
ABSTRACT
Introduction
Clinical trials of personalized cancer vaccines have shown that on-demand therapies that are manufactured for each patient, result in activated T cell responses against individual tumor neoantigens. However, their use has been traditionally restricted to adjuvant settings and late-stage cancer therapy. There is growing support for the implementation of PCV earlier in the cancer therapy timeline, for reasons that will be discussed in this review.
Areas covered
The efficacy of cancer vaccines may be to some extent dependent on treatment(s) given prior to vaccine administration. Tumors can undergo radical immunoediting following treatment with immunotherapies, such as checkpoint inhibitors, which may affect the presence of the very mutations targeted by cancer vaccines. This review will cover the topics of neoantigen cancer vaccines, tumor immunoediting, and therapy timing.
Expert opinion
Therapy timing remains a critical topic to address in optimizing the efficacy of personalized cancer vaccines. Most personalized cancer vaccines are being evaluated in late-stage cancer patients and after treatment with checkpoint inhibitors, but they may offer a greater benefit to the patient if administered in earlier clinical settings, such as the neoadjuvant setting, where patients are not facing T cell exhaustion and/or a further compromised immune system.
1. Introduction
Significant advances have been made in cancer immunotherapy with checkpoint inhibitors (CPIs) receiving approval for use in many cancer indications [Citation1], and with personalized cancer vaccines (PCVs) advancing to Phase 3 clinical trials [Citation2]. The success of CPIs has prompted an abundance of preclinical and clinical studies evaluating their efficacy in combination with other cancer treatments, including PCVs [Citation3]. CPIs have the potential to unleash the adaptive immune response [Citation4], while PCVs can activate and expand tumor-specific T cells [Citation5], providing strong rationale in exploring the combined potential of these immunotherapies.
CPI and PCV combination therapy has been evaluated for several tumor indications, including melanoma [Citation6–9], glioblastoma [Citation10], non-small cell lung cancer [Citation11], bladder cancer [Citation11], hepatocellular carcinoma [Citation12], and pancreatic cancer [Citation13]. In most clinical trials, PCVs have been administered in the adjuvant setting (post-surgery) and after treatment with CPIs [Citation3]. Few studies have explored treatment with PCVs in the neoadjuvant setting (before surgery) and before treatment with CPIs due to cost concerns and limitations in timely manufacturing of PCVs. Recent advances in manufacturing have significantly improved the production timeline for PCVs [Citation9,Citation14], enabling their potential assessment in earlier clinical settings.
Moreover, studies have shown that tumor genomes are shaped by their interaction with the immune system, a phenomenon known as immunoediting [Citation15]. Given the potential of CPI therapy to accelerate immunoediting, administering CPIs ahead of PCVs may result in modifications of the tumor and the tumor microenvironment (TME) that may subsequently impact PCV effectiveness. Clinical trials that evaluate the appropriate sequence of intervention (PCVs before, during, or after CPIs) and setting (neoadjuvant or adjuvant) should be explored to optimize the clinical benefits of PCVs. Our review will highlight why PCVs may be more effective if given ahead of CPIs and in the neoadjuvant setting, focusing on concepts, challenges, and solutions irrespective of PCV formulation (RNA, DNA, peptides). In addition to covering the topics of personalized neoantigen cancer vaccines, neoadjuvant cancer therapies, and tumor immunoediting, this review will introduce a case study of nivolumab-treated melanoma samples supporting the need to carefully assess the timing and sequence of administering PCVs and CPIs as combination therapy.
2. Optimizing the efficacy of personalized cancer vaccines
2.1. Neoantigen cancer vaccines
Following the introduction of CPIs, studies demonstrating T cell-mediated tumor control have firmly established the clinical significance of immunotherapeutic approaches that harness T cells [Citation16–22]. A critical conclusion that can be inferred from many of these clinical studies is that endogenous T cells are able to recognize mutated cancer-specific epitopes presented by major histocompatibility complex (MHC) molecules, also known in humans as human leukocyte antigens (HLA). T cells recognizing these neoantigens are unaffected by central tolerance and can mediate anti-tumor activity. Results of neoantigen-based PCV clinical trials conducted in advanced melanoma patients, first reported in 2015 and 2017 [Citation6–8], showed that PCVs can activate both pre-existing immunity and naïve T cell responses. Subsequent trials have extended these observations to other indications, including cancers with low tumor mutational burden (TMB), and have highlighted PCVs’ ability to shift the TME towards a more proinflammatory profile [Citation10–13]. While PCVs have yielded encouraging clinical results, their development has been hindered by lengthy manufacturing time, logistical complexity, and high cost compared to more traditional (non-personalized) therapies. Improvements in artificial intelligence (AI) and computational vaccinology techniques have alleviated some of these hurdles, with now fully automated vaccine design pipelines enabling expedited PCV development (see [Citation23] for a comprehensive review). However, production and formulation of PCVs using peptide, RNA, or DNA platforms have yet to be fully scaled for on-demand manufacturing. Strategies that avoid some of these hurdles and that focus on shared antigens, such as shared mutations or tumor associated antigens (TAAs), are being explored as alternatives [Citation5] but lack the multivalency offered by PCVs.
