
ABSTRACT
Background
Recombinant protein vaccines are vital for broad protection against SARS-CoV-2 variants. This study assessed ReCOV as a booster in two Phase 2 trials.
Research design and methods
Study-1 involved subjects were randomized (1:1:1) to receive 20 μg ReCOV, 40 μg ReCOV, or an inactivated vaccine (COVILO®) in the United Arab Emirates. Study-2 participating individuals were randomized (1:1:1) to receive 20 μg ReCOV (pilot batch, ReCOV HA), 20 μg ReCOV (commercial batch, ReCOV TC), or 30 μg BNT162b2 (COMIRNATY®) in the Philippines. The primary immunogenicity objectives was to compare the geometric mean titer (GMT) and seroconversion rate (SCR) of neutralizing antibodies induced by one ReCOV booster dose with those of inactivated vaccine and BNT162b2, respectively, at 14 days post-booster.
Results
Heterologous ReCOV booster doses were safe and induced comparable immune responses to inactivated vaccines and BNT162b2 against Omicron variants and the prototype. They showed significant advantages in cross-neutralization against multiple SARS-CoV-2 variants, surpassing inactivated vaccines and BNT162b2, with good immune persistence.
Conclusions
Heterologous ReCOV boosting was safe and effective, showing promise in combating COVID-19. The study highlights ReCOV’s potential for enhanced protection, supported by strong cross-neutralization and immune persistence.
Clinical Trial Registration
Study-1, www.clinicaltrials.gov, identifier is NCT05323435; Study-2, www.clinicaltrials.gov, identifier is NCT05084989.
1. Introduction
Over the past three years since the emergence of the coronavirus disease 2019 (COVID-19) outbreak in Wuhan, China, the deployment of vaccines targeting the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has played a crucial role in mitigating the burden of COVID-19. These vaccines have been instrumental in achieving robust neutralization against various SARS-CoV-2 variants. Heterologous prime-boost vaccination, in addition to demonstrating excellent safety profiles, has exhibited indisputable superiority in immune response, particularly in cross-neutralization against Omicron variants, compared to primary vaccination or homologous boosting [Citation1]. Diverse COVID-19 vaccines, utilizing various platforms such as inactivated, vector-based, mRNA, and recombinant protein, have been developed. As of January 2024, the World Health Organization (WHO) has approved 14 vaccines based on the ancestral strain or Omicron variants, developed using different technologies [Citation2]. WHO advocates for a heterologous strategy over a homologous one due to its superior immunogenicity [Citation3]. Given that inactivated vaccines constitute more than 45% of the globally distributed vaccine doses, especially to low- and middle-income countries during the early stages of the COVID-19 pandemic [Citation4], an enhanced strategy involving a heterologous booster dose based on inactivated vaccines could prove highly beneficial for billions of individuals, offering protection against both current and potentially emerging SARS-CoV-2 variants [Citation5]. The WHO’s Strategic Advisory Group of Experts recommends the use of COVID-19 WHO EUL vectored or mRNA vaccines as a booster following the primary series of COVID-19 inactivated vaccines [Citation6].
In contrast to vaccines focusing solely on the receptor-binding domain (RBD) or full-length spike sequences, ReCOV integrates the amino acid sequence from position 14 to 541, inclusive of both the N-terminal domain (NTD) and RBD of the spike protein. This strategic composition, encompassing a spectrum of neutralizing epitopes, is fortified by its conjugation with the foldon sequence from phage T4. The incorporation of the foldon sequence aims to authentically mimic the trimeric structure of the SARS-CoV-2 Spike protein, thereby enhancing the antigen’s potential efficacy [Citation7]. The antigen is adjuvated with BFA03, an oil-in-water emulsion squalene adjuvant containing two immunostimulants (squalene, α-tocopherol). With this structure-based design and combination with the potent adjuvant, ReCOV has demonstrated robust immunogenic and protective effects against SARS-CoV-2 in animal models [Citation8].
Prior to this study, we conducted Phase 1 (NCT04818801) and Phase 2 (NCT05084989) studies on the safety and immunogenicity of primary vaccination with ReCOV. These studies demonstrated that two intramuscular injections of 20 µg or 40 µg ReCOV with a 21-day interval were well-tolerated and elicited robust neutralizing antibodies (NAbs) against the prototype, Omicron BA.2, and BA.4/5 (data unpublished). The Phase 3 study (NCT05398848) evaluating the efficacy of ReCOV primary vaccination is currently underway.
