
Abstract
A series of novel benzimidazole derivatives were designed and synthesised based on the structures of reported oral available ALK inhibitor and HDAC inhibitor, pracinostat. In enzymatic assays, compound 3b, containing a 2-acyliminobenzimidazole moiety and hydroxamic acid side chain, could inhibit both ALK and HDAC6 (IC50 = 16 nM and 1.03 µM, respectively). Compound 3b also inhibited various ALK mutants known to be involved in crizotinib resistance, including mutant L1196M (IC50, 4.9 nM). Moreover, 3b inhibited the proliferation of several cancer cell lines, including ALK-addicted H2228 cells. To evaluate its potential for treating cancers in vivo, 3b was used in a human A549 xenograft model with BALB/c nude mice. At 20 mg/kg, 3b inhibited tumour growth by 85% yet had a negligible effect on mean body weight. These results suggest a attracting route for the further research and optimisation of dual ALK/HDAC inhibitors.
GRAPHICAL ABSTRACT
Keywords:
Introduction
Anaplastic lymphoma kinase (ALK), also known as CD246, is a receptor tyrosine kinase (RTK) classified as a member of the insulin receptor superfamily and expressed by the ALK gene. Involved in development of the central nervous system, ALK is expressed at high levels in the human foetus yet its expression is significantly diminished in normal adults.Citation1 Aberrant overexpression of ALK has been found in various cancers, including anaplastic large-cell lymphoma,Citation2 non–small cell lung cancer (NSCLC),Citation3 neuroblastoma,Citation4 renal cell carcinoma,Citation5,Citation6 breast cancer,Citation3 and thyroid cancer.Citation7 The oncogenic mechanisms of ALK involve increasing gene-copy number, activating point mutations (mainly in neuroblastoma),Citation8 and formation of fusion-ALK proteins. Fusion-ALK proteins are formed by chromosomal translocation or inversion and can activate ALK kinase abnormally. The best-known example is the nucleophosmin-ALK fusion protein (NPM-ALK), which is created by a 2;5-chromosomal translocation. NPM-ALK is recognised as an important factor resulting in a subset of anaplastic large-cell lymphoma. Studies also indicate that half of all inflammatory myofibroblastic tumours have an ALK gene rearrangement, particularly in young patients.Citation9 The echinoderm microtubule-associated protein-like 4–ALK fusion protein (EML4-ALK) is another well-studied oncogenic fusion-ALK protein that was first associated with NSCLC in 2007.Citation10,Citation11 Non-smoking NSCLC patients in particular show an enrichment in EML4-ALK and limited mutation of the epidermal growth factor receptor (EGFR), which restricts the efficacy of traditional EGFR-targeted therapeutics such as gefitinib and erlotinib.Citation12,Citation13 ALK-targeted agents are a promising strategy for such patients. Crizotinib, approved in 2011 by the U.S. Food and Drug Administration (), is the first drug targeting EML4-ALK for first-line treatment of EML4-ALK-expressing NSCLC patients.Citation14,Citation15
Figure 1. FDA-approved ALK and HDAC inhibitors and reported 2,4-pyrimidinediamine derivative ALK/HDAC dual inhibitor.

However, resistance to crizotinib due to EML4-ALK mutations has been reported.Citation16,Citation17 Mutations in ALK kinase domain are the most common resistance mechanisms. These mutations, including L1196M, G1269A, and G1202R, lead to the change of the domain structure, which identified to hamper the binding of ALK inhibitors.Citation18 Even though second-generation and third-generation inhibitors such as ceritinib, brigatinib, and lorlatinib () have been developed to overcome mutations, additional resistance mechanisms were characterised.Citation19,Citation20 For instance, the activation of bypass signalling pathways and epithelial-to-mesenchymal transition (EMT) were identified to be involved in the resistance development.Citation20–22 EMT can lead to functional changes in cell migration and play an important role in cancer progression. Several transcriptional factors or signalling pathway such as ZEB1, ZEB2, and TGF-ß induced signalling were reported to induce EMT.Citation23–25 The induction contributed by these factors can be inverted by miRNA 200 family. It was reported that miR-200 can decrease the expression of TGF-ß and ZEB1 can be down-regulated by miR-200c or miR-141.Citation24,Citation26 Furthermore, a study discovered that quisinostat, a HDAC inhibitor, could overcome the resistance to crizotinib via the induction of miR-200c promotor activity.Citation23
Histone deacetylases (HDACs) are a group of enzymes that regulate gene expression and many cellular functions by deacetylating histones and other proteins. Histone deacetylation represses transcription in most cases by removing the acetyl groups from histones, making the chromatin compact tightly. Inhibition of HDACs leads to reversal of tumour-suppressor gene silencing, cell cycle arrest or apoptosis,Citation27 disruption of association between non-histone proteins (e.g., HSP90, α-tubulin) and oncogenic client proteins (e.g., Akt, Raf, BCR-Abl), induction of ubiquitinylation enzymes (Ubc8) to degrade oncogenic proteins,Citation28 and modulation of cell signalling (inactivation of STAT1).Citation29 These mechanisms indicated that HDAC inhibitors have promising antitumor activity. The first approved HDAC inhibitor, vorinostat, is used to treat cutaneous T-cell lymphoma,Citation30 and panobinostat has been approved for treatment of multiple myeloma ().Citation31 HDACs are also promising targets in solid tumours such as NSCLCCitation32 and other ALK-related cancers such as neuroblastoma and lymphoma. Most importantly, the synergistic effect of HDAC inhibitors and ALK inhibitors was proved to be effective to overcome resistance.Citation23,Citation33 Due to the involvement of histone deacetylases (HDACs) in the regulation of various pathways, such as TGF-ß signalling and microRNA activities, the observed synergistic effect may be attributed to the re-sensitization of ALK inhibitors through the down-regulation of epithelial-mesenchymal transition (EMT).Citation20,Citation33
In a recent study, Gan and co-workers designed a single molecule 2,4-pyrimidinediamine derivative as a dual inhibitor of ALK and HDAC, combining the structure of ceritinib and vorinostat.Citation34 The inhibitor showed balanced inhibition of both HDACs and ALKs, including ALKL1196M and ALKG1202R. Moreover, it possessed anti-proliferative activity on several solid tumour cell lines and ALK-resistance cell lines.Citation34 However, probably due to the aliphatic linker of vorinostat, it was reported to have poor PK properties such as oral bioavailability. Based on these observations, we proposed a new design concept of integrating the structure of reported oral bioavailable ALK inhibitor compound 1 and HDAC inhibitor compound 2 (pracinostat) to develop novel dual inhibitors as potential antitumor agents (). Compound 1, bearing a benzimidazole core, was developed by Amgen, Inc. to be a potent, selective, and orally bioavailable ALK inhibitor.Citation35 Its X-ray crystal structure suggested that the C-5 or C-6 side chain of benzimidazole was outside the enzyme pocket and tolerates various groups.Citation35 Pracinostat (compound 2, ), which shows strong HDAC inhibition, good ADME properties, and potent anti-tumour activity, is a promising HDAC inhibitor undergoing clinical trials.Citation36 The benzimidazole core connected to aliphatic side chains and hydroxamic acid as the zinc-binding group. The vinyl linker would help the hydroxamic acid moiety bind to the zinc ion, which located in the deep pocket of HDACs. In our study, we preserved the pharmacophore of both compound 1 and compound 2 and adopted side chains on the core scaffold to develop a series of hybrid compounds as dual ALK/HDAC inhibitors and studied their structure-activity relationships ().
Figure 2. Design of benzimidazole dual ALK/HDAC inhibitors. The dashed boxes in the lead compounds indicated the preserved pharmacophore structure.

Results and discussion
Synthesis of hybrid compounds
First, the cis-1,4-cyclohexanecarboxamide side chain at N-1 (R2 substitution) was replaced by simple butyramide to see if ALK inhibition activity was retained. Commercially available 5-bromo-2-fluoronitrobenzene and ethyl 4-aminobutyrate hydrochloride were used to synthesise 4a through aromatic substitution reaction (Scheme 1). Following hydrolysis with sodium hydroxide and amide coupling with N-isopropylamine, 5a was obtained. 5a was then reduced with iron and the diamine intermediate was reacted with benzoyl isothiocyanate without purification. The benzimidazole ring closure was completed by treating with 1-ethyl-3–(3-dimethylaminopropyl)carbodiimide (EDCI) to yield compound 6 (Scheme 2). The hybrid compound 3a was synthesised from 5a as the starting material (Scheme 1). Heck reaction of 5a with ethyl acrylate was carried out to give 7a, and then reduction of the nitro group was conducted with iron to yield the diamine intermediate. Because iron is a mild reductant, only the nitro group was reduced, and the alkenyl on the side chain was not affected. The diamine intermediate was reacted with benzoyl isothiocyanate to give benzimidazole 8a. The ester compound 8a was hydrolysed to the acid by lithium hydroxide and coupled with NH2OTHP to give the THP-protected compound. Finally, after deprotection with TFA, the hydroxamic acid compound 3a was obtained. Compounds 3b and 3c, with chiral cyclohexanecarboxamide, were also synthesised (Schemes 1 and 3), and similar synthetic routes were adopted to obtain compounds 3d and 3e with a different R1 group (Scheme 1). Synthesis of target compound 3f, with a SAHA-like side chain B at C-5, is shown in Scheme 4. Amination with sodium azide was conducted to convert the bromide of compound 5b into the amine, and then amide coupling with monomethyl suberate was performed to give 14. Compound 15 was synthesised following similar condition of step d and e in Scheme1 starting from compound 14. The ester compound 15 was hydrolysed and coupled with NH2OTHP to give the THP-protected intermediate. Removal of the THP group with TFA was performed to obtain compound 3f.
Scheme 1. Synthesis of compounds 3a, 3b, 3d, 3e. Reagents and conditions: (a) NH2XCO2Me or NH2XCO2Et, DIPEA, ACN, 80 °C, 12 h, 89–98%; (b) (i) NaOH, H2O, MeOH, rt, 12 h; iPrNH2, EDCI, HOBt, TEA, DMF, rt, 20 h, 88%; (c) ethyl acrylate, Herrmann’s palladacycle, [(tBu)3PH]BF4, Cy2NMe, DMF, MW, 30 min, 58–69%; (d) Fe, NH4Cl, H2O, EtOH, 80 °C, 6 h; (e) (i) benzoyl isothiocyanate, THF, 0 °C, 15 min; (ii) EDCI, DIPEA, 60 °C, 2 h, then rt, 16 h, 36–75% (for step d and e); (f) (i) CNBr, EtOH, 25 °C, 16 h; (ii) RCOCl, TEA, DCM, 0 °C to rt, 2.5 h, 47% (for step d and f); (g) (i) LiOH, H2O, MeOH, rt, 20 h; (ii) NH2OTHP, EDCI, HOBt, TEA, DMF, rt, 20 h; (h) TFA, MeOH, rt, 6 h, 36–74% (for step g and h).