2.2. Neoadjuvant immunotherapies
Neoadjuvant immunotherapies (defined as treatment prior to tumor resection) aim to reduce tumor burden and activate the adaptive immune response prior to tumor resection, thus improving the odds of a successful surgery and survival [Citation24,Citation25]. Priming and activating T cells while the primary tumor is still present offers advantages in terms of breath of immune response, better response to therapy, and improved clinical outcomes [Citation26–29]. An important consideration for developers of neoadjuvant therapies is to avoid disruption of standard treatment plans. As such, neoadjuvant PCV development has to adhere to a strict turnaround time to be manufactured and administered well in advance of tumor resection, providing ample time for proper activation of the immune system and tumor killing. If a PCV cannot be produced within a timeframe of 4–5 weeks, the delay may jeopardize the utility of PCVs in the neoadjuvant setting. In addition to reduced manufacturing timeline, the cost of PCVs must be within the range of currently approved treatment options to justify their administration as first-line therapies after cost-effectiveness analyses [Citation30,Citation31].
Recent preclinical [Citation32] and clinical oncolytic virus (OV) studies [Citation33,Citation34] have illustrated the advantages of neoadjuvant cancer therapy. Increase in local inflammation due to OV therapy appears to enhance T cell infiltration in the TME and activate the overall immune system, providing added benefits in preclinical models when combined with CPIs [Citation33]. Administering PCVs in the neoadjuvant setting may provide similar benefits, while offering more practical routes of delivery compared to in situ delivery of OV therapy. PCVs may also offer less off-target effects compared to other immunotherapies, and have remained well tolerated in clinical trials. Studies are however required to validate the potential benefits of neoadjuvant PCV therapy.
2.3. Immunoediting
Immunoediting corresponds to the complex and various mechanisms by which tumors become less immunogenic, avoid immune detection, and evade killing by the immune system. For example, tumors may delete or suppress the expression of neoantigen sequences, or may alternatively promote the development of an immunosuppressive TME [Citation15]. The introduction of cancer therapy may accelerate this natural phenomenon, which is thought to be one of the main drivers of resistance to therapy [Citation16]. Immunotherapies, such as CPIs, are powerful immunoediting agents due to their direct interactions with the immune system. Examples of tumor immunoediting observed in the clinic following immunotherapy include loss of the target antigen(s) [Citation35] or alteration of the antigen presentation machinery, either via deleterious mutation of the β2-microglobulin gene [Citation36] or chromosomal deletion of the HLA gene [Citation37]. A more comprehensive review of immunoediting and its impact on T cell-based therapies has been recently published [Citation16].
PCV contribution to immunoediting remains unclear as most clinical trials have evaluated them in combination with CPIs. However, activation of cancer-specific T cells by PCVs is likely to accelerate the immunoediting process, similarly to observations made with cell therapies [Citation35,Citation37]. Results from PCV clinical trials have highlighted the association between response to therapy and decreased levels of circulating tumor DNA (ctDNA) in the periphery [Citation38,Citation39], suggesting active shedding of targeted neoantigens.
PCV effectiveness is dependent on the presentation of specific mutations selected during the vaccine design process at the time of the biopsy. As such, any immunoediting processes disrupting the presence, expression, or presentation of the selected neoantigens, and occurring in between the tumor biopsy and PCV dosing may have a reverberating impact on efficacy and patient outcomes. The effect of introducing CPIs before PCVs must hence be thoroughly studied to assess the benefits, or drawbacks, of current PCV clinical trial designs.
2.4. Timing of cancer therapies
Cancer drugs shape tumors, their TME, and the host’s immune system [Citation16]. First-line therapies will affect the context in which subsequently administered therapies will operate, which may in turn impact their efficacy. The timing, or ordering, of cancer therapies is a critical topic to address in order to unleash the full potential of PCV and CPI combination therapy. Beyond the sequencing of PCVs with CPIs, it is important to evaluate their timing with respect to other treatments, including surgery (adjuvant versus neoadjuvant) and other non-immune related therapies such as chemotherapy and radiation therapy.
Timing of CPI and radiation therapy has been studied in both preclinical and clinical settings (reviewed in [Citation40]), suggesting improved outcomes when radiation therapy is followed by PD-1 or PD-L1 blocking antibodies. Similar observations have been made when administering chemotherapy drugs prior to CPIs (reviewed in [Citation41]), although the majority of combination trials have only evaluated concurrent administration.
Strong evidence points toward the benefit of giving a combination therapy of cancer vaccine and CPI [Citation42], although the timing of administering such combinations remains to be thoroughly evaluated. An advantage to providing PCVs ahead of CPIs is the potential to apply the power of CPI to a ‘primed’ immune system that has been conditioned to target neoantigens in the tumor. Preclinical studies have highlighted the benefit of co-administering TAA-based cancer vaccines with CPI therapy, as opposed to administering vaccines after CPIs [Citation43]. Clinical evaluation of CPI naïve and pre-treated patients dosed with a peptide cancer vaccine in combination with pembrolizumab have confirmed the same observation, showing both improved T cell responses and extended survival in the anti-PD-1 naïve group [Citation44], suggesting that pre-treatment with a CPI can inhibit the therapeutic benefit of cancer vaccines. Other TAA vaccine studies have showed improved clinical benefits when co-administering vaccines and CPIs compared to sequential administration [Citation45].