In this paper, we present safety and immunogenicity data from two Phase 2 trials of ReCOV administered as a booster dose among healthy adults who previously received two or three doses of inactivated vaccines.
2. Methods
2.1. Study design and subjects
We conducted two randomized, observer-blinded, active-controlled Phase 2 trials to assess the immunogenicity and safety of ReCOV as a booster following the primary series of inactivated vaccines in adults. This evaluation involved a comparison with INACTIVATED VACCINE and BNT162b2, respectively.
Study-1 ([NCT05323435], study protocol refers to supplementary 1) was conducted in AI Kuwait Hospital in Dubai, United Arab Emirates, involving adults aged 18 to 65 years who had received two or three doses of inactivated COVID-19 vaccine manufactured by Sinopharm or Sinovac. Eligible subjects were equally randomized to receive 20 μg ReCOV, 40 μg ReCOV, or 0.5 mL INACTIVATED VACCINE.
Study-2 ([NCT05084989], study protocol refers to supplementary 2) was conducted in Health Centrum, St. Paul’s Hospital, The Medical City Iloilo, University of the Philippines-Philippine General Hospital in the Philippines. Eligible subjects aged ≥18 years old who had primed with two doses of inactivated COVID-19 vaccine manufactured by Sinopharm or Sinovac were randomized to receive 20 μg ReCOV (pilot batch, ReCOV HA), 20 μg ReCOV (commercial batch, ReCOV TC), or 30 μg BNT162b2 (an mRNA vaccine produced by Pfizer Inc.) [Citation9].
In both trials, participants were required to have received their last inactivated COVID-19 vaccine within 90 to 365 days before enrollment. Exclusion criteria included a history of COVID-19 or known SARS-CoV-2 infection within the past 6 months (Study-1) or 12 months (Study-2), a positive result by SARS-CoV-2 reverse transcriptase polymerase chain reaction at screening, or acute or uncontrolled medical conditions increasing the risk of participation.
For Study-1, the selection of low dose (20 μg) and high dose (40 μg) of ReCOV was based on a preliminary analysis from the Phase I study. Both doses had demonstrated a good safety profile and a strong humoral immune response against the SARS-CoV-2 prototype. The 40 μg ReCOV induced slightly more seroconversions and neutralizing antibodies (NAbs) after the first dose but tended to have lower NAb levels after the second dose. In Study-2, the dose level (20 μg ReCOV) was decided after extensive Phase I data analysis, indicating higher NAb titers compared to the 40 μg dose.
An interim analysis was planned for both trials after all subjects completed 28 days of follow-up, evaluating primary immunogenicity endpoints, safety, and reactogenicity. Additionally, subjects were followed up for safety and immunogenicity for 6 months post-boosting in both studies, with final analyses conducted accordingly [Citation9,Citation10].
2.2. Ethics
Both studies were conducted in strict adherence to the International Council for Harmonization Good Clinical Practice guidelines and the ethical principles outlined in the Declaration of Helsinki. Prior to the initiation of the studies, comprehensive approval for the study protocols and informed consent forms was obtained from local Ethics Committees. For Study-1, the initial protocol was approved by the Ministry of Health and Prevention Research Ethics Committee, United Arab Emirates, on 22 March 2022 (approval reference No: MOHAP/DXB-REC/F.M.M/No.15/2022), and the protocol amendment got approval on 16 February 2023. For Study-2, the initial protocol was approved by the Single Joint Research Ethics Board, Republic of the Philippines, on 7 December 2021 (SJREB protocol No: SJREB-2021-106), and the protocol amendment got approval on 23 January 2023. In both studies, written informed consent was obtained from each participant before the commencement of any study procedures. This meticulous adherence to ethical guidelines and regulatory approvals ensures the protection of participants’ rights, safety, and well-being throughout the duration of the studies.
2.3. Randomization and masking
In each study, the randomization statistician used SAS statistical software (Version 9.4) to generate both subject and vaccine random code lists using the stratified block randomization method. The allocation ratio among vaccination groups was 1:1:1 with a block size of six. The random stratification factors were previous doses (two doses or three doses) and the duration since the last vaccination (90 ~ 180 days vs. 181 ~ 365 days) in Study-1, age (18 ~ 59 years vs. ≥60 years) and the duration since the last primary vaccination (90 ~ 180 days vs. 181 ~ 365 days) in Study-2. Unique randomization number was assigned by the Interactive Web Response System (IWRS). This ensured the confidentiality and integrity of the randomization process.