![Scheme 1. Synthesis of compounds 3a, 3b, 3d, 3e. Reagents and conditions: (a) NH2XCO2Me or NH2XCO2Et, DIPEA, ACN, 80 °C, 12 h, 89–98%; (b) (i) NaOH, H2O, MeOH, rt, 12 h; iPrNH2, EDCI, HOBt, TEA, DMF, rt, 20 h, 88%; (c) ethyl acrylate, Herrmann’s palladacycle, [(tBu)3PH]BF4, Cy2NMe, DMF, MW, 30 min, 58–69%; (d) Fe, NH4Cl, H2O, EtOH, 80 °C, 6 h; (e) (i) benzoyl isothiocyanate, THF, 0 °C, 15 min; (ii) EDCI, DIPEA, 60 °C, 2 h, then rt, 16 h, 36–75% (for step d and e); (f) (i) CNBr, EtOH, 25 °C, 16 h; (ii) RCOCl, TEA, DCM, 0 °C to rt, 2.5 h, 47% (for step d and f); (g) (i) LiOH, H2O, MeOH, rt, 20 h; (ii) NH2OTHP, EDCI, HOBt, TEA, DMF, rt, 20 h; (h) TFA, MeOH, rt, 6 h, 36–74% (for step g and h).](/cms/asset/38b5bd12-ab0b-4eb6-8192-14f8e2de40cb/ienz_a_2318645_sch0001_b.jpg)
Scheme 2. Synthesis of compound 6. Reagents and conditions: (a) (i) Fe, NH4Cl, H2O, EtOH, 80 °C, 6 h; (ii) benzoyl isothiocyanate, THF, 0 °C, 15 min; EDCI, DIPEA, 60 °C, 2 h, then rt, 16 h, 33%.

Scheme 3. Synthesis of compound 3c. Reagents and conditions: (a) methyl cis-1,4-aminocyclohexanecarboxylate hydrochloride, DIPEA, I, 80 °C, 12 h, 99%; (b) (i) NaOH, H2O, MeOH, rt, 12 h; (ii) iPrNH2, EDCI, HOBt, TEA, DMF, rt, 20 h, 41%; (c) ethyl acrylate, Herrmann’s palladacycle, [(tBu)3PH]BF4, Cy2NMe, DMF, MW, 30 min, 68%; (d) Fe, NH4Cl, H2O, EtOH, 80 °C, 6 h; (e) (i) benzoyl isothiocyanate, THF, 0 °C, 15 min; (ii) EDCI, DIPEA, 60 °C, 2 h, then rt, 16 h, 63% (for step d and e); (f) (i) LiOH, H2O, MeOH, rt, 20 h; (ii) NH2OTHP, EDCI, HOBt, TEA, DMF, rt, 20 h; (g) TFA, MeOH, rt, 6 h, 75% (for step f and g).
![Scheme 3. Synthesis of compound 3c. Reagents and conditions: (a) methyl cis-1,4-aminocyclohexanecarboxylate hydrochloride, DIPEA, I, 80 °C, 12 h, 99%; (b) (i) NaOH, H2O, MeOH, rt, 12 h; (ii) iPrNH2, EDCI, HOBt, TEA, DMF, rt, 20 h, 41%; (c) ethyl acrylate, Herrmann’s palladacycle, [(tBu)3PH]BF4, Cy2NMe, DMF, MW, 30 min, 68%; (d) Fe, NH4Cl, H2O, EtOH, 80 °C, 6 h; (e) (i) benzoyl isothiocyanate, THF, 0 °C, 15 min; (ii) EDCI, DIPEA, 60 °C, 2 h, then rt, 16 h, 63% (for step d and e); (f) (i) LiOH, H2O, MeOH, rt, 20 h; (ii) NH2OTHP, EDCI, HOBt, TEA, DMF, rt, 20 h; (g) TFA, MeOH, rt, 6 h, 75% (for step f and g).](/cms/asset/76c9c166-cf27-4932-9c01-bf8e1ba994f5/ienz_a_2318645_sch0003_b.jpg)
Scheme 4. Synthesis of compound 3f. Reagents and conditions: (a) NaN3, CuI, N,N-dimethylethylenediamine, NaCO3, DMSO, 110 °C, 5 h; (b) monomethyl suberate, EDCI, HOBt, TEA, DMF, rt, 20 h, 58% (for step a and b); (c) (i) Fe, NH4Cl, H2O, EtOH, 80 °C, 6 h; (ii) benzoyl isothiocyanate, THF, 0 °C, 15 min; (iii) EDCI, DIPEA, 60 °C, 2 h, then rt, 16 h, 65%; (d) (i) LiOH, H2O, MeOH, rt, 20 h; (ii) NH2OTHP, EDCI, HOBt, TEA, DMF, rt, 20 h; (e) TFA, DCM, 0 °C to rt, 12 h, 71% (for step d and e).

In vitro ALK and HDAC inhibition activity of hybrid compounds
The results of in vitro enzymatic assays of the target compounds are shown in . Compound 6, with a simple butyramide chain at N-1 (R2 position) and a bromide on C-5 (R3 position), had only 18.8% inhibition at a concentration of 10 µM against ALK, whereas compound 3a, with an additional hydroxamate side chain at C-5 (R3 position), possessed an increased IC50 values to 5.75 µM. It was indicated that the side chain at R3 position is important for ALK inhibition. In addition, because hydroxamate group was a necessary zinc-binding moiety for HDAC, only compound 3a had HDAC inhibition instead of bromo-substituted compound 6. For compound 3d and 3e, the phenyl ring at R1 position was replaced with 3,5-difluoro phenyl or n-propyl group, respectively, and the result showed that both compounds exhibited lower ALK inhibition activity compared to phenyl-substituted compound 3a, with the n-propyl-substituted compound 3e even losing its potency. The HDAC inhibition ability of compound 3d and 3e did not show significant difference compared to compound 3a. It demonstrates that the functional groups at R1 position have greater impact on ALK inhibition rather than HDAC inhibition.
Table 1. Structure-activity relationships for test compounds (3a–3f and 6).
To explore compounds with better ALK inhibition, we replaced the pracinostat-like butyramide chain with compound 1-like cis-1,4-cyclohexancarboxamide chain to give compound 3b, 3c, and 3f. All of these compounds had significant increased ALK IC50 values to nanomolar level (16 nM and 15 nM for compound 3b and 3c, respectively) and compound 3c even exhibited 3-fold greater inhibition ability (5.2 nM) once the hydroxamate group was moved from C-5 position to C-6 position. For the HDAC inhibition of compound 3b and 3c, both of them had similar micromolar level IC50 values and compound 3b had slightly greater inhibition ability than that of compound 3c. Interestingly, the HDAC IC50 values of compound 3f, introduced with a vorinostat-like side chain at R3 position, exhibited significantly increased HDAC IC50 values, up to 10-fold higher for HDAC6 compared to compound 3b. This suggests that the longer length of side chain may enhance compounds to occupy the catalytic pocket of HDACs. In summary, compounds with phenyl group at R1 position, cis-1,4-cyclohexancarboxamide chain at R2 position, and hydroxamate group at R3 position had potential to inhibit ALK and HDACs at the same time. Proof of concept, compound 3b and 3c were further chose for biological evaluation. Albeit compound 3f demonstrated ideal HDAC inhibition compared to compound 3b or 3c, the vorinostat-like side chain may cause some pharmacokinetic issues such as solubility or bioavailability. The HDAC inhibitory activities of compound 6 and ALK inhibitors, including compound 1, Crizotinib, Ceritinib, and Staurosporine, were not assayed due to the absence of zinc-binding groups, which are essential for HDAC affinity, in their structures.
Antiproliferation activity of compound 3b and 3c against cancer cell lines
As shown in , compound 3b, 3c, and 3f can ideally inhibit both ALK and HDACs in vitro. Therefore, SRB assay was carried out to further determine their antiproliferative activities against various cancer cell lines including ALK-addicted H2228 cell line (). Compared to compound 3c and 3f, compound 3b exhibited generally greater anti-tumour activity with sub-micromolar level IC50 values in solid tumour cell lines, especially in A549 and U87MG cells. To our surprise, the potent enzyme inhibition activity of compound 3f didn’t correspond to its antiproliferative effects. Probably due to the vorinostat-like aliphatic chain, compound 3f would lose its activity at the cellular level. Furthermore, the molecule weight of compound 3f approached 600, which exceeded the rule of five threshold which might decrease its cell permeability. In the case of ALK-addicted H2228 cells, both compound 3b and 3c demonstrated mild anti-proliferative activity. Compared to pracinostat or SAHA alone, the hybrid compound 3b and 3c have better activity. This aligned with the result that the combination of ALK inhibitor (crizotinib) and HDAC inhibitor (pracinostat or SAHA) can improve the antiproliferative activity in H2228 cells (supplemental Figure S4). Notably, compound 3c had two-fold less IC50 values than that of compound 3b, aligned with the outcome of the in vitro enzymatic assays. In general, compound 3b can inhibit the growth of these solid tumour cell lines and showed to be the most potential compound for further development.
Table 2. Antiproliferative activity of compound 3b and 3c in cancer cell lines.
In vitro enzymatic activity of compound 3b against ALK mutants and various kinases
Due to the balanced ALK/HDAC inhibition in vitro and mild anti-proliferative activity, the selectivity of compound 3b was further evaluated across several RTKs as well as ALK mutants in vitro (). In the preliminary screening, compound 3b only slightly inhibited IGF1R and c-Src with IC50 values of 4.84 and 3.38 µM but not the RTKs of EGFR, VEGFR2, c-MET, or JAK2. Furthermore, five ALK mutants (C1156Y, L1196MCitation17, F1174LCitation16, G1202R, G1269ACitation37) that have been detected clinically and reported as key factors in crizotinib resistance were selected to assess the inhibition ability of compound 3b. Apparently, compound 3b was able to retain full potency against all these mutants with nanomolar level IC50 values, which displayed its potential to overcome crizotinib resistance ( and ).
Figure 3. In vitro enzymatic activity of 3b against ALK mutants (IC50 in nM). No inhibitor control as 100% enzyme activity. The assay was tested in duplicated, and the results were indicated separately as black squares and hollow inverted triangles.

Table 3. In Vitro Enzymatic Activity of 3b Against Various Receptor Tyrosine Kinases (IC50 in μM).Table Footnotea
Table 4. In Vitro enzymatic activity of 3b against various ALK Mutants (IC50 in nM).Table Footnotea
Effect of compound 3b and 3c on phosphorylation level on ALK and acetylation level of HDAC substrates in cells
Phosphorylation level on ALK is a critical indicator for assess the ALK kinase activity and thus we further evaluated the effect of compound 3b or 3c on ALK phosphorylation level as well as the acetylation level of HDAC substrates in ALK-addicted H2228 cell. Treatment with either compound significantly decreased the phosphorylation level of Y1604 on ALK at the comparable extent of ceritinib, an anticancer drug targeting EML4-ALK activity ().Citation39 Furthermore, the acetylation level of α-tubulin, a substrate of HDAC6, was obviously elevated by 3b, while the acetylated histone H3, a substrate of class I HDAC, was slightly increased (). In contrast to SAHA, a pan-HDAC inhibitor, 3b showed its selectivity on HDAC6 inhibition, as evidenced by the higher level of acetylated α-tubulin compared to acetylated histone H3, and inhibition of HDAC6 has been proven to cause G2 arrest and induce apoptosis in NSCLCs.Citation40 Consistent with the enzymatic assays, compound 3b showed strong ALK inhibition ability and higher potency over compound 3c in terms of inhibiting HDAC activity (). These observations, again, proved the potential of compound 3b as the dual inhibitor to suppress the activity of ALK kinase and HDAC as that for cell growth. Interestingly, compound 2 (pracinostat), a class I and IIa HDAC inhibitor,Citation41 enhanced the ALK kinase activity, accompanied by the highest level of acetylated histone (). This supports the notion that selective inhibition of HDAC6 (a class IIb HDAC) is preferable when combining with ALK inhibitory activity as an ALK-HDAC dual inhibitor, as opposed to SAHA or pracinostat. In addition, the protein expression level of HDAC1, HDAC6, or HDAC8 were not perturbed by the treatment of compound 3b and 3c indicating that these two compounds have little to no effect on other unwanted regulation mechanisms (supplemental Figure S3).
Figure 4. Effect of test compounds on H2228 cells. Human NSCLC H2228 cell line was treated with 3b (2 µM), 3c (4 µM), or positive controls, SAHA as pan-HDAC inhibitor, pracinostat as class I and IIa HDAC inhibitor, and crizotinib as ALK inhibitor, with DMSO (as negative control) for 6 h. (A) Western blots of phosphorylated tyrosine residue 1640 on ALK as well as acetylated (Ac) α-tubulin and histone H3 with their corresponding total proteins for normalisation. GAPDH was used as a loading control. (B) Quantification of western blotting results using Image J. Error bars indicate ± S.D.E. *p-Value ≤ 0.05, **p-value ≤ 0.01, ***p-value ≤ 0.001.

Molecular modelling studies
In our study, it is also important to understand the binding mode for our dual inhibitors in the binding site of ALK and HDAC. Therefore, molecule modelling studies of compound 3b in ALKwt (PDB ID: 4MKC) and HDAC6 (PDB ID:5EDU) were performed and the predicted binding modes were illustrated in and Citation6. In , compound 3b was observed to bind to the ATP-binding pocket of ALK, which is similar to the binding modes for ceritinib. The hydrogen of the benzimidazole core and the amide moiety can interact with M1199 via hydrogen bonding (). Furthermore, an additional hydrogen bond interaction between A1200 and hydroxamic acid moiety was shown and this interaction could lead to the improvement of binding affinity to ALKwt. This phenomenon was also reported by Gan and co-workers that their dual ALK/HDAC inhibitor also possessed this additional interaction due to the hydroxamic acid tail.Citation34
Figure 5. The predicted binding mode for 3b in the binding site of ALKwt (PDB ID: 4MKC). (A) The yellow lines indicated hydrogen bonds and the estimated lengths of bonds was shown in purple number. (B) The purple arrows indicated hydrogen bonds.

In , the binding mode for compound 3b in the catalytic pocket of HDAC6 was shown. It indicated that compound 3b can occupy the hydrophobic tunnel and the ALK-binding moiety was exposed to solvent. The hydroxamic acid group can generate metal coordinating interaction with zinc ion in the deep pocket and several hydrogen bonding with surrounding amino acid residues. () The benzimidazole core also possessed π-π interactions with F680 and H651. Taken together, the studies showed that the design of the dual ALK/HDAC inhibitor 3b can bind to the binding site of both ALK and HDAC6.
Figure 6. The predicted binding mode for 3b in the binding site of HDAC6 (PDB ID: 5EDU). (A) The yellow lines indicated hydrogen bonds and the cyan lines indicated pi-pi interactions. (B) The purple arrows indicated hydrogen bonds, the green arrows indicated π–π interactions, and the gray line indicated metal interaction.

In vivo effects of compound 3b in xenografts model
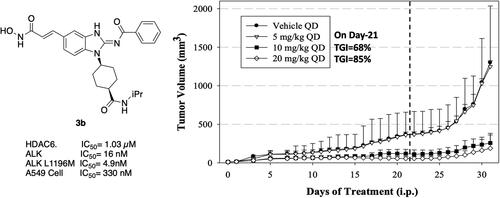
To ascertain if the observed in vitro effects correlate with in vivo tumour growth inhibition (TGI), 3b was used to treat human A549 tumour xenografts in BALB/c nude mice. Xenografts were implanted by subcutaneous injection of A549 cells and then treatment with various dosages of 3b (0, 5, 10, 20 mg/kg by intraperitoneal injection once daily for 21 days) was initiated when tumour size was 10–20 mm3. As shown in , A549 tumour growth was greatly suppressed in groups treated with 10 and 20 mg/kg 3b (68% and 85% decrease in mean tumour volume, respectively, on day 21 of treatment [dashed line]). Moreover, tumour suppression continued for an additional 10 days after treatment was suspended. (80% and 86%, respectively on day 31) The 5 mg/kg dose had no significant effect on tumour volume. Mean body weight was not significantly affected in any treatment group () and there was no animal death during the total 31-day period, suggesting that 3b had no significant acute toxicity.
Figure 7. Tumour volume and body weight following treatment of human cancer A549 xenografts in mice with 3b. Indicated doses were administered daily up to day 21 (dashed line). Data are mean ± SEM.

In vitro inhibition of CYP450 enzymes
Finally, compound 3b was profiled against CYP450 enzymes and the hERG potassium channel (). CYP450 is a large family of enzymes that catalyse oxidation and are responsible for ∼75% of drug metabolism reactions. Among them, CYP3A4 is the major player, and CYP2C9, 2C19, 2D6, and 1A2 are also important. If CYP450 enzymes are inhibited, drugs can accumulate and cause adverse effects. Thus, inhibition of CYP450 enzymes is a main cause for drug-drug interactions. With the exception of CYP2C9, 3b was a less potent inhibitor these major CYP450 enzymes compared to ALK and HDACs, indicating a low potential for drug-drug interactions. 3b also had no inhibitory affect against hERG. These observations suggested that preferred safety properties for further development of 3b as a therapeutic drug.
Table 5. Inhibition of CYP450 Enzymes and hERG by 3b (IC50 in μM).Table Footnotea
Conclusion
In this study, we aimed to design a dual function small molecule targeting both ALK and HDAC. The core structures of orally available ALK inhibitor and HDAC inhibitor, pracinostat, were adopt as the fundamental concept to develop a series of benzimidazole-based compounds. Amongst, compounds 3b with phenyl group at the R1 position, cis-1,4-cyclohexancarboxamide chain at the R2 position, and hydroxamate group at the R3 position had potential to possess balanced ALK/HDAC inhibition enzymatic inhibition assay. Consistent with enzymatic assay, the molecule modelling revealed that compound 3b can occupy both of the binding site of ALKwt and HDAC6 and the hydroxamic acid moiety created additional interaction with A1200 in ALKwt. In line with these observations, compound 3b further exhibited antiproliferative activity against various tumour cell lines, including H2228 cells, and also possessed potency of suppressing the activity of ALK and HDAC in cellular level. Of noted, compound 3b was a broad-spectrum ALK inhibitors enabling to inhibit various ALK mutants, suggesting good potential for overcoming crizotinib resistance. In agreement, compound 3b treatment significantly decreased tumour growth in the A549 xenograft model and showed prolonged antitumor activity after dosing ceased. Although the HDAC inhibition potency of compound 3b was not comparable to compound 3f, the design and preliminary evaluation of the ALK/HDAC inhibitor provide us an attractive route for further research. In the future, we would put focus on the optimisation of HDAC inhibition potency and evaluation of pharmacokinetic properties.
Experimental
Chemistry
All solvents used were purchased from Merck, ECHO, or Mallinckrodt; they were ACS grade and used without further purification. Anhydrous solvents were prepared using an SP-1 stand-alone solvent purification system (LC Technology Solution) and contained <10 ppm water as determined by Karl-Fischer moisture analysis. Chemicals were purchased from Acros, Aldrich, Alfa Aesar, Carbosynth, Fluka, KM3, Matrix, Ryss, and TCI and used as supplied. Reaction progress was monitored by TLC on Merck Kieselgel 60 F254 plates. Microwave reactions were carried out using the CEM Discover platform. Flash column chromatography was carried out on Merck silica gel 40 (40–63 μm).1H and 13C NMR spectra were obtained on a Bruker DPX-200, AV-400, or AVIII-600 operating at 200, 400, or 600 MHz. Chemical shifts are reported as ppm downfield shifted against DMSO-d6, for which the central peak was assigned as 2.49 in 1H NMR and 39.52 in 13C NMR. Multiplicity of peaks in 1H NMR are defined by the abbreviations s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Melting points were determined using a MEL-TEMP instrument (Laboratory Devices, Inc.) and are uncorrected. Mass spectroscopy was performed on a Bruker Esquire 2000 using electrospray ionisation (ESI) and analysed by quadrupole detector. High-resolution mass spectroscopy was performed on a Bruker microTOF using ESI and a time-of-flight (TOF) detector. High-performance liquid chromatography was carried out on High-performance liquid chromatography was carried out on a Shimadzu HPLC system and a Hitachi system. Shimadzu system equipped with an SPD-M20A UV − vis detector and a Kinetex XB- C18 column (5 μm pore size, 100 × 4.60 mm column dimensions) with flow rate of 1.5 mL/min. Hitachi D-7000 interface with L-7100 pump, L-7420 UV-VIS detector (λ = 254 nm), and Merck LiChrospher 100 RP-18 column (5 μm, 4 mm × 250 mm). The HPLC purities of all biological tested compounds was >95%. Elemental analysis for C, H, and N were carried out on a HERAEUS VarioEL-III or Thermo Flash 2000 and were within ±0.4% of theoretical values.
Ethyl 4-((4-bromo-2-nitrophenyl) amino) butanoate (4a)
5-Bromo-2-fluoronitrobenzene (0.37 mL, 3.00 mmol) was added to a suspension of ethyl 4-aminobutyrate hydrochloride (0.66 g, 3.83 mmol) and DIPEA (2.2 mL, 12.60 mmol) in 15 mL anhydrous ACN and the reaction mixture was stirred at 75 °C for 20 h. After being cooled to rt, the reaction mixture was concentrated and purified by flash column chromatography (silica gel: φ 4 × 12 cm; EtOAc/Hex = 1:7). The fraction with Rf = 0.21 was collected to give 4a (0.89 g, 89%) as an orange oil. 1H NMR (200 MHz, DMSO-d6) δ 8.19 (t, J = 6.0 Hz, 1 H, NH), 8.06 (d, J = 2.4 Hz, 1, H, ArH), 7.55 (dd, J = 2.4, 9.2 Hz, 1 H, ArH), 7.00 (d, J = 9.4 Hz, 1 H, ArH), 4.01 (q, J = 7.0 Hz, 2 H, CH2), 3.35 (q, J = 6.4 Hz, 2 H, CH2), 2.37 (t, J = 7.4 Hz, 2 H, CH2), 1.82 (m, 2 H, CH2), 1.14 (t, J = 7.0 Hz, 3 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 173.4, 115.0, 139.5, 132.2, 128.6, 117.5, 105.9, 60.8, 42.5, 31.6, 12.3, 14.9. HR-MS (ESI) m/z for [M + Na]+; calcd: 353.0107; found: 353.0109.
(1S,4S)-Methyl 4-((4-bromo-2-nitrophenyl) amino) cyclohexane carboxylate (4b)
5-Bromo-2-fluoronitrobenzene (0.62 mL, 5.03 mmol) was added to a suspension of methyl cis-1,4-aminocyclohexanecarboxylate hydrochloride (1.06 g, 5.49 mmol) and DIPEA (2.9 mL, 16.60 mmol) in 20 mL anhydrous can, and the reaction mixture was stirred at 75 °C for 20 h. After being cooled to rt, the reaction mixture was concentrated and purified by flash column chromatography (silica gel: φ 4 × 12 cm; EtOAc/Hex = 1:7). The fraction with Rf = 0.19 was collected to give 4b (1.76 g, 98%) as a red solid. mp = 123–124 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.15 (d, J = 2.0 Hz, 1 H, ArH), 8.07 (d, J = 8.0 Hz, 1 H, NH), 7.63 (dd, J = 2.0, 10.0 Hz, 1 H, ArH), 7.11, (d, J = 10.0 Hz, 1 H, ArH), 3.85–3.83 (m, 1 H, CH), 3.61 (s, 3 H, CH3), 2.57 (s-broad, 1 H, CH), 1.82–1.55 (m, 8 H, CH2); 13C NMR (50 MHz, DMSO-d6) δ 175.5, 144.2, 139.8, 132.2, 128.9, 118.2, 106.2, 52.4, 48.8, 29.3, 25.1. HR-MS (ESI) m/z for [M + Na]+; calcd: 379.0264; found: 379.0264.
(1S,4S)-Methyl 4-((5-bromo-2-nitrophenyl) amino) cyclohexane carboxylate (9)
4-Bromo-2-fluoronitrobenzene (1.31 g, 5.94 mmol) was added to a suspension of methyl cis-1,4-aminocyclohexanecarboxylate hydrochloride (1.21 g, 6.23 mmol) and DIPEA (3.0 mL, 17.17 mmol) in 20 mL anhydrous ACN, and the reaction mixture was stirred at 75 °C for 20 h. After being cooled to rt, the reaction mixture was concentrated and purified by flash column chromatography (silica gel: φ 4 × 13 cm; EtOAc/Hex = 1:7). The fraction with Rf = 0.35 was collected to give 9 (2.11 g, 99%) as an orange oil. 1H NMR (200 MHz, DMSO-d6) δ 8.09 (d, J = 7.8 Hz, 1 H, NH), 7.93 (d, J = 9.0 Hz, 1 H, ArH), 7.26 (d, J = 2.0 Hz, 1 H, ArH), 6.77 (dd, J = 2.0, 9.2 Hz, 1 H, ArH), 3.87–3.84 (m, 1 H, CH), 3.60 (s, 3 H, CH3), 2.56–2.51 (m, 1 H, CH), 1.80–1.57 (m, 8 H, CH2); 13C NMR (50 MHz, DMSO-d6) δ 175.5, 145.5, 132.1, 131.0, 129.0, 119.1, 117.6, 52.3, 48.6, 29.3, 25.1. HR-MS (ESI) m/z for [M + Na]+; calcd: 379.0269; found: 379.0277.
(E)-Ethyl 3–2-((3,5-difluorobenzoyl) imino)-1–(4-(isopropylamino)-4-oxobutyl)-2,3-dihydro-1H-benzo[d]imidazol-5-yl) acrylate (8d)
Iron powder (0.17 g, 3.08 mmol) and 1.5 mL saturated aq. ammonium chloride were added to a suspension of 7a (0.44 g, 1.21 mmol) in 18 mL EtOH/H2O (5:1), and the mixture was stirred at 80 °C for 6 h. After being cooled to rt, the reaction mixture was filtered through a short path of Celite and the residue was washed with EtOAc. The filtrate was concentrated and then partitioned with DCM/H2O. The organic portion was dried over MgSO4 and concentrated under reduced pressure to give the reductive intermediate. Crude diamine intermediate was dissolved in 20 mL EtOH, cyanogen bromide (0.23 g, 2.21 mmol) was added, and the reaction mixture was stirred at rt for 16 h. The reaction mixture was then treated with 10 mL saturated aq. NaHCO3 and concentrated under reduced pressure. The residue was partitioned with DCM/H2O and the organic portion dried over MgSO4 and concentrated to give the 2-aminobenzimidazole intermediate. The suspension of crude intermediate in 15 mL anhydrous DCM was cooled to 0 °C on an ice bath, then 3,5-difluorobenzoyl chloride (0.18 mL, 1.45 mmol) was added dropwise followed by TEA (0.67 mL, 4.85 mmol), and the reaction mixture was stirred at 0 °C for 30 min. The reaction mixture was partitioned with saturated aq. NH4Cl and the organic portion was purified by flash column chromatography (silica gel: φ 3 × 17 cm; EtOAc/DCM = 1:1 → 2:1). The fraction with Rf = 0.57 (EtOAc/DCM = 2:1) was collected to give 8d (0.28 g, 47%) as a white solid. MP 236–238 °C. 1H NMR (200 MHz, DMSO-d6) δ 12.85 (s, 1 H, NH), 7.81–7.35 (m, 7 H, NH, CH, ArH), 7.39 (t, J = 9.0 Hz, 1 H, ArH), 6.50 (d, J = 16.0 Hz, 1 H, CH), 4.28 (t, J = 6.0 Hz, 1 H, CH), 4.18 (q, J = 8.0 Hz, 2 H, CH2), 3.87–3.71 (m, 1 H, CH), 2.10–1.97 (m, 4 H, CH2), 1.26 (t, J = 8.0 Hz, 3 H, CH3), 0.95 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 171.6, 171.1, 167.1, 163.2 (d, JC-F = 243.5 Hz), 162.9 (d, JC-F = 244.5 Hz), 153.5, 145.5, 143.2, 143.0, 132.0, 130.3, 129.8, 124.7, 117.5, 112.7, 112.5, 112.2, 111.2, 60.9, 42.4, 41.0, 32.8, 24.8, 23.2, 15.1. HR-MS (ESI) m/z for [M + Na]+; calcd: 521.1971; found: 521.1976.
(E)-Ethyl 3–2-(butyrylimino)-1–(4-(isopropylamino)-4-oxobutyl)-2,3-dihydro-1H-benzo[d]imidazol-5-yl) acrylate (8e)
Iron powder (0.20 g, 3.51 mmol) and 1.5 mL saturated aq. ammonium chloride were added to a suspension of 7a (0.49 g, 1.35 mmol) in 18 mL EtOH/H2O (5:1), and the mixture was stirred at 80 °C for 6 h. After being cooled to rt, the reaction mixture was filtered through a short path of Celite and the residue was washed with EtOAc. The filtrate was concentrated and partitioned with DCM/H2O. The organic portion was dried over MgSO4 and concentrated under reduced pressure to give the reductive intermediate. Crude diamine intermediate was dissolved in 20 mL EtOH, cyanogen bromide (0.23 g, 2.16 mmol) was added, and the reaction mixture was stirred at rt for 16 h. The reaction mixture was then treated with 10 mL saturated aq. NaHCO3 and concentrated under reduced pressure. The residue was partitioned with DCM/H2O and the organic portion was dried over MgSO4 and concentrated to give the 2-aminobenzimidazole intermediate. The suspension of crude intermediate in 15 mL anhydrous DCM was cooled to 0 °C on an ice bath, then butyryl chloride (0.20 mL, 1.95 mmol) was added dropwise followed by TEA (0.80 mL, 5.76 mmol), and the reaction was stirred at 0 °C for 30 min. The reaction mixture was partitioned with saturated aq. NH4Cl and the organic portion was purified by flash column chromatography (silica gel: φ 3 × 14 cm; EtOAc/DCM = 1:1). The fraction with Rf = 0.28 was collected to give 8e (0.21 g, 36%) as a white solid. mp = 233–236 °C. 1H NMR (200 MHz, DMSO-d6) δ 10.52 (s, 1 H, NH), 7.91–7.52 (m, 5 H, NH, CH & ArH), 6.59 (d, J = 16.0 Hz, 1 H, CH), 4.18 (q, J = 8.0 Hz, 2 H, CH2), 4.07 (t, J = 6.0 Hz, 2 H, CH2), 3.89–3.72 (m, 1 H, CH), 2.40 (s-broad, 2 H, CH2), 2.03–1.86 (m, 4 H, CH2), 1.72–1.54 (m, 2 H, CH2), 1.26 (t, J = 8.0 Hz, CH3), 1.01 (d, J = 6.0 Hz, 6 H, CH3), 0.97 (t, J = 8.0 Hz, 3 H, CH3); 13C NMR (150 MHz, CDCl3-d) δ 171.3, 171.2, 167.1, 144.53, 144.50, 129.8, 123.9, 117.5, 109.7, 60.7, 41.7, 41.4, 32.8, 32.1, 29.8, 24.7, 22.9, 19.6 14.5, 14.1, 1.15. HR-MS (ESI) m/z for [M + H]+; calcd: 429.2502; found: 429.2506.
Methyl 8-((4-(((1S,4S)-4-(isopropylcarbamoyl) cyclohexyl) amino)-3-nitrophenyl) amino)-8-oxooctanoate (14)
N,N-Dimethylenediamine (0.46 mL, 4.26 mmol) was added to a solution of 5b (1.09 g, 2.84 mmol), sodium azide (0.55 g, 8.53 mmol), and copper(I) iodide (0.28 g, 1.49 mmol) in DMSO (8 mL), and the reaction mixture was stirred at 110 °C under Ar pressure for 5 h. After the reaction mixture cooled to rt, it was partitioned with DCM/NaHCO3 and the organic portion was dried over MgSO4 and concentrated under reduced pressure to give crude 13. Crude 13 was added to a solution of monomethyl suberate (0.60 mL, 3.34 mmol), EDCI (0.66 g, 3.44 mmol), and HOBt (0.51 g, 3.32 mmol) in anhydrous DMF (20 mL). The mixture was then treated with TEA (1.20 mL, 8.63 mmol) and stirred at rt for 20 h. After the solvent was removed under reduced pressure, the residue was partitioned with DCM/H2O and the organic portion was purified by flash column chromatography (silica gel: φ 4 × 13 cm; EtOAc/DCM = 1:3 → 1:1). The fraction with Rf = 0.37 (EtOAc/DCM = 1:1) was collected to give 14 (0.80 g, 58%) as a dark solid. mp = 164–166 °C. 1H NMR (200 MHz, DMSO-d6) δ 9.88 (s, 1 H, NH), 8.47 (d, J = 2.4 Hz, 1 H, ArH), 8.17 (d, J = 7.4 Hz, 1 H, NH), 7.66 (dd, J = 2.4, 9.2 Hz, 1 H, ArH), 7.56 (d, J = 7.6 Hz, 1 H, NH), 7.07 (d, J = 9.4 Hz, 1 H, ArH), 3.92–3.89 (m, 1 H, CH), 3.86–3.72 (m, 1 H, CH), 3.56 (s, 3 H, CH3), 2.31–2.21 (m, 4 H, CH2), 1.80–1.44 (m, 12 H, CH2), 1.30–1.24 (m, 4 H, CH2), 1.02 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.3, 174.2, 171.9, 142.0, 130.69, 130.66, 128.8, 116.1, 115.9, 52.0, 47.5, 42.6, 37.0, 34.1, 29.4, 29.2, 29.1, 25.8, 25.2, 25.1, 23.3. HR-MS (ESI) m/z for [M + Na]+; calcd: 513.2689; found: 513.2690.
General procedure a for preparation of compound 5a, 5b, 10
4-((4-Bromo-2-nitrophenyl) amino)-N-isopropyl butanamide (5a)
NaOH (1 N, 25.0 mL, 25.0 mmol) was added to a solution of 4a (1.68 g, 5.07 mmol) in 7 mL MeOH, and the reaction mixture was stirred at rt for 20 h. The mixture was acidified to pH 4–5 with CH3COOH (10% v/v) and concentrated to remove the organic solvent. The residue was partitioned with DCM/H2O and the organic portion was dried over MgSO4 and concentrated. The residue plus EDCI (1.1280 g, 5.88 mmol) and HOBt (0.7881 g, 5.83 mmol) were dissolved in 20 mL anhydrous DMF, and then isopropylamine (0.54 mL, 6.30 mmol) and triethylamine (2.55 mL, 18.34 mmol) were added and the reaction mixture was stirred at rt for 20 h. The reaction mixture was then concentrated under reduced pressure and partitioned with DCM/H2O, and the organic portion was dried over MgSO4 and concentrated to give 5a (1.54 g, 88%) as an orange solid. Rf = 0.19 (EtOAc/Hex = 1:1). mp = 157–159 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.22 (t, J = 5.0 Hz, 1 H, NH), 8.14 (d, J = 2.0 Hz, 1 H, NH), 7.69 (d, J = 8.0 Hz, 1 H, ArH), 7.63 (dd, J = 2.0, 8.0 Hz, 1 H, ArH), 7.05 (d, J = 8.0 Hz, 1 H, ArH), 3.89–3.72 (m, 1 H, CH), 3.40–3.28 (m, 2 H, CH2), 2.15–2.08 (m, 2 H, CH2), 1.86–1.72 (m, 2 H, CH2), 1.00 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 171.4, 145.1, 139.6, 132.1, 128.7, 117.8, 105.8, 42.9, 41.1, 33.3, 25.0, 23.3. HR-MS (ESI) m/z for [M + Na]+; calcd: 366.0424; found: 366.0429.
(1S,4S)-4-((4-Bromo-2-nitrophenyl) amino)-N-isopropylcyclohexanecarboxamide (5b)
General procedure A was followed starting from 4b (1.29 g, 3.60 mmol) to give 5b (1.22 g, 88%) as an orange solid. Rf = 0.24 (EtOAc/Hex = 1:1). mp = 161–163 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.21 (d, J = 8.0 Hz, 1 H, ArH), 8.16 (d, J = 2.0 Hz, 1 H, NH), 7.66–7.56 (m, 2 H, NH & ArH), 7.07 (d, J = 10.0 Hz, 1 H, ArH), 3.91–3.71 (m, 2 H, CH), 2.22–2.15 (m, 1 H, CH), 1.85–1.55 (m, 8 H, CH2), 1.01 (d, J = 8.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.3, 144.3, 139.9, 132.2, 128.9, 118.2, 106.1, 47.8, 42.5, 29.3, 25.1, 23.3. HR-MS (ESI) m/z for [M + Na]+; calcd: 406.0737; found: 406.0736.
(1S,4S)-4-((5-Bromo-2-nitrophenyl) amino)-N-isopropylcyclohexanecarboxamide (10)
General procedure A was followed starting from 9 (2.01 g, 5.62 mmol) to give 10 (0.90 g, 41%) as a yellow solid. Rf = 0.68 (EtOAc/DCM = 1:6). mp = 171–172 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.24 (d, J = 7.8 Hz, 1 H, NH), 7.98 (d, J = 9.2 Hz, 1 H, ArH), 7.57 (d, J = 7.0 Hz, 1 H, NH), 7.26 (s-broad, 1 H, ArH), 6.81 (dd, J = 1.8, 9.2 Hz, 1 H, ArH), 3.99–3.89 (m, 1 H, CH), 3.85–3.72 (m, 1 H, CH), 2.23–2.15 (m, 1 H, CH), 1.77–1.59 (m, 8 H, CH2), 1.02 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.3, 145.6, 132.2, 131.1, 129.2, 119.2, 117.7, 47.6, 42.5, 29.3, 25.0, 23.3. HR-MS (ESI) m/z for [M + Na]+; calcd: 406.0742; found: 406.0756.
General procedure B for preparation of compound 7a, 7b, 11
(E)-Ethyl 3–(4-((4-(isopropylamino)-4-oxobutyl) amino)-3-nitrophenyl) acrylate (7a)
Ethyl acrylate (0.35 mL, 3.22 mmol) and Cy2NMe (0.66 mL, 3.12 mmol) were added to a solution of 5a (0.71 g, 2.08 mmol), palladacycle (0.094 g, 0.10 mmol), and [(t-Bu)3PH]BF4 (0.062 g, 0.21 mmol) in 20 mL anhydrous DMF, and the reaction mixture was irradiated with microwaves (100 W, open-vessel) for 30 min. After being cooled to rt, the reaction mixture was concentrated and purified by flash column chromatography (silica gel: φ 3 × 15 cm; EtOAc/Hex = 3:1). The fraction with Rf = 0.32 was collected and washed with hexane to give 7a (0.44 g, 58%) as a yellow solid. mp = 179–180 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.48 (t, J = 6.0 Hz, 1 H, NH), 8.32 (d, J = 2.0 Hz, 1 H, NH), 7.95 (dd, J = 2.0, 8.0 Hz, 1 H, ArH), 7.70 (d, J = 8.0 Hz, 1 H, ArH), 7.59 (d, J = 16.0 Hz, 1 H, CH), 7.11 (d, J = 10.0 Hz, 1 H, ArH), 6.48 (d, J = 16.0 Hz, 1 H, CH), 4.15 (q, J = 6.0 Hz, 2 H, CH2), 3.90–3.73 (m, 1 H, CH), 3.44–3.33 (m, 2 H, CH2), 2.13 (t, J = 7.0 Hz, 2 H, CH2), 1.88–1.74 (m, 2 H, CH2), 1.23 (t, J = 6.0 Hz, 3 H, CH3), 1.01 (d, J = 8.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 171.3, 167.3, 146.9, 143.8, 135.4, 131.6, 128.8, 122.1, 116.4, 116.1, 60.7, 41.9, 41.1, 33.3, 25.2, 23.3, 15.1. HR-MS (ESI) m/z for [M + Na]+; calcd: 386.1686; found: 386.1695.
(E)-Ethyl 3–(4-(((1S,4S)-4-(isopropylcarbamoyl) cyclohexyl) amino)-3-nitrophenyl) acrylate (7b)
General procedure B was followed starting from 5b (1.03 g, 2.68 mmol) to give 7b (0.74 g, 69%) as an orange solid. Rf = 0.16 (EtOAc/Hex = 1:1). mp = 142–144 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.47 (d, J = 6.0 Hz, 1 H, NH), 8.33 (s, 1 H, ArH), 7.95 (d, J = 8.0 Hz, 1 H, ArH), 7.63–7.55 (m, 2 H, ArH & CH), 7.11 (d, J = 8.0 Hz, 1 H, ArH), 6.48 (d, J = 16.0 Hz, 1 H, CH), 4.15 (q, J = 8.0 Hz, 2 H, CH2), 3.99 (s-broad, 1 H, CH), 3.89–3.72 (m, 1 H, CH), 2.20 (s-broad, 1 H, CH), 1.80–1.63 (m, 8 H, CH2), 1.23 (t, J = 8.0 Hz, 3 H, CH3), 1.02 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.3, 167.2, 146.0, 143.7, 135.6, 131.5, 128.9, 122.4, 116.6, 116.4, 60.7, 47.9, 42.4, 29.4, 25.1, 23.3, 15.1. HR-MS (ESI) m/z for [M + H]+; calcd: 404.2180; found: 404.2178.
(E)-Ethyl 3–(3-(((1S,4S)-4-(isopropylcarbamoyl) cyclohexyl) amino)-4-nitrophenyl) acrylate (11)
General procedure B was followed starting from 10 (0.78 g, 2.04 mmol) to give 11 (0.56 g, 68%) as a red solid. Rf = 0.61 (EtOAc/DCM = 1:9). mp = 158–159 °C. 1H NMR (200 MHz, CDCl3-d) δ 8.35 (d, J = 7.2 Hz, 1 H, NH), 8.15 (d, J = 9.0 Hz, 1 H, ArH), 7.56 (d, J = 16.0 Hz, 1 H, CH), 6.88 (s-broad, 1 H, ArH), 6.77 (dd, J = 1.8, 9.0 Hz, 1 H, ArH), 6.46 (d, J = 16.0 Hz, 1 H, CH), 4.26 (q, J = 7.0 Hz, 2 H, CH2), 4.15–3.98 (m, 1 H, CH), 3.83–3.81 (m, 1 H, CH), 2.27–2.21 (m, 1 H, CH), 1.98–1.79 (m, 8 H, CH2), 1.32 (t, J = 7.0 Hz, 3 H, CH3), 1.13 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, CDCl3-d) δ 173.7, 166.3, 144.6, 142.8, 141.7, 132.0, 127.8, 122.3, 114.7, 113.1, 61.0, 47.9, 42.9, 41.2, 29.2, 25.0, 22.9, 14.3. HR-MS (ESI) m/z for [M + Na]+; calcd: 426.2005; found: 426.2021.
General procedure C for preparation of compound 8a, 8b, 12, 15
(E)-Ethyl 3–2-(benzoylimino)-1–(4-(isopropylamino)-4-oxobutyl)-2,3-dihydro-1H-benzo[d]imidazol-5-yl) acrylate (8a)
Iron powder (0.15 g, 2.75 mmol) and 1.5 mL saturated aq. ammonium chloride were added to a suspension of 7a (0.41 g, 1.12 mmol) in 18 mL EtOH/H2O (5:1), and the mixture was stirred at 80 °C for 6 h. After being cooled to rt, the reaction mixture was filtered through a short path of Celite and the residue was washed with EtOAc. The filtrate was concentrated and then partitioned with DCM/H2O, and the organic portion was dried over MgSO4 and concentrated under reduced pressure to give the reductive intermediate. Crude diamine intermediate was dissolved in 15 mL anhydrous THF and the solution was cooled to 0 °C on an ice bath. Benzoyl isothiocyanate (0.21 mL, 1.56 mmol) was added and the mixture was stirred at 0 °C for 15 min. EDCI (0.3600 g, 1.88 mmol) and DIPEA (0.35 mL, 2.00 mmol) were added and the mixture was stirred at 60 °C for 2 h followed by rt for additional 16 h. The reaction mixture was concentrated and partitioned with DCM/H2O, and the organic portion was purified by flash column chromatography (silica gel: φ 3 × 14 cm; EtOAc/Hex = 1:1 → 3:1). The fraction with Rf = 0.41 (EtOAc/Hex = 3:1) was collected to give 8a (0.39 g, 75%) as a white solid. mp = 227–230 °C. 1H NMR (200 MHz, DMSO-d6) δ 12.81 (s, 1 H, NH), 8.25 (d, J = 6.0 Hz, 2 H, ArH), 7.75–7.46 (m, 8 H, NH, CH & ArH), 6.50 (d, J = 16.0 Hz, 1 H, CH), 4.27–4.13 (m, 4 H, CH2), 3.79 (tt, J = 6.0 Hz, 1 H, CH), 2.10–2.02 (m, 4 H, CH2), 1.26 (t, J = 7.0 Hz, 3 H, CH3), 0.96 (d, J = 8.0 Hz, 6 H, CH3);13C NMR (50 MHz, DMSO-d6) δ 174.6, 171.0, 167.1, 153.8, 145.6, 138.9, 132.2, 132.0, 130.4, 129.7, 129.5, 128.8, 124.5, 117.3, 112.2, 110.9, 42.3, 41.1, 33.0, 24.9, 23.2, 15.1. HR-MS (ESI) m/z for [M + Na]+; calcd: 485.2159; found: 485.2155.
(E)-Ethyl 3–2-(benzoylimino)-1-((1S,4S)-4-(isopropylcarbamoyl)cyclohexyl)-2,3-dihydro-1H-benzo[d]imidazol-5-yl)acrylate (8b)
General procedure C was followed starting from 7b (0.61 g, 1.51 mmol) to give 8b (0.50 g, 66%) as a white solid. Rf = 0.40 (EtOAc/DCM = 1:7). mp = 144–145 °C. 1H NMR (200 MHz, DMSO-d6) δ 12.87 (s, 1 H, NH), 8.27 (d, J = 6.0 Hz, 2 H, ArH), 7.77–7.45 (m, 8 H, NH, CH & ArH), 6.48 (d, J = 16.0 Hz, 1 H, CH), 4.91 (t, J = 13.0 Hz, 1 H, CH), 4.17 (q, J = 8.0 Hz, 2 H, CH2), 4.05–3.92 (m, 1H, CH), 2.84–2.66 (m, 2 H, CH2), 2.11–1.60 (m, 7 H, CH2 & CH), 1.24 (t, J = 8.0 Hz, 3 H, CH3), 1.08 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.7, 174.6, 167.1, 153.4, 145.5, 138.9, 132.0, 131.0, 130.6, 129.8, 129.2, 128.8, 124.2, 117.4, 112.4, 112.4, 112.0, 60.8, 60.6, 53.8, 40.9, 37.3, 28.2, 25.9, 23.3, 21.6, 15.1. HR-MS (ESI) m/z for [M + Na]+; calcd: 525.2478; found: 525.2498.
(E)-Ethyl 3–2-(benzoylimino)-3-((1S,4S)-4-(isopropylcarbamoyl)cyclohexyl)-2,3-dihydro-1H-benzo[d]imidazol-5-yl)acrylate (12)
General procedure C was followed starting from 11 (0.56 g, 1.39 mmol) to give 12 (0.44 g, 63%) as a white solid. Rf = 0.71 (EtOAc/DCM = 1:7). mp = 200 °C. 1H NMR (200 MHz, DMSO-d6) δ 12.88 (s, 1 H, NH), 8.36 (s, 1 H, NH), 8.26 (dd, J = 1.8, 7.8 Hz, 2 H, ArH), 7.80 (d, J = 7.6 Hz, 1 H, ArH), 7.70 (d, J = 15.8 Hz, 1 H, CH), 7.55–7.42 (m, 5 H, ArH), 6.90 (d, J = 15.8 Hz, 1 H, CH), 5.02–4.90 (m, 1 H, CH), 4.23–4.07 (m, 3 H, CH2 & CH), 2.91–2.72 (m, 2 H, CH2), 2.51 (s-broad, 1 H, CH), 2.12–2.06 (m, 2 H, CH2), 1.81–1.62 (m, 4 H, CH2), 1.25 (t, J = 7.0 Hz, 3 H, CH3), 1.10 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.8, 174.7, 167.4, 153.4, 145.5, 138.9, 132.1, 132.0, 129.8, 129.6, 129.4, 128.8, 125.8, 117.5, 113.0, 110.1, 60.7, 52.9, 40.9, 37.3, 28.0, 25.3, 23.4, 15.1. HR-MS (ESI) m/z for [M + Na]+; calcd: 525.2478; found: 525.2488.
Methyl 8-(((E)-2-(benzoylimino)-1-((1S,4S)-4-(isopropylcarbamoyl)cyclohexyl)-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)-8-oxooctanoate (15)
General procedure C was followed starting from 14 (0.78 g, 1.59 mmol) to give 15 (0.61 g, 65%) as a white solid. Rf = 0.45 (EtOAc/DCM = 1:1). mp = 102–104 °C. 1H NMR (200 MHz, DMSO-d6) δ 12.75 (s, 1 H, NH), 9.94 (s, 1 H, NH), 8.25 (d, J = 6.0 Hz, 2 H, ArH), 7.92 (s, 1 H, ArH), 7.69 (d, J = 8.0 Hz, 1 H, NH), 7.58–7.35 (m, 5 H, ArH), 4.95–4.81 (m, 1 H, CH), 4.09–3.92 (m, 1 H, CH), 3.56 (s, 3 H, CH3), 2.81–2.63 (m, 2 H, CH2), 2.31–2.25 (m, 4 H, CH2), 2.13–1.98 (m, 2 H, CH2), 1.78–1.52 (m, 8 H, CH2), 1.28 (s-broad, 4 H, CH2), 1.09 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (200 MHz, DMSO-d6) δ 174.6, 174.22, 174.16, 171.8, 152.8, 139.2, 135.4, 131.7, 130.2, 129.7, 128.7, 124.9, 115.2, 111.5, 104.4, 53.5, 52.0, 40.9, 37.4, 37.2, 34.1, 29.2, 29.1, 28.2, 26.0, 25.9, 25.2, 23.3. HR-MS (ESI) m/z for [M + Na]+; calcd: 612.3162; found: 612.3163.
General procedure D for preparation of compound 3a–3f
(E)-N-(5-((E)-3-(Hydroxyamino)-3-oxoprop-1-en-1-yl)-1–(4-(isopropylamino)-4-oxobutyl)-1H-benzo[d]imidazol-2(3H)-ylidene)benzamide (3a)
LiOH (2.5 M, 1.5 mL, 3.75 mmol) was added to a suspension of 8a (0.31 g, 0.66 mmol) in 7 mL MeOH, and the mixture was stirred at rt for 20 h. The mixture was concentrated under reduced pressure to remove the organic solvent and then partitioned with DCM/H2O. The aqueous portion was cooled on an ice bath and then acidified to pH 4–5 with CH3COOH (10% v/v). The white precipitate was collected by filtration, then dissolved with EDCI (0.17 g, 0.90 mmol) and HOBt (0.12 g, 0.86 mmol) in 20 mL anhydrous DMF. NH2OTHP (0.13 g, 1.07 mmol) and triethylamine (0.40 mL, 2.88 mmol) were added and the mixture was stirred at rt for 20 h. The mixture was concentrated under reduced pressure and purified by flash column chromatography (silica gel: φ 3 × 15 cm, MeOH/DCM = 3:47). The fraction with Rf = 0.15 was collected to give the THP-protected intermediate as a white solid. A suspension of the intermediate (0.26 g, 0.49 mmol) was prepared in 20 mL MeOH and then 8 mL trifluoroacetic acid/MeOH (1:1) was added dropwise over 15 min. The reaction mixture was stirred at rt for 6 h. DCM (20 mL) was added and then the pH was adjusted to 7 with saturated aq. NaHCO3. The precipitate was collected and washed with DCM and water to give 3a (0.078 g, 36%) as a white solid. mp = 209–211 °C. 1H NMR (200 MHz, DMSO-d6) δ 12.79 (s, 1 H, NH), 10.80 (s, 1 H, NH), 9.05 (s, 1 H, NH), 8.25 (d, J = 6.0 Hz, 2 H, ArH), 7.71 (s, 1 H, NH), 7.66–7.45 (m, 7 H, CH & ArH), 6.41 (d, J = 14.0 Hz, 1 H, CH), 4.27 (t, J = 8.0 Hz, 2 H, CH2), 3.79 (tt, J = 6.0 Hz, 1 H, CH), 2.11–1.99 (m, 4 H, CH2), 0.96 (d, J = 8.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.5, 171.1, 163.8, 153.7, 139.6, 138.9, 132.0, 131.5, 130.5, 130.4, 129.7, 128.8, 124.3, 118.3, 110.8, 42.3, 41.1, 33.0, 24.9, 23.2. MS (ESI) m/z 447.8 [M–H]–. Anal. (C24H27N5O4·0.8H2O) C, H, N; calcd: 62.14, 6.21, 15.10; found: 62.47, 6.02, 15.03. HPLC purity: 97.9%.
(E)-N-(5-((E)-3-(Hydroxyamino)-3-oxoprop-1-en-1-yl)-1-((1S,4S)-4-(isopropylcarbamoyl)cyclohexyl)-1H-benzo[d]imidazol-2(3H)-ylidene)benzamide (3b)
General procedure D was followed starting from 8b (0.33 g, 0.58 mmol) to give 3b (0.15 g, 53%) as a white solid. mp = 200–201 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.23 (dd, J = 1.6, 7.6 Hz, 2 H, ArH), 7.77–7.70 (m, 3 H, ArH & NH), 7.56–7.43 (m, 5 H, ArH & CH), 6.42 (d, J = 16.0 Hz, 1 H, CH), 4.93–3.81 (m, 1 H, CH), 4.09–3.92 (m, 1 H, CH), 2.84–2.65 (m, 2 H, CH2), 2.53–2.47 (m, 1 H, CH), 2.11–2.05 (m, 2 H, CH2), 1.79–1.64 (m, 4 H, CH2), 1.09 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 174.7, 173.6, 163.7, 159.6, 151.9, 139.3, 138.0, 132.3, 131.0, 130.5, 130.3, 129.8, 128.9, 124.3, 118.8, 112.4, 111.5, 54.2, 40.9, 37.3, 28.12, 25.9, 23.3. MS (ESI) m/z 487.8 [M–H]–. Anal. (C27H31N5O4·H2O) C, H, N; calcd: 63.89, 6.55, 13.90; found: 63.79, 6.54, 13.78. HPLC purity: 98.5%.
(E)-N-(6-((E)-3-(Hydroxyamino)-3-oxoprop-1-en-1-yl)-1-((1S,4S)-4-(isopropylcarbamoyl)cyclohexyl)-1H-benzo[d]imidazol-2(3H)-ylidene)benzamide (3c)
General procedure D was followed starting from 12 (0.13 g, 0.23 mmol) to give 3c (0.085 g, 75%) as a white solid. mp = 166–167 °C. 1H NMR (200 MHz, DMSO-d6) δ 8.25 (dd, J = 1.8, 6.2 Hz, 2 H, ArH), 7.96 (s, 1 H, NH), 7.71 (d, J = 7.8 Hz, 1 H, ArH), 7.59–7.44 (m, 6 H, ArH & CH), 6.5 (d, J = 16.0 Hz, 1 H, CH), 4.90–4.77 (m, 1 H, CH), 4.17–4.00 (m, 1 H, CH), 2.95–2.72 (m, 2 H, CH2), 2.54 (s-broad, 1 H, CH), 2.15–2.09 (m, 2 H, CH2), 1.79–1.64 (m, 4 H, CH2), 1.04 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.5, 173.8, 164.0, 159.6, 158.8, 152.3, 139.5, 138.3, 132.2, 131.4, 130.6, 129.9, 129.8, 128.9, 122.8, 119.2, 113.7, 111.6, 54.1, 40.9, 37.4, 28.1, 25.9, 23.4. HR-MS (ESI) m/z for [M + Na]+; calcd: 512.2274; found: 512.2267. HPLC purity: 96.8%.
(E)-3,5-Difluoro-N-(5-((E)-3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-1–(4-(isopropylamino)-4-oxobutyl)-1H-benzo[d]imidazol-2(3H)-ylidene)benzamide (3d)
General procedure D was followed starting from 8d (0.21 g, 0.37 mmol) to give 3d (0.13 g, 74%) as a white solid. mp = 248 °C (dec.). 1H NMR (200 MHz, DMSO-d6) δ 12.85 (s, 1 H, NH), 10.79 (s, 1 H, NH), 9.08 (s, 1 H, OH), 7.83–7.35 (m, 8 H, NH, CH & ArH), 6.40 (d, J = 16.0 Hz, 1 H, CH), 4.29 (t, J = 6.0 Hz, 2 H, CH2), 3.87–3.70 (m, 1 H, CH), 2.13–1.97 (m, 4 H, CH2), 0.95 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 171.5, 171.1, 163.7, 163.2 (d, JC-F = 244.5 Hz), 162.9 (d, JC-F = 244.5 Hz), 153.4, 143.1, 142.9, 139.5, 131.3, 130.7, 130.4, 124.4, 118.6, 112.6, 112.2, 111.1, 107.2. MS (ESI) m/z 483.7 [M–H]–. Anal. (C24H25F2N5O4·0.5H2O) C, H, N; calcd: 58.29, 5.30, 14.16; found: 58.30, 5.24, 14.03. HPLC purity: 96.7%.
4-((E)-2-(butyrylimino)-5-((E)-3-(hydroxyamino)-3-oxoprop-1-en-1-yl)-2,3-dihydro-1H-benzo[d]imidazol-1-yl)-N-isopropylbutanamide (3e)
General procedure D was followed starting from 8e (0.10 g, 0.21 mmol) to give 3e (0.034 g, 39%) as a white solid. mp = 92–193 °C. 1H NMR (200 MHz, DMSO-d6) δ 10.73 (s, 1 H, NH), 10.53 (s, 1 H, OH), 9.03 (s, 1 H, NH), 7.70–7.42 (m, 5 H, NH, CH, & ArH), 6.42 (d, J = 16.0 Hz, 1 H, CH), 4.06 (s-broad, 2 H, CH2), 3.89–3.72 (m, 1 H, CH), 2.38 (s-broad, 2 H, CH2), 2.03–1.90 (m, 4 H, CH2), 1.72–1.54 (m, 2 H, CH2), 1.00 (d, J = 8.0 Hz, 6 H, CH3), 0.92 (t, J = 7.0 Hz, 3 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 173.9, 171.1, 164.0, 147.1, 142.0, 140.2, 135.9, 129.8, 122.5, 118.9, 117.9, 111.4, 43.6, 38.0, 32.9, 25.5, 23.2, 19.2, 14.6. MS (ESI) m/z 413.8 [M–H]–. Anal. (C21H29F2N5O4·0.8H2O) C, H, N; calcd: 58.67, 7.17, 16.29; found: 58.81, 7.01, 16.19. HPLC purity: 95.1%.
N-((E)-2-(benzoylimino)-1-((1S,4S)-4-(isopropylcarbamoyl)cyclohexyl)-2,3-dihydro-1H-benzo[d]imidazol-5-yl)-N-hydroxymalonamide (3f)
General procedure D was followed starting from 15 (0.31 g, 0.66 mmol) until the THP-protected intermediate was obtained. The intermediate (0.37 g, 0.55 mmol) was suspended in DCM (10 mL) on an ice bath, TFA (0.5 mL) was added dropwise over 15 min, and the reaction mixture was stirred at rt for 12 h. The pH of the mixture was adjusted to 7 with saturated aq. NaHCO3 and the precipitate was collected by filtration to give 3f (0.23 g, 71%) as a white solid. mp = 147–150 °C. 1H NMR (200 MHz, DMSO-d6) δ 10.00 (s, 1 H, NH), 8.21 (d, J = 6.4 Hz, 2 H, ArH), 7.99 (s-broad, 1 H, NH), 7.72 (d, J = 7.6 Hz, 1 H, ArH), 7.62 (d, J = 9.0 Hz, 1 H, ArH), 7.54–7.38 (m, 4 H, ArH), 4.90–4.78 (m, 1 H, CH), 4.08–3.91 (m, 1 H, CH), 2.82–2.62 (m, 2 H, CH2), 2.50–2.49 (m, 1 H, CH), 2.31 (t, J = 7.0 Hz, 2 H, CH2), 2.16–2.05 (m, 2 H, CH2), 1.94 (t, J = 7.0 Hz, 2 H, CH2), 1.77–1.42 (m, 8 H, CH2), 1.29 (s-broad, 4 H, CH2), 1.09 (d, J = 6.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 175.4, 174.6, 173.1, 172.0, 170.0, 159.5, 158.8, 151.2, 138.1, 135.8, 132.2, 130.5, 129.7, 128.9, 124.9, 115.6, 112.0, 104.6, 54.0, 40.9, 37.3, 37.2, 34.5, 33.1, 29.3, 28.1, 25.9, 23.3. HR-MS (ESI) m/z for [M + Na]+; calcd: 613.3114; found: 613.3114. HPLC purity: 99.8%.
(E)-N-(5-Bromo-1–(4-(isopropylamino)-4-oxobutyl)-1H-benzo[d]imidazol-2(3H)-ylidene)benzamide (6)
Iron powder (0.34 g, 6.06 mmol) and 2 mL saturated aq. ammonium chloride were added to a suspension of 5a (0.86 g, 2.36 mmol) in 18 mL EtOH/H2O (5:1), and the mixture was stirred at 80 °C for 6 h. After being cooled to rt, the reaction mixture was filtered through a short path of Celite and the residue was washed with EtOAc. The filtrate was concentrated and then partitioned with DCM/H2O. The organic portion was dried over MgSO4 and concentrated under reduced pressure to give the reductive intermediate. Crude diamine intermediate was dissolved in 15 mL anhydrous THF and the solution was cooled to 0 °C on an ice bath. Benzoyl isothiocyanate (0.40 mL, 2.98 mmol) was added and the mixture was stirred at 0 °C for 15 min. After treatment with EDCI (0.68 g, 3.55 mmol) and DIPEA (0.64 mL, 3.66 mmol), the mixture was stirred at 60 °C for 2 h and then at rt for an additional 16 h. The reaction mixture was concentrated and partitioned with DCM/H2O, and the organic portion was purified by flash column chromatography (silica gel: φ 3 × 16 cm; EtOAc/DCM = 1:7). The fraction with Rf = 0.33 was collected to give 6 (0.3497, 33%) as a white solid. mp = 220–221 °C. 1H NMR (200 MHz, DMSO-d6) δ 12.80 (s, 1 H, NH), 8.24 (dd, J = 2.0, 8.0 Hz, 2 H, ArH), 7.67–7.37 (m, 7 H, NH & ArH), 4.25 (t, J = 6.0 Hz, 2 H, CH2), 3.87–3.70 (m, 1 H, CH), 2.13–1.97 (m, 4 H, CH2), 0.96 (d, J = 8.0 Hz, 6 H, CH3); 13C NMR (50 MHz, DMSO-d6) δ 174.6, 171.0, 153.4, 138.9, 132.0, 131.3, 129.7, 128.8, 126.1, 115.3, 115.1, 112.3, 42.3, 41.1, 33.0, 24.8, 23.2. MS (ESI) m/z 442.7 [M–H]–. Anal. (C21H23BrN4O2) C, H, N; calcd: 56.89, 5.23, 12.64; found: 57.25, 5.27, 12.54. HPLC purity: 99.1%.
HDAC activity assays
HDAC inhibition assays were performed by Reaction Biology Corporation using a baculovirus expression system in SF9 cells. Test compounds were dissolved in DMSO at 30 μM and then tested in ten-dose IC50 mode with three-fold serial dilutions starting at 10 or 30 μM. The enzyme was diluted in reaction buffer (50 nM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mg/mL BSA, 1% DMSO) and then the test compound and specific substrate were added in sequence. For HDAC1, 6, and 11, a fluorogenic peptide from p53 residues 379–382 (RHKKAc, 50 μM) was used as the substrate. For HDAC8, a fluorogenic diacylpeptide based on p53 residues 379–382 (RHKAcKAc, 50 μM) was used. The reaction was stopped after 2 h at 30 °C. Trichostatin A was used as a positive control.
ALK, ALK mutants, and RTK activity assays
RTK inhibition assays were performed by Reaction Biology Corporation using a radioisotope-based P81 filter-binding assay. Test compounds were dissolved in DMSO at 10 μM and tested either in single dose mode at 1 μM or in ten-dose IC50 mode with three-fold serial dilutions starting at 10 μM. Enzyme and specific substrates were diluted in reaction buffer (20 mM HEPES, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/mL BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO) and then test compound was added to the reaction mixture. Reactions were initiated by addition of 33P-ATP (20 μM, final concentration 10 μM) and stopped after 2 h at rt. Reactions were spotted onto P81 ion-exchange paper (Whatman # 3698–915) and unreacted free 33P-ATP was washed away using phosphoric acid (0.75%) before detection. Staurosporine was used as a positive control. IC50 values were reported as mean of two independent reactions or one duplicate reaction with coefficient of variation (CV) <30%. Results of kinase activity in the presence of 1 μM compound were reported as percentage of inhibition with vehicle defined as 0% inhibition. Substrate for ALK, ALK mutants, c-SRC, JAK2, VEGFR-2, and EGFR was poly[Glu:Tyr] (4:1) (0.2 mg/mL); substrate for c-MET and IGF1R was [KKKSPGEYVNIEFG] (20 μM).
Cell culture and antiproliferation assays
To determine the ability of test compounds to affect the growth of adherent cells, the following cancer cell lines were used: A549 (Bioresource Collection and Research Centre, Taiwan, 60074), HepG2 (BCRC 60025), MCF7 (BCRC 60436), U87MG (BCRC 60360), and H2228 (ATCC CRL-5935). Cells were maintained at 37 °C in a 5% CO2, humidified incubator. Culture media were: RPMI-1640 for A549 and H2228 cells; Dulbecco’s modified Eagle’s medium for HepG2 and MCF7 cells; 90% Minimum essential medium Eagle with 2 mM l-glutamine and Earle’s BSS, 1.5 g/L sodium bicarbonate, 0.1 mM non-essential amino acids, 1.0 mM sodium pyruvate, and 10% foetal bovine serum for U87MG cells. Cells were seeded in 96-well plates at a density of 5 × 103 cells/well with appropriate control medium. They were treated for 48 h with test compounds at various concentration in medium containing 5% serum. Antiproliferative activity was determined by sulforhodamine B (SRB) assay. Cells treated with vehicle were used as control (100%). Antiproliferative activity was reported as percentage decrease of optical density at 510 nm for the SRB-protein complex. IC50 values were obtained on the basis of relative antiproliferation under different concentrations of compounds by regression analysis. Cell viability of compound combination was performed using MTS assay using CellTiter 96® AQueous One Solution Cell Proliferation Assay kit (Promega). In brief, 1000 cells were seeded in each well in 96-well plate and incubated for overnight. DMSO or indicated compounds were added and incubated for another 3 days. 20 µL of MTS reagent was added and incubated for 2 h followed by recording the optical density at 490 nm.
Western blot analysis
H2228 cells were treated with DMSO or target compounds at their IC50 dosage for 6 h. Treated cells were washed three times with ice-cold phosphate-buffered saline and lysed in lysis buffer containing 100 mM Tris-HCl, 12 mM sodium deoxycholate, 12 mM sodium lauroylsarcosinate, and protease inhibitor cocktail (BioTools). Cells were heated in lysis buffer at 95 °C for 5 min and then the cell membrane was broken up by sonication (Bioruptor). Cellular debris and unbroken cells were removed by centrifugation (12,500 × g for 30 min, 4 °C). Protein concentration was determined using the bicinchoninic acid assay (Thermo). Cell extracts (20 μg) were resolved by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to PVDF membranes, and probed separately with antibodies for Ac-histone H3, Ac-α-tubulin, histone H3, α-tubulin, HDAC1, HDAC6, HDAC8 and GAPDH. The antibodies of GAPDH, HDAC1, and HDAC8 were purchased from ABclonal and all other antibodies were from Cell Signalling Technology. Antibody signals were detected using an enhanced chemiluminescence system.
Tumour suppression in human cancer xenograft model
The procedure for establishing tumour xenografts and the dosing of 3b were carried out in accordance with the National Taiwan University College of Medicine Laboratory Animal Centre (NTUCMLAC) care and use committee. Female nude mice (BALB/cAnN.Cg-Foxn1nu/CrlNarl, 6–8 weeks old) were purchased from the National Laboratory Animal Centre (Taiwan). Mice were implanted subcutaneously in the right flank with 1 × 107 A549 cells (human NSCLC, in 1:1 PBS/Geltrex, 200 μL). Treatments were initiated when tumours were 10–20 mm3 and continued for 21 consecutive days. Mice were randomly assigned to cohorts (n = 5 per group). Compound 3b (10 mg/kg) or vehicle (PBS + 5% DMSO + 10% Pharmasolve) was administered once daily (qd) via intraperitoneal (i.p.) injection. Tumour volume and body weight were assessed daily for 31 days from initiation of treatment. Calliper measurements of tumours were converted to mean tumour volume using the formula: 0.5 × (length × widthCitation2).
CYP450 and hERG inhibition assays
Assays were performed by Reaction Biology Corporation. Test compounds were dissolved in DMSO at 10 or 30 μM and used for testing in ten-dose IC50 mode with three-fold serial dilutions starting at 10 μM. CYP assays used human recombinant CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 expressed in CYP cDNA baculovirus expression systems. Furafylline (for CYP1A2) and ketoconazole (all other CYPs) served as positive controls. Substrates used were 3-cyano-7-ethoxycoumarin for CYP1A2, 2C19, and 2D6; Vivid OOMR Substrate for CYP2C9; and Vivid BOMR Substrate for CYP3A4. hERG inhibition assays involved a binding competition between the tested compounds and a fluorescently labelled compound tracer (Predictor hERG Tracer Red) for membrane-bound hERG (Predictor hERG Membrane). E-4031 served as the positive control.
Molecular docking
The cocrystal structure of wild-type ALK complexed with ceritinib (PDB: 4MKC) and HDAC6 complexed with trichostatin A (PDB: 5EDU) were taken from the Protein Data Bank (http://www.rcsb. org). Molecular modelling was performed using the Schrödinger small molecule drug discovery suite 2021–4. The receptors were prepared by Protein Preparation Wizard (Schrödinger Release 2021–4: Protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2021; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2021.). The centroid of the original ligands was selected to be the site of the grid box to cover the surrounding residues of the binding pockets. The 2D ligand structures were prepared using Chemdraw 8, and the 3D structures were generated by LigPrep (Schrödinger Release 2021–4: LigPrep, Schrödinger, LLC, New York, NY, 2021.). The molecular docking simulations were performed using Glide (Schrödinger Release 2021– 4: Glide, Schrödinger, LLC, New York, NY, 2021.) and the results indicated that the original ligands (ceritinib and trichostatin A) can be reproduced. The binding pose with the lowest score in each case is selected to represent the predicted binding mode. The 3D and 2D protein − ligand interaction plots were presented using Maestro (Schrödinger Release 2021–4: Maestro, Schrödinger, LLC, New York, NY, 2021.).
Author contributions
The research work was done through the contributions of all authors. All authors have approved the final version of the manuscript.
Supplemental Material
Download PDF (4.7 MB)Supplemental Material
Download PDF (3.5 MB)Acknowledgements
The authors thank Hsuan-Chun Huang for help with HPLC sample analysis. The authors thank Pei-Shan Wu,Ting-An Chen, and Zong-Yu Yang for antiproliferation study, western blot assay, and IC50 analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of alk, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14(4):1–16.
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, npm, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284.
- Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, Pandita A, Tang J, Modrusan Z. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res. 2009;7(9):1466–1476.
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–935.
- Sugawara E, Togashi Y, Kuroda N, Sakata S, Hatano S, Asaka R, Yuasa T, Yonese J, Kitagawa M, Mano H, et al. Identification of anaplastic lymphoma kinase fusions in renal cancer. Cancer. 2012;118(18):4427–4436.
- Sukov WR, Hodge JC, Lohse CM, Akre MK, Leibovich BC, Thompson RH, Cheville JC. ALK alterations in adult renal cell carcinoma: frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod Pathol. 2012;25(11):1516–1525.
- Murugan AK, Xing MZ. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011;71(13):4403–4411.
- George RE, Sanda T, Hanna M, Fröhling S, Luther W, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–978.
- Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol. 2008;61(4):428–437.
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566.
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203.
- Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253.
- Yano T, Haro A, Shikada Y, Maruyama R, Maehara Y. Non-small cell lung cancer in never smokers as a representative ‘non-smoking-associated lung cancer’: epidemiology and clinical features. Int J Clin Oncol. 2011;16(4):287–293.
- Cui JJ, Tran-Dubé M, Shen H, Nambu M, Kung P-P, Pairish M, Jia L, Meng J, Funk L, Botrous I, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–6363.
- Shaw AT, Yasothan U, Kirkpatrick P. Crizotinib. Nat Rev Drug Discov. 2011;10(12):897–898.
- Sasaki T, Okuda K, Zheng W, Butrynski J, Capelletti M, Wang L, Gray NS, Wilner K, Christensen JG, Demetri G, et al. The neuroblastoma-associated f1174l ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70(24):10038–10043.
- Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739.
- Pan Y, Deng C, Qiu Z, Cao C, Wu F. The resistance mechanisms and treatment strategies for ALK-rearranged non-small cell lung cancer. Front Oncol. 2021;11:713530. (
- Huang WS, Liu S, Zou D, Thomas M, Wang Y, Zhou T, Romero J, Kohlmann A, Li F, Qi J, et al. Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J Med Chem. 2016;59(10):4948–4964.
- Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo-Jack I, Gadgeel S, Schultz K, Singh M, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–1133.
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra17.
- Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS, Sabnis A, et al. Ras-mapk dependence underlies a rational polytherapy strategy in EML4-ALK–positive lung cancer. Nat Med. 2015;21(9):1038–1047.
- Fukuda K, Takeuchi S, Arai S, Katayama R, Nanjo S, Tanimoto A, Nishiyama A, Nakagawa T, Taniguchi H, Suzuki T, et al. Epithelial-to-mesenchymal transition is a mechanism of ALK inhibitor resistance in lung cancer independent of ALK mutation status. Cancer Res. 2019;79(7):1658–1670.
- Pan G, Liu Y, Shang L, Zhou F, Yang S. Emt-associated micrornas and their roles in cancer stemness and drug resistance. Cancer Commun (Lond). 2021;41(3):199–217.
- Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–172.
- Zhang J, Ma L. Microrna control of epithelial-mesenchymal transition and metastasis. Cancer Metastasis Rev. 2012;31(3-4):653–662.
- Glaser KB, Li J, Staver MJ, Wei R-Q, Albert DH, Davidsen SK. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun. 2003;310(2):529–536.
- Kim H-J, Bae S-C. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3(2):166–179.
- Krämer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Gührs K-H, Stauber RH, Böhmer FD, Heinzel T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009;23(2):223–235.
- Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25(1):84–90.
- Atadja P. Development of the pan-dac inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009;280(2):233–241.
- Neal JW, Sequist LV. Exciting new targets in lung cancer therapy: ALK, IGF-1R, HDAC, and HH. Curr Treat Options Oncol. 2010;11(1–2):36–44.
- Stockhammer P, Ho CSL, Hegedus L, Lotz G, Molnár E, Bankfalvi A, Herold T, Kalbourtzis S, Ploenes T, Eberhardt WEE, et al. HDAC inhibition synergizes with ALK inhibitors to overcome resistance in a novel ALK mutated lung adenocarcinoma model. Lung Cancer. 2020;144(:20–29.
- Pan T, Dan Y, Guo D, Jiang J, Ran D, Zhang L, Tian B, Yuan J, Yu Y, Gan Z. Discovery of 2,4-pyrimidinediamine derivatives as potent dual inhibitors of ALK and HDAC. Eur J Med Chem. 113672;224:113672.
- Lewis RT, Bode CM, Choquette DM, Potashman M, Romero K, Stellwagen JC, Teffera Y, Moore E, Whittington DA, Chen H, et al. The discovery and optimization of a novel class of potent, selective, and orally bioavailable anaplastic lymphoma kinase (alk) inhibitors with potential utility for the treatment of cancer. J Med Chem. 2012;55(14):6523–6540.
- Wang H, Yu N, Chen D, Lee KCL, Lye PL, Chang JWW, Deng W, Ng MCY, Lu T, Khoo ML, et al. Discovery of (2e)-3-{2-butyl-1-[2-(diethylamino)ethyl]-1h-benzimidazol-5-yl}-n-hydroxyacrylamide (sb939), an orally active histone deacetylase inhibitor with a superior preclinical profile. J Med Chem. 2011;54(13):4694–4720.
- Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non–small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482.
- Alam MW, Borenäs M, Lind DE, Cervantes-Madrid D, Umapathy G, Palmer RH, Hallberg B. Alectinib, an anaplastic lymphoma kinase inhibitor, abolishes ALK activity and growth in ALK-positive neuroblastoma cells. Front Oncol. 2019;9:579. (
- Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197.
- Deskin B, Yin Q, Zhuang Y, Saito S, Shan B, Lasky JA. Inhibition of HDAC6 attenuates tumor growth of non-small cell lung cancer. Transl Oncol. 2020;13(2):135–145.
- Novotny-Diermayr V, Sangthongpitag K, Hu CY, Wu X, Sausgruber N, Yeo P, Greicius G, Pettersson S, Liang AL, Loh YK, et al. SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol Cancer Ther. 2010;9(3):642–652.