Overall, these studies highlight the need to optimize the sequence and timing of CPIs and cancer vaccines to enhance molecular and clinical responses. Additional studies are required to confirm these observations in the context of PCVs and understand how patient outcomes are affected by other parameters such as type of antigen(s) (shared/private), vaccine platform (peptide/DNA/RNA), dosing, frequency, or cancer indication/stage.
2.5. Case study: loss of immunogenic neoantigens follows nivolumab immunotherapy
As most patients enrolled in PCV clinical trials are treated with a CPI ahead of the PCV, it is critical to understand how CPIs affect the tumor, the TME, and how they may alter the PCV’s potential to effectively target the tumor. To exemplify the importance of the order of PCV and CPI therapies, and the detrimental effect that incorrect timing may have, we evaluated a published dataset of melanoma tumors collected from the same site before (Pre) and while on (On) nivolumab immunotherapy [Citation46]. This cohort of patients was selected based on the public availability of the full sequencing dataset.
Whole Exome Sequencing (WES) data for normal tissues, ‘Pre’ therapy tumor tissues, and ‘On’ therapy tumor tissues collected from 68 patients was downloaded from the Sequence Read Archive (SRA) database, as described in [Citation46] (SRA: SRP095809; BioProject: PRJNA359359). Tumor and normal FASTQ data were processed through an internal bioinformatics pipeline following GATK’s Best Practices [Citation47]. Sequencing reads were aligned to the reference GRCh38/hg38 human genome to obtain BAM files. Normal DNA sequencing data was analyzed with the Kourami algorithm to predict the HLA class I and HLA class II haplotypes of each patient [Citation48]. Tumor-specific mutations were identified using the Mutect2 and Strelka2 variant callers [Citation49,Citation50] and subsequently annotated using Variant Effect Predictor (VEP) [Citation51]. Paired tumor/normal amino acid sequences were extracted for each mutation modifying protein coding regions. Mutations derived from paired tumor samples collected from the same patients (n=41) were compared to identify mutations detectable in the ‘Pre’ therapy sample only, in the ‘On’ therapy sample only, and identified in both ‘Pre’ and ‘On’ therapy samples ().
Figure 1. Analysis of melanoma tumors collected before (Pre Tx) and after (On Tx) one nivolumab infusion. Next-generation sequencing data from paired (Pre/On) tumor samples was analyzed to identify mutations lost (blue), maintained (orange), and newly acquired (green) over the course of the treatment (a). Mutations were subsequently analyzed with the Ancer platform to determine each mutation’s immunogenic and tolerance potential with respect to the patients’ HLAs using the EpiMatrix and JanusMatrix algorithms, respectively (b).
Each individual patient’s mutated and normal sequences were analyzed with the Ancer platform to identify neoantigens binding to their HLAs and evaluate their immunogenic and tolerogenic potential () [Citation23]. The Ancer platform integrates two well-validated algorithms, EpiMatrix and JanusMatrix. EpiMatrix identifies MHC class I and MHC class II restricted neoantigens from mutanome analyses. EpiMatrix accuracy ranges from 77% to 88% for HLA class II predictions, and from 93% to 99% for HLA class I, depending on the allele [Citation52]. JanusMatrix compares neoantigens to ‘self’ sequences capable of binding MHC, by analyzing the TCR facing residues of the selected HLA class I and class II neoantigens, and eliminates any that might be tolerated or tolerogenic based on their cross-conservation with self-like sequences. Studies have confirmed that these self-like epitopes stimulate Tregs [Citation53–55], suppress immunogenicity [Citation56–58], and hamper vaccine efficacy [Citation59].
As described in [Citation46], an overall contraction of the TMB was observed in this cohort after nivolumab therapy, with a total of 15,208 and 12,329 non-synonymous mutations identified in ‘Pre’ and ‘On’ therapy samples, respectively. Mutanome analysis with the Ancer platform revealed that mutations from ‘On’ therapy samples had a lower immunogenic potential (lower EpiMatrix score) than mutations found in ‘Pre’ therapy samples (, Mann-Whitney test, p = 0.0001). These results are suggestive of the immune pressure exerted on the tumor by CPIs, with a loss of immunogenic mutations and/or an accrual of mutations with low immunogenic potential after CPI therapy. After further distinguishing mutations deleted (n = 6,217), maintained (n = 8,991), and acquired (n = 3,338) while on therapy, we found that newly acquired mutations had a significantly lower immunogenic potential compared to other mutations (Kruskal-Wallis test, adjusted for multiple comparisons, p< 0.0001). This decrease in immunogenic potential suggests that mutations found after administering CPIs have fewer neoantigens than mutations found prior to the start of the therapy.
Figure 2. Mutations gained after nivolumab therapy are less immunogenic (a) and more tolerogenic (b). Mutations found in melanoma tumors collected before (Pre) and while on (On) nivolumab were analyzed with Ancer. Mutations only detected after CPI therapy (On Tx only) have lower immunogenic (a) and higher tolerance (b) potential compared to mutations found prior to therapy. Pre Tx vs On Tx comparisons: Mann-Whitney test. Pre Tx only vs Shared vs On Tx only comparisons: Kruskal-Wallis test. ***p-value < 0.001, ****p-value < 0.0001.