The investigational products (IP)s related management processes were implemented by study unblinded team from sponsor, CRO, investigational sites, and IPs depot, and would not be disclosed to any blinded individual according to Blinding Management Plans for both studies, including IP labeling and packaging, IP storage and management, IP dispense and vaccination, unblinding documents recording, monitoring, quality control and management. Blinding was maintained until interim analysis completed, i.e. after all subjects completing the visit at 28 days post boosting. After that, the study CORE team, including Medical Lead, PV Physician, Statistician, DM Lead and Project Lead, would be unblinded, while the investigators and study subjects remained blinded till end of the study.
2.4. Procedures
All subjects who received the booster vaccination were diligently monitored for safety and reactogenicity in both studies. Following vaccination, subjects were observed for 30 minutes at the study site to assess immediate adverse events (AEs). Solicited local and systemic AEs within 7 days post-boosting and unsolicited AEs within 28 days post-boosting were diligently recorded by subjects in diaries. Serious adverse events (SAEs) and adverse events of special interest (AESIs) were continuously monitored throughout the entire study period. AESIs were defined as potential immune-mediated diseases and COVID-19-specific AEs, in accordance with the Coalition for Epidemic Preparedness Innovations Safety Platform for Emergency Vaccines Project guidelines [Citation11]. AE severity evaluation adhered to the China National Medical Product Administration Guidelines for Adverse Event Classification Standards for Clinical Trials of Preventive Vaccines [Citation12].
Blood samples were collected at various time points: before boosting, and on days 14, 28, 3 months, and 6 months post-boosting. A validated SARS-CoV-2 plaque reduction neutralization test (PRNT) was employed at the central laboratory, G42 laboratory LLC in the United Arab Emirates. The results were reported as PRNT50 for live-virus neutralizing antibodies (NAbs) against SARS-CoV-2 viruses. The sequence of the prototype was represented by the original strain with the spike D614G mutation, and sequences for Omicron BA.2 and BA.5 were confirmed as part of the national SARS-CoV-2 sequencing program. In Study-1, live-virus NAbs for the prototype and BA.2 were assessed at baseline, 14 days, 28 days, 3 months, and 6 months post-study vaccination. In Study-2, live-virus NAbs for the prototype and BA.5 were assessed at baseline and 14 days post-study vaccination.
Additionally, a validated VZV-based pseudovirus neutralization assay was employed at the central laboratory, Gobond Testing Technology (Beijing) Co., Ltd in China. Results were reported as ND50 for NAbs against the Omicron variants and prototype. In Study 1, all subjects were assessed pseudovirus NAbs for the prototype, Omicron BA.2, BA.4/5, and BA.2.75 at baseline, 14 days, 28 days, 3 months, and 6 months post boosting. In Study-2, all subjects were assessed pseudovirus NAbs for the prototype, Omicron BA.4/5, BA.2.75, and BF.7 at baseline and 14 days post boosting, while only the first 100 randomized subjects (50 each of ReCOV TC group and BNT162b2 group, respectively) were assessed for immune durability at 28 days, 3 months, and 6 months post boosting. Furthermore, pseudovirus NAbs against emergent Omicron variants BQ.1.1, XBB, and CH.1.1 were assessed among the first 200 randomized subjects (100 each for ReCOV TC and BNT162b2 groups) at baseline and 14 days post-boosting. In the ReCOV TC group, the first 30 subjects with seroconversion for BQ.1.1 and XBB at 14 days post-boosting were explored for neutralizing activities against XBB.1.5 at 14 days post-boosting and the persistence of immunity against BQ.1.1 and XBB.1.5 at 3 months post-boosting.
2.5. Outcomes
The primary immunogenicity endpoints for both studies were the Seroconversion Rates (SCRs) and Geometric Mean Titers (GMTs) of live-virus Neutralizing Antibodies (NAbs) against the SARS-CoV-2 prototype at 14 days post-boost vaccination. Additionally, secondary endpoints included the evaluation of cross-neutralizing antibodies (cross-NAbs) against Omicron variants, as described in the Procedures section. The assessment of immune persistence until the end of the studies was also considered a secondary endpoint. Seroconversion was defined as the proportion of subjects exhibiting at least a 4-fold increase in post-vaccination antibody titers compared to baseline. Furthermore, the Geometric Mean Increase (GMI) for all tested strains was evaluated as a secondary endpoint to assess the overall immune response in both studies.
