
ABSTRACT
Introduction
During the clinical development of a vaccine, study participants are monitored for the occurrence of adverse events (AEs) over a defined period post-vaccination to assess the safety of prophylactic vaccines. Among the safety data collected, a standard practice in prophylactic vaccine clinical trials involves collecting reactogenicity data through daily AE solicitation of pre-defined sets of symptoms (i.e. solicited AEs).
Areas covered
This paper aims to propose recommendations to improve and harmonize the collection of active AE solicitation in prophylactic vaccine clinical trials.
Expert opinion
We recommend using limited lists of solicited AEs adapted to the vaccine technology and target population. While the US Food and Drug Administration toxicity grading scale is commonly used in adolescents/adults, harmonizing grading criteria in infants/children would facilitate the comparison of vaccines’ safety profiles. Solicited systemic AEs should not systematically be considered causally related to vaccination. Collection of solicited AEs should occur in cohorts of a maximum of 1,000 vaccinated participants, as larger cohort sizes do not improve substantially the precision of AE incidence. The incidence of daily solicited AEs should be compared with a control group for improved interpretations of their clinical relevance. These suggestions would improve the characterization of safety profiles of vaccines.
1. Introduction
Prophylactic vaccines are typically administered to healthy individuals to prevent a disease. While vaccination brings many benefits to public health, its benefits for an individual depend on their exposure to a specific disease. Therefore, from a public health perspective, the tolerance for risks following vaccination is very low. To assess this risk during the clinical development of a vaccine, reactogenicity and safety are actively monitored following vaccination of trial participants.
Reactogenicity refers to the physical manifestations that are expected to occur following an inflammatory response to vaccination [Citation1]. A standard practice for the evaluation of reactogenicity in prophylactic vaccine clinical trials involves questioning study participants daily about the occurrence of pre-defined sets of symptoms (i.e. collection of solicited adverse events [AEs]) over a defined period post-vaccination. The period of collection and types of symptoms depend on the vaccine and study objectives.
Other symptoms (e.g. unsolicited AEs, AEs of special interest [AESIs; i.e. significant AEs that are judged to be of special interest because of clinical importance, known or suspected class effects, or based on nonclinical signals]), are also collected in all clinical vaccine trials. These events are usually recorded by the investigator at scheduled contacts or visits. In this paper, this mode of collection is referred to as passive collection [Citation2].
The added process of active solicitation of reactogenicity data in the evaluation of prophylactic vaccines contrasts with the passive collection of safety data through reporting of unsolicited AEs in pre-defined periods, which is the safety standard in clinical trials evaluating other types of medicinal products, typically used to treat ill patients.
The recent Center for Biologics Evaluation and Research (CBER) Guidance for Industry on the Emergency Use Authorization for Vaccines to Prevent Coronavirus disease 2019 (COVID-19) [Citation3] requires phase III studies to collect local and systemic solicited AEs in an adequate number of participants to characterize the reactogenicity profile in each age group participating in the trial.
Nevertheless, solicited AEs are usually reported at higher frequencies than if participants report them as unsolicited symptoms. In fact, the active solicitation of AEs influences the incidence of events and leads to reporting events otherwise perceived as normal. This may result in biased conclusions, especially when incidences in the vaccine group are presented separately from incidences in the control group [Citation1].
This paper aims to provide specific recommendations and advocate for regulatory guidelines on the solicitation of AEs linked to vaccine reactogenicity in clinical trials investigating prophylactic vaccines.
2. Limitations of soliciting AEs
Soliciting AEs is useful to make informed decisions on the selection of better tolerated vaccines, e.g. in phase I/II trials evaluating reactogenicity profiles across different vaccines and/or populations, in comparison with standard of care, in co-administration studies (with a single vaccine as control), or age de-escalation trials.
However, soliciting AEs also leads to a focused collection of events well known to be associated with vaccination and that may be less clinically relevant since they are mostly mild-to-moderate in severity [Citation1]. Additionally, in phase III clinical trials, more clinically relevant AEs can be collected at dedicated visits/contacts through a passive collection process.
2.1. Recording solicited AEs favors noise collection
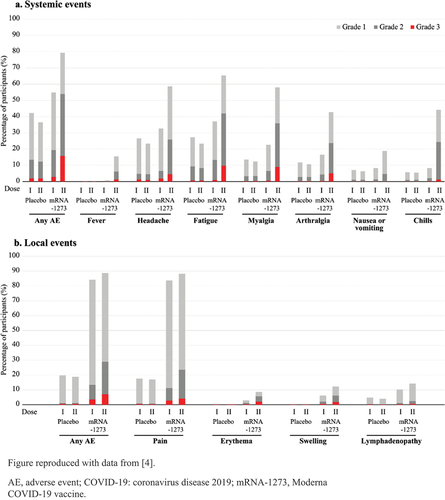
A recent Moderna COVID-19 vaccine (Spikevax, Moderna Biotech Spain S.L.) phase III, 2-dose, randomized clinical trial (NCT04470427) with more than 30,000 participants, showed that 36.5%–42.2% of placebo (saline) recipients reported solicited systemic AEs after each vaccination [Citation4] (). This suggests that many reported solicited AEs may not be related directly to the vaccine but could be due to other factors; such as the vaccination process and its potential nocebo effect (especially in a pandemic), underlying medical conditions, co-incidental illness, or the method of collection through active solicitation. Similar findings were observed in phase III studies with other COVID-19 vaccines [Citation5]. The crude rates of solicited AEs can therefore give an overly negative picture of the reactogenicity profile, and they need to be understood and interpreted in the context of the underlying incidence of those symptoms.