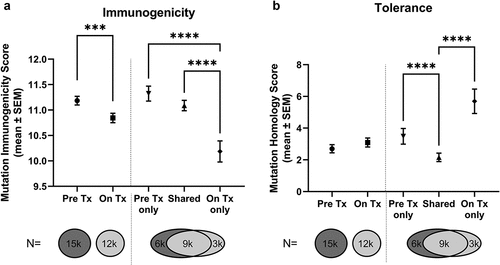
In addition to immunogenicity, the tolerance potential, or degree of homology with self sequence, of each mutation was assessed with the JanusMatrix component of the Ancer platform. A trend towards increased tolerance potential was observed in ‘On’ therapy mutations but did not meet statistical significance. However, a strong tolerance signal was identified in mutations acquired after CPI therapy compared to the other mutation subsets (, Kruskal-Wallis test, adjusted for multiple comparisons, p< 0.0001). The greater tolerance potential, or homology with self, observed with newly acquired mutations suggests an increased likelihood for these mutations to be tolerated by the immune system, or actively recognized by regulatory T cells (Tregs) and thus dampening immune responses, as described in other studies involving sequences with elevated degree of homology with self [Citation54,Citation56,Citation57].
Overall, our analysis indicates that CPI therapy reduces the immunogenic potential and increases the tolerance potential of mutations found in the tumor, thus altering its immunogenicity profile and mutational landscape during the time that PCVs would be manufactured.
2.6. The case for neoadjuvant cancer vaccines
The case study detailed above indicates that CPI therapy can radically affect the immunogenicity and tolerance profiles of mutations found in residual tumor in a manner that is consistent with the concepts of immunoediting discussed previously. This phenomenon also parallels the concept of immune camouflage observed with viral sequences and contributing to reduced vaccine efficacy [Citation52,Citation59,Citation60]. These results highlight that identifying relevant mutations for PCVs will become more difficult once patients are treated with a CPI, providing further information to optimize the timing of their administration. CPI therapy prior to PCV dosing may not be beneficial, as 1) fewer immunogenic neoepitopes would remain, 2) immunogenic neoantigens selected for PCVs may be edited out of the tumor by the time PCVs are provided to CPI-treated patients, and 3) accumulation of mutations favorable to tumor immune avoidance may curtail PCV effectiveness. Of course, more clinical studies will be required to confirm these hypotheses and to further elucidate the optimal timing of PCVs and CPIs.
It is worth noting that as an alternative to PCVs, off-the-shelf neoadjuvant therapy with ‘shared epitope’-based vaccines, or with allogenic vaccines, have proven to be effective in preclinical models [Citation61,Citation62] and to a certain degree these strategies are also working in the clinic [Citation63,Citation64]. Given these observations, administering a more tailored vaccine that is specific to the mutations in an individual patient’s tumor (PCV) in the neoadjuvant setting, and ahead of CPIs, may prove to be even more beneficial. However, this paradigm shift still faces some practical limitations, such as adequately shortening the turnaround time for PCV manufacture and reducing development costs to warrant PCVs as first line therapies. These are some of the same hurdles that are being addressed in pandemic responsiveness efforts, such as advancing the development of novel vaccine platform technologies against ‘Disease X’ through support from the Coalition for Epidemic Preparedness Innovations (CEPI) [Citation65]. We view these manufacturing issues likely to be addressed by engineering and process-related solutions and likely to be easily adaptable to the PCV field. Once engineering and process-related delays are overcome, patients are highly likely to benefit from vaccines that are tailored to improve their tumor-specific adaptive immune responses in the period prior to tumor resection, which may also improve their tumor-free duration and quality of life after surgical intervention.
Implementing neoadjuvant PCV therapy in the clinic will require significant changes. Drug development and manufacturing has historically been highly centralized, and current PCV strategies depend on centralized laboratories for sequencing biopsies, manufacturing drug substances, and formulating the final product. Peptide-based PCVs is a noteworthy exception where final formulation is often performed at bedside (at the clinical site) [Citation8,Citation10,Citation11]. Overall, a more modular, automated, and decentralized approach to drug development may be of great benefit to reduce both time and cost to manufacture PCVs, where sequencing, manufacture, and formulation could directly occur at clinical sites. This approach, analogous to CAR-T manufacture in hospital pharmacy, would however require significant upfront investments to purchase the necessary material and equipment and to secure bioprocessing certifications. As no PCV have been approved to date, additional guidelines from the regulatory agencies are necessary to further help streamline and optimize the PCV production process while ensuring compliance with regulatory requirements that remain undetermined to date.
Despite any improvements in manufacturing and cost, neoadjuvant PCVs will remain highly dependent on the disease status of patients and their overall health. Advanced cancer patients may not have the time to wait for personalized therapies, even if turnaround times are reduced to a couple of weeks, and immunocompromised patients may benefit from alternative, non-immune related therapies, such as surgery, chemotherapy, or radiation therapy.
3. Conclusion
Optimizing the sequence and timing of PCV and CPI combination therapy remains a critical aspect to address in maximizing PCV efficacy and enhancing patient outcomes. Practical limitations have restricted PCVs to the adjuvant clinical setting and after treatment with CPIs [Citation9,Citation12,Citation13]. As exemplified in our case study, tumors may undergo accelerated immunoediting after the start of CPI therapy. As such, neoantigens targeted by PCVs may no longer be valid targets by the time PCVs are administered, hindering their effectiveness. Strategies to overcome this challenge include administering PCVs before or concomitantly with CPIs. Additionally, favoring neoadjuvant clinical settings, prior to removal of the tumor, may be of benefit as patients’ immune systems have not been affected by other therapies. Indirect evidence generated with TAA-based cancer vaccines suggests that PCV effectiveness may be compromised when given after CPIs [Citation43]. Moreover, clinical examples have highlighted the advantages of neoadjuvant immunotherapy [Citation33,Citation34], further suggesting the potential benefit of PCVs in this setting.
Additional studies are necessary to fully understand optimal timing of PCV therapy with respect to surgery and other immunomodulating treatments such as CPIs. First, observations made with off-the-shelf cancer vaccines must be expanded to PCVs to confirm if concurrent or sequential administration with CPIs enhances molecular and clinical responses. Comparative studies with PD-1/PD-L1 and CTLA-4 blocking antibodies will also determine which CPI(s) are best paired with PCVs. Second, neoadjuvant PCV studies are needed to clarify the benefit of moving vaccines to earlier clinical settings. Results from these studies will provide much needed data to inform future PCV clinical trials. If supported by positive clinical observations, then efforts must be undertaken to enhance PCV production and affordability and to render them more easily available to patients.
4. Expert opinion
PCVs have regained momentum with the recent release of clinical data [Citation9,Citation12,Citation13] and their advancement to Phase 3 clinical trial in Q3 2023 [Citation2]. While progress has been made in streamlining many of the steps required for PCV design and manufacturing, significant hurdles remain to unleash their full potential. However, many of these limitations can be alleviated with technological improvements. For example, exclusion of patients with non-resectable tumors can be overcome with advances in de novo neoantigen identification from Next Generation Sequencing (NGS) of circulating tumor DNA (ctDNA), target selection can be improved with enhancements in AI techniques, and lengthy turnaround times and high manufacturing costs can be reduced with enhanced streamlining, automation, and small-batch production optimization. However, large investments in these technologies and PCV research will only materialize if cancer vaccines prove to be effective and significantly improve patient outcomes in large-scale Phase 2/3 clinical trials. Some may argue that PCVs may be subjected to increased scrutiny regarding their efficacy given their high cost and logistical complexity compared to off-the-shelf immunotherapies and large-batch drug manufacturing standards. Given these heightened expectations and the biopharmaceutical industry’s reticence to overhaul existing production pipelines, questions surrounding PCV timing and setting must be addressed to ensure maximum benefits to the patients.
PCVs effectiveness is dependent on T cell activity and the overall status of patients’ immune system. As such, providing these immunotherapies in late-stage cancer patients with severely exhausted T cells or with compromised immune system due to prior systemic chemotherapy or checkpoint inhibitor therapy, as it is currently being implemented, may limit their true potential. This hypothesis may in part explain the mixed reviews that PCV trials have received over the years. While activation of neoantigen-specific T cell responses has been observed after repeated PCV dosing [Citation7,Citation8,Citation13], limited clinical effect has been observed, perhaps due to terminal T cell exhaustion or immunoediting. Our case study evaluating tumors treated with nivolumab raises an additional concern with the current sequencing of cancer immunotherapies. Administering CPIs before PCVs may result in potential loss of immunogenic neoantigens before vaccination. This observation, and preclinical studies by others [Citation43], highlight the potential benefit of modifying current clinical trial designs and dosing patients with PCVs before or with CPIs. Moreover, we argue that PCVs may better fit in earlier clinical settings, such as the neoadjuvant setting, when patients’ T cells and immune system has not yet been undermined by other lines of therapies, when the primary tumor is still present to enhance T cell activation and breadth of the immune response, and when given prior to or co-administered with CPIs to avoid resistance issues arising by blocking the PD-1 pathway in suboptimally primed T cells.
We acknowledge bringing PCVs to the neoadjuvant setting will be a challenging endeavor given current practical manufacturing and cost constraints. The logistical complexity of PCVs has for long restricted their usage to small patient cohorts or N-of-1 studies, however, recent Phase 2 clinical trials have shown this limitation is softening [Citation9]. PCVs production cost has remained an obstacle for their acceptance as commercially and clinically relevant cancer drugs. While some reduction in manufacturing cost can be expected in the future, perhaps stemming from enhancements in synthetic DNA/RNA synthesis or automation, the price of customized therapies such as PVCs will likely remain above the one of readily available, off-the-shelf, drugs. In this context, strong clinical evidence will be required from PCVs to justify their usage in early clinical settings.
In the next five years, clinical results from Phase 3 PCV trials will become available and regulatory agencies will review requests for their approval in the adjuvant setting, if backed by robust clinical data. Improvements in liquid biopsies and sequencing technology will enable de novo identification of tumor variants and vaccine design from blood draws, allowing patients with unresectable or hard to access tumors to benefit from PCVs. Additionally, initiatives to enhance mapping of alternative vaccine targets, such as dark matter antigens, will enable the treatment of cancer patients with low TMB. In the next ten years, PCVs will be evaluated in an increasing number of cancer indications and will further establish their relevance in the treatment of cancer if landmark preclinical and clinical studies assess optimal strategies for their administration and combination with other therapies. In the long-term, improvements in AI, NGS, and manufacturing may offer the potential for developing affordable, prophylactic (preventative) PCVs by collecting mutational information from circulating precancerous cells during routine medical examination, allowing treatment and eradication of cancerous clones before tumor establishment and immunoediting. While these advancements may never materialize, overcoming these challenges may support PCVs to become a fundamental armamentarium for cancer treatment and prevention.
Article highlights
Personalized cancer vaccines have regained interest given recent positive clinical trial results.
Personalized cancer vaccines are currently being evaluated in the adjuvant setting and after treatment with checkpoint inhibitors.
Tumors may radically undergo immunoediting after treatment with checkpoint inhibitors, affecting the presence of mutations targeted by cancer vaccines.
Personalized cancer vaccines may be most effective in earlier clinical settings before surgery and checkpoint inhibitor therapy.
Declaration of interest
A De Groot and WD Martin are senior officers and majority shareholders of EpiVax, Inc, a privately owned immunoinformatics and vaccine design company. G Richard is a senior officer, N Ruggiero was a senior officer of EpiVax Therapeutics, Inc., a precision immunotherapy company and subsidiary of EpiVax, Inc. G Richard and N Ruggiero have equity in EpiVax Therapeutics. GD Steinberg is a member of Clinical Trial Protocol Committees for the following companies: Merck, BMS, Janssen, Cold Genesys, Pfizer, PhotoCure, Fidia, is or has been a scientific advisor/consultant within the past 5 years for the following companies: Heat Biologics, Cold Genesys, PhotoCure, Merck, Roche/Genentech, Ciclomed, Taris Biomedical, MDxHealth, Fidia Farmaceuticals, Urogen, Ferring, Aduro, Boston Scientific, Bristol Myers Squibb, Astra Zeneca, Pfizer, Janssen, EpiVax Therapeutics, Natera, FKD, Ferring, EnGene Bio, SesenBio, BioCanCell, Nucleix, Ipsen, Combat Medical, Astellas, Fergene, Dendreon, Abbvie, Seattle Genetics, and has equity stock/options in EpiVax Therapeutics and Urogen. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Reviewers on this manuscript have received honorarium for their review work. Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose.
Author contributions
G Richard and A De Groot drafted the manuscript. All authors made substantial edits, reviewed, and approved the manuscript for submission.
Additional information
Funding
References
- Vaddepally RK, Kharel P, Pandey R, et al. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers (Basel). 2020;12(3):1–19. doi: 10.3390/cancers12030738
- Merck and moderna initiate phase 3 study evaluating V940 (mRNA-4157) in combination with KEYTRUDA® (pembrolizumab) for adjuvant treatment of patients with resected high-risk (stage IIB-IV) melanoma [Internet]. Available from: https://www.merck.com/news/merck-and-moderna-initiate-phase-3-study-evaluating-v940-mrna-4157-in-combination-with-keytruda-pembrolizumab-for-adjuvant-treatment-of-patients-with-resected-high-riskstage-iib-iv-melanom/
- Shemesh CS, Hsu JC, Hosseini I, et al. Personalized cancer vaccines: clinical landscape, challenges, and opportunities. Mol Ther. 2021;29(2):555–570. doi: 10.1016/j.ymthe.2020.09.038
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239
- Lin MJ, Svensson-Arvelund J, Lubitz GS, et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. 2022;3:911–926.
- Carreno BM, Magrini V, Becker-Hapak M, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. doi: 10.1126/science.aaa3828
- Sahin U, Derhovanessian E, Miller M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–226. doi: 10.1038/nature23003
- Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–221. doi: 10.1038/nature22991
- Khattak A, Carlino M, Meniawy T, et al. Abstract CT001: a personalized cancer vaccine, mRNA-4157, combined with pembrolizumab versus pembrolizumab in patients with resected high-risk melanoma: efficacy and safety results from the randomized, open-label phase 2 mRNA-4157-P201/Keynote-942 trial. Cancer Res. 2023;83(8_Supplement):CT001. doi: 10.1158/1538-7445.AM2023-CT001
- Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–239. doi: 10.1038/s41586-018-0792-9
- Ott PA, Hu-Lieskovan S, Chmielowski B, et al. A phase ib trial of personalized neoantigen therapy plus anti-PD-1 in patients with advanced melanoma, non-small cell lung cancer, or bladder cancer. Cell. 2020;183(2):347–362.e24. doi: 10.1016/j.cell.2020.08.053
- Gane E, Yarchoan M, Marron T, et al. 693 personalized DNA neoantigen vaccine (GNOS-PV02) in combination with plasmid IL-12 and pembrolizumab as second-line (2L) treatment for advanced hepatocellular carcinoma (HCC). J Immunother Cancer. 2022;10:A724.
- Rojas LA, Sethna Z, Soares KC, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618(7963):144–150. doi: 10.1038/s41586-023-06063-y
- Carvalho T. Personalized anti-cancer vaccine combining mRNA and immunotherapy tested in melanoma trial. Nat Med. 2023;29(10):2379–2380. doi: 10.1038/d41591-023-00072-0
- Gubin MM, Vesely MD. Cancer immunoediting in the era of immuno-oncology. Clin Cancer Res. 2022;28(18):3917–3928.
- O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–167.
- Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466
- Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257(1):56–71. doi: 10.1111/imr.12132
- Dudley ME, Gross CA, Somerville RPT, et al. Randomized selection design trial evaluating CD8±enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma. J Clin Oncol. 2013;31:2152–2159. doi: 10.1200/JCO.2012.46.6441
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172
- Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954
- Massari F, Di Nunno V, Cubelli M, et al. Immune checkpoint inhibitors for metastatic bladder cancer. Cancer Treat Rev. 2018;64:11–20. doi: 10.1016/j.ctrv.2017.12.007
- Richard G, Princiotta MF, Bridon D, et al. Neoantigen-based personalized cancer vaccines, the emergence of precision cancer immunotherapy. Expert Rev Vaccines. 2021;21:173–184. doi: 10.1080/14760584.2022.2012456
- Bilusic M, Gulley JL. Neoadjuvant Immunotherapy: An Evolving Paradigm Shift? J Natl Cancer Inst. 2021;113(7):799–800. doi: 10.1093/jnci/djaa217
- Mittendorf EA, Burgers F, Haanen J, et al. Neoadjuvant immunotherapy: leveraging the immune system to treat early-stage disease. Am Soc Clin Oncol Educ B. 2022;(42):189–203. doi: 10.1200/EDBK_349411
- Rozeman EA, Hoefsmit EP, Reijers ILM, et al. Survival and biomarker analyses from the OpACIN-neo and OpACIN neoadjuvant immunotherapy trials in stage III melanoma. Nat Med. 2021;27(2):256–263. doi: 10.1038/s41591-020-01211-7
- Rozeman EA, Menzies AM, van Akkooi ACJ, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol. 2019;20(7):948–960. doi: 10.1016/S1470-2045(19)30151-2
- Menzies AM, Amaria RN, Rozeman EA, et al. Pathological response and survival with neoadjuvant therapy in melanoma: a pooled analysis from the international neoadjuvant melanoma consortium (INMC). Nat Med. 2021;27(2):301–309. doi: 10.1038/s41591-020-01188-3
- Patel SP, Othus M, Chen Y, et al. Neoadjuvant–Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N Engl J Med. 2023;388(9):813–823. doi: 10.1056/NEJMoa2211437
- Kasztura M, Richard A, Bempong NE, et al. Cost-effectiveness of precision medicine: a scoping review. Int J Public Health. 2019;64:1261–1271. doi: 10.1007/s00038-019-01298-x
- Vellekoop H, Versteegh M, Huygens S, et al. The net benefit of personalized medicine: a systematic literature review and regression analysis. Value Heal. 2022;25(8):1428–1438. doi: 10.1016/j.jval.2022.01.006
- Bourgeois-Daigneault M-C, Roy DG, Aitken AS, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018;10(422):10. doi: 10.1126/scitranslmed.aao1641
- Samson A, Scott KJ, Taggart D, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10(422):10. doi: 10.1126/scitranslmed.aam7577
- Li R, Zhang J, Gilbert SM, et al. Using oncolytic viruses to ignite the tumour immune microenvironment in bladder cancer. Nat Rev Urol. 2021;18(9):543–555. doi: 10.1038/s41585-021-00483-z
- Orlando EJ, Han X, Tribouley C, et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med. 2018;24(10):1504–1506. doi: 10.1038/s41591-018-0146-z
- Benitez R, Godelaine D, Lopez-Nevot MA, et al. Mutations of the β2-microglobulin gene result in a lack of HLA class I molecules on melanoma cells of two patients immunized with MAGE peptides. Tissue Antigens. 1998;52(6):520–529. doi: 10.1111/j.1399-0039.1998.tb03082.x
- Tran E, Robbins PF, Lu Y-C, et al. T-Cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016;375(23):2255–2262. doi: 10.1056/NEJMoa1609279
- Perales R, Perales-Puchalt A, Bartha G, et al. 692 circulating tumor DNA analysis of advanced hepatocellular cancer (HCC) patients treated with neoantigen targeted personalized cancer DNA vaccine (GNOS-PV02) in combination with plasmid IL-12 (pIL12) and anti-PD1 (pembrolizumab). J Immunother Cancer. 2022;10:A723.
- Carlino MS, Khattak A, Weber JS, et al. Minimal residual disease by circulating tumor DNA as a biomarker of recurrence free survival in resected high-risk melanoma patients treated with mRNA-4157/V940, a personalized cancer vaccine, and pembrolizumab. J Clin Oncol. 2023;41(17_suppl):LBA9515. doi: 10.1200/JCO.2023.41.17_suppl.LBA9515
- Williamson CW, Sherer MV, Zamarin D, et al. Immunotherapy and radiation therapy sequencing: state of the data on timing, efficacy, and safety. Cancer. 2021;127:1553–1567. doi: 10.1002/cncr.33424
- Heinhuis KM, Ros W, Kok M, et al. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol. 2019;30(2):219–235. doi: 10.1093/annonc/mdy551
- Collins JM, Redman JM, Gulley JL. Combining vaccines and immune checkpoint inhibitors to prime, expand, and facilitate effective tumor immunotherapy. Expert Rev Vaccines. 2018;17(8):697–705. doi: 10.1080/14760584.2018.1506332
- Verma V, Shrimali RK, Ahmad S, et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat Immunol. 2019;20(9):1231–1243. doi: 10.1038/s41590-019-0441-y
- Vavolizza RD, Petroni GR, Mauldin IS, et al. Phase I/II clinical trial of a helper peptide vaccine plus PD-1 blockade in PD-1 antibody-naïve and PD-1 antibody-experienced patients with melanoma (MEL64). J Immunother Cancer. 2022;10(9):1–14. doi: 10.1136/jitc-2022-005424
- McNeel DG, Eickhoff JC, Wargowski E, et al. Concurrent, but not sequential, PD-1 blockade with a DNA vaccine elicits anti-tumor responses in patients with metastatic, castration-resistant prostate cancer. Oncotarget. 2018;9(39):25586–25596. doi: 10.18632/oncotarget.25387
- Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934–949.e15. doi: 10.1016/j.cell.2017.09.028
- van der Auwera G, O’Connor BD. Genomics in the cloud: using docker, GATK, and WDL in terra. Sebastopol, CA: O’Reilly Media, Incorporated; 2020. https://www.oreilly.com/library/view/genomics-in-the/9781491975183/
- Lee H, Kingsford CK. Kourami: graph-guided assembly for novel human leukocyte antigen allele discovery. Genome Biol. 2018;19(1):1–16. doi: 10.1186/s13059-018-1388-2
- Benjamin D, Sato T, Cibulskis K, et al. Calling somatic SNVs and indels with Mutect2. bioRxiv. 2019;861054:1–8. https://www.biorxiv.org/content/10.1101/861054v1
- Kim S, Scheffler K, Halpern AL, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods. 2018;15(8):591–594. doi: 10.1038/s41592-018-0051-x
- McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17(1):1–14. doi: 10.1186/s13059-016-0974-4
- De Groot AS, Moise L, Terry F, et al. Better epitope discovery, precision immune engineering, and accelerated vaccine design using Immunoinformatics tools. Front Immunol. 2020;11:1–13. doi: 10.3389/fimmu.2020.00442
- Losikoff PT, Mishra S, Terry F, et al. HCV epitope, homologous to multiple human protein sequences, induces a regulatory T cell response in infected patients. J Hepatol. 2015;62(1):48–55. doi: 10.1016/j.jhep.2014.08.026
- Liu R, Moise L, Tassone R, et al. H7N9 T-cell epitopes that mimic human sequences are less immunogenic and may induce treg-mediated tolerance. Hum Vaccines Immunother. 2015;11(9):2241–2252. doi: 10.1080/21645515.2015.1052197
- Dembele M, Tao S, Massoud AH, et al. Tregitopes improve asthma by promoting highly suppressive and Antigen-Specific Tregs. Front Immunol. 2021;12:12. doi: 10.3389/fimmu.2021.634509
- De Groot AS, Rosenberg AS, Miah SMS, et al. Identification of a potent regulatory T cell epitope in factor V that modulates CD4+ and CD8+ memory T cell responses. Clin Immunol. 2021;224:108661. doi: 10.1016/j.clim.2020.108661
- Miah SMS, Lelias S, Gutierrez AH, et al. A SARS-CoV-2 NSP7 homolog of a treg epitope suppresses CD4+ and CD8+ T cell memory responses. Front Immunol. 2023;14:14. doi: 10.3389/fimmu.2023.1290688
- Wada Y, Nithichanon A, Nobusawa E, et al. A humanized mouse model identifies key amino acids for low immunogenicity of H7N9 vaccines. Sci Rep. 2017;7(1):1283. doi: 10.1038/s41598-017-01372-5
- Jang H, Meyers LM, Boyle C, et al. Immune-engineered H7N9 influenza hemagglutinin improves protection against viral influenza virus challenge. Hum Vaccines Immunother. 2020;16(9):2042–2050. doi: 10.1080/21645515.2020.1793711
- Khan S, Parrillo M, Gutierrez AH, et al. Immune escape and immune camouflage may reduce the efficacy of RTS,S vaccine in Malawi. Hum Vaccines Immunother. 2020;16(2):214–227. doi: 10.1080/21645515.2018.1560772
- Grinshtein N, Bridle B, Wan Y, et al. Neoadjuvant vaccination provides superior protection against tumor relapse following surgery compared with adjuvant vaccination. Cancer Res. 2009;69(9):3979–3985. doi: 10.1158/0008-5472.CAN-08-3385
- Fisher SA, Cleaver A, Lakhiani DD, et al. Neoadjuvant anti-tumor vaccination prior to surgery enhances survival. J Transl Med. 2014;12(1):1–9. doi: 10.1186/s12967-014-0245-7
- Ogino H, Taylor JW, Nejo T, et al. Randomized trial of neoadjuvant vaccination with tumor-cell lysate induces T cell response in low-grade gliomas. J Clin Invest. 2022;132(3):132. doi: 10.1172/JCI151239
- Heumann T, Judkins C, Li K, et al. A platform trial of neoadjuvant and adjuvant antitumor vaccination alone or in combination with PD-1 antagonist and CD137 agonist antibodies in patients with resectable pancreatic adenocarcinoma. Nat Commun. 2023;14(1):1–12. doi: 10.1038/s41467-023-39196-9
- Gouglas D, Christodoulou M, Plotkin SA, et al. CEPI: driving progress toward epidemic preparedness and response. Epidemiol Rev. 2019;41:28–33. doi: 10.1093/epirev/mxz012