Safety endpoints included the occurrence of solicited local and systemic AEs within 7 days post boosting, unsolicited AEs within 28 days post boosting, SAEs and AESI till 6 months post boosting. All subjects were required to undergo a 30-minute observation period at the site for the assessment of immediate AEs. Solicited local AEs included injection site pain, pruritus, redness, swelling, skin rash, induration, and cellulitis. Solicited systemic AEs comprised fatigue/asthenia, constipation, pyrexia, cough, dyspnea, syncope, gastrointestinal symptoms, headache, myalgia, arthritis, arthralgia, non-inoculation site pruritus, non-inoculation site pain, abnormal cutaneous mucosa, acute bronchospasm, acute allergic reactions, new onset seizure, insomnia, and agitation or inhibition. For a comprehensive overview of study objectives and endpoints, refer to Table S1.
2.6. Statistics
For Study-1, we assumed SCR in INACTIVATED VACCINE group was 60%, and the coefficient of variation for the NAb titers was 1.2, and the drop-out rate was around 10%. A total number of 300 eligible subjects with 100 per group was sufficient to provide 80% power to detect SCR difference of 20% and GMT ratio of 1.5 between each ReCOV dose group and INACTIVATED VACCINE group for live-virus NAb against the prototype at 14 days post boosting, at one-sided type I error of 0.025.
In Study-2, 200 subjects per group was able to provide at least 85% power to detect non-inferiority for both SCR and GMT between ReCOV TC group and BNT162b2 group for live-virus NAb against the prototype at 14 days post boosting, assuming the SCR in BNT162b2 group was 90%, the coefficient of variation for the NAb concentration was 1.2, one-side type I error was 0.025, and the drop-out rate was around 10%. The non-inferiority was established if the lower limit of 95% CI for the SCR difference was greater than −10% and the lower limit of 95% CI for the GMT ratio was greater than 0.67.
In both studies, analysis of covariance (ANCOVA) was used for testing intergroup differences of GMTs with adjustment of baseline values and stratification factors. Stratification factors in Study-1 included previous doses with inactivated vaccines (two doses, three doses) and duration since the last vaccination (90 ~ 180 days, 181 ~ 365 days), while those in Study-2 included age (18 ~ 59 years, ≥60 years) and since the last vaccination (90 ~ 180 days, 181 ~ 365 days). For comparing intergroup differences on SCR, Logistic model adjusted with stratification factors and Chi-square test/Fisher’s exact test, were applied in Study-1 and Study-2, respectively. Immunogenicity analyses were conducted among all randomized subjects who received study vaccination had at least one valid immunogenicity result prior and after vaccination (immunogenicity analysis set) for data collected at baseline and 14 days post boosting. Analyses for immune persistence were conducted among immunogenicity analysis set with valid results at each corresponding time point. Safety analyses were performed among all randomized subjects who received study vaccination (safety analysis set). All analyses were conducted using SAS Version 9.4 or higher (SAS Institute).
3. Results
3.1. Study subjects
Between May 2022 and January 2023, a total of 301 adults aged 18 to 65 years, who had previously received two or three doses of inactivated COVID-19 vaccine, were randomized in Study-1. This cohort comprised 99 individuals in the 20 μg ReCOV group, 99 in the 40 μg ReCOV group, and 103 in the INACTIVATED VACCINE group (refer to , Panel A). The majority of participants were of Asian descent (99.3%), with a mean age of 27.5 years. Notably, 99.7% of the participants were male. All but one subject received the study vaccination, and a total of 295 subjects, who had at least one valid immunogenicity test result before and after the study vaccination, were included in the corresponding immunogenicity analysis set.
Figure 1. Flow diagram of study-1 (panel A) and study-2 (panel B).
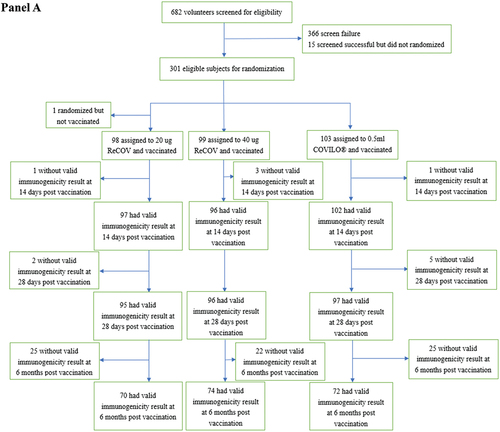
Figure 1. (Continued).