Figure 1. Incidences and severity of solicited systemic or local AEs within 7 days following each of the 2 doses of either placebo or the Moderna COVID-19 vaccine.
2.2. Soliciting AEs focuses on clinically insignificant events rather than clinically relevant AEs
The acceptability of the reactogenicity profile may vary across vaccine products depending on the target population, indication, and expected benefits (e.g. children vs adults; mild vs severe symptoms/outcome; prophylactic vs therapeutic vaccine). Additionally, the acceptance of reactogenicity is different for an epidemic disease (e.g. seasonal influenza) versus a pandemic (e.g. COVID-19) or outbreak (e.g. Ebola) situation. However, reactogenicity usually represents common minor reactions following vaccination, i.e. they have little or no impact on daily activities, and infrequently require treatment or medical attention (mild-to-moderate in intensity), and their active solicitation leads participants to focus on signs and symptoms they would likely not report if their attention was not drawn to them [Citation2,Citation6] (see examples below). The effect of such heightened reporting practices could lead to undue concern or an expectation by individuals that they will experience side-effects after vaccination. On the other hand, this information could ‘normalize’ these AE experiences for the end-user reading the package insert and wondering what to expect.
Example 1: About 80% of the 30,000 Moderna COVID-19 vaccine recipients in the phase III study mentioned in Section 2.1 reported solicited local AEs within 7 days after each dose; most local AEs were transient and of mild-to-moderate severity [Citation4] ().
Example 2: A Pfizer COVID-19 vaccine (Comirnaty, BioNTech Manufacturing GmbH) phase II/III study (NCT04368728) [Citation7] enrolled a reactogenicity cohort where solicited AEs were collected over 7 days following each dose, and a separate safety cohort where only unsolicited AEs were collected. In the reactogenicity cohort (N=8,183), around 66%–83% of the vaccine recipients reported pain at the injection site vs 8%–14% of the placebo recipients (depending on age and dose). Among the 43,252 total enrolled participants, 11.3% of the vaccine recipients reported unsolicited ‘pain at the injection site’ vs 1.5% of placebo recipients, up to 1 month post-vaccination [Citation8]. Despite the longer follow-up post-vaccination, the rate of this unsolicited AE was substantially lower than the rate of solicited ‘pain at the injection site’ within 7 days post-vaccination. Similar findings were observed for other solicited AEs reflecting the overreporting of minor, primarily mild-to-moderate AEs when solicited: for instance, the incidence of solicited ‘headache’ ranged from 25%–52% vs 14%–34% in the vaccine and placebo groups, respectively (1.4-fold increase), while 5.2% vs 1.6% of the total enrolled participants reported unsolicited ‘headache’ (3-fold increase) [Citation7]. This possibly means that soliciting AEs from participants leads to a higher reporting of common and mild events which might not have been reported without daily solicitation, as they are typically considered ‘acceptable’ by study participants. In addition, a control group allows putting incidences in perspective [Citation8].
Example 3: In a recombinant zoster vaccine (RZV; Shingrix, GSK) placebo-controlled study (Zoster-006 study, NCT01165177) [Citation9], solicited AEs were collected in a cohort of 8,747 adults aged ≥50 years. While there was a clear difference in the reporting of any systemic solicited AEs in the vaccine group vs control group, it was more pronounced for grade 3 AEs. However, most of these systemic grade 3 AEs did not lead to medical consultation, since solicited AEs leading to medical attended visits were infrequent (<1%) and comparable in the vaccine and control groups. This suggests that most of these systemic solicited AEs, even when the participants graded them as severe, could be self-managed ().
Table 1. Incidences of solicited systemic adverse events within 7 days following vaccination with the recombinant zoster vaccine (RZV) or placebo.
2.3. Any increase in observed risk may be detected with passive reporting of unsolicited AEs
Publications comparing the rates of unsolicited AEs vs solicited AEs in three randomized clinical trials concluded that active solicitation of AEs does not lead to a better detection of potential adverse reactions (ARs) to the vaccine [Citation2,Citation6]. This has also been illustrated in the Pfizer COVID-19 vaccine study [Citation6] (presented in Section 2.2).
The Zoster-006 study [Citation9] (presented in Section 2.2) is another illustration of this observation. The study involved active and passive surveillance cohorts for reactogenicity. presents a summary of the same AEs from both cohorts over the same window post-vaccination. Comparison of incidences in vaccine and placebo groups shows that both passive and active surveillance allows to identify potential ARs and their severity [Citation2].
Table 2. Incidences of solicited and unsolicited systemic adverse events within 7 days of the recombinant zoster vaccine (RZV) or placebo.
