
Abstract
In this article, a new series of 2-((3,5-disubstituted-2-thioxo-imidazol-1-yl)imino)acenaphthylen-1(2H)-ones were synthesized. Imidazole-2-thione with acenaphthylen-one gave a hybrid scaffold that integrated key structural elements essential for DNA damage via direct DNA intercalation and inhibition of the topoisomerase II enzyme. All the synthesized compounds were screened to detect their DNA damage using a terbium fluorescent probe. Results demonstrated that 4-phenyl-imidazoles 5b and 5e in addition to 4-(4-chlorophenyl)imidazoles 5h and 5j would induce detectable potent damage in ctDNA. The four most potent compounds as DNA intercalators were further evaluated for their antiproliferative activity against HepG2, MCF-7 and HCT-116 utilizing the MTT assay. The highest anticancer activity was recorded with compounds 5b and 5h against the breast cancer cell line MCF-7 which were 1.5- and 3- folds more active than doxorubicin, respectively. Therefore, imidazole-2-thione tethered acenaphthylenone derivatives can be considered as promising scaffold for the development of effective dual DNA intercalators and topoisomerase II inhibitors.
Introduction
Cancer is regarded as the second leading cause of death worldwide in the 21st centuryCitation1. The synthesis of new potent and effective anticancer agents is of constant and growing interest in cancer treatment. As for the discovery of new drug candidates, researchers are committed to take a leap to discover new chemotherapeutic agents that interfere with DNACitation2. Numerous medications including doxorubicin and mitoxantrone are FDA-approved DNA intercalators; nevertheless, during clinical use, various adverse effects, including cardiotoxicity and the development of secondary malignancies, have been reported. Therefore, one of the main goals of modern medicinal chemistry is to create new safer DNA intercalators for the treatment of cancer. The role of DNA-interfering compounds has different approaches to exert their anticancer activities involving intercalating with the DNA and/or enzymes necessary for relevant DNA functions giving rise to a cellular response change which leads to cell deathCitation3. Typical DNA intercalators are compounds characterized by their ability to be inserted perpendicularly into DNA to form a DNA-intercalator complex stabilized by different hydrophobic stackings without forming covalent bondsCitation4. Polycyclic aromatic hydrocarbons have become one of the key molecular objectives in anticancer activity owing to their ability to be inserted between stacked base pairs of DNACitation5.
DNA topoisomerase enzyme plays crucial roles in employing the topology of DNA and controls many vital functions in the cell cycleCitation6. Topoisomerase enzyme is present in both mammalian and bacterial cells which makes it a conspicuous target as antibacterial drugs and as first-line anticancer agentsCitation7. Inhibition of topoisomerase enzyme has been the focus of anticancer drug discovery. In eukaryotic cells and according to its mechanism of action, topoisomerase is classified into topoisomerase I (Topo I) and II (Topo II)Citation6,Citation8. It has been found that mammalian Topo II worked through two different modes of action and thus categorised into: (a) topo II poisons and (b) topo II catalytic inhibitors;Citation9 (a) topo II poisons irreversibly bind to DNA-protein complex via covalent bonds and induce DNA double strand breaks (DSBs) which finally results in an apoptotic fateCitation2,Citation10. The second class is (b) catalytic inhibitors which act by binding non-covalently to the Topo II protein complex, preventing Topo II from binding to DNA or to ATPCitation11. Many well-known anticancer drugs act as topo II poisons via inter-calation with DNA such as: doxorubicin and mitoxantrone. Although these drugs have worked successfully as anticancer agents on a clinical basis, their use has been limited due to some toxicity and resistance consequences as mentionedCitation3,Citation12.
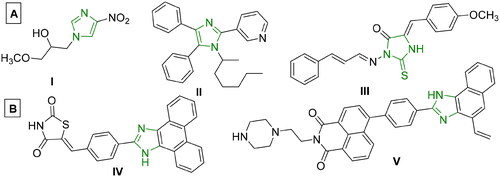
Imidazoles are an important class of heterocyclic molecules that are widely used to design medicinally important structural components with various pharmacological applicationsCitation13,Citation14. Various drugs bearing the imidazole moiety have been reported to have anticancer activityCitation5,Citation15,Citation16. Imidazole derivatives have also been proven to bind with DNACitation17, and exhibit good anticancer activity against MCF-7, HepG-2 and HCT-116 cell linesCitation18,Citation19. Some imidazole derivatives displayed strong DNA intercalating activityCitation19,Citation20, while other reported compounds demonstrated cytotoxicity at micromolar doses, as seen in I-III ()Citation21–23,Citation26,Citation27. Moreover, acenaphthequinone (acenaphthylene-1,2-dione) is a quinone derivative, widely used as significant starting material for the synthesis of various heterocycles, and as a key intermediate in organic synthesis for different reactions and pharmaceutical applicationsCitation28–30. Some derivatives of acenaphthequinone have been used as biologically active compounds, dyes, pharmaceuticals, drugs, pesticides, and therapeutic agentsCitation31–33. It was reported that tethering different poly aromatic chromophores with imidazole exhibited significant anticancer activity as in IV-V ()Citation5,Citation24,Citation25.
Figure 1. (A) Reported imidazole-containing drugs I-III as anticancer agentsCitation21–23, (B) Reported drugs bearing imidazole- polynuclear scaffold hybrids IV-V as anticancer agentsCitation5,Citation24,Citation25.
Rational design
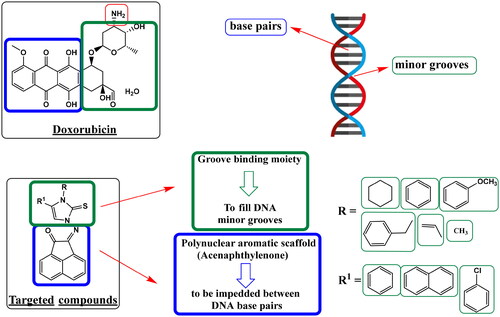
Literature has discovered three key pharmacophoric components shared by the DNA intercalator and the Topo II toxin doxorubicin. The first need is a polynuclear aromatic chromophore that can be placed in between DNA base pairs. Also, a cationic site that may attach to the phosphate group of DNA at physiological pH is the second structural feature. This might be a heterocyclic group that is nitrogen-containing or nitrogen-linked. Moreover, to fill DNA minor grooves, a groove-binding moiety (non-intercalator) is needed as the third componentCitation34. In addition, as reported, acenaphthoquinone-imidazole hybrids increased overall anticancer activity and yielded promising anticancer agents as topo II inhibitorsCitation24. Owing to the previously mentioned findings, the current study was carried out as a continuation of our earlier researchCitation35,Citation36 and to discover possible ligands with strong anticancer activity and consequently anti-topoisomerase II activityCitation37 along with a better safety profile than doxorubicin.
In the conducted study, series of 2-((3,5-disubstituted-2-thioxo-imidazol-1-yl)imino)acenaphthylen-1(2H)-ones were synthesized. The design of newly synthesized compounds took in consideration the hybridization of two structural features for DNA intercalation, acenaphthylenone (the polynuclear aromatic scaffold) and imidazoline-thione (the groove-binding moiety). These hybrids may significantly improve the anticancer activity by dual DNA intercalation and Topo II inhibition (). Therefore, newly synthesized compounds were tested for their DNA damage and their antiproliferative effect. Additionally, topoisomerase inhibition assessment was carried out, and molecular docking studies were employed for analysing the findings. Pharmacokinetic profiles and in-silico predictions of physiochemical attributes were also performed.
Figure 2. Work approach describes a hypothesis for designing targeted compounds based on structure features required for DNA damage through double helix intercalation and topoisomerase II inhibition, as in standard DNA intercalator Doxorubicin.
Results and discussion
Chemistry
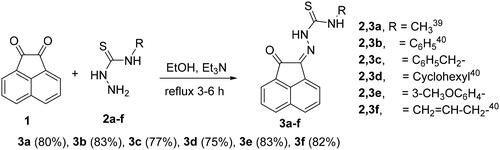
Preparation of N-substituted-2-(2-oxoacenaphthylen-1(2H)-ylidene)-hydrazinecarbothioamide derivatives 3a-f
Condensation of thiosemicarbazide derivatives 2a-f with acenaphthalene-1,2-dione (1) in refluxing ethanol and in the presence of triethylamine (Et3N), gave the corresponding N-substituted-2-(2-oxoacenaphthylen-1(2H)-ylidene)hydrazinecarbothioamide derivatives 3a-fCitation38,Citation39 (Scheme 1).
Scheme 1. Synthesis of N-substituted-2-(2-oxoacenaphthylen-1(2H)-ylidene)hydrazine-carbothioamide derivatives 3a-f.
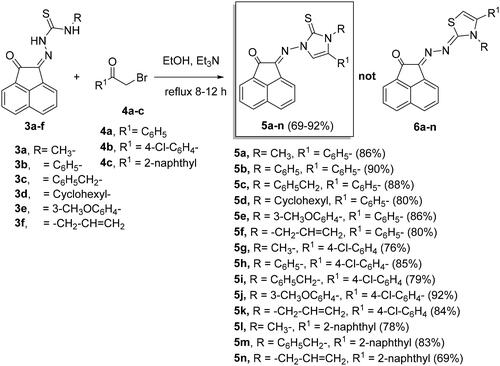
Synthesis of imidazole-2-thiones 5a-n
Reaction of compounds 3a-f with 2-bromo-1-substituted-ethan-1-ones 4a-c in ethanol and catalyzed by triethylamine (Et3N), produced 2-((3,5-disubstituted-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)acenaphthylen-1(2H)-ones 5a-n in 69%–92% yields (Scheme 2).
Scheme 2. Synthesis of imidazole-2-thiones 5a-n.
The structure of the newly prepared derivatives of N-substituted-2-(2-oxoacenaphthylen-1(2H)-ylidene)hydrazinecarbothioamides 3a-f was proved by IR, mass and NMR spectra in addition to elemental analysis. As for example, the structure of compound 3c was identified as (Z)-N-benzyl-2-(2-oxoacenaphthylen-1(2H)-ylidene)hydrazine-1-carbothioamide. IR spectrum showed absorptions at ν = 3314 (for ΝΗ str), 1607 (for C = N str), and 1023 cm−1 (for C = S str). The 1H NMR spectrum revealed two singlets at δH = 12.70 and 9.94 ppm for the two protons of the NH groups, whereas the benzyl-CH2 protons were resonated at δH = 4.94 ppm (J = 6.2 Hz). The 13C NMR spectrum showed the C = O, C = S and CH2- carbon signals at δC =188.5, 178.0 and 47.3 ppm, respectively (see the experimental).
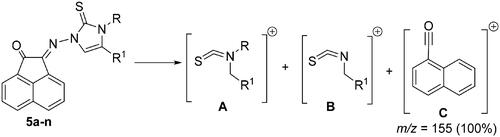
Also IR, NMR, mass spectra in addition to elemental analysis were used to elucidate the structure of the obtained imidazole-2-thiones 5a-n. Generally, in the most mass spectra of compounds 5a-n, two distinctive fragmentation patterns A and B were noted (). Mass spectra of most compounds also showed permanent fragmentation patterns of C at m/z = 155 (30%–50%) and 154 (100%) as shown in .
Figure 3. Fragmentation patterns A-C for mass spectra of compounds 5a-n.
In 1H NMR spectra of 5a-n, the disappearance of the two NH signals indicating their removal during the reaction pathway. The appearance of the C = S carbon signals at nearly δC = 175.8–178.4 ppm in the 13C NMR spectra of the formed products, excluded the formation of thiazoles 6a-n (Scheme 2).
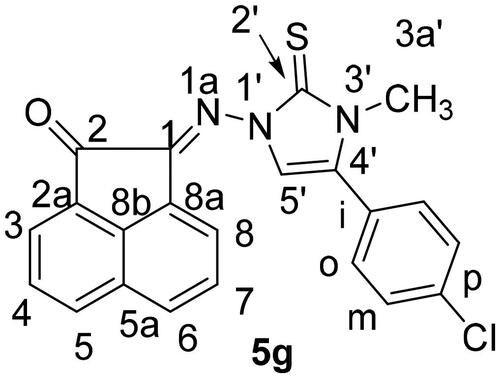
It is noteworthy that the expected thiazole derivatives 6a-n were not obtained. NMR spectra proved the imidazole structure of 5a-n. As for example, the 1H NMR spectrum of 5c revealed the CH2-Ph and the imidazole-H-5 as two singlets at δH = 5.35 and 6.94 ppm, respectively. The 13C NMR spectrum showed the CH2-Ph and imidazole-CH-5′ at δC = 49.4 and 104.0 ppm, respectively. The 1H NMR spectrum of compound 5f, as another example, showed some distinctive protons for aromatic protons as four double-doublets at δH = 8.25 (1H, J = 8.3, 1.2 Hz), 8.03 (1H, J = 8.0, 1.2 Hz) and 7.78 ppm (2H, J = 7.8, 1.2 Hz). In addition, two multiplets appeared at δH = 7.59–7.49 (2H) and 7.33–7.26 ppm (5H) for the remaining aromatic protons. The imidazole-H-5′ appeared as a singlet at δH = 6.62, whereas the allyl protons resonated as a multiplet at δH = 5.80–5.75 ppm for the allyl-CH = proton. In addition, two double-doublets appeared at δH = 4.97 (J = 7.5, 1.2 Hz) and 4.79 ppm (J = 7.5, 1.2 Hz) for allyl-CH2= protons. Another double-triplet for allyl-CH2 protons appeared at δH = 4.47 ppm (J = 7.5, 1.2 Hz). In 13C NMR spectrum of 5f, the allyl-CH2, allyl-CH2 = and allyl-CH = protons resonated at δC = 48.6, 104.5 and 129.8 ppm, respectively. The C = O, C = S and exc-C = N carbon signals resonated in the 13C NMR spectrum at δC = 189.1, 175.8 and 146.6 ppm, respectively. The 1H NMR spectrum of 5g () revealed the six aromatic of acenaphythylenone as four doublets; each for one proton, at δH = 8.57 (J = 6.9 Hz, H-5), 8.28 (J = 8.2 Hz, H-6), 8.06 (J = 8.4 Hz, H-3) and 8.00 ppm (J = 7.0 Hz, H-8). In addition, two double-doublets, each for one proton, were also resonated at δH = 7.82 (J = 7.9, 7.4 Hz, H-7) and 7.78 ppm (J = 8.1, 7.4 Hz, H-4). Whereas, the di-substituted phenyl protons appeared at δH = 7.66–7.62 ppm (4H). Imdazole-CH-5 (C-5′) and H-3a’ protons resonated as two singlets at δH = 6.93 and 3.63 ppm, respectively. The 13C NMR spectrum of 5g showed the carbon signals of N-CH3 and imidazole-CH-5 at δC = 34.3 and 104.7 ppm, respectively.
Figure 4. Distinctive carbons of compound 5 g.
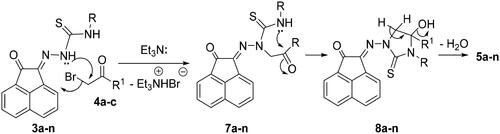
The reaction mechanism describes the formation of 5a-n was based upon an initial nucleophilic attack of hydrazone nitrogen lone-pair in 3a-n to the active methylene of 4a-c enhancing by the presence of Et3N. Thereafter, elimination of triethylamine hydrobromide molecule would then give the intermediates 7a-n (Scheme 3). Further nucleophilic attack of the other free NH-lone pair to the electrophilic carbonyl carbon would give intermediates 8a-n. Finally, elimination of water molecule from 8a-n would give compounds 5a-n (Scheme 3).
Scheme 3. Mechanism describes the formation of 5a-n.
Biological screening
DNA interaction studies
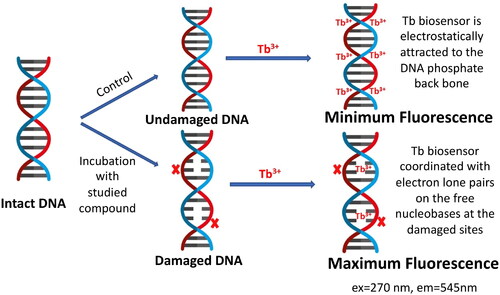
Luminescence studies were accomplished in order to examine the possible DNA interaction with the studied compounds. Many luminescent biosensors are reported in literature for studying the possibility of DNA interaction with small moleculesCitation40–43. In this article, we used the luminescent terbium (III) chloride (Tb) biosensor for probing such interaction due to its superior sensitivity, selectivity over many other reported biosensors, in addition to being inexpensive with simple mix-and-read procedureCitation44. presents a schematic diagram for the fluorometric detection of the induced DNA damage using Tb biosensor. Tb3+ ion in the Tb luminescent probe is a trivalent lanthanide cation having low intrinsic luminescence in aqueous solutionsCitation45,Citation46. When Tb biosensor is added to a solution of DNA, it interacts differently with double-stranded DNA (dsDNA) and single-stranded DNA (ssDNA). With dsDNA, Tb3+ ions are electrostatically attracted to the negatively charged phosphate backbone of the DNA. On the other hand, with ssDNA, it coordinates with the lone pairs of electrons of the free nucleobases, where upon DNA excitation, energy is transferred to the Tb biosensor causing significant enhancement of its luminescenceCitation47. Therefore, the assessed compound damages dsDNA, ssDNA sites would be formed opposite to the site of damage and Tb3+ would directly coordinate to the unpaired nucleobases in this single-stranded site. Upon excitation at λmax = 270 nm, energy will be transferred to Tb3+ with significant enhancement in its luminescence proportional to the amount of damage induced to dsDNA.
Figure 5. Schematic diagram for the fluorimetric detection of the induced DNA damage using terbium chloride (Tb3+) luminescent biosensor.
For investigating the DNA interaction potential of the fourteen studied compounds, calf thymus DNA (ctDNA) was incubated with each compound for 24 h and then Tb was added to each mixture. Doxorubicin (DOX), a commonly used reference for DNA damaging agent was used and treated the same condition as the studied compounds. Tb luminescent intensity was measured at λmax = 545 nm after excitation at λmax = 270 nm. Tb luminescence enhancement of each compound was compared with the luminescence of ctDNA-Tb mixture and Tb solution alone (results are demonstrated in ). A significant enhancement of Tb luminescence appeared with ctDNA samples incubated with doxorubicin and compounds 5b, 5e, 5h and 5j.
Figure 6. Tb3+ luminescence enhancement at λmax = 545 nm after excitation at λmax = 270 nm of the studied compounds compared to reference doxorubicin (DOX). Controls such as DNA-Tb3+ mixture, and Tb3+ alone are also presented.
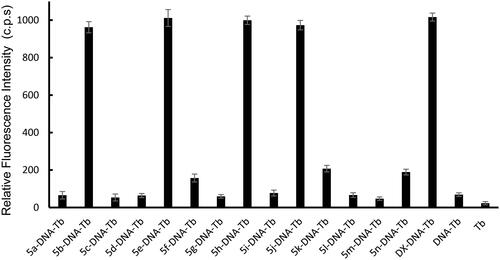
Compounds 5f, 5k and 5n showed minimum enhancement of Tb luminescence. While, the entire remaining compound did not any significant change in Tb luminescence from that of ctDNA-Tb mixture. The significant increase in Tb luminescence is related to the coordination of Tb with free nucleobases at the damage sites in ct DNA that were induced by compounds 5b, 5e, 5h, and 5j. While the slight increase in the biosensor luminescence indicates that compounds 5f, 5k, and 5n induced minimum DNA damage with the formation of very small number of damaged sites in DNA and thus, less potent to cause DNA damage. The remaining seven compounds did not show any significant interaction with DNA and thus the recorded luminescence was comparable to that of ctDNA-Tb with no compounds added.
The potency of DNA damage induction for each of the four most potent compounds, 5b, 5e, 5h, and 5j, was further assessed on natural ctDNA and compared to doxorubicin. Several concentrations within the range of 0.1 pM–200.0 μM of each of the studied compounds were incubated with ctDNA for 24 h. After which, Tb solution was added to aliquots of such mixtures and the luminescence intensity was recorded. presents Tb luminescence intensity as a function of the compound concentration for various compound/ctDNA/Tb mixtures. At minimum concentration of the studied compounds, minimum Tb luminescence was recorded. This reflects that the DNA molecule is undamaged, intact and acquiring a double helical structure. In this conformation, Tb ions are electrostatically attracted to the negatively charged DNA-phosphate backbone showing no increase in its luminescence. For solutions with higher concentrations of compounds 5b, 5h, and 5j (). Tb fluorescence increased proportionally with the concentration until a concentration of 100 and 60 μM for compounds 5b, 5h, and 5j, respectively, where no further increase in the fluorescence occurs. This reflects that more DNA damage is induced with increasing concentrations of the three compounds. At concentrations higher than 100 and 60 μM for compounds 5b, 5h, and 5j, respectively, Tb luminescence remains constant at high fluorescence levels. That indicates that maximum DNA damage has occurred. On the other hand, for compound 5e, no enhancement of Tb luminescence intensity with increasing compound 5e concentration until 20 μM (), after which proportional increase in Tb luminescence was recorded with the increase of 5e concentration until 80 μM, whereas the Tb luminescence remains constant at high luminescence indicating that maximum DNA damage has been attained. illustrates the analytical parameters elucidated as shown in for the quantification of the induced DNA damage by the studied compounds. The linearity parameters were calculated from the calibration curve in -inset in order to obtain the minimum concentration of the studied compounds that would induce DNA damage. Results show that a concentration as low as 7.2,7,14,78.14 and 12.7 nM of 5b, 5e, 5h, and 5j, respectively, are enough to induce detectable damage in ctDNA which comparable to doxorubicin () indicating their potency to induce DNA damage in natural DNA with compound 5h being the most potent to induce direct DNA damage.
Figure 7. Effect of increasing concentration of (a) 5b, (b) 5e, (c) 5h, (d) 5j, and (e) doxorubicin (DOX) as a reference of the luminescence of Tb- ctDNA complex. The insets show the fit to the linear region of the constructed plots.
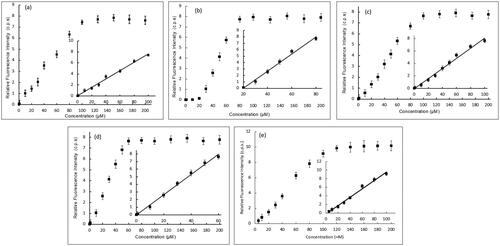
Table 1. Analytical parameters for the fluorescence measurements of 5b, 5e, 5h, 5j, and doxorubicin as a reference with ctDNA mixtures.
In vitro antitumor evaluation
In vitro cytotoxic activity against three cancer cell lines: The most potent DNA intercalating compounds 5b, 5e, 5h, and 5j were in vitro evaluated against their anticancer activities against three cancer cell lines namely; hepatocellular carcinoma (HepG2), human breast adenocarcinoma (MCF-7) and human colon cancer (HCT-116) utilising MTT assayCitation48 and the most active compounds were evaluated against normal breast cell line (MCF-10a). The IC50 values of the newly synthesized compounds were compared to doxorubicin as a positive control as shown in . The obtained IC50 values of the tested compounds displayed different levels of anticancer activity ranging from high to weak activities against all tested cell lines.
Figure 8. In vitro anticancer activity, IC50 (μM) of 5b, 5e, 5h, and 5j in comparison to Doxorubicin.
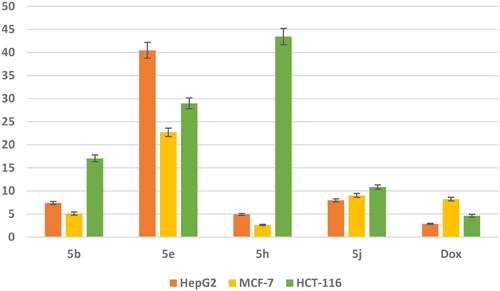
Data represented in revealed that compounds 5b and 5h exhibit superior anticancer activity against MCF-7 cell line with IC50 values 5.105 and 2.65 μM, respectively compared with doxorubicin as a reference drug with IC50 value of 8.240 μM. Meanwhile, they showed moderate activity against HepG2 cell line with IC50 values 7.397 and 4.929 μM, respectively, and weak anticancer activity against HCT-116 cell line with IC50 values 17.063 and 43.45 μM, respectively. Moreover, the safety of compounds 5b and 5h was assessed against a normal breast cell line (MCF-10a) using the MTT assay. The results revealed that compounds 5b and 5h showed a remarkably less cytotoxic effect towards MCF-10a with IC50 value of 89.759 and 130.576 μM, respectively compared with Dox with IC50 value of 32.416 μM. In addition, compound 5j showed moderate anticancer activity against three cancer cell lines HepG2 and MCF-7 and HCT-116 with IC50 value of 7.9, 964.030 and 10.847 μM, respectively. While compound 5e exhibited weak anticancer activity against three cancer cell lines.
The selectivity index (SI)Citation49 was calculated for the most active compounds 5b and 5h against breast cancer cell line and the results revealed that, compounds 5b and 5h showed high selectivity index (17.58 and 49.27, respectively) against MCF-7 cell line compared with doxorubicin with value of 3.9 (). Consequently, compounds 5b and 5h were the least cytotoxic and highest anticancer activity with high SI value compared to the Dox. These results indicated a successful discovery of new anticancer drug candidates and confirmed that they could be safe drugs against tumours.
Table 2. Selectivity Index (SI) of compounds 5b, 5h, and doxorubicin against human breast adenocarcinoma (MCF-7).
The initiation of apoptosis and the stages of the cell cycle are important in the era of cancer treatment. Several imidazole-2-thiones molecules have been reported as apoptosis inducers in several cancer cell lines. Therefore, the two most active compounds 5b and 5h, were chosen to evaluat their effect on apoptosis and the cell-cycle profile.
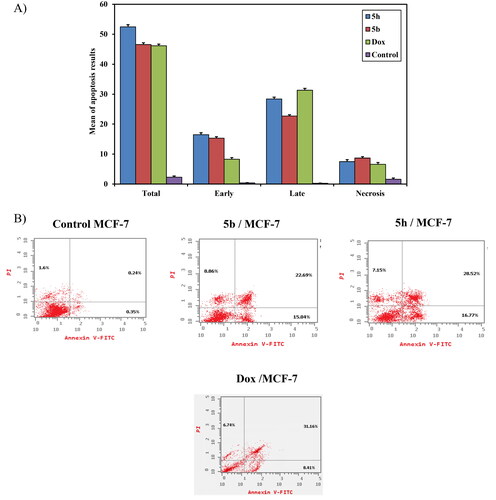
Annexin V-FITC/PI apoptosis induction analysis: The breast carcinoma cell line (MCF-7) was used for revealing the induction of apoptosis and cell-cycle profile of compounds 5b, 5h, and doxorubicin as a reference drug owing to its significant sensitivity towards those compounds. Flow cytometric annexin V/propidium iodide analysisCitation50 was used for detecting the apoptotic effects of the most active and non-toxic anticancer compounds 5b, 5h, and doxorubicin and applied to MCF-7 cells at concentrations equal to their IC50 values (5.105, 2.65 and 8.240 μM, respectively) on the MCF-7 cell line. The results indicated that the selected candidates induce apoptosis of MCF-7 cells at both early and later stages. As shown in , the percentage of early apoptosis increased from 0.37 ± 0.11% (control) to 15.3 ± 0.50% and 16.5 ± 0.64% for compounds 5b and 5h, respectively. Whereas doxorubicin showed an increase in the percentage of early apoptosis by 8.3 ± 0.53%. In addition, the percentage of late apoptosis was increased from 0.26 ± 0.06% (control) to 22.7 ± 0.43% and 28.4b ± 0.62% upon exposure to compounds 5b and 5h, respectively. At the same time, doxorubicin induced the late cell apoptosis by 31.3 ± 0.62%. So, these findings confirm that compounds 5b and 5h induce apoptosis in MCF‐7 cells by total cell apoptosis higher than doxorubicin as shown in . It can be concluded that the anticancer mechanism of action for compounds 5b and 5h is attributed to their apoptosis inducing activity in MCF-7 cells.
Figure 9. (A) A graphical representation of apoptosis induction analysis of compounds 5b, 5h and doxorubicin as reference against MCF-7 at their IC50(μM). (B) Apoptosis induction analysis of compounds 5b, 5h, and doxorubicin as reference against MCF- 7 at their IC50 (μM).
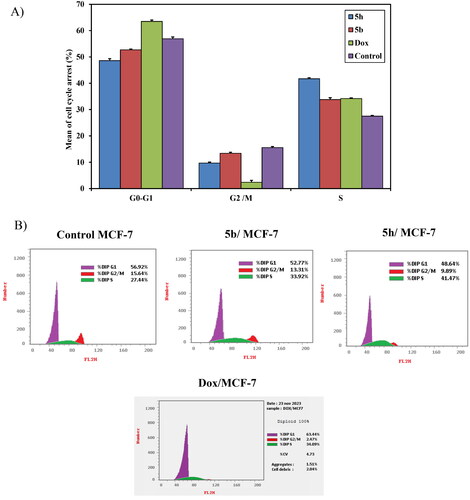
Cell cycle analysis: The most active compounds 5b and 5h were investigated for their cell cycle profile using DNA flow cytometric analysis on MCF-7 in comparison with doxorubicin as a reference drugCitation22,Citation51. Treatment of MCF-7 cells with IC50 concentration dose value of those compounds resulted in a significant alteration in cell cycle profile. The results revealed significant increase in the percentage of cell population at the S phase from (27.5 ± 0.28%) control to 33.8 ± 0.70% and 41.7 ± 0.39% upon treatment with compounds 5b and 5h, respectively. In addition, doxorubicin induced cell cycle arrest at G1/S phase in comparison with the untreated control. Therefore, it can be concluded that compounds 5b and 5h inhibited the cell proliferation of MCF-7 cells via cell cycle arrest at the S phase similar to doxorubicin as reference drug (). For compounds with a mechanism of action similar to doxorubicin, it can be expected an arrest at the G2/M phase. However, compounds 5b and 5h arrested at the S phase which may be due to the presence of acenaphythylenone, the poly aromatic hydrocarbon (PAH). It was found that the large adducts of PAH to DNA bases can result in a range of chromosomal changes, including frameshift mutations, deletions, S-phase arrest, and chromosomal alterationsCitation52. According to the literature, it was reported that the polyaromatic hydrocarbons only suppressed the S phase of the cell cycleCitation53. A probable explanation for the observed cell cycle arrest at the S phase is the production of DNA damage as a result of DNA intercalation within the cells. During the mitotic S phase, DNA replication and Topo II levels riseCitation54,Citation55. Therefore, those compounds that destroy cells in the S phase aid in stopping DNA and topo II production.
Figure 10. (A) A graphical representation of cell cycle analysis of compounds 5b, 5h, and doxorubicin as a reference against MCF-7 at their IC50 (μM). (B) Cell cycle analysis of compounds 5b, 5h, and doxorubicin as a reference against MCF-7 at their IC50 (μM).
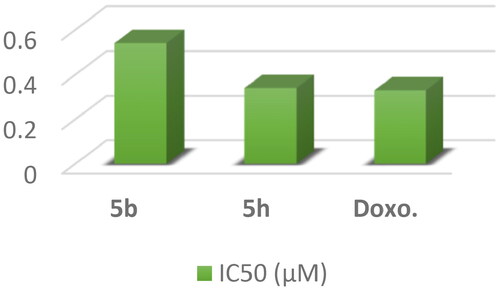
Topoisomerase II inhibitory activity: The most active compounds 5b and 5h were further examined as topoisomerase II inhibitors using doxorubicin as a positive control to expect the possible molecular mechanism underlying the potent anticancer activity of compoundsCitation56. The results showed that compound 5h revealed potent inhibitory activity against topo II enzyme with IC50 value of 0.34 μM compared with doxorubicin (IC50 = 0.33 μM). Moreover, compound 5b showed moderate inhibitory activity against topo II enzyme with IC50 value of 0.54 μM ().
Figure 11. A graphical representation of 5b, 5h and DOX against Topo II inhibitory activity.
Structure activity relationship (SAR)
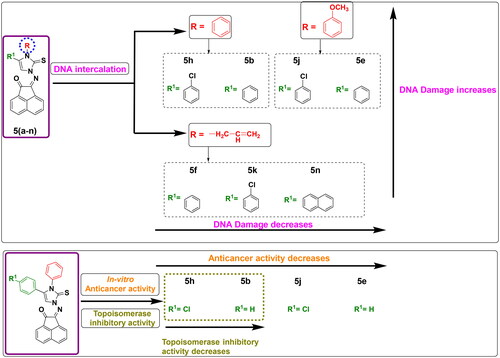
The findings from biological screening offered a useful profile for the structure-activity relationship and demonstrated a positive influence on the structure optimization of newly synthesized compounds. Among the study, anticancer screening was significantly impacted by the substitution of the imidazole ring at its N-atom or at position 4 with diverse aliphatic and aromatic moieties. The interpreted findings of induction of DNA damage using terbium fluorescent probe were varied in which seven compounds out of fourteen tested ones exhibited damage to DNA. These seven compounds shared an aromatic scaffold at imidazole carbon-4. It was noticed that compounds bearing N-phenyl or N-methoxy phenyl moiety of imidazole showed the most potent induction of DNA damage as found in 5b, 5e, 5h, and 5j. It was also noticed that compounds bearing N-allyl moiety of imidazole ring showed moderate induction of DNA damage as in 5f, 5k, and 5n. This observation demonstrates that replacing the cyclic aromatic scaffold into an open unsaturated chain moiety decreases the activity as DNA intercalator, while aliphatic substitution (methyl or cyclohexyl) showed no significant activity. On the other hand, the antiproliferative activity was examined the highest in compounds 5b and 5h that bearing N-phenyl moiety of imidazole-2-thione ring, while methoxy phenyl derivatives 5j and 5e exhibited less anticancer potency against selected cell lines. This finding may explain why less bulkiness at imidazole nitrogen is favourable. In addition, the substitution at position 4 of imidazole-2-thione with 4-chlorophenyl moiety (5h) instead of unsubstituted phenyl (5b) significantly duplicated the topoisomerase inhibitory activity. This may be attributed to the importance of substituted atoms in enhancing the fitting of the protein active site ().
Figure 12. Structure-Activity relationship of DNA damage testing, antiproliferative activity and topoisomerase inhibitory activity for tested compounds.
In silico studies
Molecular docking
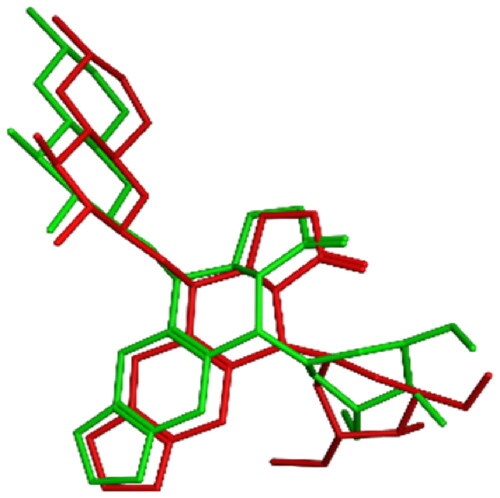
To gain further comprehension about the binding mode of topoisomerase II as a potential anticancer target, in-silico docking experiments were established for all tested compounds, and comparisons with the in-vitro assay results were visualised and anticipated. Studies employing molecular docking were performed on the binding site of human topoisomerase II beta enzyme complexed with DNA (PDB ID: 3QX3) using Molecular operating Environment software (MOE 2022.02)Citation57. The purpose was to give an account about the mechanism of action of the most active compounds, their inhibitory activities and their binding affinities to the enzyme active site. The outcomes were contrasted with the co-crystallized ligand: etoposide (EVP) and to reference drug doxorubicin (DOX). The proposed docking algorithm was validated by re-docking of the co-crystallized ligand etoposide into the active site of human topoisomerase IIβ DNA complex. The initial poses generated from PDB were retrieved with RMSD of 1.06 Å and with significant overlay of re-docked ligand to the co-crystallized ligand EVP (). Appropriate RMSD indicated that MOE docking can reliably predict docking poses for the studied compounds and assure the proposed docking protocolCitation58.
Figure 13. 3D representation of the overlay of re-docked ligand (green) to the co-crystallized ligand EVP (red) (RMSD =1.06 Å).
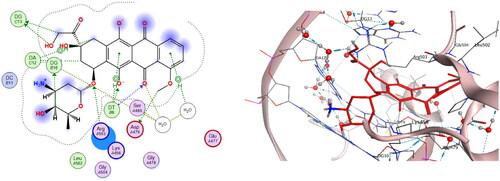
It was recorded that interactions of etoposide EVP in DNA-Topo IIβ complex encompass some aminoacid residues as; GLU477, ASP479, Arg503, GLN778 and Met782, in addition to some nucleotides such as: guanine13, adenine12 and thymidine 9Citation2,Citation3. Consequently, the re-docking results of the co-crystallized ligand EVP to the active site of DNA-Topo IIβ complex showed binding score = −10.092 Kcal/mol. EVP displayed many hydrogen bonds interactions between its sugar moiety and Gln778, Met782, in addition to other hydrogen bonds interactions with DT9, DG13 and a water molecule. The cyclohexyl ring in EVP also showed two hydrogen bonds with Arg503 and DT9, while the planar aromatic ring of EVP exhibited a hydrogen bond with DG13 besides other H-pi interactions with DT9 and DA12 ().
Figure 14. The binding interaction of EVP (green) at the pocket of the active site of DNA-Topo IIβ complex in 2D (left panel) and 3D representation (right panel).
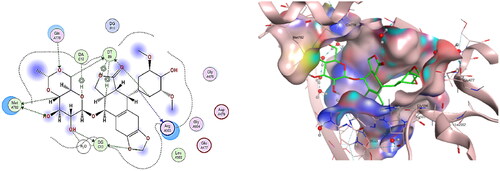
Docking interpretation was performed on doxorubicin as reference drug at the active site of DNA-Topo IIβ complex and it showed binding score = −6.41 Kcal/mol and it shared many interactions with crucial aminoacids and nucelobases all with distances less than 4.5 Å as shown in . It was observed that methoxy group at C-4 of tetracene ring binds with Asp479 through a hydrogen bond. Also, the carbonyl group at C-5 of tetracene forms two hydrogen bonds with DT9 and water molecule as well as the hydroxy group at C-6. Another hydrogen bond was observed between O-atom at C-7 of tetracene and Arg503. Moreover, the hydroxy acetyl moiety shared two hydrogen bonds with DG13. Also, C-4 of pyran ring binds to DG10 via hydrogen bonding. In addition, tetracene ring exhibited four H-pi interactions with DT9, DA12 and with a water molecule.
Figure 15. The binding interaction of doxorubicin (red) at the active site of DNA-Topo IIβ complex in 2D (left panel) and 3D representation (right panel).
All newly synthesized compounds underwent docking assessments, and their binding scores and binding modalities were evaluated. As shown in , compounds 5a-n were successfully occupied in the active site of topoisomerase II and displayed favourable binding scores between −6.59 and −8.07 Kcal/mol. The former examination was done together with doxorubicin and co-crystallized ligand etoposide (EVP). With crucial essential amino acids and nucleobases, they demonstrated various hydrophilic and hydrophobic interactions with accepted distances (less than 4.5 Å). It was evident that the sulphur atom and imidazole ring displayed the most frequent hydrogen bond interactions in the protein pocket. Likewise, Numerous arene-hydrogen interactions were shared by the polyaromatic acenaphthylenone moiety.
Table 3. Molecular docking scoring and interactions of tested compounds at the active site of DNA-Topo IIβ (PDB ID: 3QX3) in comparison to reference drugs EVP and DOX.
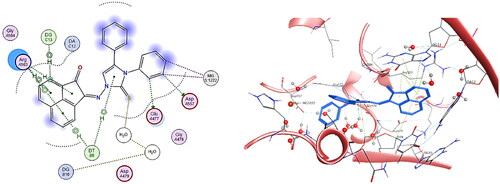
Regarding the findings of biological screening as DNA intercalation, anticancer activity and topoisomerase inhibition, it was observed that the most active compounds 5b and 5h were shown to have excellent binding modes at the active site of the DNA-Topo II complex with a variety of interactions that are highly similar to those identified with co-crystallized ligand EVP. The binding pattern of compound 5b was examined and its binding score to DNA-Topo IIβ complex active site was found equal to −6.59 Kcal/mol. It was investigated that compound 5b shared key aminoacids and nucleobases as found in EVP as follows: two hydrogen bonds were observed between phenyl ring substituted on N-atom of imidazole ring and Glu477, Asp557, in addition to two Mg metal interactions with the same phenyl moiety. Moreover, sulphur atom of imidazole ring binds to a water molecule through a hydrogen bond whereas imidazole ring binds to DT9 via H-pi interaction. It was observed that acenaphthylenone ring involves many H-pi interactions with Arg503, with DG13 and with DT9 with distances less than 4.5 Å ().
Figure 16. The binding interaction of compound 5b (blue) at the active site of DNA-Topo IIβ complex in 2D (left panel) and 3D representation (right panel).
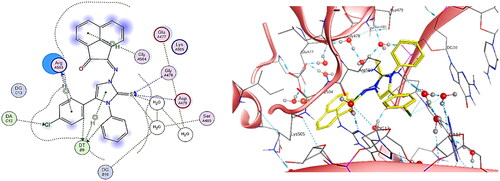
By observing the binding pattern of compound 5h, it was crystal clear that it showed excellent binding mode at DNA-Topo IIβ complex active site, with binding score = −7.00 Kcal/mol. The docking results showed both hydrogen bond and H-pi interactions between chloro-phenyl ring substituted at C-4 of imidazole ring and Arg503 and another hydrogen bond was observed with DT9. Similarly, hydrogen bonds were examined between chlorine atom and DA12 as well as between the phenyl ring substituted on N-atom of imidazole ring and DT9. Likewise, four hydrogen bonds were observed between sulphur atom of imidazole ring and Gly478, Asp479 and two water molecules all with distances less than 4.5 Å. In addition, two H-pi interactions were detected between acenaphthylenone ring and Gly504, and between imidazole ring and DT9 ().
Figure 17. The binding interaction of compound 5h (yellow) at the active site of DNA-Topo IIβ complex in 2D (left panel) and 3D representation (right panel).
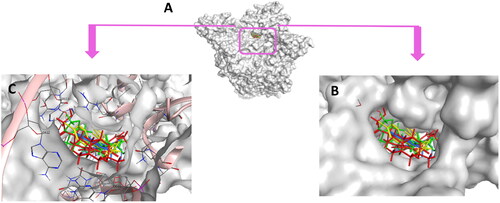
Conclusively, docking of 5b and 5h along with the reference control doxorubicin to DNA-Topo IIβ complex was accurately achieved at the pocket of the active site of target protein in comparison to the co-crystallized ligand etoposide EVP (). Thus, support the biological screening findings and declare the mechanism of action of these new candidates as topoisomerase inhibitors and as DNA intercalators.
Figure 18. (A) Cartoon representation of the molecular surface of DNA-Topo IIβ complex (PDB:3QX3) showing EVP (green), 5b (blue), 5h (yellow), doxorubicin (red) in the binding pocket. (B) Focused view of the active site. (C) Transparent mode of the cartoon representation illustrating the binding pocket with the essential aminoacids and nucleotides.
In silico predication of physiochemical properties, pharmacokinetic profile and drug likeness optimization measures
The drug-likeness and pharmacokinetic properties were explicated for the active four compounds 5b, 5e, 5h, and 5j which can be supportive in the context of medicinal chemistry. We employed the automated SwissADMECitation59 for the pharmacokinetics and drug-likeness predictions. The evaluations against passive human gastrointestinal absorption (GIA) and blood-brain barrier (BBB) permeation which give some parameters for the pharmacokinetics predictions were extracted from the BOILED-Egg modelCitation59,Citation60. The compounds 5b and 5e are presented high as seen in . Also, all molecules were suggested to fail to permeate through BBB, and hence, they are expected to show low incidence for central nervous system (CNS) adverse effects. Permeability glycoprotein (P-gp) is proposed to be the most important member among ATP-binding cassette transporters or ABC-transportersCitation59, since it is part of the resistance mechanisms against drugsCitation59,Citation62. All compounds were predicted to escape the P-gp indicating low incidence of developing resistance against human biological membranes.
Table 4. In silico predictions of the pharmacokinetics and drug-likeness properties for 5b, 5e, 5h, and 5j.
Commonly, 50%–90% of therapeutic molecules are judged to be a substrate of at least one of the five major isoforms of Cytochrome P (CYP) enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4)Citation59,Citation63,Citation64. Inhibition of these isoenzymes is unquestionably one cause of pharmacokinetics-related drug-drug interactions leading to toxic or adverse effectsCitation65,Citation66. As shown in (), all molecules were predicted to display inhibition of at least two of the CYP isoforms. This recommends administering these candidates in a sole regime and not in combination with other therapeutic agents. Fortunately, all molecules showed no alert to be a possible PAINS (pan-assay interference compounds)Citation67, as shown in . This stresses that their chemical structures would not interfere in protein assays denoting the in vitro results to be robust with minimum artefacts. Lastly, drug-likeness properties underlined that all molecules obey Veber rules Citation68 and highlighting that these candidates would demonstrate good bioavailability profiles.
Conclusion
In summary, a new series of imidazole‐2‐thiones derived by acenaphythylenones were synthesized and screened for their activity as anticancer agents by detection of their DNA damage using terbium fluorescent probe. Compounds 5b, 5e, 5h, and 5j pronouncedly induced DNA damage at different concentrations. Further, those compounds were evaluated for their in vitro anticancer activity against three cancer cell lines namely; hepatocellular carcinoma (HepG2), human breast adenocarcinoma (MCF-7) and human colon cancer (HCT-116) aided by doxorubicin as reference drug owing to its role in DNA damage. Using tumour management utilising microculture MTT assay, compounds 5b and 5h demonstrated the highest anticancer activity, with IC50 values of 5.105 and 2.65 μM, respectively, compared to the reference drug doxorubicin with IC50 value of 8.240 μM. In addition, compounds 5b and 5h showed a great safety margin against normal breast cell line (MCF-10a) with IC50 = 89.759 and 130.576 μM, respectively, better than doxorubicin (IC50 = 32.416 μM). Furthermore, those compounds exhibited high selectivity on the HepG2 and MCF-7 cell lines higher than doxorubicin (safer than doxorubicin). In addition, The DNA flow cytometry analysis of compounds 5b and 5h indicated that MCF‐7 cells revealed cell cycle arrest at S phase of the cell cycle profile with induction of apoptosis. It was observed that the percentage of late apoptosis was increased from 0.24% (control) to 22.69% and 28.52% upon exposure to compounds 5b and 5h, respectively. According to these results, the mechanism of action of the anticancer activity for compounds 5b and 5h is attributed to its apoptosis inducing activity in MCF-7 cells. Furthermore, the most active compounds examined as topoisomerase II inhibitors using doxorubicin as a positive control and the results showed that compound 5h exhibited a potent inhibitory activity against topo II enzyme with IC50 value of 0.34 μM like that afforded by doxorubicin (IC50 = 0.33 μM). Analysing the molecular docking results on the active site of DNA-Topo IIβ complex showed fabulous binding mode and scoring values. Whereas the in silico physiochemical interpretations were consistent and robust. As a result, compounds 5b and 5h emerged as valuable lead compounds with a fruitful matrix that might be useful as antitumor agents, particularly as dual DNA intercalator topoisomerase inhibitors with safety profile.
Experimental section
Chemistry
Reagents and materials
All materials were obtained from commercial suppliers and used without further purification. Reactions were monitored by TLC (Kieselgel 60 F254 precoated plates, E. Merck, Germany), the spots were detected by exposure to UV lamp at 254 nm. Melting points were determined on an electrothermal melting point apparatus (Stuart Scientific Co.) and were uncorrected. NMR spectra were measured in DMSO-d6 on a Bruker AV-400 spectrometer (Bruker Bio Spin Corp., Billerica, MA, USA) (400 MHz for 1H, 101 MHz for 13C, and 40.15 MHz for 15N) at Florida Institute of Technology, USA. The 1H and 13C chemical shifts are given relative to internal standard TMS = 0, and external liquid ammonia = 0 for 15N. Coupling constants are stated in Hz. Correlations were established using 1H-1H COSY, and 1H-13C and 1H-15N HSQC and HMBC experiments. Vario EL III German CHN Elemental analyser model was used for Elemental analysis.
Synthesis of thiosemicarbazones 3a-f
An ethanolic solution of N-substituted-hydrazinecarbothioamides 2a-f (0.01 mol) was added to an ethanolic solution (50 ml) containing acenaphthylene-1,2-dione (1, 0.01 mol) with 0.5 ml of triethylamine (Et3N). The mixture was refluxed for 3–6 h during which time a yellow precipitate separated out. The reaction mixture was then cooled to room temperature and the precipitate was filtered off and the obtained products 3a-f were recrystallized.
(Z)-N-Methyl-2-(2-oxo-1,2-dihydroacenaphthylen-1-ylidene)hydrazinecarbothioamide (3a): Yield: 80%; yellow crystals (EtOH); m.p. = 295 °C (m.p. = 293 °C, lit.Citation38).
(2-oxoacenaphthylen-1-(2H)-ylidene)-N-phenylhydrazinecarbothioamide (3b): Yield: 83%; yellow crystals (MeOH); m.p. 212–214 °C (m.p.: 208–210 °C, lit.Citation39).
(Z)-N-Benzyl-2-(2-oxoacenaphthylen-1(2H)-ylidene)hydrazine-1-carbothioamide (3c): Yield: 84%, yellow crystals, (EtOH), m.p.: 226–228 °C. IR (KBr, cm−1): ν = 3314 (str ΝΗ), 1607 (str C = N), 1023 (str C = S); 1H NMR (400 MHz, DMSO-d6): δH = 12.70 (s, 1H, NH), 9.94 (bs 1H, NH), 8.37 (d, J = 8.2; 1H), 8.16 (d, J = 8.3; 1H), 8.10 (d, J = 7.0; 1H), 8.00 (d, J = 7.0; 1H), 7.88 (dd, J = 8.0,7.2; 1H); 7.83 (dd, J = 8.2,7.2; 1H), 7.42 (d, J = 7.2; 2H), 7.37 (dd, J = 7.7.3; 2H), 7.28 (t, J = 7.1; 1H), 4.94 (d, J = 6.2; 2H); 13C NMR (100.1 MHz, DMSO-d6): δC = 188.5 (C-2), 178.0 (C-1c), 139.2 (C-8b), 138.4 (C-i), 137.5 (C-1), 132.8 (C-5), 130.4,130.0,129.9 (C-2a, 5a, 8a), 128.8 (C-7), 128.6 (C-4), 128.3 (C-m), 127.4 (C-o), 127.1 (C-p), 127.0 (C-6), 122.4 (C-3), 118.3 (C-8), 47.3 (C-1e). 15N NMR: (DMSO-d6): δN = 168.2 (N-1b), 122.3 (N-1d). C20H15N3OS (345.42): Calcd: C, 69.54; H, 4.38; N, 12.17; S, 9.28. Found: C, 69.67; H, 4.42; N, 12.25; S, 9.32.
(Z)-N-cyclohexyl-2-(2-oxoacenaphthylen-1-(2H)-ylidene)hydrazinecarbothioamide (3d): Yield: 75%; yellow crystals (CH3CN); m.p.: 202–204 °C (m.p.: 202–204 °C, lit.Citation40).
(Z)-N-(3'-Methoxyphenyl)-2-(2-oxoacenaphthylen-1(2H)-ylidene)hydrazine-1-carbothioamide (3e): Yield: 84%, yellow crystals (EtOH), m.p.: 230–232 °C. 1H NMR (400 MHz, DMSO-d6): δH = 12.9 (s, 1H; NH), 10.91 (1H; NH), 8.41 (d, J = 8.2, 1H; H-5), 8.18 (d, J = 8.3, 1H; H-6), 8.17 (d, J = 7.2, 1H; H-3), 8.14 (d, J = 7.2, 1H; H-8), 7.92 (dd, J = 8.2,7.8, 1H; H-4), 7.88 (dd, J = 8.1,7.8, 1H; H-7), 7.36 (dd, J = 8.4,8.0, 1H), 7.34 (bs, 1H), 7.28 (bd, J = 8.0, 1H), 6.88 (dd, J = 8.2.3, 1H), 3.80 (s, 3H). 13C NMR (100.1 MHz, DMSO-d6): δC = 188.6, 176.3, 159.2, 139.6, 139.4,137.2,132.8,130.4,129.9,129.1,128.9,128.6,127.3,122.5,118.9,117.5,111.6,111.3,55.2 (OCH3). 15N NMR: (DMSO-d6): δN = 170.5 (N-1b), 132.0 (N-1d). Calcd: C, 66.47; H, 4.18; N, 11.63; S, 8.87. Found: C, 66.55; H, 4.22; N, 11.60; S, 8.9.
(Z)-N-Allyl-2-(2-oxoacenaphthylen-1-(2H)-ylidene)hydrazinecarbothioamide (3f): Yield: 82%; light orange crystals (EtOH), m.p.: 190–191 °C (m.p. 193 °C, lit.Citation39).
Synthesis of imdazole-2-thiones 5a-n
An ethanolic solution of N-substituted-thiosemicarbazones 3a-f (1 mmol) was added to an ethanolic solution (20 ml) containing a mixture of 2a-c (1 mmol) and 3a-c (1 mmol) with 0.5 ml of Et3N. The mixture was refluxed for 8–12 h during which time a yellow precipitate separated out. The reaction was followed up by TLC analysis. The reaction mixture was then cooled to room temperature and the precipitate of 5a-n was filtered off and the obtained products were recrystallized from the stated solvents.
2-((3-Methyl-4-phenyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)acenaphthylen-1(2H)-one (5a): Yellow crystals, Rf = 0.4 (Toluene: EtOAc, 10:2), yield: 86%, m.p. 220–2 °C (EtOH). 1H NMR (400 MHz, DMSO-d6): δH = 8.60 (dd, 1H, J = 8.0,1.2 Hz, Ar-H), 8.40 (dd, 1H, J = 8.0,1.2 Hz, Ar-H), 8.25–8.20 (m, 2H, Ar-H), 7.55–7.62 (m, 2H, Ar-H), 7.25–7.30 (m, 5H, Ar-H), 6.82 ppm (s, 1H, imidazole-H-5), 3.60 ppm (s, 3H, CH3). 13C NMR (100.1 MHz, DMSO-d6): δC = 189.1 (C = O, C-2), 176.1 (C = S, C-2′), 146.2 (C = N),141.5, 138.3,132.1,130.3,130.0,129.8 (Ar-C), 129.6,129.4 (Ar-CH), 129.1,129.0 (Ar-2CH), 128.5,128.3,128.2,127.9 (Ar-CH), 121.02 (Ar-CH), 104.1 ppm (imdazole-CH-5, C-5′), 34.3 ppm (CH3). MS (70 e.V., %): m/z = 370 (M + 1, 100), 269 (M+, 78), 289 (5), 180 (36), 154 (32). Calcd. For C22H15N3OS (369.44): C, 71.52; H, 4.09; N, 11.37; S, 8.68. Found: C, 71.70; H, 4.18; N, 11.50; S, 8.80.
2-((3,4-Diphenyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)acenaphthylen-1(2H)-one (5b): Yellow crystals, Rf = 0.4 (Toluene: EtOAc, 10:3), yield: 90%, m.p. 240–2 °C (CH3CN). 1H NMR (400 MHz, DMSO-d6): δH = 8.53 (dd, 1H, J = 8.0,1.2 Hz, Ar-H), 8.26 (d, 2H, J = 8.0, Ar-H), 8.00–8.04 (m, 4H, Ar-H), 7.75–7.82 (m, 4H, Ar-H), 7.52–7.70 (m, 5H, Ar-H), 6.88 ppm (s, 1H, imidazole-H-5). 13C NMR (100.1 MHz, DMSO-d6): δC = 189.0 (C = O, C-2), 178.4 (C = S, C-2′), 147.6 (C = N),140.2, 137.0,135.2,131.6,129.9,129.2,129.0 (Ar-C), 128.8,128.7,128.6,128.5,128.3,128.2,127.9 (Ar-2CH), 126.6,121.0 (Ar-CH), 104.8 ppm (imdazole-CH-5, C-5′). MS (70 e.V., %): m/z = 432 (M + 1, 100), 431 (M+, 61), 289 (5), 180 (37), 154 (57). Calcd. For C27H17N3OS (431.51): C, 75.15; H, 3.97; N, 9.74; S, 7.43. Found: C, 75.30; H, 3.80; N, 9.90; S, 7.50.
2-((3-Benzyl-4-phenyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5c): Yellow crystals, Rf = 0.50 (Toluene: EtOAc, 10:2), yield: 88%, m.p. 212–4 °C (CH3OH). 1H NMR (400 MHz, DMSO-d6): δH = 8.26 (t, 1H, J = 7.5 Hz, Ar-H), 8.11 (d, 1H, J = 7.7 Hz, Ar-H), 7.99–8.03 (m, 2H, Ar-H), 7.81 (dd, 1H, J = 7.7 Hz, Ar-H), 7.56–7.52 (m, 5H, Ar-H), 7.36–7.22 (m, 4H, Ar-H), 7.18–7.15 (m, 2H, Ar-H), 6.94 (s, 1H, imidazole-H-5), 5.35 ppm (s, 2H, CH2-Bz). 13C NMR (100.1 MHz, DMSO-d6): δC = 189.0 (C = O, C-2), 175.8 (C = S, C-2′), 164.0 (C = N), 146.9, 141.2, 138.4,136.2,131.9,131.6,129.9 (Ar-C), 129.8,129.7,129.1,129.0,128.9,128.6 (Ar-CH), 128.5,128.4,128.0,127.4 (Ar-2CH), 124.2,124.1 (Ar-CH), 104.0 (imdazole-CH-5, C-5′), 49.4 ppm (Bz-CH2). MS (70 e.V., %): m/z = 446 (M + 1, 57), 445 (M+, 27), 155 (33), 154 (100). Calcd. for C28H19N3OS (445.54): C, 75.48; H, 4.30; N, 9.43; S, 7.20. Found: C, 75.60; H, 4.40; N, 9.60; S, 7.30.
2-((3-Cyclohexyl-4-phenyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5d): Yellow crystals, Rf = 0.35 (Toluene: EtOAc, 10:1), yield: 80%, m.p. 192–4 °C (CH3OH). 1H NMR (400 MHz, DMSO-d6): δH = 8.59 (d, 1H, J = 8.0 Hz, Ar-H), 8.27 (t, 1H, J = 7.2 Hz, Ar-H), 8.09–8.15 (m, 2H Ar-H), 7.70–7.84 (m, 2H, Ar-H), 7.55–7.65 (m, 5H, Ar-H), 6.80 (s, 1H, imidazole-H-5), 3.86–3.94 (m, 1H, cyclohexyl-H), 2.6–2.8 (m, 2H, cyclohexyl-H), 1.75–1.79 (m, 4H, cyclohexyl-H),1.27–1.45 (m, 2H, cyclohexyl-H),1.10–1.25 ppm (m, 2H, cyclohexyl-H). 13C NMR (100.1 MHz, DMSO-d6): δC = 189.0 (C = O, C-2), 176.6 (C = S, C-2′), 145.6 (C = N), 138.3,133.4,133.2,132.0,130.1 (Ar-C), 129.7,129.3 (Ar-CH), 129.2 (Ar-C), 129.0,128.4,128.0,126.60 (Ar-2CH), 121.1 (Ar-CH), 105.1 (imdazole-CH-5, C-5′), 60.0 (CH), 27.7,25.4 (2CH2), 24.8 ppm (CH2). MS (70 e.V., %): m/z = 438 (M + 1, 100), 437 (M+, 53), 289 (7), 180 (44), 154 (45), 154 (100). Calcd. for C27H23N3OS (437.56): C, 74.11; H, 5.30; N, 9.60; S, 7.33. Found: C, 74.30; H, 5.40; N, 9.70; S, 7.50.
2-((3-(3-Methoxyphenyl)-4-phenyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5e): Yellow crystals, Rf = 0.35 (Toluene: EtOAc, 10:2), yield: 86%, m.p. 212–4 °C (DMF/CH3OH: 2: 10). 1H NMR (400 MHz, DMSO-d6): δH = 8.10–8.07 (m, 1H, Ar-H), 7.90–7.85 (m, 2H, Ar-H), 7.80–7.75 (m, 2H, Ar-H), 7.45–7.40 (m, 2H, Ar-H), 7.30–7.25 (m, 5H, Ar-H), 7.22 (dd, 1H, J = 7.1,8.2 Hz, 2H), 7.15–7.10 (m, 1H, Ar-H), 6.90–6.86 (m, 2H, Ar-H), 6.82 (s, 1H, imidazole-H-5), 3.76 ppm (s, 3H, OCH3). 13C NMR (100.1 MHz, DMSO-d6): δC = 189.0 (C = O, C-2), 176.3 (C = S, C-2′), 159.6 (Ar-C-OCH3), 147.7 (C = N), 140.5, 138.5,136.1,131.7,131.0,129.9 (Ar-C), 129.8,129.1 (Ar-CH), 128.9,128.6 (Ar-2CH), 128.3,127.7,126.8,124.1,121.12,120.5 (Ar-CH), 114.5 (Ar-CH-p), 114.4 (Ar-2CH), 104.7 (imdazole-CH-5, C-5′), 55.5 ppm (OCH3). MS (70 e.V., %): m/z = 462 (M + 1, 27), 461 (M+, 23), 155 (33), 154 (100). Calcd. for C28H19N3O2S (461.54): C, 72.87; H, 4.15; N, 9.10; S, 6.95. Found: C, 72.95; H, 4.30; N, 9.00; S, 7.12.
2-((3-Allyl-4-phenyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)acenaphthylen-1(2H)-one (5f): Yellow crystals, Rf = 0.45 (Toluene: EtOAc, 10:2), yield: 80%, m.p. 212–4 °C (EtOH). 1H NMR (400 MHz, DMSO-d6): δH = 8.25 (dd, 1H, J = 8.1,3.2 Hz, Ar-H), 8.03 (dd, 1H, J = 8.0,1.2 Hz, Ar-H), 7.78 (dd, 2H, J = 7.8 Hz, Ar-H), 7.59–7.49 (m, 2H, Ar-H), 7.33–7.26 (m, 5H, Ar-H), 6.62 (s, 1H, imidazole-H-5), 5.80–5.75 (m, 1H, Allyl-CH=), 4.97 (dd, 1 H, J = 7.1,5.2 Hz, Allyl-CH2=), 4.79 (dd, 1H, J = 7.1,5.2 Hz, Allyl-CH2=), 4.47 (ppm dt, 2H, J = 7.1,5.2 Hz, Allyl-CH2). 13C NMR (100.1 MHz, DMSO-d6): δC = 189.1 (C = O, C-2), 175.8 (C = S, C-2′), 146.6 (C = N), 141.1, 138.4,132.0,131.9,131.6,130.0 (Ar-C), 129.8 (Allyl-CH=), 129.3,129.1,128.8,128.70,128.3,128.0 (Ar-CH), 126.6,124.2 (Ar-2CH), 121.1 (Ar-CH), 117.0 (Allyl-CH2=), 104.5 (imdazole-CH-5, C-5′), 48.6 ppm (Allyl-CH2). Calcd. for C24H17N3OS (395.48): C, 72.89; H, 4.33; N, 10.63; S, 8.11. Found: C, 73.00; H, 4.40; N, 10.80; S, 8.00.
2-((4–(4-Chlorophenyl)-3-methyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5 g): Yellow crystals, Rf = 0.55 (Toluene: EtOAc, 10:2), yield: 76%, m.p. 262–4 °C (EtOH). 1H NMR (400 MHz, DMSO-d6): δH =8.57 (d, 1H, J = 6.9 Hz, H-5), 8.28 (d, 1H, J = 8.2 Hz, H-6), 8.06 (d, 1H, J = 8.4 Hz, H-3), 8.00 (d, 1H, J = 7.0 Hz, H-8), 7.82 (dd, 1H, J = 7.7,9.4 Hz, H-7), 7.78 (dd, 1H, J = 8.1,7.4 Hz, H-4), 7.66–7.62 (m, 4H; H-o, m), 6.93 (s, 1H; H-5′), 3.63 ppm (s, 3H; H-3a’). 13C NMR (100.1 MHz, DMSO-d6): δC =189.1 (C = O, C-2), 176.1 (C = S, C-2′), 146.4 (C = N), 140.3, 138.4,134.4,132.1,131.6,131.0,130.0 (Ar-C), 129.4 (Ar-2CH, CH-o), 128.9 (Ar-2CH, CH-m), 128.8,128.6,128.0,126.5,124.4,121.0 (Ar-CH), 104.7 (imdazole-CH-5, C-5′), 34.3 ppm (N-CH3, C-3a’). IR (KBr, cm−1): ν = 1701 (C = O), 1576 (C = N), 1056 (C = S). MS (70 e.V., %): m/z = 405 (M + 2, 12), 404 (M + 1, 24), 403 (M+, 15), 307 (40), 289 (15), 180 (8), 155 (100), 137 (66). Calcd. for C22H14ClN3OS (403.88): C, 65.43; H, 3.49; Cl, 8.78; N, 10.40; S, 7.94. Found: C, 65.60; H, 3.40; Cl, 8.90; N, 10.50; S, 8.12.
2-((4-(4-chlorophenyl)-3-phenyl-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5h): Yellow crystals, Rf = 0.25 (Toluene: EtOAc, 10:2), yield: 85%, m.p. 260–2 °C (CH3CN). 1H NMR (400 MHz, DMSO-d6): δH = 8.23 (d, J = 8.1, 1H; H-6/5), 7.97 (d, J = 6.8, 1H; H-8/3), 7.90 (d, J = 8.5, 1H; H-5/6), 7.79 (dd, J = 8.0,7.1, 1H; H-7/4), 7.74 (d, J = 7.0, 1H; H-3/8), 7.57 (“t”, 2H; H-m’), 7.51 (m, 3H; H-o’, p’), 7.44 (dd, J = 8.2,7.2, 1H; H-4/7), 7.39 (d, J = 8.6, 2H; H-o), 7.31 (d, J = 8.6, 2H; H-m), 7.12 ppm (s, 1H; H-5′). 13C NMR (100.1 MHz, DMSO-d6): δC = 189.0 (C = O, C-2), 176.3 (C = S, C-2′), 147.8 (C = N), 139.3, 138.5, 137.0 133.7,131.9,131.6 (Ar-C), 130.5(Ar-2CH-, CH-o), 129.9,129.2 (Ar-C), 129.0,128.8,128.8 (Ar-CH), 128.6(Ar-2CH-, CH-m), 128.4 (Ar-2C), 128.3 (Ar-CH-p’), 127.9,126.7,124.1 (Ar-CH), 121.1 (Ar-2CH), 105.4 ppm (imdazole-CH-5, C-5′). IR (KBr, cm−1): ν =1698 (C = O, 1082 (C = S). MS (70 e.V., %): m/z = 467 (M + 2, 6), 466 (M + 1, 12), 465 (M+, 9), 289 (13), 180 (3), 155 (32), 154 (100). Calcd. for C27H16ClN3OS (465.96): C, 69.60; H, 3.46; Cl, 7.61; N, 9.02; S, 6.88. Found: C, 69.80; H, 3.60; Cl, 7.80; N, 9.19; S, 6.94.
2-((3-Benzyl-4-(4-chlorophenyl)-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5i): Yellow crystals, Rf = 0.35 (Toluene: EtOAc, 10:2), yield: 79%, m.p. 260–2 °C (DMF: EtOH, 2:10). IR (KBr, cm−1): ν =1697 (C = O, 1070 (C = S). 1H NMR (400 MHz, DMSO-d6): δH =8.24 (d, J = 8.2, 1H; H-6/5), 8.13 (d, J = 7.0, 1H; H-3/8), 8.00 (d, J = 8.6, 1H; H-5/6), 7.98 (d, J = 8.6, 1H; H-8/3), 7.80 (dd, J = 8.0,7.2, 1H; H-7/4), 7.58 (dd, J = 8.2,7.2, 1H; H-4/7), 7.55 (d, J = 8.4, 2H; H-o), 7.49 (d, J = 8.6, 2H; H-m), 7.32 (dd, J = 7.6,7.2, 2H; H-m’), 7.24 (t, J = 7.2, 1H; H-p’), 7.15 (d, J = 7.3, 1H; H-o’), 6.98 (s, 1H; H-5′), 5.35 ppm (s, Bz-CH2). 13C NMR (100.1 MHz, DMSO-d6): δC =189.0 (C = O, C-2), 175.9 (C = S, C-2′), 147.1 (C = N), 139.9, 138.4,136.1,134.6,131.9,131.6 (Ar-C), 130.9 (Ar-2CH-o), 129.9,129.9 (Ar-C), 129.2,128.9 (Ar-CH), 128.6(Ar-2CH-m), 128.5,128.0,127.4,126.6 (Ar-CH), 126.3,126.6 (Ar-2CH), 121.1 (Ar-CH-p), 105.3 (imdazole-CH-5, C-5′), 49.5 ppm (Bz-CH2, C-3a’). MS (70 e.V., %): m/z = 481 (M + 2, 3), 480 (M + 1, 7), 479 (M+, 5), 180 (3), 155 (32), 154 (100). Calcd. for C28H18ClN3OS (479.98): C, 70.07; H, 3.78; Cl, 7.39; N, 8.75; S, 6.68. Found: C, 70.20; H, 3.82; Cl, 7.50; N, 8.90; S, 6.72.
2-((4-(4-Chlorophenyl)-3-(3-methoxyphenyl)-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)-imino)acenaphthylen-1(2H)-one (5j): Yellow crystals, Rf = 0.3 (Toluene: EtOAc, 10:1), yield: 92%, m.p. 282–4 °C (DMF: EtOH, 2:10). 1H NMR (400 MHz, DMSO-d6): δH =8.24 (d, J = 8.1, 1H; H-6/5, Ar-H), 7.99 (d, J = 8.1, 1H; H-5/6, Ar-H), 7.98 (d, J = 6.5, 1H; H-8/3, Ar-H), 7.87 (d, J = 7.0, 1H; H-3/8, Ar-H), 7.79 (dd, J = 7.7,9.3, 1H; H-7/4, Ar-H), 7.46 (dd, J = 8.1,7.3, 1H; H-4/7, Ar-H), 7.43 (dd, J = 8.1,8.0, 1H; H-5”, Ar-H), 7.41 (d, J = 8.6, 2H; H-o, Ar-H), 7.34 (d, J = 8.6, 2H; H-m, Ar-H), 7.21 (dd, J = 2.0,2.0, 1H; H-2”), 7.11 (s, imdazole-H-5, 1H; H-5′), 7.08 (dd, J = 8.2,4.1, 1H; H-6”, Ar-H), 6.98 (dd, J = 6.1,2.0, 1H; H-4”, Ar-H), 3.78 ppm (s, 3H, Ar-OCH3). 13C NMR (100.1 MHz, DMSO-d6): δC =189.0 (C = O, C-2), 176.2 (C = S, C-2′), 159.7 (Ar-C-OCH3), 147.9 (C = N), 139.2, 138.5, 138.0,133.7,131.9,131.6,130.7,130.0 (Ar-C), 129.9,129.0 (Ar-2CH), 128.4(Ar-2CH-, CH-m), 128.2,128.0,126.8,124.1,121.1,120.5,114.6,114.5 (Ar-CH), 105.3 (imdazole-CH-5, C-5′), 55.5 ppm (OCH3, C-3a”). IR (KBr, cm−1): nν = 1704 (C = O, 1577 (C = N), 1086 (C = S). MS (70 e.V., %): m/z = 496 (M + 1, 42), 495 (M+, 24), 289 (16), 180 (10), 155 (32), 154 (100). Calcd. for C28H18ClN3O (495.98): C, 67.81; H, 3.66; Cl, 7.15; N, 8.47; S, 6.46. Found: C, 67.93; H, 3.72; Cl, 7.30; N, 8.60; S, 6.60.
2-((3-Allyl-4-(4-chlorophenyl)-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5k): Yellow crystals, Rf = 0.4 (Toluene: EtOAc, 10:1), yield: 84%, m.p. 262–4 °C (DMF:EtOH, 1:10).1H NMR (400 MHz, DMSO-d6): δH =8.49 (d, 1H, J = 7.0 Hz, Ar-H), 8.28 (d, 1H, J = 8.2 Hz, Ar-H), 8.06 (d, 1H, J = 8.4 Hz, Ar-H), 8.00 (d, 1H, J = 6.9 Hz, Ar-H), 7.82 (dd, 1H, J = 7.6,7.6 Hz, Ar-H), 7.76 (dd, 1H, J = 7.8 Hz, Ar-H), 7.62 (AB, 2H, J = 8.6 Hz, Ar-H-o), 7.50 (AB, 2H, Ar-H-m), 6.94 (s, 1H, imidazole-H-5), 5.99 (ddt, H, Jd= 17.1,10.2 Hz, Jt = 5.0 Hz; Allyl-CH=), 5.21 (d, 1H, J = 10.4 Hz; Allyl-CH2), 5.03 (d, 1H, J = 17.4 Hz; allyl-CH2), 4.71 (d, 2H, J = 4.1 Hz, allyl-CH2). 13C NMR (100.1 MHz, DMSO-d6): δC =189.0 (C = O, C-2), 175.7 (C = S, C-2′), 146.7 (C = N), 139.8, 138.5,134.6,132.0,131.9,131.6 (Ar-C), 131.0(Ar-2CH-o), 130.0 (Ar-C), 129.3 (Allyl-CH=), 128.9(Ar-2CH-m), 128.7 (Ar-H), 128.6 (Ar-2CH), 126.6,124.2,121.1 (Ar-CH), 117.0 (Allyl-CH2=), 105.1 (imidazole-CH-5, C-5′), 48.6 (Allyl-CH2-). IR (KBr, cm−1): ν =1699 (C = O, 1089 (C = S). MS (70 e.V., %): m/z = 430 (M + 1, 82), 429 (M+, 40), 180 (45), 154 (100). Calcd. for C24H16ClN3OS (429.92): C, 67.05; H, 3.75; Cl, 8.25; N, 9.77; S, 7.46. Found: C, 67.15; H, 3.82; Cl, 8.30; N, 9.90; S, 7.54.
2-((3-Methyl-4-(naphthalen-2-yl)-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5 l): Yellow crystals, Rf = 0.4 (Toluene: EtOAc, 10:2), yield: 78%, m.p. 240–2 °C (EtOH: hexane, 5: 10). 1H NMR (400 MHz, DMSO-d6): δH = 8.59 (d, 1H, J = 7.0 Hz), 8.28 (d, 1H, J = 8.1 Hz), 8.20 (bs, 1H, Ar-H), 8.08–8.04 (m, 5H, Ar-H), 7.82 (dd, 1H, J = 8.0,7.3 Hz), 7.78 (dd, 1H, J = 8.2,7.2 Hz, Ar-H), 7.73 (dd, 1H, J = 8.1,4.6 Hz), 7.65–7.63 (m, 2H, Ar-H), 7.00 (s, 1H, s, 1H, imidazole-H-5), 3.70 ppm (s, 3H, (N-CH3, H-3a’). 13C NMR (100.1 MHz, DMSO-d6): δC =189.1 (C = O, C-2), 176.1 (C = S, C-2′), 146.3 (C = N), 141.5, 138.3,133.0,132.6,132.1,131.58 (Ar-CH), 130.0,129.5,128.8,128.7,128.4,128.3,128.0,127.7 (Ar-C), 127.3,127.2,127.0,126.5,126.1,124.4,121.0 (Ar-CH), 104.5 imidazole-CH-5, C-5′), 34.5 ppm (CH3(N-CH3, C-3a’). IR (KBr, cm−1): ν =1691 (C = O, 1070 (C = S). MS (70 e.V., %): m/z = 420 (M + 1, 20), 419 (M+, 15), 289 (15), 155 (33), 154 (100). Calcd. for C26H17N3OS (419.50): C, 74.44; H, 4.08; N, 10.02; S, 7.64. Found: C, 74.60; H, 4.18; N, 10.12; S, 7.80
2-((3-Benzyl-4-(naphthalen-2-yl)-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5 m): Yellow crystals, Rf = 0.3 (Toluene: EtOAc, 10:3), yield: 83%, m.p. 332–4 °C (DMF/CH3OH, 10: 3). 1H NMR (400 MHz, DMSO-d6): δH = 8.25 (d, 1H, J = 8.2 Hz, Ar-H), 8.15 (d, 1H, J = 7.0, Ar-H), 8.08 (bs, 1H, Ar-H), 8.05–8.03 (m, 4H, Ar-H), 7.93 (dd, 1H, J = 6.2, 6.0 Hz, Ar-H), 7.80 (dd, 1H, J = 7.6,7.5 Hz, Ar-H), 7.60 (m, 4H, Ar-H), 7.30 (dd, 2H, J = 7.5,7.2 Hz, Ar-H), 7.23 (t, 1H, J = 7.3 Hz, Ar-H), 7.17 (d, 2H, J = 7.4 Hz, Ar-H), 7.05 (s, 1H s, 1H, imidazole-H-5), 5.42 ppm (s, 2H, bz-CH2). 13C NMR (100.1 MHz, DMSO-d6): δC =189.0 (C = O, C-2), 175.9 (C = S, C-2′), 146.9 (C = N), 141.1, 138.4,132.9,132.4,131.9,131.5,129.9,129.0,128.9 (Ar-C), 128.5,128.4 (Ar-2CH), 128.3,128.1,127.9,127.6,127.3,127.2 (Ar-CH), 127.1,126.9,126.5 (Ar-CH), 126.2 (Ar-2CH), 125.8,124.2 (Ar-CH), 121.0 (Ar-CH-p), 105.0 (imidazole-CH-5) 49.5 ppm (bz-CH2). MS (70 e.V., %): m/z = 496 (M + 1, 37), 495 (M+, 23), 289 (16), 180 (10), 155 (32), 154 (100). Calcd. for C32H21N3OS (495.60): C, 77.55; H, 4.27; N, 8.48; S, 6.47. Found C, 77.70; H, 4.40; N, 8.60; S, 6.53.
2-((3-Allyl-4-(naphthalen-2-yl)-2-thioxo-2,3-dihydro-1H-imidazol-1-yl)imino)-acenaphthylen-1(2H)-one (5n): Yellow crystals, Rf = 0.2 (Toluene: EtOAc, 10:3), yield: 69%, m.p. 230–2 °C (EtOH). IR (KBr, cm−1): ν =1701 (C = O, 1068 (C = S). 1H NMR (400 MHz, DMSO-d6): δH = 8.51 (d, 1H, J = 7.0 Hz, Ar-H), 8.28 (d, 1H, J = 8.1 Hz, Ar-H), 8.15 (bs, 1H, Ar-H), 8.07–8.04 (m, 5H, Ar-H), 7.83 (dd, 1H, J = 8.0,7.1 Hz, Ar-H), 7.76 (dd, 1H, J = 8.2,7.2 Hz, Ar-H), 7.72–7.65 (m, 3H, Naphthyl-H), 7.01 (s, 1H, imidazole-H-5), 6.02 (ddt, 1H, Jd= 17.1,10.1, Jt = 5.0 Hz, Allyl-CH=), 5.23 (dd, 1H, J = 10.0,5.9 Hz, Allyl-CH=), 5.07 (dd, 1H, J = 17.1,2.0 Hz, Allyl-CH=), 4.78 (d, 2H, J = 4.2 Hz, Allyl-CH2). 13C NMR (100.1 MHz, DMSO-d6): δC =189.0 (C = O, C-2), 175.7 (C = S, C-2′), 146.7 (C = N), 141.0, 139.8, 138.5,134.6,132.0,131.9,131.6 (Ar-C), 131.0 (Ar-2CH), 130.0 (Ar-C), 129.3 (Allyl-CH=), 128.9,128.7 (Ar-2CH), 128.6 128.3,128.0,126.6,126.4,124.2,121.1 (Ar-CH), 117.0 (Allyl-CH2=), 105.1 (imidazole-CH-C-5′), 48.6 (Allyl-CH2). Calcd. for C28H19N3OS (445.54): C, 75.48; H, 4.30; N, 9.43; S, 7.20. Found: C, 75.60; H, 4.35; N, 9.60; S, 7.30.
Biological evaluation
All chemicals, solvents, media and kits were purchased from commercial suppliers.
DNA interaction studies
Fluorescence measurements: Fluorescence spectra were measured between 290 and 800 nm with excitation at 270 nm of 100 µL solutions in a 1 cm path length Suprasil quartz fluorescence cuvette.
The PerkinElmer LS 55 fluorescence spectrophotometer was used to measure the fluorescence. After 24 h of incubation in buffer solution (40 mM Tris, 10 mM NaCl, pH 7.5), aliquots of each DNA-Compound mixture were combined with TbCl3 to confirm DNA damage. The final concentrations were 1 g/mL ctDNA, 3 mM TbCl3, and 50 M of each compound in buffer solution (40 mM Tris, 10 mM NaCl, pH 7.5). Calf thymus DNA (ctDNA) was combined with various quantities of each substance, ranging from 0.1 pM to 200 M, for the experiment. The ultimate concentrations of 1 g/mL ctDNA and 3 mM were achieved by adding Tb3 + solution to the ctDNA/compound combinations in buffer solution (40 mM Tris, 10 mM NaCl, pH 7.5) after 24 h of incubation.Citation44
In vitro anticancer screening
The anticancer effect of the most active synthesized compounds 5b, 5e, 5h, and 5j was determined against three cancer cell lines namely: hepatocellular carcinoma (HepG2), human breast adenocarcinoma (MCF-7) and human colon cancer (HCT-116) utilising MTT assayCitation48 (Supplementary file). Compounds 5b and 5h were screened for their effects on the normal breast cell line MCF‐10a. Cells at a density of 1 × 104 were seeded in a 96‐well plate at 37 °C for 48 h under 5% CO2. After incubation, the cells were treated with different concentrations of the prepared molecules and incubated for 24 h. MTT dye was added after 24 h of drug treatment and incubated for 4 h at 37 °C. Also, 100 μl of dimethyl sulphoxide was added to each well to dissolve the purple formazan formed. The colour intensity of the formazan product, which represents the growth condition of the cells, was quantified using an enzyme‐linked immunosorbent assay (ELISA) plate reader at 570 nm. The experiments were carried out with at least three replicates and they were repeated at least three times.
Annexin V-FITC/PI apoptosis induction analysis
The BioVision® annexin-V-FITC apoptosis detection kit was used to detect apoptosis in MCf-7 cells as per the manufacturer’s instructions. Apoptosis was then quantified by flow cytometry at 488 nm using the FITC signal detector (typically FL1) and PI staining by the phycoerythrin emission signal detector (typically FL2). Briefly, 1–5 × 105 cells were were quickly centrifuged and collected. Compounds 5b and 5h were applied to cells for 24 h at their IC50 concentrations before they were resuspended in 500 μl of binding buffer. Annexin-V-FITC and PI were added. After that, they were kept at room temperature for five minutes in the dark. An exclusive signal detector was used to examine the binding of annexin-V to FITC.Citation50
Cell cycle analysis: A FACS Calibur flow cytometer was used to measure the amount of DNA in the cell cycle analysis at 488 nm in accordance with the manufacturer’s recommendations. Briefly, chosen compounds 5b and 5h were applied to 2 × 105 cells/well for 24 h at their IC50 concentrations. Cells were treated, rinsed twice, and then re-suspended in ice-cold PBS (phosphate buffer saline). After washing, 0.7 ml of 100% ethanol was added, and the mixture was then allowed to sit at 20 °C for 20 min. After washing, 500 μl of RNase was added, and then the mixture was incubated for 30 min. After that, PI was added, and the mixture was incubated without light for 30 minCitation22,Citation51.
Statistical analysis of the data
Data were fed to the computer and analysed using IBM SPSS software package version 20.0. (Armonk, NY: IBM Corp). Quantitative data were expressed as mean and standard error One way ANOVA test was used for comparing the four studied groups and followed by Post Hoc test (Tukey) for pairwise comparison. Significance of the obtained results was judged at the 5% level (Supplementary file).
In vitro topoisomerase II inhibitory assay
The most active anti-proliferative members were analyzed for their Topo II inhibitory activities. Doxorubicin was used as a positive control. Compounds 5b and 5h were selected to be evaluated against topo II [MBS#942146] using human DNA topoisomerase II-β (TOPII-β) ELISA kit according to manufacturer’s instructionsCitation56 (Supplementary file).
In silico studies
Molecular docking (Supplementary file)
Computer-aided docking experiments were performed using Molecular Operating Environment (MOE 2020.09)Citation57 software (Chemical Computing Group, Montreal, Canada). The crystallographic structure of Human topoisomerase II beta in complex with DNA and etoposide (PDB ID: 3QX3) was retrieved from Protein Data Bank.
By adding hydrogens, calculating partial charges, and minimising energy using the Amber 10: EHT Force Field with a root mean square deviation (RMSD) gradient of 0.1 kcal/mol, the database of tested compounds was obtained. The downloaded protein was further processed by removing repetitive chains while maintaining certain nucleotides and water molecules that are important for ligand binding. The MOE QuickPrep methodology was then used to optimise structural issues, perform 3D protonation, and calculate partial charges. The test compounds’ best score values and advantageous binding conformations were determined using the MOE Dock application’s default procedure, which uses the triangle matcher placement approach with London dG as the primary scoring function. To further optimise the induced fit receptor technique, a force field-based scoring function (GBVI/WSA dG) was used to choose poses that displayed the protein’s maximum hydrogen-bond, ionic, and hydrophobic interactions. The energy scores in kcal/mol for the complexes generated between the ligand conformers and the binding sites were included in the output database. Finally, visual inspection and analysis of the interactions with active site residues were performed on the generated docking poses. The default position was the one with the highest score and the best ligand-enzyme interaction. Visual inspection and analysis of interactions with active site residues were done on the docking poses that were obtained. The default position was the one with the highest score and the best ligand-enzyme interaction.
In silico predication of physiochemical properties, pharmacokinetic profile and drug likeness optimization measures
The in silico predictions of the pharmacokinetics properties were performed using SwissADMECitation59 for the pharmacokinetics and drug-likeness predictions. The evaluations against (GIA) and (BBB) permeation which give some parameters for the pharmacokinetics predictions were extracted from the BOILED-Egg modelCitation59,Citation60.
Author contributions
A. H. Mohamed: Concept, analysis, editing; M. B. Alshammari; A. Ahmad: editing; A. A. Aly: Concept, writing, editing and submit; K. U. Sadek: editing; E. A. Aziz: Methodology, A. F. Yazbi, E. J. El-Agroudy and M. E. Abdelaziz: concept, design, methodology ,analysis and writing. All authors agree to be accountable of all aspects of the work.
Supplemental Material
Download PDF (11.7 MB)Acknowledgements
The authors thank the Deanship of Scientific Research at Prince Sattam bin Abdulaziz University under research project No 2021/01/18104.
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Data availability statement
Data set generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Global cancer report, world health organization. https://www.who.int/en/news-room/fact-sheets/detail/cancer [accessed in June 2023].
- Khalifa MM, Al-Karmalawy AA, Elkaeed EB, Nafie MS, Tantawy MA, Eissa IH, Mahdy HA. Topo II inhibition and DNA intercalation by new phthalazine-based derivatives as potent anticancer agents: Design, synthesis, anti-proliferative, docking, and in vivo studies. J Enzyme Inhib Med Chem. 2022;37(1):299–314.
- Hammoud MM, Nageeb AS, Morsi MA, Gomaa EA, Elmaaty AA, Al-Karmalawy AA. Design, synthesis, biological evaluation, and SAR studies of novel cyclopentaquinoline derivatives as DNA intercalators, topoisomerase II inhibitors, and apoptotic inducers. New J Chem. 2022;46(23):11422–11436.
- Gaber AA, Sobhy M, Turky A, Eldehna WM, El-Sebaey SA, El-Metwally SA, El-Naggar AM, Ibrahim IM, Elkaeed EB, Metwaly AM, et al. New [1,2,4]triazolo[4,3-c]quinazolines as intercalative topo II inhibitors: Design, synthesis, biological evaluation, and in silico studies. PLOS One. 2023;18(1):e0274081.
- Singh I, Luxami V, Paul K. Synthesis of naphthalimide-phenanthro[9,10-d]imidazole derivatives: In vitro evaluation, binding interaction with DNA and topoisomerase inhibition. Bioorg Chem. 2020;96:103631.
- Zhao M, Yang K, Zhu X, Gao T, Yu W, Liu H, You Z, Liu Z, Qiao X, Song Y. Design, synthesis and biological evaluation of dual topo II/HDAC inhibitors bearing pyrimido[5,4-b]indole and pyrazolo[3,4-d]pyrimidine motifs. Eur J Med Chem. 2023;252:115303.
- Swedan HK, Kassab AE, Gedawy EM, Elmeligie SE. Design, synthesis, and biological evaluation of novel ciprofloxacin derivatives as potential anticancer agents targeting topoisomerase II enzyme. J Enzyme Inhib Med Chem. 2023;38(1):118–137.
- Deng X, Luo T, Zhang X, Li Y, Xie L, Jiang W, Liu L, Wang Z. Design, synthesis and biological evaluation of 3-arylisoquinoline derivatives as topoisomerase I and II dual inhibitors for the therapy of liver cancer. Eur J Med Chem. 2022;237:114376.
- Skok Ž, Durcik M, Zajec Ž, Gramec Skledar D, Bozovičar K, Pišlar A, Tomašič T, Zega A, Peterlin Mašič L, Kikelj D, et al. ATP-competitive inhibitors of human DNA topoisomerase IIα with improved antiproliferative activity based on N-phenylpyrrolamide scaffold. Eur J Med Chem. 2023;249:115116.
- Xu G, Li Z, Ding Y, Shen Y. Discovery of 1,2-diphenylethene derivatives as human DNA topoisomerase II catalytic inhibitors and antitumor agents. Eur J Med Chem. 2022;243:114706.
- Swedan HK, Kassab AE, Gedawy EM, Elmeligie SE. Topoisomerase II inhibitors design: Early studies and new perspectives. Bioorg Chem. 2023;136:106548.
- Singh V, Afshan T, Tyagi P, Varadwaj PK, Sahoo AK. Recent development of multi-targeted inhibitors of human topoisomerase II enzyme as potent cancer therapeutics. Int J Biol Macromol. 2023;226:473–484.
- Kumar M, Kumar D, Raj V. Studies on imidazole and its derivatives with particular emphasis on their chemical/biological applications as bioactive molecules/intermediated to bioactive molecule. Curr Synth and Syst Biol. 2017;05(01):1–10.
- Zhang H-B, Zhang X-F, Chai L-Q, Tang L-J, Zhang H-S. Zinc(ii) and cadmium(ii) complexes containing imidazole ring: Structural, spectroscopic, antibacterial, DFT calculations and hirshfeld surface analysis. Inorganica Chim Acta. 2020;507:119610.
- Al-Wahaibi LH, Youssif BGM, Taher ES, Abdelazeem AH, Abdelhamid AA, Marzouk AA. Design, synthesis, biological evaluation, and computational studies of novel tri-aryl imidazole-benzene sulfonamide hybrids as promising selective carbonic anhydrase IX and XII inhibitors. Molecules. 2021;26(16):4718.
- Yang YQ, Chen H, Liu QS, Sun Y, Gu W. Synthesis and anticancer evaluation of novel 1h-benzo[d]imidazole derivatives of dehydroabietic acid as pI3Kα inhibitors. Bioorg Chem. 2020;100:103845.
- Alghamdi SS, Suliman RS, Almutairi K, Kahtani K, Aljatli D. Imidazole as a promising medicinal scaffold: Current status and future direction. Drug Des Devel Ther. 2021;15:3289–3312.
- Al-Harbi RAK, Al-Sharari MAM, El-Sharief MAMS. Synthesis and anticancer evaluation of imidazolidine derivatives: Study the reaction of imidazolidine-iminothione derivatives with amino acids methyl ester. J Taibah Univ for Sci. 2020;14(1):842–848.
- Sharma P, LaRosa C, Antwi J, Govindarajan R, Werbovetz KA. Imidazoles as potential anticancer agents: An update on recent studies. Molecules. 2021;26(14):4213.
- Lungu CN, Bratanovici BI, Grigore MM, Antoci V, Mangalagiu II. Hybrid imidazole-pyridine derivatives: An approach to novel anticancer DNA intercalators. Curr Med Chem. 2020;27(1):154–169.
- Biskupiak JE, Grierson JR, Rasey JS, Martin GV, Krohn KA. Synthesis of an (iodovinyl)misonidazole derivative for hypoxia imaging. J Med Chem. 1991;34(7):2165–2168.
- Zaki I, Ramadan HMM, El-Sayed EH, Abd El-Moneim M. Design, synthesis, and cytotoxicity screening of new synthesized imidazolidine-2-thiones as VEGFR-2 enzyme inhibitors. Arch Pharm (Weinheim)). 2020;353(11):e2000121.
- Aruchamy B, Drago C, Russo V, Pitari GM, Ramani P, Aneesh TP, Benny S, Vishnu VR. Imidazole-pyridine hybrids as potent anti-cancer agents. Eur J Pharm Sci. 2023;180:106323.
- Sadula A, Gaddhe L. Synthesis, computational studies and biological evaluation of novel acenaphthoquinone-imidazole derivatives as dual inhibitors of HSP90 and topo II in cancer therapy. Results Chem. 2023;5:100796.
- Singh I, Rani R, Luxami V, Paul K. Synthesis of 5-(4-(1h-phenanthro[9,10-d]imidazol-2-yl)benzylidene)thiazolidine-2,4-dione as promising DNA and serum albumin-binding agents and evaluation of antitumor activity. Eur J Med Chem. 2019;166:267–280.
- Franchetti P, Marchetti S, Cappellacci L, Yalowitz J, Jayaram H, Goldstein B, Grifantini M. A new c-nucleoside analogue of tiazofurin. Bioorg Med Chem Lett. (1)2001;11:67–69.
- Cui B, Zheng BL, He K, Zheng QY. Imidazole alkaloids from lepidium meyenii. J Nat Prod. 2003;66(8):1101–1103.
- El Ashry ESH, Hamid HA, Kassem AA, Shoukry M. Synthesis and reactions of acenaphthenequinones-part-2. The reactions of acenaphthenequinones. Molecules. 2002;7(2):155–188.
- Ziarani G, Hajiabbasi P, Gholamzadeh P. Cheminform abstract: Development of the acenaphthenequinone reactions. Chem Inform. 2012;43(45).
- Yavari I, Khajeh Khezri A. Recent advances in the synthesis of hetero- and carbocyclic compounds and complexes based on acenaphthylene-1,2-dione. Synthesis. 2018;50(20):3947–3973.
- Hyatt JL, Wadkins RM, Tsurkan L, Hicks LD, Hatfield MJ, Edwards CC, Ross CR, Cantalupo SA, Crundwell G, Danks MK, et al. Planarity and constraint of the carbonyl groups in 1,2-diones are determinants for selective inhibition of human carboxylesterase 1. J Med Chem. 2007;50(23):5727–5734.
- Rodriguez-Argüelles MC, Belicchi Ferrari M, Gasparri Fava G, Pelizzi C, Pelosi G, Albertini R, Bonati A, Dall’Aglio PP, Lunghi P, Pinelli S. Acenaphthenequinone thiosemicarbazone and its transition metal complexes: Synthesis, structure, and biological activity. J Inorg Biochem. 1997;66(1):7–17.
- Zhang Z, Yang H, Wu G, Li Z, Song T, Li XQ. Probing the difference between BH3 groove of MCL-1 and BCL-2 protein: Implications for dual inhibitors design. Eur J Med Chem. 2011;46(9):3909–3916.
- Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56(2):185–229.
- Aly AA, Alshammari MB, Ahmad A, A. M. Gomaa H, G. M. Youssif B, Bräse S, A. A. Ibrahim M, Mohamed AH. Design, synthesis, docking and mechanistic studies of new thiazolyl/thiazolidinylpyrimidine-2,4-dione antiproliferative agents. Arab J Chem. 2023;16(4):104612.
- Alshammari MB, Aly AA, Youssif BGM, Bräse S, Ahmad A, Brown AB, Ibrahim MAA, Mohamed AH. Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E. Front Chem. 2022;10:1076383.
- Baviskar AT, Madaan C, Preet R, Mohapatra P, Jain V, Agarwal A, Guchhait SK, Kundu CN, Banerjee UC, Bharatam PV. N-fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase iiα and induce apoptosis in G1/S phase. J Med Chem. 2011;54(14):5013–5030.
- Vimala G, Govindaraj J, Haribabu J, Karvembu R, SubbiahPandi A. Crystal structure of (2e)-n-methyl-2-(2-oxo-1,2-di-hydroacenaphthylen-1-ylidene)hydrazinecarbo-thioamide. Acta Crystallogr Sect E Struct Rep Online. 2014;70(Pt 11):415–417.
- Aly AA, Mohamed NK, Hassan AA, El-Shaieb KM, Makhlouf MM, Bräse S, Nieger M, Brown AB. Functionalized 1,3-thiazolidin-4-ones from 2-oxo-acenaphtho-quinylidene- and [2.2]paracyclophanylidene-thiosemicarbazones. Molecules. 2019;24(17):3069.
- Ethics committee, faculty of medicine, Alexandria university, member of ICLAS, FWO: 00018699, sn: 0107785 (11/6/2023.
- Institutional animal care and use committee, Alexandria university, member of ICLAS, SN: 188 (21/6/2023.
- El-Wakil MH, Khattab SN, El-Yazbi AF, El-Nikhely N, Soffar A, Khalil HH. New chalcone-tethered 1,3,5-triazines potentiate the anticancer effect of cisplatin against human lung adenocarcinoma A549 cells by enhancing DNA damage and cell apoptosis. Bioorg Chem. 2020;105:104393.
- El-Yazbi AF, Loppnow GR. 2-aminopurine hairpin probes for the detection of ultraviolet-induced DNA damage. Anal Chim Acta. 2012;726:44–49.
- El-Yazbi AF, Wong A, Loppnow GR. A luminescent probe of mismatched DNA hybridization: Location and number of mismatches. Anal Chim Acta. 2017;994:92–99.
- Sabbatini N, Guardigli M, Lehn J-M. Luminescent lanthanide complexes as photochemical supramolecular devices. Coord Chem Rev. 1993;123(1-2):201–228.
- Valente P, Lincoln SF, Wainwright KP. External coordination of europium(III) prior to its encapsulation within a cyclen-based pendant donor macrocycle. Inorg. Chem. 1998;37(12):2846–2847.
- El-Yazbi AF, Khalil HA, Belal TS, El-Kimary EI. Inexpensive bioluminescent genosensor for sensitive determination of DNA damage induced by some commonly used sunscreens. Anal Biochem. 2022;651:114700.
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. (1-2)1983;65:55–63.
- Prayong P, Barusrux S, Weerapreeyakul N. Cytotoxic activity screening of some indigenous thai plants. Fitoterapia. 2008;79(7-8):598–601.
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461.
- Zaki I, Abdelhameid MK, El-Deen IM, Abdel Wahab AHA, Ashmawy AM, Mohamed KO. Design, synthesis and screening of 1,2,4-triazinone derivatives as potential antitumor agents with apoptosis inducing activity on MCF-7 breast cancer cell line. Eur J Med Chem. 2018;156:563–579.
- Abdel-Shafy HI, Mansour MSM. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egypt J Pet. 2016;25(1):107–123.
- Smith J, Neupane R, McAmis W, Singh U, Chatterjee S, Raychoudhury S. Toxicity of polycyclic aromatic hydrocarbons involves nox2 activation. Toxicol Rep. 2019;6:1176–1181.
- Srivastava S, Somasagara RR, Hegde M, Nishana M, Tadi SK, Srivastava M, Choudhary B, Raghavan SC. Quercetin, a natural flavonoid interacts with DNA, arrests cell cycle and causes tumor regression by activating mitochondrial pathway of apoptosis. Sci Rep. (1)2016;6:24049.
- Lee JH, Berger JM. Cell cycle-dependent control and roles of DNA Topoisomerase II. Genes (Basel). 2019;10(11):859.
- https://www.Mybiosource.Com/top2b-human-ELISA-kits/DNA-topoisomerase-2-beta/763112; 2023 [accessed in June 2023].
- Molecular operating environment (MOE), chemical computing group inc.:Montreal, qc, Canada. http://www.Chemcomp.Com; 2023 [last accessed 20 June 2023].
- Hevener KE, Zhao W, Ball DM, Babaoglu K, Qi J, White SW, Lee RE. Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J Chem Inf Model. 2009;49(2):444–460.
- Daina A, Michielin O, Zoete V. Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. (1)2017;7:42717.
- Daina A, Zoete V. A boiled-egg to predict gastrointestinal absorption and brain penetration of small molecules. Chem Med Chem. 2016;11(11):1117–1121.
- Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, Li Y, Wang R, Lai L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47(6):2140–2148.
- Szakács G, Váradi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (adme-tox). Drug Discov Today. 2008;13(9-10):379–393.
- Farghaly AH., Abdel‐Rahman . Synthesis, reactions and antimicrobial activity of some new indolyl-1,3,4-oxadiazole, triazole and pyrazole derivatives. J Chinese Chemical Soc. (1)2004;51:147–156.
- Di L. The role of drug metabolizing enzymes in clearance. Expert Opin Drug Metab Toxicol. 2014;10(3):379–393.
- Hollenberg PF. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab Rev. 2002;34(1–2):17–35.
- Huang S-M, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, Habet SA, Baweja RK, Burckart GJ, et al. New era in drug interaction evaluation: US food and drug administration update on cyp enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48(6):662–670.
- Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623.