Between May 2022 and January 2023, a total of 301 adults aged 18 to 65 years, who had previously received two or three doses of inactivated COVID-19 vaccine, were randomized in Study-1. This cohort comprised 99 individuals in the 20 μg ReCOV group, 99 in the 40 μg ReCOV group, and 103 in the INACTIVATED VACCINE group (refer to , Panel A). The majority of participants were of Asian descent (99.3%), with a mean age of 27.5 years. Notably, 99.7% of the participants were male. All but one subject received the study vaccination, and a total of 295 subjects, who had at least one valid immunogenicity test result before and after the study vaccination, were included in the corresponding immunogenicity analysis set.
In both studies, the demographic and baseline clinical characteristics of the subjects were comparable among the different vaccination groups (refer to ).
Table 1. Subjects demographic characteristics of study-1 and study-2.
3.2. Immunogenicity outcomes
3.2.1. Study-1
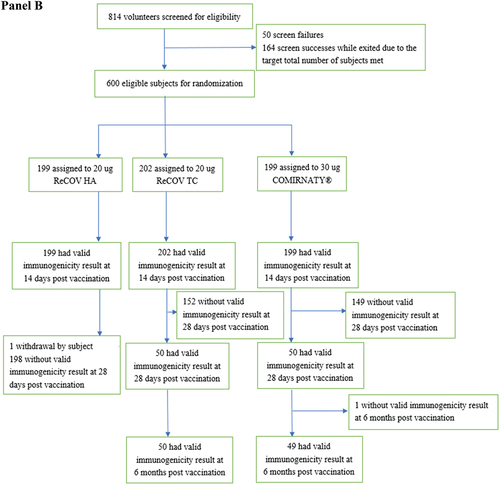
In Study-1, the 20 μg and 40 μg ReCOV groups exhibited remarkable immunogenicity outcomes at 14 days post-boosting. Seroconversion Rates (SCRs) against the live-virus Neutralizing Antibodies (NAb) for the prototype were significantly higher in the 20 μg (95.9%) and 40 μg (91.7%) ReCOV groups compared to the INACTIVATED VACCINE group (25.5%) (p values <0.0001; see Appendix 2 pp 4–6). Although baseline Geometric Mean Titers (GMTs) were similar among groups, the 20 μg and 40 μg ReCOV groups exhibited significantly elevated GMTs (95% CI) of 9199.2 (7749.4, 10920.2) and 8610.8 (7097.2, 10447.2), respectively, with corresponding Geometric Mean Increases (GMIs) (95% CI) of 36.1 (27.6, 47.3) and 35.4 (27.6, 47.3). Both GMTs and GMIs were significantly higher than those in the INACTIVATED VACCINE group (GMT: 532.7, 95% CI 442.4, 641.4; GMI: 1.9, 95% CI 1.5, 2.5) at 14 days post-boosting (p values <0.0001; , Panel A; see Appendix 2 pp 4–6). Regarding the live-virus NAb against Omicron BA.2, SCRs were 97.9% (20 μg ReCOV) and 90.6% (40 μg ReCOV), significantly surpassing the INACTIVATED VACCINE group (32.4%) at 14 days post-boosting (p values <0.0001). The GMT ratios for BA.2 between the 20 μg and 40 μg ReCOV groups and the INACTIVATED VACCINE group were 16.9 (95% CI 11.8, 24.2) and 15.5 (95% CI 10.8, 22.2), respectively, demonstrating a significant superiority in both ReCOV groups over the INACTIVATED VACCINE group (p values <0.0001). Additionally, the fold changes from baseline (GMIs) (95% CI) were 47.1 (35.3, 62.8), 44.9 (30.3, 66.6), and 2.7 (1.9, 3.8) in the 20 μg ReCOV, 40 μg ReCOV, and INACTIVATED VACCINE groups, respectively. No significant differences in SCRs, GMTs, or GMIs for both strains between the 20 μg and 40 μg ReCOV dose groups were noted, although the three immunogenicity variables of the 20 μg ReCOV group tended to be numerically higher than those of the 40 μg ReCOV group (see Appendix 2 pp 4–6).
Figure 2. GMTs of live-virus neutralizing antibody against SARS-CoV-2 prototype in study-1 (panel A) and study-2 (panel B).
High levels of pseudovirus Neutralizing Antibodies (NAbs) were elicited by ReCOV boosting against the prototype and Omicron BA.2 (see Appendix 2 pp 4–6). Furthermore, strong correlations between live-virus and pseudovirus NAbs were established at all tested time points (baseline, 14 days, 28 days, 3 months, and 6 months post-boosting) for both the prototype and BA.2, with overall correlation coefficients of 0.94 for the prototype and 0.86 for BA.2, respectively (p values <0.0001; see Appendix 9 pp 14–15, Panel A and Panel B).
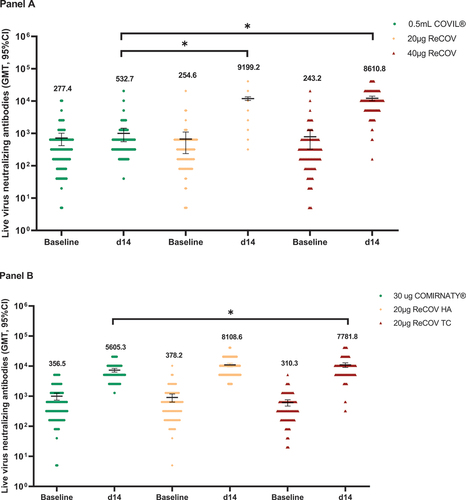
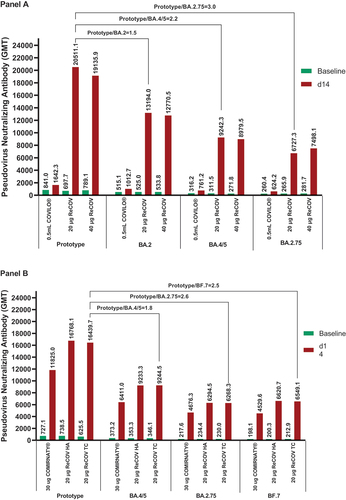
Pseudovirus NAbs against Omicron BA.4/5 and BA.2.75 were further evaluated. At 14 days post-boosting, SCRs were consistently higher than 90% in ReCOV groups compared to the INACTIVATED VACCINE group (p values <0.0001), except for 88.5% in the 40 μg ReCOV group, which was numerically higher than that in the INACTIVATED VACCINE group (SCRs <30%). Additionally, SCRs in the 20 μg ReCOV group tended to be higher than those in the 40 μg ReCOV group (see Appendix 2 pp 4–6). Baseline GMTs for all these strains were similar among vaccination groups. After vaccination, the 20 μg and 40 μg ReCOV groups exhibited significantly higher levels of NAbs than the INACTIVATED VACCINE group for all tested strains (, Panel A, all p values <0.0001). GMTs and GMIs seemed to be numerically higher in the 20 μg ReCOV group than the 40 μg ReCOV group. In both ReCOV groups, GMTs were only around 2.2- to 3-fold lower for BA.4/5 and BA.2.75 compared with that of the prototype.
Figure 3. GMTs of pseudovirus neutralizing antibody against SARS-CoV-2 prototype and omicron variants in study-1 (panel A) and study-2 (panel B).
Moreover, NAbs against the Omicron variants and prototype persisted at 28 days, 3 months, and 6 months (study end) post-boosting in all ReCOV groups. In the 20 μg and 40 μg ReCOV groups, around 3.4- to 6.0-fold reductions in NAbs against the prototype and Omicron BA.2, BA.4/5, and BA.2.75 were observed, respectively, at the end of the study compared with the peaking levels at 14 days post-boosting. However, the levels of NAbs post-vaccination in ReCOV groups were consistently higher than those in the INACTIVATED VACCINE group during the 6-month study period (see Appendix 8 p 13, Panel A).
3.2.2. Study-2
In Study-2, the live-virus Neutralizing Antibody (NAb) seroconversion rates (SCRs) against the prototype were robust, with values of 91.5% (ReCOV HA), 96.0% (ReCOV TC), and 91.0% (BNT162b2) at 14 days post-boosting. Notably, the SCR difference between the ReCOV TC and BNT162b2 groups was 5.08% (95% CI 0.27%, 10.35%), indicating non-inferiority as the lower limit of the 95% CI for the SCR difference exceeded the pre-defined margin (−10%). Moreover, statistical superiority was achieved in the ReCOV TC group compared to the BNT162b2 group, with a lower limit of the 95% CI for the SCR difference above 0 and a p-value of 0.039 (see Appendix 3 pp 7–8). At 14 days post-boosting, the Geometric Mean Titers (GMTs) (95% CI) in the ReCOV HA, ReCOV TC, and BNT162b2 groups were 8108.6 (7341.3, 8956.2), 7781.8 (6932.46, 8735.2), and 5605.3 (5094.4, 6167.4), respectively. The adjusted GMT ratio between the ReCOV TC and BNT162b2 groups was 1.4 (95% CI 1.2, 1.7), confirming non-inferiority as the lower limit of the 95% CI for the ratio exceeded the pre-defined value of 0.67. Furthermore, statistical superiority of the ReCOV TC group versus BNT162b2 was demonstrated, with the lower limit of the 95% CI for the adjusted GMT ratio above 1 (p value <0.0001; , Panel B; see Appendix 3 pp 7–8). The statistical equivalence in the GMTs was observed between the ReCOV HA and ReCOV TC groups, with an adjusted GMT ratio of 1.0 (95% CI 0.9, 1.2), well within the equivalence bound of (0.67, 1.5). Additionally, the Geometric Mean Increases (GMIs) (95% CI) for the ReCOV HA, ReCOV TC, and BNT162b2 groups were 21.44 (17.55, 26.19), 25.08 (20.61, 30.52), and 15.72 (12.91, 19.16), respectively. Similar GMIs were observed in the ReCOV HA and TC groups at 14 days post the booster vaccination, markedly higher than that in the BNT162b2 group.
