdulaziz University (KAU), Jeddah, Saudi Arabia for technical and financial support.
Disclosure statement
The authors report no conflicts of interest.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249.
- Henley SJ, Ward EM, Scott S, Ma J, Anderson RN, Firth AU, Thomas CC, Islami F, Weir HK, Lewis DR, et al. Annual report to the nation on the status of cancer, part I: National Cancer Statistics. Cancer. 2020;126(10):2225–2249.
- Buckup M, Rice MA, Hsu E-C, Garcia-Marques F, Liu S, Aslan M, Bermudez A, Huang J, Pitteri SJ, Stoyanova T. Plectin is a regulator of prostate cancer growth and metastasis. Oncogene. 2021;40(3):663–676.
- Fujita K, Hayashi T, Matsushita M, Uemura M, Nonomura N. Obesity, inflammation, and prostate cancer. J Clin Med. 2019;8(2):201.
- Hennrich U, Eder M. [177Lu] Lu-PSMA-617 (PluvictoTM): the first FDA-approved radiotherapeutical for treatment of prostate cancer. Pharmaceuticals. 2022;15(10):1292.
- Fallah J, Agrawal S, Gittleman H, Fiero MH, Subramaniam S, John C, Chen W, Ricks TK, Niu G, Fotenos A, et al. FDA approval summary: lutetium Lu 177 vipivotide tetraxetan for patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2023;29(9):1651–1657.
- Chen Y, Zhou Q, Hankey W, Fang X, Yuan F. Second generation androgen receptor antagonists and challenges in prostate cancer treatment. Cell Death Dis. 2022;13(7):632.
- Ma C-C, Liu Z-P. Design and synthesis of coumarin derivatives as novel PI3K inhibitors. Anticancer Agents Med Chem. 2017;17(3):395–403.
- Liu X, Xu Y, Zhou Q, Chen M, Zhang Y, Liang H, Zhao J, Zhong W, Wang M. PI3K in cancer: its structure, activation modes and role in shaping tumor microenvironment. Future Oncol. 2018;14(7):665–674.
- Noser AA, Shehadi IA, Abdelmonsef AH, Salem MM. Newly synthesized pyrazolinone chalcones as anticancer agents via inhibiting the PI3K/Akt/ERK1/2 signaling pathway. ACS Omega. 2022;7(29):25265–25277.
- Wu Y, Xu J, Liu Y, Zeng Y, Wu G. A review on anti-tumor mechanisms of coumarins. Front Oncol. 2020;10:592853.
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–1322.
- Mafi S, Mansoori B, Taeb S, Sadeghi H, Abbasi R, Cho WC, Rostamzadeh D. mTOR-mediated regulation of immune responses in cancer and tumor microenvironment. Front Immunol. 2021;12:774103.
- Makhov PB, Golovine K, Kutikov A, Teper E, Canter DJ, Simhan J, Uzzo RG, Kolenko VM. Modulation of Akt/mTOR signaling overcomes sunitinib resistance in renal and prostate cancer cells. Mol Cancer Ther. 2012;11(7):1510–1517.
- Yasumizu Y, Miyajima A, Kosaka T, Miyazaki Y, Kikuchi E, Oya M. Dual PI3K/mTOR inhibitor NVP-BEZ235 sensitizes docetaxel in castration resistant prostate cancer. J Urol. 2014;191(1):227–234.
- Croisy-Delcey M, Croisy A, Mousset S, Letourneur M, Bisagni E, Jacquemin-Sablon A, Pierre J. Genistein analogues: effects on epidermal growth factor receptor tyrosine kinase and on stress-activated pathways. Biomed Pharmacother. 1997;51(6–7):286–294.
- Day KC, Lorenzatti Hiles G, Kozminsky M, Dawsey SJ, Paul A, Broses LJ, Shah R, Kunja LP, Hall C, Palanisamy N, et al. HER2 and EGFR overexpression support metastatic progression of prostate cancer to bone. Cancer Res. 2017;77(1):74–85.
- Tseng J-C, Wang B-J, Wang Y-P, Kuo Y-Y, Chen J-K, Hour T-C, Kuo L-K, Hsiao P-J, Yeh C-C, Kao C-L, et al. Caffeic acid phenethyl ester suppresses EGFR/FAK/Akt signaling, migration, and tumor growth of prostate cancer cells. Phytomedicine. 2023;116:154860.
- Wang T, Peng T, Wen X, Wang G, Sun Y, Liu S, Zhang S, Wang L. Design, synthesis and preliminary biological evaluation of benzylsulfone coumarin derivatives as anti-cancer agents. Molecules. 2019;24(22):4034.
- Shen P, Wang H-G, Li M-M, Ma Q-Y, Zhou C-W, Pan F, Xie R. Isofraxidin inhibited proliferation and induced apoptosis via blockage of Akt pathway in human colorectal cancer cells. Biomed Pharmacother. 2017;92:78–85.
- García-Vilas JA, Quesada AR, Medina MÁ. 4-Methylumbelliferone inhibits angiogenesis in vitro and in vivo. J Agric Food Chem. 2013;61(17):4063–4071.
- Kishk SM, Eltamany EE, Nafie MS, Khinkar RM, Hareeri RH, Elhady SS, Yassen AS. Design and synthesis of coumarin derivatives as cytotoxic agents through PI3K/AKT signaling pathway inhibition in HL60 and HepG2 cancer cells. Molecules. 2022;27(19):6709.
- Turkekul K, Colpan RD, Baykul T, Ozdemir MD, Erdogan S. Esculetin inhibits the survival of human prostate cancer cells by inducing apoptosis and arresting the cell cycle. J Cancer Prev. 2018;23(1):10–17.
- Jeon YJ, Cho JH, Lee SY, Choi YH, Park H, Jung S, Shim JH, Chae JI. Esculetin induces apoptosis through EGFR/PI3K/Akt signaling pathway and nucleophosmin relocalization. J Cell Biochem. 2016;117(5):1210–1221.
- Ghany LMA, El-Dydamony NM, Helwa AA, Abdelraouf SM, Abdelnaby RM. Coumarin-acetohydrazide derivatives as novel antiproliferative agents via VEGFR-2/AKT axis inhibition and apoptosis triggering. New J Chem. 2022;46(36):17394–17409.
- Sairam KV, Gurupadayya B, Vishwanathan BI, Chandan R, Nagesha DK. Cytotoxicity studies of coumarin analogs: design, synthesis and biological activity. RSC Adv. 2016;6(101):98816–98828.
- Takla FN, Bayoumi WA, El-Messery SM, Nasr MN. Developing multitarget coumarin based anti-breast cancer agents: synthesis and molecular modeling study. Sci Rep. 2023;13(1):13370.
- Eliwa EM, Frese M, Halawa AH, Soltan MM, Ponomareva LV, Thorson JS, Shaaban KA, Shaaban M, El-Agrody AM, Sewald N. Metal-free domino amination-Knoevenagel condensation approach to access new coumarins as potent nanomolar inhibitors of VEGFR-2 and EGFR. Green Chem Lett Rev. 2021;14(4):578–599.
- BioRender; 2023. Available from: https://app.biorender.com/biorender-templates
- Shawish I, Barakat A, Aldalbahi A, Malebari AM, Nafie MS, Bekhit AA, Albohy A, Khan A, Ul-Haq Z, Haukka M, et al. Synthesis and antiproliferative activity of a new series of mono- and bis(dimethylpyrazolyl)-s-triazine derivatives targeting EGFR/PI3K/AKT/mTOR signaling cascades. ACS Omega. 2022;7(28):24858–24870.
- Tian Q, Wang G, Ma X, Shen Q, Ding M, Yang X, Luo X, Li R, Wang Z, Wang X, et al. Riboflavin integrates cellular energetics and cell cycle to regulate maize seed development. Plant Biotechnol J. 2022;20(8):1487–1501.
- Kattan SW, Nafie MS, Elmgeed GA, Alelwani W, Badar M, Tantawy MA. Molecular docking, anti-proliferative activity and induction of apoptosis in human liver cancer cells treated with androstane derivatives: implication of PI3K/AKT/mTOR pathway. J Steroid Biochem Mol Biol. 2020;198:105604.
- Hussar P. Apoptosis regulators bcl-2 and caspase-3. Encyclopedia. 2022;2(4):1624–1636.
- Lossi L. The concept of intrinsic versus extrinsic apoptosis. Biochem J. 2022;479(3):357–384.
- MOE. Montreal, Canada: Molecular Operating Environment (MOE) CCGU; 2023.
- Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–46272.
- Furet P, Guagnano V, Fairhurst RA, Imbach-Weese P, Bruce I, Knapp M, Fritsch C, Blasco F, Blanz J, Aichholz R, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23(13):3741–3748.
- El-Ansary SL, Hussein MM, Rahman DEA, Ghany LMA. Synthesis, docking and in vitro anticancer evaluation of some new benzopyrone derivatives. Bioorg Chem. 2014;53:50–66.
- Canter FW, Curd FH, Robertson A. CLXVI.—Hydroxy-carbonyl compounds. Part III. The preparation of coumarins and 1: 4-benzopyrones from phloroglucinol and resorcinol. J Chem Soc (Resumed). 1931;1255–1265.
- Yu X, Wen Y, Liang C-G, Liu J, Ding Y-B, Zhang W-H. Design, synthesis and antifungal activity of psoralen derivatives. Molecules. 2017;22(10):1672.
- Shilin S, Garazd M, Khilya V. Synthesis of dipeptide derivatives of 3,4-substituted 7-hydroxycoumarins. Chem Nat Compd. 2008;44(3):301–305.
- Timonen J, Vuolteenaho K, Leppänen T, Nieminen R, Moilanen E, Aulaskari P, Jänis J. 7‐(2‐Oxoalkoxy) coumarins: synthesis and anti‐inflammatory activity of a series of substituted coumarins. J Heterocycl Chem. 2015;52(5):1286–1295.
- El-Ansary SL, Rahman DEA, Ghany LMA. Synthesis and anticancer evaluation of some new 3-benzyl-4,8-dimethylbenzopyrone derivatives. Open Med Chem J. 2017;11(1):81–91.
- Van de Loosdrecht A, Beelen R, Ossenkoppele G, Broekhoven M, Langenhuijsen M. A tetrazolium-based colorimetric MTT assay to quantitate human monocyte mediated cytotoxicity against leukemic cells from cell lines and patients with acute myeloid leukemia. J Immunol Methods. 1994;174(1–2):311–320.
- Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988;48(3):589–601.
- Kvastad L, Werne Solnestam B, Johansson E, Nygren AO, Laddach N, Sahlén P, Vickovic S, Bendigtsen SC, Aaserud M, Floer L, et al. Single cell analysis of cancer cells using an improved RT-MLPA method has potential for cancer diagnosis and monitoring. Sci Rep. 2015;5(1):16519.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCT method. Methods. 2001;25(4):402–408.
- Eldehna WM, Abo-Ashour MF, Ibrahim HS, Al-Ansary GH, Ghabbour HA, Elaasser MM, Ahmed HY, Safwat NA. Novel [(3-indolylmethylene) hydrazono] indolin-2-ones as apoptotic anti-proliferative agents: design, synthesis and in vitro biological evaluation. J Enzyme Inhib Med Chem. 2018;33(1):686–700.
- Alkhaldi AAM, Al-Sanea MM, Nocentini A, Eldehna WM, Elsayed ZM, Bonardi A, Abo-Ashour MF, El-Damasy AK, Abdel-Maksoud MS, Al-Warhi T, et al. 3-Methylthiazolo[3,2-a]benzimidazole-benzenesulfonamide conjugates as novel carbonic anhydrase inhibitors endowed with anticancer activity: design, synthesis, biological and molecular modeling studies. Eur J Med Chem. 2020;207:112745.
- El-Naggar M, Eldehna WM, Almahli H, Elgez A, Fares M, Elaasser MM, Abdel-Aziz HA. Novel thiazolidinone/thiazolo[3,2-a]benzimidazolone–isatin conjugates as apoptotic anti-proliferative agents towards breast cancer: one-pot synthesis and in vitro biological evaluation. Molecules. 2018;23(6):1420.
- Eldehna WM, Nocentini A, Elsayed ZM, Al-Warhi T, Aljaeed N, Alotaibi OJ, Al-Sanea MM, Abdel-Aziz HA, Supuran CT. Benzofuran-based carboxylic acids as carbonic anhydrase inhibitors and antiproliferative agents against breast cancer. ACS Med Chem Lett. 2020;11(5):1022–1027.
- Sanduja M, Gupta J, Rawat R, Singh U, Verma SM. Designing, molecular docking, and dynamics simulations studies of 1,2,3-triazole clamped uracil–coumarin hybrids against EGFR tyrosine kinase. J Appl Pharm Sci. 2020;10(3):1–11.
- Hevener KE, Zhao W, Ball DM, Babaoglu K, Qi J, White SW, Lee RE. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009;49(2):444–460.