2.4. Collection of solicited AEs is not standardized
The pre-defined list of the solicited AEs collected in phase III studies and the sample size of the reactogenicity cohort vary across vaccines and age groups but also among different vaccine companies. For instance, solicited AEs were collected for more than 30,000 participants in the Moderna phase III COVID-19 vaccine clinical trials [Citation4], while in other phase III COVID-19 vaccine clinical trials, solicited AEs were collected in smaller reactogenicity cohorts [Citation10,Citation11]. Additionally, while solicited AEs collected for injectable vaccines generally include local signs and symptoms, such as pain, erythema/redness, and swelling at the injection site, solicited systemic AEs vary across vaccine trials. For example, although chills were not an AE solicited for the Janssen COVID-19 vaccine (Jcovden, Janssen Pharmaceuticals), it appears as common AR in its prescribing information (PI), affecting 1%–10% of participants [Citation12]. The same symptom was solicited for the AstraZeneca COVID-19 vaccine (Vaxzevria, AstraZeneca) and consequently appears in its PI as a very common AR, affecting more than 10% of participants, even though post-marketing surveillance data for both vaccines indicate that it is unlikely that one vaccine would induce chills more frequently than the other [Citation13].
3. Recommendations for the collection of solicited AEs in vaccine clinical trials
3.1. A standardized list of AEs is preferred to document vaccine reactogenicity and events of special interest
In clinical trials, a limited list of local and systemic solicited AE terms is generally collected to characterize the reactogenicity profile in the protocol-defined population. As trial participants usually self-record solicited AEs daily, they should be able to easily recognize and assess them without in-depth medical knowledge. For instance, trial participants can conveniently track their body temperature to assess and grade potential fever with a thermometer, or their injection site erythema and injection site swelling using a ruler. In contrast, ‘lymphadenopathy’ requires ascertainment by trained medical staff and would not be appropriate to consider as a solicited AE.
Furthermore, the United States Food and Drug Administration (FDA)’s guidance on toxicity grading for vaccine clinical trials recommends restricting the list of solicited AEs to signs or symptoms for which causality can be reasonably attributed to vaccination. The standard list should be also kept short, and consistent within the vaccine program [Citation14]. The choice of solicited AEs to include in the list may be based on causality assessment factors including:
biological plausibility of relatedness, considering the vaccine technology (e.g. some live-attenuated vaccines may be associated with modified/milder manifestations of natural infection, such as rashes), the population concerned (e.g. age group), and the route of administration;
whether the AE is known to be related to vaccine(s) or is a recognized class effect;
the list of solicited systemic AEs historically collected within the vaccine program, provided these are based on relevant individual symptoms (e.g. nausea, vomiting, diarrhea) and not group of symptoms, like ‘gastrointestinal symptoms’;
any correlation between rates of any AE and dose of antigenic components or adjuvant, or changes in rates of any AE with sequential doses;
common labelled events (if the vaccine or its comparators are already marketed with the event identified as an AR) [Citation15].
For injectable vaccines, solicited local AEs to be reported usually include, as a minimum, pain, erythema/redness and swelling at the injection site in all age groups. For most vaccines, fever, assessed by measuring body temperature, is typically collected as solicited systemic AE, as it has been commonly reported for all vaccines and all age groups, and it is associated with the induction of inflammatory and innate immune responses. Furthermore, collection of fever in all vaccine programs brings added value and quality to the reactogenicity measure, as temperature is objectively measurable. Other solicited systemic AEs are considered according to the vaccine candidate specificities: the list of AEs historically collected within the vaccine program, labelled ARs if the vaccine is marketed, vaccine technology, study population (e.g. different age groups, route of administration, and/or a recognized class effect).
The evolving knowledge on vaccines’ mechanism of action, the increasing experience in tolerability profiles, and the vaccine platform/technology are key to adapting the solicited AEs list by class of vaccines (e.g. arthralgia for adjuvanted vaccines in adults) or route of administration (e.g. nausea/vomiting and diarrhea for vaccines administered orally, and respiratory tract symptoms for intranasal vaccines).
Active surveillance of AESIs is also recommended for rare events that reflect vaccine-specific immune mechanisms (e.g. type II hypersensitivity with tuberculosis vaccines, or large swelling reactions in repeated diphtheria-containing vaccines). Standard collection tools are available that facilitate AESI characterization, and interpretation. For example, AESI lists and case definitions with associated publications can be accessed from the Brighton collaboration [Citation16,Citation17].
3.2. Use of eDiaries allows fast/remote recording of solicited AEs
In most vaccine clinical trials, the vaccine recipients, caregivers, or study staff daily record solicited AEs using paper or electronic diary cards (eDCs). Diary cards (DCs) completion level should be checked during a face-to-face or telephone contact between the trial participant/caregiver and study staff, who may provide further instructions to improve compliance and data quality.
The use of electronic methods such as eDCs allows fast, remote collection of solicited AEs [Citation18], compliance checks and follow-up, and it is particularly useful in a pandemic context where visits to physicians are not recommended except for medical emergencies. However, the use of eDCs may not be feasible in resource-limited settings or in populations with low levels of technical expertise.
AE recording has to be Attributable, Legible, Contemporaneous, Original, Accurate and Complete (ALCOAC) [Citation19], preventing post-hoc recording and correction. To mitigate the risk of missing potentially severe AEs if a vaccine recipient fails to timely or accurately complete the diary, the investigator should be able to complete or correct the evaluation of solicited AEs, either at the next planned visit or after a phone contact prompted by a missed daily recording alert. This should be allowed either using a dedicated electronic case report form (e.g. in a separate investigator assessment of reactogenicity) or using a data correction process and associated tools (e.g. correction form) ensuring traceability. However, to avoid potential recall bias, the investigator should ideally collect any missing solicited AE information as soon as possible and not beyond a pre-specified timeframe (e.g. 24-48 hours) after the solicited AE collection period.