At 14 days post-boosting, the SCRs for Omicron BA.2 and BA.5 were 92.0% and 92.0% in the ReCOV HA group, 91.6% and 92.1% in the ReCOV TC group, and 89.4% and 88.4% in the BNT162b2 group, respectively (see Appendix 3 pp 7–8). The adjusted GMT ratios for BA.2 and BA.5 between the ReCOV TC and BNT162b2 groups were 1.4 (95% CI 1.2, 1.8) for both, demonstrating higher Neutralizing Antibody (NAbs) levels in the ReCOV TC group compared to BNT162b2 (p values <0.0001). Similar to Study-1, a strong correlation between live-virus and pseudovirus NAbs was established at baseline and 14 days post-boosting for both the prototype and BA.5, with an overall Pearson’s correlation coefficient of 0.94 for the prototype and 0.94 for BA.5, respectively (p values <0.0001; see Appendix 9 pp 14–15, Panel C and Panel D).
Pseudovirus NAbs against Omicron BA.2.75 and BF.7 were evaluated, with >90% SCRs at 14 days post-boosting in the ReCOV TC group, while 88.4% in the BNT162b2 group, respectively. Moreover, non-inferiority in the ReCOV TC group versus BNT162b2 group was achieved for both variants (see Appendix 3 pp 7–8). At 14 days post-boosting, the GMTs of pseudovirus NAbs in the ReCOV TC group were statistically superior to those in the BNT162b2 group, with p values of 0.0029 (BA.2.75) and <0.0001 (BF.7). Compared with the GMT of pseudovirus NAbs against the prototype, GMTs reduced by around 2.6-fold and 2.5-fold for BA.2.75 and BF.7, respectively (, Panel B). At 6 months post-boosting, approximately 3.7- to 5.0-fold reductions for NAbs against the prototype, BA.4/5, BA.2.75, and BF.7 from the relevant peaking were observed in the ReCOV TC group, respectively (see Appendix 8 p 13, Panel B). The equivalent GMTs were further demonstrated between the two batches of ReCOV groups (see Appendix 3 pp 7–8). Additionally, at 14 days post-boosting, for Omicron BQ.1.1, XBB, and CH.1.1 variants, the ReCOV TC group showed SCRs of 92.0%, 85.0%, 93.0%, and GMTs (95% CI) of 2378.1 (1931.0, 2928.8), 631.2 (506.5, 786.7), and 1791.0 (1405.8, 2281.7), respectively. Meanwhile, for these variants, the BNT162b2 group had SCRs of 88.0%, 89.9%, 88.0%, and GMTs of 2162.3 (1830.7, 2554.0), 672.2 (551.2, 819.9), and 1301.4 (1056.3, 1603.3), respectively (see Appendix 4 p 9). The comparable trends of GMIs were also evident between the ReCOV TC and BNT162b.
3.3. Safety outcomes
3.3.1. Study-1
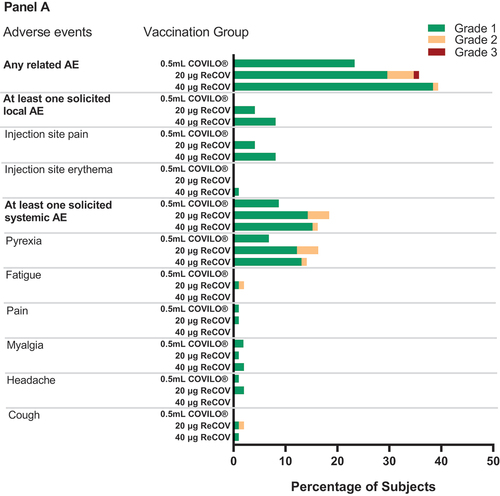
In Study-1, the incidence rates of overall adverse reactions were comparable between the 20 μg ReCOV and 40 μg ReCOV groups, standing at 35.7% and 39.4%, respectively. These rates were slightly higher than the corresponding rate of 23.3% in the INACTIVATED VACCINE group (see , Panel A; Appendix 5 p 10). Similarly, solicited Adverse Events (AEs) within 7 days and unsolicited AEs within 28 days post-vaccination showed similar trends, with frequencies of 20.4% and 21.4% for the 20 μg ReCOV group, 20.2% and 28.3% for the 40 μg ReCOV group, and 8.7% and 15.5% for the INACTIVATED VACCINE group. Notably, the majority (99.7%) of reported AEs were of mild or moderate severity. Only one subject in the 20 μg ReCOV group reported severe pyrexia, which resolved within 3 days. Importantly, there were no reported deaths, Serious Adverse Events (SAEs), Adverse Events of Special Interest (AESI), or AEs leading to study discontinuation.
Figure 4. (Continued).
The common solicited AEs (≥5%) in ReCOV groups included injection site pain (20 μg ReCOV 4.1%, 40 μg ReCOV 8.1%) and pyrexia (20 μg ReCOV 15.3%, 40 μg ReCOV 14.1%), and all solicited AEs were transient with the median duration of 1.0 day to 7.5 days (appendix 6 p 11). Blood triglycerides as an unsolicited AE was reported among subjects receiving ReCOV (20 μg ReCOV 15.3%, 40 μg ReCOV 22.2%), which were all mild and considered as probably related to vaccination by investigator.
3.3.2. Study-2
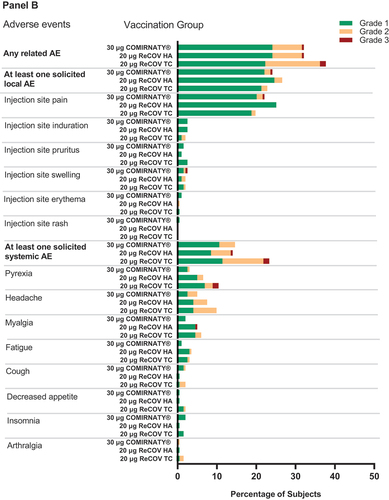
In Study-2, the incidences of adverse reactions post-vaccination were comparable across all groups, with rates of 32.2%, 37.6%, and 32.2% in the ReCOV HA, ReCOV TC, and BNT162b2 groups, respectively. Solicited local and unsolicited adverse events also demonstrated comparability among vaccination groups, with rates for solicited local AEs at 26.6%, 22.8%, and 24.1%, and unsolicited AEs at 5.0%, 5.4%, and 8.0% in the ReCOV HA, ReCOV TC, and BNT162b2 groups, respectively. However, the incidence of solicited systemic AEs tended to be higher in the ReCOV TC group (23.3%) compared to the ReCOV HA group (14.1%) and the BNT162b2 group (14.6%) (see , Panel B; Appendix 5 p 10). Importantly, the majority (96.4%) of AEs were of mild or moderate severity.
Figure 4. Frequency of any related adverse events, solicited local and systemic adverse eveIn study-1 (panel A) and study-2 (panel B).
Notably, seven subjects reported eight severe AEs, including one subject with myalgia (judged as related to study vaccination) in the ReCOV HA group, four subjects (including one case of cerebrovascular disorder, judged as unrelated to study vaccination, and three cases of pyrexia, judged as related to study vaccination) in the ReCOV TC group, and two subjects (one case of injection site pain and swelling, judged as related to study vaccination, and one case of pyrexia, judged as unrelated to study vaccination) in the BNT162b2 group. All severe AEs were recovered within 7 days. Additionally, one fatal Serious Adverse Event (SAE) of cerebrovascular disorder was documented in a subject in the ReCOV TC group, and another SAE involving suicide was reported in the BNT162b2 group. Importantly, both of these events were determined to be unrelated to the study vaccination.
In line with observations from Study-1, common solicited AEs (≥5%) in the ReCOV groups included injection site pain (ReCOV HA 25.1%, ReCOV TC 19.8%), headache (ReCOV HA 7.5%, ReCOV TC 9.9%), pyrexia (ReCOV HA 6.5%, ReCOV TC 10.4%), and myalgia (ReCOV HA 5.0%, ReCOV TC 5.9%), with median durations ranging from 1.0 day to 4.5 days (see Appendix 7 p 12). