
Abstract
A novel series of hydantoins incorporating phthalimides has been synthesised by condensation of activated phthalimides with 1-aminohydantoin and investigated for their inhibitory activity against a panel of human (h) carbonic anhydrase (CA, EC 4.2.1.1): the cytosolic isoforms hCA I, hCA II, and hCA VII, secreted isoform hCA VI, and the transmembrane hCA IX, by a stopped-flow CO2 hydrase assay. Although all newly developed compounds were totally inactive on hCA I and mainly ineffective towards hCA II, they generally exhibited moderate repressing effects on hCA VI, VII, and IX with KIs values in the submicromolar to micromolar ranges. The salts 3a and 3b, followed by derivative 5, displayed the best inhibitory activity of all the evaluated compounds and their binding mode was proposed in silico. These compounds can also be considered interesting starting points for the development of novel pharmacophores for this class of enzyme inhibitors.
GRAPHICAL ABSTRACT
Introduction
The carbonic anhydrases (CAs, EC 4.2.1.1) are superfamily of metallo-enzymes that are found predominantly in both prokaryotes and eukaryotes and categorised into eight major groups (α, β, γ, δ, ζ, η, θ, and ι) based on their amino acid sequence similarity.Citation1 Human (h) CAs belong exclusively to the group of α-CAs and comprise 15 different isoforms (CA I, II, III, VA, VB, VI, VII, VIII, IX, X, XI, XII, XIII, XIV, and XV) Citation2. There is increasing evidence that abnormal levels and/or activities of these enzymes lead to a variety of diseases (e.g. glaucoma, oedema, epilepsy, obesity, sterility, and cancer) Citation3. Therefore, selective inhibition of hCA isozymes is an important approach for discovery and development of safe and effective medicines.Citation4
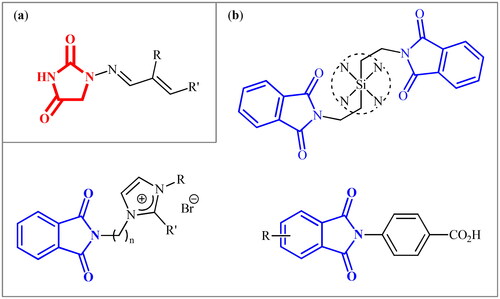
The hydantoin (or glycolylurea, imidazolidine-2,4-dione) moiety is a N-heterocyclic structural scaffold frequently present in various natural productsCitation5 and synthetic pharmaceuticalsCitation6 with diverse nature of activities from anti-bacterial to anti-androgen or anticonvulsant action. Similarly, phthalimide (or isoindoline-1,3-dione) is a ubiquitous structural motif in various natural productsCitation7 and biologically active pharmaceuticals.Citation8 Due to their diverse properties, many researchers have been working to explore these 1,2-dicarboxamides to their maximum potential against several diseases or disorders.Citation9–13 In this context, recently, we disclosed that the clinically used antibiotic Furagin () and its derivatives, which contain a hydantoin core, display effective inhibitory activity on several human (h) CAs (EC 4.2.1.1)Citation14. Several phthalimide-based compounds were developed by various research groups (), which showed appreciable activity against various hCA isozymes.Citation15
Figure 1. (a) General structure of furagin derivatives developed by our group as isoform-selective CAIs; (b) selected examples of phthalimide-based CAIs.
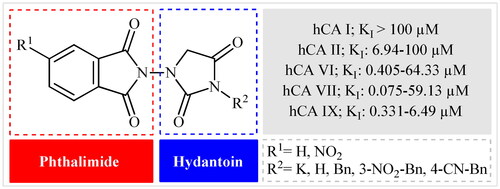
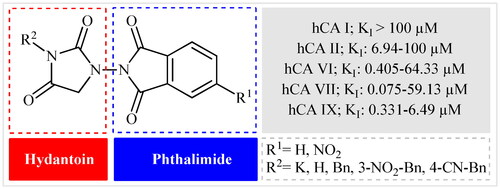
Keeping all the above facts in mind, and with consideration that the incorporation of different pharmacophores usually leads to the formation of more active compounds,Citation13 in connection with our works on the field of drug design of CAs inhibitors, herein, we decided to synthesise a series of hitherto unknown phthalimide–hydantoin hybrids and investigate their inhibitory capability against five hCAs isozymes: the cytosolic isoforms hCA I, II, and VII as well as the secreted isoform hCA VI and trans-membrane tumour-associated isoform hCA IX ().
Figure 2. General structure of phthalimide–hydantoin hybrids investigated in the paper.
Results and discussion
Chemistry
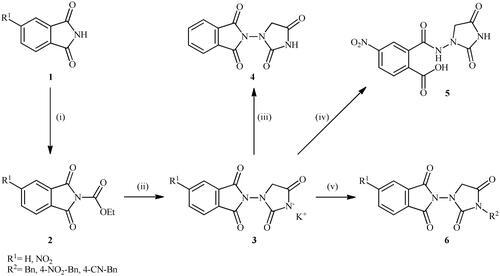
The synthesis of the target phthalimide–hydantoin hybrids is shown in Scheme 1. The synthesis started from phthalimides 1, which were converted to the corresponding N-ethoxycarbonylphthalimides 2 via treatment with ethyl chloroformate (ClCO2Et) in a basic medium. The phthalimide–hydantoin hybrid salts 3 were then prepared through the reaction of appropriate N-ethoxycarbonylphthalimide 2 with commercially available 1-aminohydantoin hydrochloride in the presence of 2 equiv. K2CO3 in NMP at 100 °C. Neutralisation of unsubstituted intermediate 3a led to the formation of the expected phthalimide–hydantoin hybrid 4, while in the case of NO2-substituted one 3b, a ring opening took place to give 5 as the sole product. There is no need to say that strongly electron-withdrawing nitro functionality make the carbonyl groups of phthalimide core in compound 3b more electrophilic and increase their reactivity towards hydrolysis. In parallel, acidic medium may also increase the rate of this ring opening reaction by acting as a catalyst. Finally, potassium 2,5-dioxoimidazolidin-1-ides 3 were reacted with a series of benzyl bromide derivatives in the absence of any additive, leading to the desired N-benzyl substituted derivatives 6.
Scheme 1. Reagents and conditions: (i) ClCO2Et (1 equiv.), 0–20 °C, 4 h, 31%; (ii) 1-aminohydantoin hydrochloride (1 equiv.), K2CO3 (2 equiv.), 100 °C, 20 h; (iii) H2O/HCO2H (10:1), 70 °C, 2 h; (iv) H2O/HCO2H (10:1), 50 °C, 30 min; (v) (a) K2CO3, DMF, 100 °C, 1.5 h; (b) Bn-Br (1.5 equiv.), DMSO, 100 °C, 5 h.
Carbonic anhydrase inhibition
All of the newly synthesised compounds (3–6) are tested in vitro for their inhibitory activities against five hCAs isozymes: the cytosolic isoforms hCA I, II, and VII as well as the trans-membrane tumour-associated isoform hCA IX and secreted isoform hCA VI, by means of the stopped-flow carbon dioxide hydration assay.Citation16 Their inhibition profiles are compared to the acetazolamide (AAZ; standard reference inhibitor for hCA) as outlined in .
Table 1. Inhibition data of human CA I, II, VI, VII, and IX with phthalimide–hydantoin hybrids (3–6) and the reference drug (AAZ) by a stopped-flow CO2 hydrase assay [Citation16].
The following structure–activity relationship (SAR) can be observed regarding the inhibition data of :
The ubiquitous isoform hCA I was not inhibited at all by the newly developed hydantoin derivatives. On the other hand, the second widely expressed isoform hCA II was poorly inhibited by three derivatives (3a, 3b, and 5) with inhibition constants in the range of 6.94–58.58 μM, all of which possess the free –NH/K group.
The unique secreted isoform hCA VI was on the other hand moderately to poorly inhibited by the phthalimide–hydantoin hybrids reported here, with KIs in the submicromolar to micromolar ranges, more precisely 0.405–64.33 μM (). Again, the best inhibitors were salts 3a and 3b; albeit, being 37- and 64-fold, respectively, inferior to AAZ whose KI value is 0.011 μM. High inhibitory activity of these salts indicated that this class of compounds bind to the CA Zn ion via the imidic nitrogen of the hydantoin nucleus.
The other cytosolic isoform, hCA VII, was affected efficiently by the newly developed compounds, as compounds 3a and 3b showed potent activity inhibition constants in the sub-micromolar ranges (0.075 and 0.163 μM, respectively). However, compounds 5 and 6a–f showed weak to moderate activity against hCA VII (KIs = 13.88–59.13 μM). It should be mentioned that the electronic effects of substituents on the phenyl ring periphery of N-benzyl substituted derivatives (6a–f) almost did not influence their inhibitory capability.
The extracellular, tumour-associated isoform hCA IX was moderately to poorly inhibited by the tested compounds with inhibition constants in the range of 0.331–6.49 μM. Like all other isoforms, in this case, potassium 3-(1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-ide 3a demonstrated the best activity, with a KI value of 0.331 μM but was still 13-fold less potent than AAZ (KI of 0.025 μM).
Computational chemistry studies
Predictions on the binding mode and studies on the ligand/target interactions for the most active compounds 3a, 3b, and 5 against the isoforms I, II, VI, VII, and IX of human carbonic anhydrases () were carried out by docking calculations. Solutions were found for all the isoenzymes studied, except for hCA I; in this case, residues H67, F91, and H200 prevent ligand binding and the inhibition profiles are actually very poor in this isoform. In the other isoforms, the hydantoin scaffold deeply bound to the zinc ion with the deprotonated imidic nitrogen atom (CON−CO) for ligands 3a and 3b, according to the literature Citation14 (). The binding of ligands completed the tetrahedral coordination sphere of the metal and the overall stabilisation is reinforced by an H-bond between the C=O in position 4 of the hydantoin scaffold and the backbone NH of T199 and by van der Waals (vdW) contacts occurring between the heterocycle and the residues H94, H96, H119, L198, T200, and W209.
Figure 3. Predicted binding mode of ligands 3a (cyan) and 3b (orange) within the human: (A) CA II, (B) CA VI, (C) CA VII, and (D) CA IX active site. H-bonds and π–π stacking interactions are represented as black and cyan dashed lines, respectively.
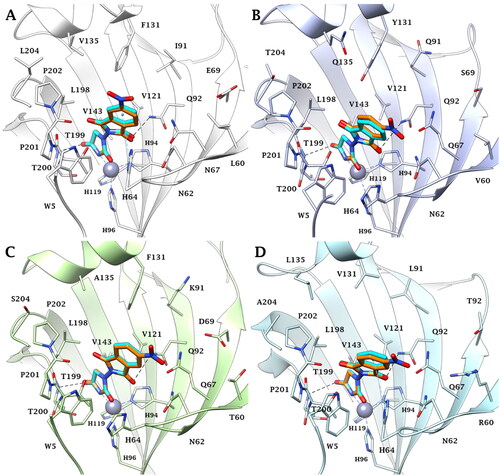
The isoindoline-1,3-dione scaffold is able to engage π–π stacking interactions with the proton shuttle residue H64 () and the C=O group is in H-bond distance with the side chain NH2 of the conserved Q92 () in the active sites of all investigated isozymes.
Because of the mutation N67/Q97 (hCA II/hCA VI, VII, and IX), the improved ligand/isoforms matching makes the investigated compounds selective for hCA VI, VII, and IX compared to the hCA II (). Other mutations, such as Y131/F131 (hCA VI/hCA VII) or Y131/V131 (hCA VII/hCA IX), may lead to decreased inhibitory activity of ligands 3a and 3b in hCA VI due to the polar character of Y131 and to steric hindrance associated to its phenolic OH ().
Instead, the presence of V131 in hCA IX active site decreases the ability of the target to stabilise the ligands binding mode by vdW interactions ().
Several literature dataCitation4 reporting the CA inhibition mechanism of ligands bearing the carboxylic group support the hypothesis that the binding of compound 5 to the metal ion occurs via a Zn-bonded water molecule (). The carboxylate group interacts with the oxygen HOH lone-pairs forming, in addition, a hydrogen bond with the side chain OH of the conserved T200. Complete the stabilisation pattern of the H-bond occurring between the water molecule and the side chain OH of T199.
Figure 4. Predicted binding mode of ligand 5 (green) within the human: (A) CA II, (B) CA VI, (C) CA VII, and (D) CA IX active site. H-bonds and π–π stacking interactions are represented as black and cyan dashed lines, respectively.
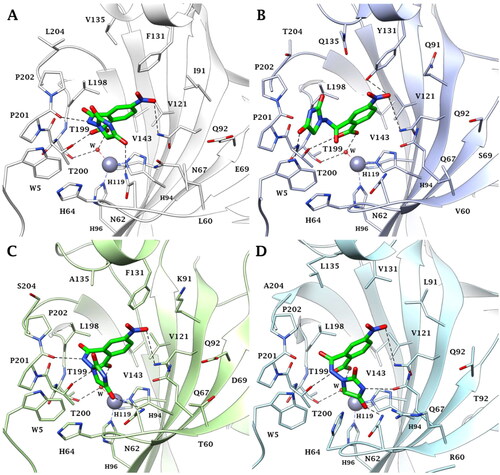
In hCA II and hCA VII active sites, the amidic NH, imidic C=O and NO2 group are in H-bond distance with the backbone C=O of P201, and with the N62 and Q92 side chain NH2, respectively (). Because of the mutations N67/Q67 and I91/K91 (hCA II/hCA VII) and the resulting change in the network of hydrophobic contacts or steric hindrance, the inhibitory profile of 5 is modulated differently in these isoforms.
In the case of the secreted-isoform hCA VI, while the imidic C=O engages an H-bond with the indolic NH of conserved W5, the NO2 group is in H-bond distance with the side chain NH2 of Q92 and with the side chain OH of the peculiar Y131 (). It is likely that the specificity of this latter interaction plays a role in the better KI value of compound 5 for hCA VI over hCA II and VII.
In conclusion, the binding of compound 5 within the tumour-associated hCA IX is favoured by residues Q67 and V131, peculiar to this isoform. In fact, due to the lower bulky nature of V131, the compound is able to accommodate well within the binding cavity and to establish direct interactions with Q67. This could explain the better hCA IX inhibition profile of derivative 5 over the hCA II, VI, and VII isoforms ().
Experimental
Chemistry methods
All the chemicals used were of analytical grade purity. Thin-layer chromatography was performed on silica gel, spots were visualised with UV light (254 and 365 nm).1H and 13C NMR spectra were registered in DMSO-d6 using Bruker 500 MHz instrument (Billerica, MA). NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartette, m = multiplet, and br = broad signal. Chemical shifts (δ) are given in parts per million (ppm) and are referenced to TMS (1H,13C). High-resolution mass spectra (HRMS) were recorded on a mass spectrometer with a Q-TOF micro mass analyser using the ESI technique.
Synthesis
General procedure (A) for preparation of ethyl 1,3-dioxoisoindoline-2-carboxylates (2)
To an ice-cooled solution of the appropriate isoindoline-1,3-dione (50 mmol, 1.0 equiv.) and Et3N (60 mmol, 1.2 equiv.) in dry DMF (100 ml) was dropwise added ethyl chloroformate (50 mmol, 1.0 equiv.). After 1 h of stirring at 0 °C, the reaction flask was allowed to warm to 20 °C and the mixture was stirred at the indicated temperature for 4 h. Then, the mixture was quenched with water (200 ml) and the formed precipitate was filtered, washed with water (500 ml) and Et2O (50 ml) and dried under vacuum to give the final desired products as off-white solids.
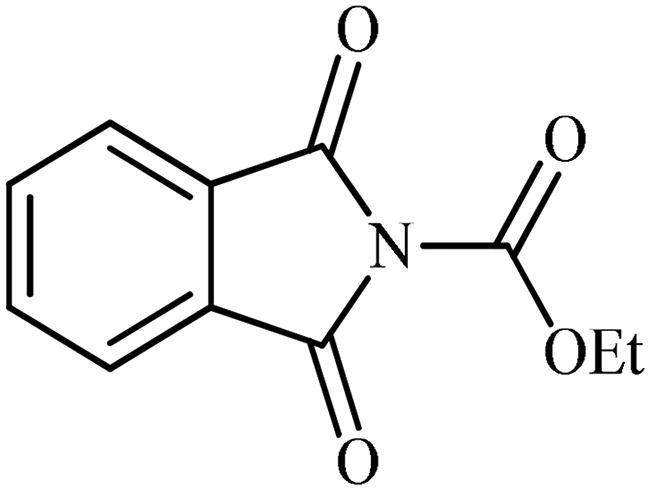
Ethyl 1,3-dioxoisoindoline-2-carboxylate (2a)
Following the general procedure A, 2a was obtained as a white solid. (6.80 g, 48% yield). 1H NMR (500 MHz, DMSO-d6) δ = 1.36 (t, 3H, J = 7.1 Hz), 4.40 (q, 2H, J = 7.1 Hz), 7.95–8.00 (m, 4H) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 14.9, 64.2, 124.9, 131.8, 136.4, 149.0, and 164.6 ppm.
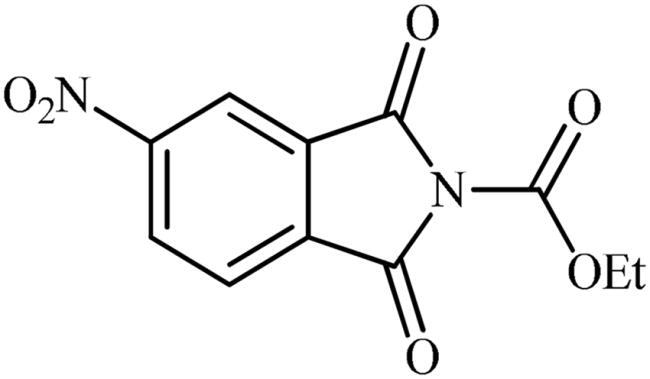
Ethyl 5-nitro-1,3-dioxoisoindoline-2-carboxylate (2b)
Following the general procedure A, 2b was obtained as a white solid (9.56 g, 72% yield). 1H NMR (500 MHz, DMSO-d6) δ = 1.37 (t, 3H, J = 7.0 Hz), 4.44 (q, 2H, J = 7.0 Hz), 8.24 (d, 1H, J = 8.2 Hz), 8.60 (s, 1H), 8.71 (d, 1H, J = 8.2 Hz) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 14.9, 64.5, 119.7, 126.5, 131.2, 133.4, 136.5, 148.6, 152.8, 163.0, and 163.2 ppm.
General procedure (B) for preparation of potassium 3-(1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-ides (3)
The appropriate ethyl 1,3-dioxoisoindoline-2-carboxylate (10 mmol, 1.0 equiv.) and 1-aminohydantoin hydrochloride (10 mmol, 1.0 equiv.) were added to NMP (40 ml) at room temperature. Then, K2CO3 (20 mmol, 2.0 equiv.) was slowly added to the above reaction mixture and the reaction mixture was stirred for 20 h at 100 °C. After completion of the reaction, the mixture was quenched with tBuOMe (400 ml) and the formed precipitate was filtered, washed with tBuOMe (100 ml) and MeOH (200 ml) and dried under vacuum to give the final desired products.
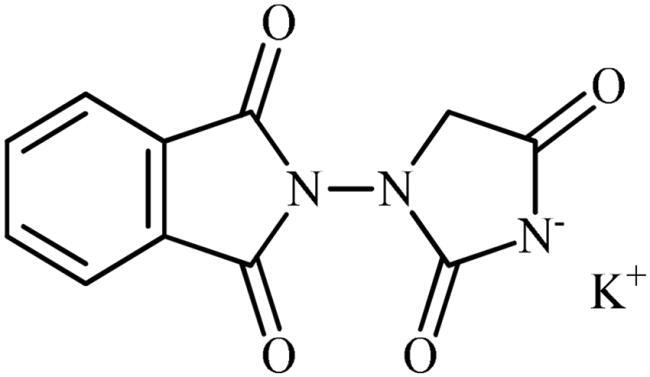
Potassium 3-(1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-ide (3a)
Following the general procedure B, 3a was obtained as a white solid (2.35 g, 83% yield). Notably, 0.74 g of KCl was also participated with the product. 1H NMR (500 MHz, D2O) δ = 4.06 (s, 2H), 7.45–7.53 (m, 3H), 7.59–7.61 (m, 1H) ppm. 13C NMR (125 MHz, D2O) δ = 53.8, 127.5, 128.3, 129.6, 131.1, 131.7, 137.9, 172.3, 173.2, 175.2, and 187.8 ppm. HRMS (ESI) [M + H]+: m/z calcd. for (C11H8N3O4) 246.0515. Found 246.0513.
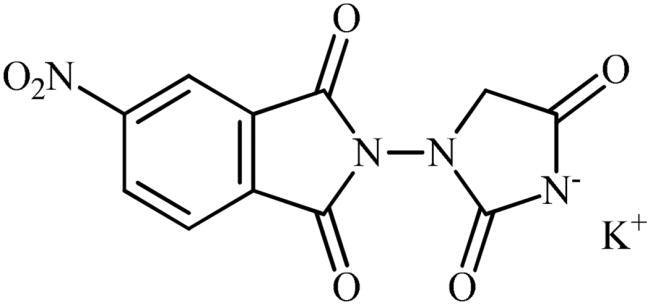
Potassium 3-(5-nitro-1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-ide (3b)
Following the general procedure B, 3b was obtained as a white solid. (1.59 g, 48% yield). Notably, 0.74 g of KCl was also participated with the product. 1H NMR (500 MHz, D2O) δ = 4.16 (s, 2H), 7.73–7.78 (m, 1H), 8.29–8.35 (m, 1H), 8.45 and 8.46 (s, 1H) ppm. 13C NMR (125 MHz, D2O) δ = 153.1, 53.2, 123.0, 123.7, 124.7, 126.2, 129.0, 129.5, 132.4, 138.1, 139.2, 144.3, 147.4, 148.7, 167.3, 167.4, 169.7, 170.9, 171.8, 173.0, 181.7, and 181.8 ppm. HRMS (ESI) [M + Na]+: m/z calcd for (C11H6N4NaO6) 313.0185. Found 315.0352.
2-(2,4-dioxoimidazolidin-1-yl)isoindoline-1,3-dione (4)
To a solution of potassium 3-(1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-ide (3a) (300 mg, 1.06 mmol) in H2O (3 ml) under stirring, HCO2H (0.3 ml) was slowly added and the mixture was heated at 70 °C for 2 h. After cooling to room temperature, the formed precipitate was filtered, washed with water (30 ml) and Et2O (10 ml) and dried under vacuum to afford 4 (140 mg, 54%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 4.34 (s, 2H), 7.99–8.02 (m, 2H), 8.03–7.06 (m, 2H), 11.81 (s, 1H) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 54.4, 124.9, 130.2, 136.5, 157.5, 165.3, and 170.4 ppm. HRMS (ESI) [M + H]+: m/z calcd for (C11H8N3O4) 246.0515. Found 246.0517.

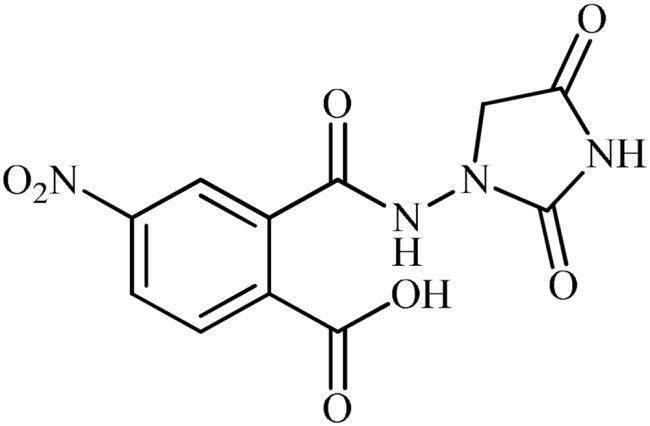
2-((2,4-Dioxoimidazolidin-1-yl)carbamoyl)-4-nitrobenzoic acid (5)
To a solution of potassium 3-(5-nitro-1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-ide (3b) (300 mg, 0.91 mmol) in H2O (3 ml) under stirring, HCO2H (0.3 ml) was slowly added and the mixture was heated at 50 °C for 30 min. After cooling to room temperature, the formed precipitate was filtered, washed with water (30 ml) and Et2O (10 ml) and dried under vacuum to afford 5 (135 mg, 48%) as a white powder.
1H NMR (500 MHz, DMSO-d6) δ = 4.17 (s, 2H), 7.76 (s, 1H), 8.58 (d, 2H, J = 39.6 Hz), 10.87 (s, 1H), 11.37 (s, 1H), 14.06 (s, 1H) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 52.5, 125.5, 127.9, 130.8, 132.6, 141.8, 149.1, 158.5, 166.1, 167.8, and 171.0 ppm. HRMS (ESI) [M + Na]+: m/z calcd for (C11H8N4NaO7) 331.0291. Found 331.0294.
General procedure (C) for preparation of 2-(3-benzyl-2,4-dioxoimidazolidin-1-yl)isoindoline-1,3-diones (6a–f)
To a suspension of appropriate potassium 3-(1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-ide (1.0 mmol, 1.0 equiv.) in DMSO (4 ml) was added the appropriate benzyl bromide (1.5 mmol, 1.5 equiv.) and the mixture was stirred at 100 °C for 5 h. The mixture was cooled to room temperature and water (80 ml) and Et2O (10 ml) were added to the mixture and the solution was vigorously stirred for 30 min. Subsequently, the solids formed in the organic phase were filtered and washed with water and Et2O to afford the desired 2-(3-benzyl-2,4-dioxoimidazolidin-1-yl)isoindoline-1,3-dione derivatives. Notably, in the case of 6b, after the reaction was completed, the mixture was poured into ethyl acetate, washed with brine, extracted with ethyl acetate, dried over Na2SO4, filtered, and dried under vacuum, and finally the oily residue was crystallised in iPrOH.
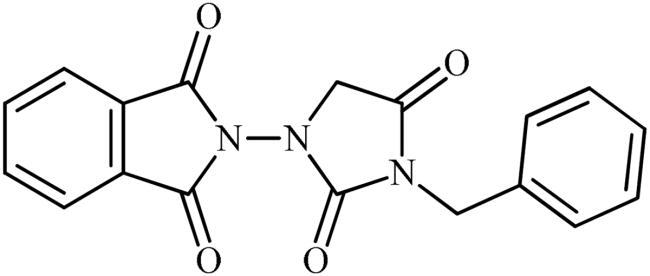
2-(3-Benzyl-2,4-dioxoimidazolidin-1-yl)isoindoline-1,3-dione (6a)
Following the general procedure C, 6a was obtained as a white solid (57 mg, 17% yield). 1H NMR (500 MHz, DMSO-d6) δ = 4.48 (s, 2H), 4.74 (s, 2H), 7.34–7.44 (m, 5H), and 7.98–8.08 (m, 4H) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 43.0, 52.6, 125.0, 128.4, 128.6, 129.6, 130.2, 136.5, 136.7, 157.1, 165.2, and 168.9 ppm. HRMS (ESI) [M + H]+: m/z calcd for (C18H14N3O4) 336.0984. Found 336.0998.
2-(3-Benzyl-2,4-dioxoimidazolidin-1-yl)-5-nitroisoindoline-1,3-dione (6b)
Following the general procedure C, 6b was obtained as a white solid (103 mg, 27% yield). 1H NMR (500 MHz, DMSO-d6) δ = 4.49 (s, 2H), 4.75 (s, 2H), 7.34–7.38 (m, 3H), 7.40–7.44 (m, 2H), 8.31 (d, 1H, J = 7.9 Hz), 8.69 (s, 1H), and 8.76 (d, 2H, J = 7.9 Hz) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 43.0, 52.5, 119.9, 126.6, 128.4, 128.7, 129.6, 131.5, 131.6, 134.8, 136.6, 152.9, 156.9, 163.4, 163.7, and 168.8 ppm. HRMS (ESI) [M + H]+: m/z calcd for (C18H13N4O6) 381.0835. Found 381.0836.
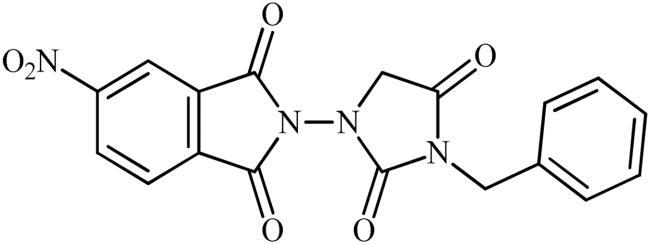
2-(3-(4-Nitrobenzyl)-2,4-dioxoimidazolidin-1-yl)isoindoline-1,3-dione (6c)
Following the general procedure C, 6c was obtained as a yellow solid (61 mg, 16% yield). 1H NMR (500 MHz, DMSO-d6) δ = 4.51 (s, 2H), 4.90 (s, 2H), 7.64 (d, 2H, J = 4.7 Hz), 7.99–8.08 (m, 4H), and 8.29 (d, 2H, J = 4.7 Hz) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 42.4, 52.8, 124.8, 125.0, 129.6, 130.2, 136.6, 144.2, 148.0, 156.9, 165.1, and 169.0 ppm. HRMS (ESI) [M + H]+: m/z calcd for (C18H13N4O6) 381.0835. Found 381.0816.
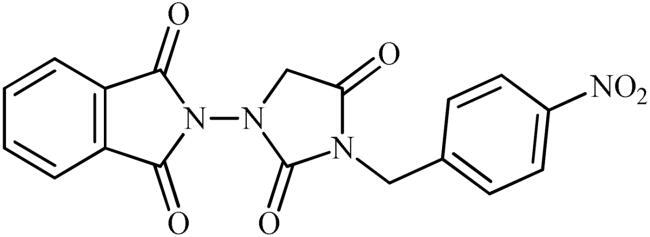
5-Nitro-2-(3-(4-nitrobenzyl)-2,4-dioxoimidazolidin-1-yl)isoindoline-1,3-dione (6d)
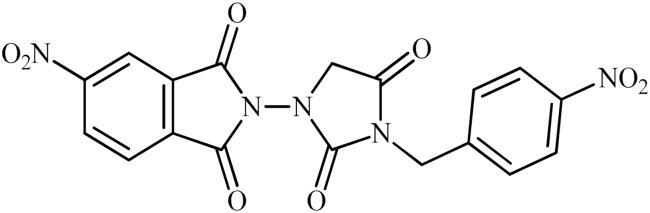
Following the general procedure C, 6d was obtained as a white solid (178 mg, 42% yield). 1H NMR (500 MHz, DMSO-d6) δ = 4.52 (s, 2H), 4.90 (s, 2H), 7.64 (d, 2H, J = 7.7 Hz), 8.29 (d, 2H, J = 7.7 Hz), 8.31 (d, 1H, J = 7.5 Hz), 8.69 (s, 1H), and 8.76 (d, 1H, J = 7.5 Hz) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 42.5, 52.6, 119,9, 124.8, 126.6, 129.6, 131.5, 131.6, 134.8, 144.1, 148.0, 152.9, 156.7, 163.4, 163.6, and 168.9 ppm. HRMS (ESI) [M + H]+: m/z calcd for (C18H12N5O8) 426.0686. Found 426.0691.
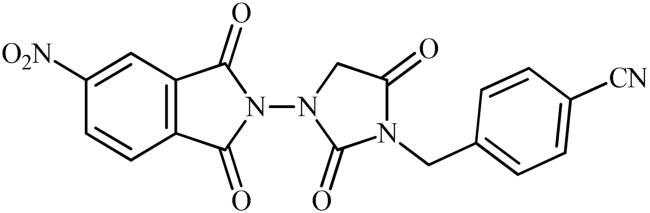
4-((3-(1,3-Dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-yl)methyl)benzonitrile (6e)
Following the general procedure C, 6e was obtained as a white solid (68 mg, 19% yield). 1H NMR (500 MHz, DMSO-d6) δ = 4.50 (s, 2H), 4.84 (s, 2H), 7.55 (d, 2H, J = 7.3 Hz), 7.91 (d, 2H, J = 7.3 Hz), and 7.99–8.08 (m, 4H) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 42.6, 52.7, 111.5, 119.5, 125.0, 129.1, 130.2, 133.6, 136.5, 142.2, 156.9, 165.1, and 169.0 ppm. HRMS (ESI) [M + H]+: m/z calcd for (C19H13N4O4) 361.0937. Found 361.0935.

4-((3-(5-Nitro-1,3-dioxoisoindolin-2-yl)-2,5-dioxoimidazolidin-1-yl)methyl)benzonitrile (6f)
Following the general procedure C, 6f was obtained as a white solid (259 mg, 64% yield). 1H NMR (500 MHz, DMSO-d6) δ = 4.52 (s, 2H), 4.85 (s, 2H), 7.55 (d, 2H, J = 7.3 Hz), 7.92 (d, 2H, J = 7.3 Hz), 8.31 (d, 2H, J = 7.6 Hz), 8.69 (s, 1H), and 8.76 (d, 2H, J = 7.6 Hz) ppm. 13C NMR (125 MHz, DMSO-d6) δ = 42.7, 52.6, 111.6, 119.6, 119.9, 126.6, 129.2, 131.5, 131.6, 133.6, 134.9, 142.2, 152.9, 156.8, 163.4, 163.7, and 168.9 ppm. HRMS (ESI) [M + H]+: m/z calcd for (C19H12N5O6) 406.0788. Found 406.0785.
CA inhibitory assay
An applied photophysics stopped-flow instrument was employed to determine the CA catalysed CO2 hydration activity,Citation16 as described earlier by our groupCitation17,Citation18. The tested CA enzymes were in-house produced recombinant proteins, that their preparation procedures have previously been reportedCitation18,Citation19. Briefly, cDNAs were expressed in Escherichia coli strain BL21 (DE3) from the plasmids pACA/hCA I and pACA/hCA II (for hCA I and II) and of the catalytic domains of hCA IX and XII.Citation14 The constructs were inserted in the pCAL-n-FLAG vector and then cloned and expressed in Escherichia coli strain BL21-GOLD(DE3). The bacterial cells were lysed and homogenated in a buffered solution (pH 8) of 4 M urea and 2% Triton X-100. The homogenates thus obtained were centrifuged (11 000 × g) for removing soluble and membrane associated proteins and cellular debris. The pellets were washed by repeated homogenisation and centrifugation in water, in order to remove the remaining urea and Triton X-100. Enzymes were thereafter purified by column chromatography on sulphonamide based columns.Citation4 The amount of proteins was determined by spectrophotometric measurements and their activities by stopped-flow measurements, with CO2 as substrate.Citation16
Docking studies
The crystal structures CA II (PDB 3K34)Citation18, CA VI (alphafold model) Citation20 CA VII (PDB 6H38) Citation21, and CA IX (PDB 5FL4)Citation22 were downloaded by Protein Data Bank (RCSB.org)Citation23 and prepared using the Protein Preparation module implemented in Maestro Schrödinger suite,Citation24 assigning bond orders, adding hydrogens, deleting water molecules, and optimising H-bonding networks. Finally, energy minimisation with a root mean square deviation (RMSD) value of 0.30 was applied using an optimised potential for liquid simulation (OPLS4) force field.Citation25 The 3D ligand structures were prepared by MaestroCitation23b and evaluated for their ionisation states at pH 7.3 ± 1.0 with EpikCitation23c. The conjugate gradient method in MacromodelCitation24 was used for energy minimisation (maximum iteration number: 2500; convergence criterion: 0.05 kcal/mol/Å2). Grids for docking were centred in the centroid of the complexed ligand. Docking studies were carried out with the program GlideCitation24 using the standard precision (SP) mode. Figures were generated with Maestro and ChimeraCitation24,Citation26.
Conclusions
We have synthesised a small series of hitherto unknown phthalimide–hydantoin hybrids and screened them against five human CA isoforms: the cytosolic isoforms hCA I, II, and VII as well as the unique secreted isoform hCA VI and the trans-membrane tumour-associated isoform hCA IX. All compounds showed potent inhibitory activity against hCA II, VI, VII, and IX whereas they did not display any inhibitory activity towards ubiquitous cytosolic isoform hCA I. Among them, ionic compounds 3a and 3b exhibited superior inhibitory activity, followed by derivative 5, and their binding mode was proposed by in silico studies. The SAR indicated that blocking the N3-position of the hydantoin core resulted in a strong decrease in activity. Interestingly, hydantoin salt 3a (an NH-free hydantoin derivative) exhibited much better inhibitory against tumour-associated CA IX compared to all newly developed Furagin derivatives (KIs = 0.35–7.3 μM, average: 2.84 μM). Importantly, this compound also showed excellent selectivity towards CA IX over the dominant cytosolic isoforms CA I and II, far better than the reference drug, AAZ. Therefore, it may be considered as a promising starting point for the development of novel anti-cancer agents.
Author contributions
All authors contributed to the writing and revision of the manuscript. MA synthesised the compounds. MA and AB tested the in vitro enzyme activity. AB and PG performed the computational work. CTS and RZ supervised the entire research and were responsible for the funding of the research.
Disclosure statement
CT Supuran is Editor-in-Chief of the Journal of Enzyme Inhibition and Medicinal Chemistry. He was not involved in the assessment, peer review, or decision-making process of this paper. The authors have no relevant affiliations of financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Data availability statement
Additional data may be requested from the authors.
Additional information
Funding
References
- Serbian I, Schwarzenberger P, Loesche A, Hoenke S, Al-Harrasi A, Csuk R. Ureido benzenesulfonamides as efficient inhibitors of carbonic anhydrase II. Bioorg Chem. 2019;91:103123.
- Hassan MI, Shajee B, Waheed A, Ahmad F, Sly WS. Structure, function and applications of carbonic anhydrase isozymes. Bioorg Med Chem. 2013;21(6):1570–1582.
- Aspatwar A, Tolvanen MEE, Barker H, Syrjänen L, Valanne S, Purmonen S, Waheed A, Sly WS, Parkkila S. Carbonic anhydrases in metazoan model organisms: molecules, mechanisms, and physiology. Physiol Rev. 2022;102(3):1327–1383.
- Mishra CB, Tiwari M, Supuran CT. Progress in the development of human carbonic anhydrase inhibitors and their pharmacological applications: where are we today? Med Res Rev. 2020;40(6):2485–2565.
- Barve IJ, Dalvi PB, Thikekar TU, Chanda K, Liu YL, Fang CP, Liu CC, Sun CM. Design, synthesis and diversification of natural product-inspired hydantoin-fused tetrahydroazepino indoles. RSC Adv. 2015;5(89):73169–73179.
- Hassanin MA, Mustafa M, Abourehab MA, Hassan HA, Aly OM, Beshr EA. Design and synthesis of new hydantoin acetanilide derivatives as anti-NSCLC targeting EGFRL858R/T790M mutations. Pharmaceuticals. 2022;15(7):857.
- Konnert L, Lamaty F, Martinez J, Colacino E. Recent advances in the synthesis of hydantoins: the state of the art of a valuable scaffold. Chem Rev. 2017;117(23):13757–13809.
- Sharma U, Kumar P, Kumar N, Singh B. Recent advances in the chemistry of phthalimide analogues and their therapeutic potential. Mini Rev Med Chem. 2010;10(8):678–704.
- Das S. Beyond conventional construction of the phthalimide core: a review. New J Chem. 2021;45(44):20519–20536.
- Patel VC, Samresh PR, Dipayan T, Nidhi C. Investigation into anti-Alzheimer activity of newly developed phthalimide derivatives in experimental animals. Pharm Chem J. 2023;57(1):60–69.
- Upadhyay N, Tilekar K, Loiodice F, Anisimova NY, Spirina TS, Sokolova DV, Smirnova GB, Choe J-Y, Meyer-Almes F-J, Pokrovsky VS, et al. Pharmacophore hybridization approach to discover novel pyrazoline-based hydantoin analogs with anti-tumor efficacy. Bioorg Chem. 2021;107:104527.
- Tabatabaei Rafiei LS, Asadi M, Hosseini FS, Amanlou A, Biglar M, Amanlou M. Synthesis and evaluation of anti-epileptic properties of new phthalimide-4,5-dihydrothiazole-amide derivatives. Polycycl Aromat Compd. 2022;42(4):1271–1281.
- Prasad H, Ananda A, Mukarambi A, Gaonkar N, Sumathi S, Spoorthy H, Mallu P. Design, synthesis, and anti-bacterial activities of piperazine based phthalimide derivatives against superbug-methicillin-resistant Staphylococcus aureus. Curr Chem Lett. 2023;12(1):65–78.
- Pustenko A, Nocentini A, Gratteri P, Bonardi A, Vozny I, Žalubovskis R, Supuran CT. The antibiotic furagin and its derivatives are isoform-selective human carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020;35(1):1011–1020.
- Yiğit M, Demir Y, Barut Celepci D, Taskin-Tok T, Arınç A, Yiğit B, Aygün M, Özdemir İ, Gülçin İ. Phthalimide-tethered imidazolium salts: synthesis, characterization, enzyme inhibitory properties, and in silico studies. Arch Pharm. 2022;355(12):e2200348.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem. 1971;246(8):2561–2573.
- Nocentini A, Angeli A, Carta F, Winum JY, Zalubovskis R, Carradori S, Capasso C, Donald WA, Supuran CT. Reconsidering anion inhibitors in the general context of drug design studies of modulators of activity of the classical enzyme carbonic anhydrase. J Enzyme Inhib Med Chem. 2021;36(1):561–580.
- Supuran CT. Carbon-versus sulphur-based zinc binding groups for carbonic anhydrase inhibitors? J Enzyme Inhib Med Chem. 2018;33(1):485–495.
- Martin DP, Cohen SM. Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors. Chem Commun. 2012;48(43):5259–5261.
- Behnke CA, Le Trong I, Godden JW, Merritt EA, Teller DC, Bajorath J, Stenkamp RE. Atomic resolution studies of carbonic anhydrase II. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 5):616–627.
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50(D1):D439–D444.
- Leitans J, Kazaks A, Balode A, Ivanova J, Zalubovskis R, Supuran CT, Tars K. Efficient expression and crystallization system of cancer-associated carbonic anhydrase isoform IX. J Med Chem. 2015;58(22):9004–9009.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242.
- Schrödinger suite release 2022-4. New York (NY): Schrödinger, LLC; 2022 ((a) Prime, v.5.5; (b) Maestro v.13.2; (c) Epik, v.6.0; (d) Macromodel, v.13.6; (e) Glide, v.9.5; (f) Impact, v.9.5).
- Lu C, Wu C, Ghoreishi D, Chen W, Wang L, Damm W, Ross GA, Dahlgren MK, Russell E, Von Bargen CD, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021;17(7):4291–4300.
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612.