3.3. A standard grading system for the solicited AEs is needed
The intensity of each symptom occurring after vaccination is graded (usually by the subject, or the investigator in pre-defined reassessment of the AE) using protocol-defined scales of intensity (toxicity grading scale). Severity grading scales differ among vaccine programs and between individual clinical trials, and exceptional adjustments to specific populations can be considered when appropriate. For instance, grade 3 (severe) vomiting can be assigned when any treatment is given for the vomiting, and in practice, ‘treatment’ can mean one of a variety of situations (which may be unrelated to standards of care), for example: antiemetic taken orally, antiemetic administered intravenously, or the need for hydration. A grade 3 episode can also be assigned according to the number of vomiting episodes in 24 hours, or if the patient has no significant food intake (not able to eat). However, within a vaccine program, all clinical trials should preferably use standard severity grading scales to allow comparisons and pooling of data from different studies. Uniform criteria for categorizing and grading (solicited) AEs can improve comparisons of safety data among populations within the same study and between different studies or vaccines.
3.3.1. FDA’s grading system for adults and adolescents is adopted
The FDA’s Guidance for Industry recommends a core list of solicited AEs and severity grading for healthy adults and adolescents [Citation14], which may be adapted for specific populations (e.g. participants with comorbidities). This grading system is now adopted by most vaccine manufacturers, except for the use of grade 4. Indeed, grade 4 reflects seriousness (i.e. hospitalization, life-threatening or important medical event) that is assessed separately by the investigators.
3.3.2. A harmonized pediatric AE grading system is needed
The grading systems used for infants/children differ greatly between vaccine manufacturers. In 2007, the Division of Microbiology and Infectious Diseases (DMID) developed a draft guidance on grading of AEs in infants and children, but it is no longer available online [Citation20]. The National Institute of Allergy and Infectious Diseases proposed an alternative grading system, where grading criteria have been modified and integrated in more recent toxicity scales such as the Division of AIDS Table for Grading the Severity of Adult and Pediatric AEs [Citation21], which proposes significantly different grading values as compared to the DMID (Table S1). The scale used in a recent Pfizer COVID-19 vaccine trial in children [Citation22] further illustrates the inconsistency in grading systems used across vaccine clinical trials: for swelling and redness, ≥0.5–2 cm was used for grade 1 and >7 cm for grade 3, while the DMID scale used <1 cm for grade 1 and >2.5 cm for grade 3 or 4 and the AIDS scale uses ≤2.5 cm for grade 1, and ≥50% surface area of the upper arm or thigh for grade 3.
Regulatory authorities should propose a validated list of pediatric solicited AEs for vaccine manufacturers, with specific grading criteria adapted to younger age groups, considering that symptoms are usually assessed and reported by parents or caretakers (via observation of the child rather than self-reporting as in adults or adolescents). For instance, scales to evaluate pain at injection site may be based on description of different reactions (resistance, withdrawal, crying) upon contact when mobilizing the limb in young children.
Furthermore, the core list of solicited systemic AEs to be collected and documented in pediatric studies is determined by the age range in the study. Typically, an age limit of 6 years (switching point from an observational-based collection to a self-reporting-based approach) is used for certain solicited systemic AEs such as irritability/fussiness, loss of appetite, and drowsiness. However, the age limit may be adjusted depending on vaccine-specific requirements.
Ideally, pediatric toxicity grading should be harmonized through a sole reference guidance, based on the example of the CBER’s guidance for adults and adolescents.
3.4. Follow-up periods to document reactogenicity need to be adapted for inactivated and live vaccines
Duration of the follow-up of reactogenicity should be adapted in vaccine clinical trials. For most live-attenuated (replicating) vaccines, solicited AEs are typically recorded daily for at least 4–7 days after each vaccine dose. The World Health Organization (WHO) recommends a recording period of 5–7 days [Citation23], while a recent Chinese guidance suggests 14 days [Citation24].
The duration of the recording period of solicited AEs depends on the biological mechanism and vaccine platform technology. As a precaution, it is usually longer in phase I/II than in phase III studies. Characteristics including the type of vaccine, vaccine composition, and its mechanism of action are also key to understand the onset timeframes of AEs associated with the vaccine administered. Common systemic AEs (e.g. systemic inflammatory reactions such as fever, irritability, nausea, vomiting, or myalgia) generally occur within 24–48 hours following the administration of inactivated vaccines, while for live vaccines such reactions can arise 14–21 days following administration.
We recommend to record solicited AEs daily for 7 days after each dose for non-live/non-replicating vaccines. Longer periods (i.e. 10–14 days) are appropriate for live-attenuated vaccines, as their replication kinetics tends to peak a few days post-vaccination (e.g. fever peaks 4–7 days after the first dose of measles-containing vaccines).
3.5. Causality assessment of AEs should be performed
According to the WHO’s Global manual on surveillance of AEs following immunization [Citation25], investigators should assess all solicited and unsolicited AEs occurring in clinical trials and judge whether they are causally related to vaccination. Although for regulatory reporting purposes in a clinical trial setting, the FDA [Citation26,Citation27] and the European Medicines Agency [Citation28,Citation29] require investigators and sponsors to perform an assessment of causality only for serious AEs, some sponsors require investigators’ causality assessment also for non-serious AEs (including solicited and unsolicited non-serious AEs). For example, while a reasonable possibility of causal relationship for injection site reactions can be assumed for injectable vaccines, recall bias may occur when participants report any AE to the investigator at a later stage, because they may not remember experiences accurately or omit details. Therefore, the investigator’s causality assessment for non-serious AEs, including solicited AEs, should be pursued to increase the quality of the documentation of the AE, because solicitation leads to an increased frequency of reporting events occurring in normal life (refer to Section 2). A suspected causal association must be supported by factors that can be based either on aggregate data or individual participant data, including:
increased AE incidence, as compared to a randomized control group or to a historical placebo group;
dose-response, when multiple doses have been assessed;
pharmacological plausibility: the event is coherent according to the known facts in the natural history/biology of the disease (i.e. not related to the underlying disease). It should be consistent with the pharmacology of the vaccine and known class effects/caused by related vaccines;
temporal relationship: a plausible temporal relationship is observed between vaccine exposure and the timing of the event;
specificity: the absence of alternative causes or confounding factors such as underlying medical conditions, concomitant therapy, and other risk factors provides more evidence that the event is causally related. For AEs typically associated with product/therapy/vaccination (e.g. liver failure, rhabdomyolysis, anaphylaxis, intussusception), even a very low number of cases may provide sufficient evidence of a causal relationship between the event and the product;
consistency: an increased frequency is systematically observed across identified subgroups after exposure to the vaccine such as age, gender, country. Consistency according to clinically relevant attributes such as seriousness, severity/toxicity grade or outcome provides further confidence on causality [Citation15].
In summary, the causality assessment for non-serious AEs, including solicited non-serious AEs, would ensure the quality of the documentation of the observation of suspected relatedness.
3.6. Safety of co-administered vaccines should be compared to the estimated safety of cumulative vaccinations
When two or more vaccines are administered at the same time, the reactogenicity profile may be influenced by one of the vaccines; therefore, a common list of solicited AEs should be used to compare with similar criteria the reactogenicity data of all vaccines. The DC should also ensure that separate data are recorded for each vaccine’s injection site.
When comparing the incidence of solicited AEs after sequential vs co-administration, it is not recommended to present the percentage of participants with solicited AEs throughout the clinical trial because of the differences in the duration of follow-up between groups. Additionally, such presentation does not reflect the burden associated with repeated occurrence of solicited AEs after each single dose. Similarly, the proportion of vaccination visits resulting in the recording of solicited AEs in each group will not allow a fair comparison due to the difference in the number of vaccination visits between groups. To determine whether co-administration enhances the occurrence of a solicited AE, the incidence expected with the co-administration of vaccines can be derived from the incidences observed after each single administration, assuming there is no interaction between the vaccines. In , data are presented for co-administration of two vaccines at the same visit (investigational group) and for each of the two single vaccines. The incidence of a solicited AE after the co-administration of vaccines should be compared with its expected incidence estimated from the AE incidences after each single vaccine administration. The incidence of both fatigue and shivering was higher after co-administration than after administration of each single vaccine; however, expected incidence estimated from the AE incidences after each single vaccine administration showed no clinically relevant increase in the incidence of fatigue after co-administration, while the risk of shivering was more substantially increased. The incidence of solicited AEs occurring repeatedly after the two single vaccines is also useful as it quantifies the risk of repeated occurrences and validates the independence assumption (if independence is true, the observed incidence of a solicited AE occurring after both administrations should be close to the multiplication of incidences from separate administrations). The choice of co-administration vs sequential vaccinations should be further assessed based on the benefit associated with the reduced number of vaccination visits and the reduced duration of the period in which an AE would occur.
Table 3. Estimating adverse event (AE) incidence after co-administration vs incidence after single administration of each vaccine, in the absence of enhancement due to co-administration.
3.7. A cohort size of 1,000 exposed participants is recommended for the assessment of reactogenicity in phase III vaccine clinical trials
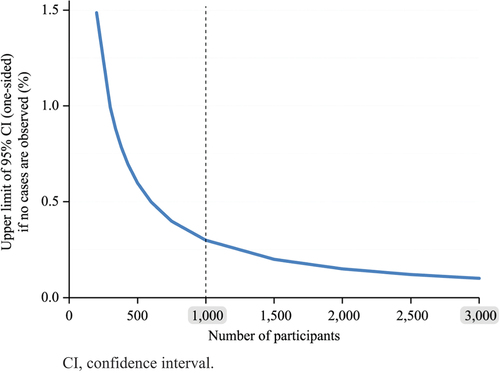
The sample size needed to show the true rate of an AE increases exponentially with the precision of this rate. For rare events, shows that the observation of 0 reactions in a database would be associated with a 95% probability that the true rate of the AE is <0.3% with 1,000 vaccinated participants, as compared to <0.1% with 3,000 participants. This is a relative gain in precision of 3-fold for a 3-fold increase of the sample size. Reaching a precision <0.033% (3-fold further increase) would require a sample size of 10,000 participants (i.e. a 3.3-fold increase). The use of 3,000 exposed participants is typically accepted by authorities for passive surveillance of infrequent events in phase III studies and allows detecting the occurrence of an event associated to 0.1% incidence.
Figure 2. Upper limit of the one-sided 95% CI of the true incidence of an event according to the number of participants in the absence of reported cases.
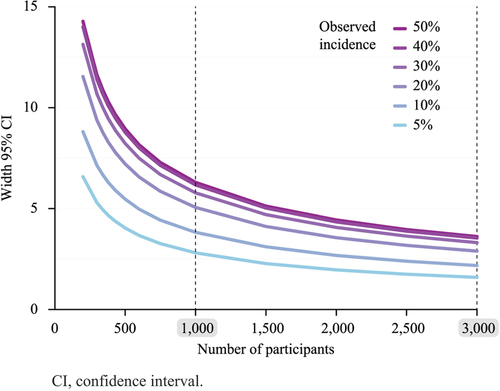
For active surveillance of more frequent AEs, there is no substantial increase in precision of AE incidences when a clinical trial sample size exceeds 1,000 vaccinated participants in the investigational group. In fact, shows that a reactogenicity cohort of 1,000 active vaccinated individuals with an observed AE incidence of 5% and 50% displays a two-sided 95% CI width for the true incidence of the event of 2.8% and 6.3%, respectively. Enlarging the cohort has a minimal effect on the gain in precision: when the cohort size increases from 1,000–3,000 participants, for observed event incidences of 5% or 50%, the width becomes 1.66% and 2.76%, which corresponds to a 1.8 and 2.3-fold gain in precision, respectively. Accordingly, a reactogenicity cohort of 1,000 participants/arm is deemed appropriate to estimate the true incidence of solicited AEs.
Figure 3. Width of the two-sided 95% confidence interval of the true incidence of an event according to the number of participants and for different observed incidences.
4. Recommendations for consistent presentation of solicited AE incidences in the labels
4.1. Incidences of solicited AEs should be compared head-to-head with those of a control group
Publications highlight the importance of designing studies with an appropriate control group to untangle the proportion of signs or symptoms caused by vaccination [Citation1]. Using such study design allows putting safety incidences in perspective.
Reactogenicity information presented in vaccine PIs should also be compared with a control group. However, PI documents are not presented in a uniform way across countries. While in the United States PIs display reactogenicity AE incidences in the investigational group together with the control group, in Europe, only incidences in the investigational group are included. Cultural, seasonal, or geographical context of clinical studies lead to variability of observed incidences of solicited AEs therefore, without comparison to a control group, practitioners cannot be well informed on the true reactogenicity induced by a vaccine.
Vaccine labels should include incidences of AEs solicited in the vaccine group presented head-to-head with those observed either in a control group, ideally a placebo, or in cohorts where AEs are not solicited.
4.2. Presentation of incidence of solicited AE derived from a passive surveillance cohort allows putting incidences in perspective
The complementary presentation of frequencies derived from passive surveillance allows putting in perspective the overreporting of solicited AEs with the incidences of AEs occurring in normal life.
Since unsolicited AEs are commonly reported by System Organ Class (SOC) and preferred terms (PTs), grouping AE terms by PTs may be needed for a more accurate reflection of the frequency of conditions and strengthen the evidence of causal association. To allow for the presentation of more clinically meaningful expected frequencies in vaccine labels, terms can be grouped using the MedDRA High-Level Term (HLT). They gather PTs with similar medical context and facilitate event evaluation, while limiting over-representation of similar or synonymous PTs. For instance, irritability and irritability post-vaccinal should be grouped. Likewise, PT ‘redness’ could be represented together with: ‘administration or injection or vaccination site erythema’ and ‘administration, injection, or vaccination site erythema’. Terms can also be grouped using a Standardized MedDRA Query (SMQ), if a suitable one is available for the condition, or a customized MedDRA query. However, to date, standardized MedDRA queries exist only for unsolicited and not for reactogenicity AEs [Citation4]. In addition, harmonizing the grouping of PTs linked to reactogenicity would allow a better comparability of safety data across vaccines. This recommendation to group similar clinical terms is also applicable to unsolicited AEs from clinical trials and from post-marketing data as it allows clinically meaningful presentation while limiting over-representation of similar or synonymous PTs.
5. Conclusion
Vaccine safety is monitored through the close observation of AEs following vaccination. AEs can be daily solicited or collected as unsolicited. We provide recommendations to ensure solicited AEs are collected and reported consistently to improve the definition of safety objectives at protocol stages and the safety assessments in vaccine clinical trials. Steps to consider during study design, analysis and when updating the label with AE data are summarized in . As next steps, we call for specific regulatory guidelines to provide harmonization in the reporting of solicited AEs.
Table 4. Steps to consider during study design, analysis and when updating the label with AE data.
6. Expert opinion
This paper provides the authors’ recommendations to harmonize the collection of solicited AEs in prophylactic vaccine clinical trials:
A standard list of solicited AEs that can be easily understood by trial participants who self-record these AEs should be developed for use in vaccine clinical trials. We recommend tailoring the list to the target population, the vaccine technology/indication and the potentially causally associated AEs. In addition, the number of pre-defined solicited AEs should be limited to facilitate compliance.
The use of eDCs, when available, favors the centralized monitoring of daily solicited AEs. Additionally, the eDC technology allows to set up alerts that notify investigators when participants miss recording their daily entries. This facilitates compliance in recording. Regardless of the use of paper or electronic recording, investigator reviews should be planned as close as possible to the AE solicitation time, to complement the information, confirm the absence of a solicited AE over the designated period, and correct erroneous entries/missed reporting of a solicited AE.
In all studies, a standard toxicity/severity grading scale should be used to compare and pool safety data. The need of reference grading scales for pediatric clinical trials is highlighted.
For non-live/non-replicating vaccines, solicited AEs should be recorded daily within 4–7 days after dose administration. For live-attenuated/replicating vaccines, a longer period (e.g. 10–14 days) post-vaccination should be used to account for the peak in replication kinetics occurring several days after vaccination.
Solicited AEs should not be considered related to vaccination per se. Like any AE, relatedness of solicited AEs should be supported by factors including vaccine dose-response or pharmacological plausibility, based on knowledge of the disease or known class effects.
When investigating the effect of co-administration of multiple vaccines vs separate administration of vaccines, the authors propose to study the enhancement in incidence of systemic solicited AEs. We do not recommend comparing the incidence after co-administration of vaccines with the incidence after sequential administrations of vaccines, since the latter cumulates incidences from each separate vaccine. On the contrary, presenting the solicited AE incidence of repeated occurrence estimated from incidences after a single vaccine administration in each vaccine group allows assessing the risk-benefit of co-administration vs sequential vaccines.
A maximum of 1,000 vaccinated participants per treatment arm is adequate to collect solicited AEs, since larger study groups do not substantially improve precision in the recording of their incidence. In contrast, unsolicited AE recording in phase III studies requires larger groups of participants considering their lower occurrence.
Finally, requirements for PI presentation are currently not harmonized between marketing authorization bodies. The authors recommend that incidences of solicited AEs described in the PI should be presented head-to-head with incidences obtained in a control group (ideally a placebo) or in a cohort where recorded AEs are all unsolicited. The latter allows putting in perspective the overreporting of AEs occurring in normal life (e.g. headache).
We believe these suggestions would help categorizing the safety profile of vaccines.
Article highlights
Solicited adverse events (AEs) are reported at higher rates than unsolicited AEs
Vaccine programs should use limited and clinically relevant lists of solicited AEs
Severity grading criteria should be harmonized in pediatric vaccine trials
A sample of 1,000 participants is optimal to assess the incidence of solicited AEs
Vaccine product information should include reactogenicity data from the vaccine vs a control group (ideally a placebo)
Declaration of interests
B Cheuvart is an employee of GSK and holds shares in GSK. F Tavares-Da-Silva was an employee of the GSK at the time of the Zoster-006 study (NCT01165177) and is currently an employee of Organon. B Spiessens and R van Heesbeen are employees of Johnson & Johnson. D Hung and C Andrade are employees of Seqirus. J Korejwo-Peyramond is an employee of Sanofi. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or material discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Authors contribution
BC and FTDS coordinated the conception of the paper. BC, BS, RvH, DH, CA, JKP and FTDS contributed to the development of proposals, reviewed, and edited the manuscript. All authors have read and approved the final manuscript and attest that they meet the ICMJE criteria for authorship.
Trademark statement
Comirnaty is a trademark of BioNTech Manufacturing GmbH; Jcovden is a trademark of the Janssen group of companies; Shingrix is a trademark of GSK; Spikevax is a trademark of the Moderna Biotech group of companies; Vaxzevria is a trademark of the AstraZeneca group of companies.
Data sharing statement
The datasets generated during and/or analyzed during the current study are available in:
[dataset] Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine, Supplementary Materials, 2021, https://doi.org/10.1056/NEJMoa2035389.
[dataset] Lal H, Cunningham AL, Godeaux O, Chlibek R, Diez-Domingo J, Hwang SJ, et al. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. : Adverse Events and Reactogenicity, 2015, https://doi.org/10.1056/NEJMoa1501184.
Supplemental Material
Download MS Word (476.1 KB)Acknowledgments
The authors would like to thank Paula Barbosa from IFPMA who kindly coordinated the review of the position paper within the IFPMA vaccine subgroup. The authors also thank the Akkodis Belgium platform, on behalf of GSK: Lucienne Duru and Joanne Wolter provided writing support and Carlos Marin coordinated manuscript development.
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14760584.2023.2262571.
Additional information
Funding
References
- Hervé C, Laupèze B, Del GG, et al. The how’s and what’s of vaccine reactogenicity. NPJ Vaccines. 2019 Sep 24;4(1):39. doi: 10.1038/s41541-019-0132-6
- Crowe B, Brueckner A, Beasley C, et al. Current practices, challenges, and statistical issues with product safety labeling. Stat Biopharm Res. 2013;5(3):180–193. doi: 10.1080/19466315.2013.791640
- CBER. Emergency use authorization for vaccines to prevent COVID-19; Guidance for Industry. 2022 [cited 2022 Dec]. Available from: https://www.fda.gov/media/142749/download.
- Baden LR, El Sahly HM, Essink B, et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021 Feb 4;384(5):403–416. doi: 10.1056/NEJMoa2035389
- Haas JW, Bender FL, Ballou S, et al. Frequency of adverse events in the placebo arms of COVID-19 vaccine trials: a systematic review and meta-analysis. JAMA Netw Open. 2022 Jan 4;5(1):e2143955. doi: 10.1001/jamanetworkopen.2021.43955
- Wernicke JF, Faries D, Milton D, et al. Detecting treatment emergent adverse events in clinical trials : a comparison of spontaneously reported and solicited collection methods. Drug Saf. 2005;28(11):1057–1063. doi: 10.2165/00002018-200528110-00006
- Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020 Dec 31;383(27):2603–2615. doi: 10.1056/NEJMoa2034577
- FDA. Vaccines and related biological products advisory committee meeting December 10, 2020. [cited 2022 Dec]. Available from: https://www.fda.gov/media/144245/download.
- Lal H, Cunningham AL, Godeaux O, et al. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med. 2015 May 28;372(22):2087–2096. doi: 10.1056/NEJMoa1501184
- Voysey M, Clemens SAC, Madhi SA, et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet. 2021 Jan 9;397(10269):99–111. doi: 10.1016/S0140-6736(20)32661-1
- Sadoff J, Gray G, Vandebosch A, et al. Safety and efficacy of single-dose Ad26.COV2.S vaccine against Covid-19. N Engl J Med. 2021 Jun 10;384(23):2187–2201. doi: 10.1056/NEJMoa2101544
- EMA. COVID-19 vaccine (Ad26.COV2-S [recombinant]); Summary of product characteristics. 2021 [cited 2022 Dec]. Available from: https://www.ema.europa.eu/en/documents/product-information/jcovden-previously-covid-19-vaccine-janssen-epar-product-information_en.pdf.
- EMA. COVID-19 vaccine (ChAdOx1-S [recombinant]); Summary of product characteristics. 2021 [cited 2022 Dec]. Available from: https://www.ema.europa.eu/en/documents/product-information/covid-19-vaccine-astrazeneca-product-information-approved-chmp-29-january-2021-pending-endorsement_en.pdf.
- CBER. Toxicity grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. 2007. [cited 2022 Dec]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/toxicity-grading-scale-healthy-adult-and-adolescent-volunteers-enrolled-preventive-vaccine-clinical.
- UMC. The use of the WHO-UMC system for standardised case causality assessment. 2018. [cited 2022 Jun]. Available from: https://who-umc.org/media/164200/who-umc-causality-assessment_new-logo.pdf.
- Gidudu JF, Walco GA, Taddio A, et al. Immunization site pain: case definition and guidelines for collection, analysis, and presentation of immunization safety data. Vaccine. 2012 Jun 22;30(30):4558–4577. doi: 10.1016/j.vaccine.2012.03.085
- Marcy SM, Kohl KS, Dagan R, et al. Fever as an adverse event following immunization: case definition and guidelines of data collection, analysis, and presentation. Vaccine. 2004 Jan 26;22(5–6):551–556. doi: 10.1016/j.vaccine.2003.09.007
- FDA. Computerized systems used in clinical investigations. 2007. [cited 2022 Dec]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/computerized-systems-used-clinical-investigations.
- ALCOA+. ALCOA+ principles. 2021. [cited 2022 Dec]. Available from: https://pharmaguidesop.com/2020/08/what-is-alcoa-plus-alcoa-details-alcoa-principle.html.
- DMID. Pediatric toxicity tables 2007. [cited 2017 Oct]. Available from: http://www.niaid.nih.gov/LabsAndResources/resources/DMIDClinRsrch/Documents/dmidpedtox.pdf.
- DAIDS. DAIDS table for grading the severity of adult and pediatric adverse events, corrected version 2.1. 2017. [cited 2022 Dec]. Available from: https://rsc.niaid.nih.gov/clinical-research-sites/daids-adverse-event-grading-tables.
- CBER. Vaccines and related biological products advisory committee meeting. 2021 October 26. [cited 2022 Dec]. Available from: https://www.fda.gov/media/153447/download.
- WHO. Guidelines on clinical evaluation of vaccines: regulatory expectations. 2017 [cited 2022 Dec]. Available from: https://cdn.who.int/media/docs/default-source/prequal/vaccines/who-trs-1004-web-annex-9.pdf?sfvrsn=9c8f4704_2&download=true.
- NMPA. Announcement of the State Food and Drug Administration on issuing technical guidelines for clinical comparability research of preventive vaccines (2019 No 94). 2019 [cited 2021 Dec]. Available from: https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20191224104601789.html.
- WHO. Global manual on surveillance of adverse events following immunization. 2016 [cited 2022 Dec]. Available from: https://www.who.int/publications/i/item/10665206144.
- FDA CaC. Guidance for Industry and Investigators. Safety reporting requirements for INDs and BA/BE studies (US). 2012 [cited 2022 Dec]. Available from: https://www.fda.gov/files/drugs/published/Safety-Reporting-Requirements-for-INDs-%28Investigational-New-Drug-Applications%29-and-BA-BE-%28Bioavailability-Bioequivalence%29-Studies.pdf.
- Regulations CoF. Part 312—Investigational new drug application. 1987 [cited 2021 Dec]. Available from: https://www.ecfr.gov/cgi-bin/text-idx?SID=841d69f34da8bdb3b0b1f7fa59e8157a&mc=true&node=pt21.5.312&rgn=div5%20-%20se21.5.312_164.
- EMA. ICH Topic E 2 A: Clinical safety data management: definitions and standards for expedited reporting. 1995 [cited 2022 Dec]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-15.pdf.
- EMA. Regulation (EU) no 536/2014 of the European parliament and of the council of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC. 2014 [cited 2022 Dec]. Available from: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf.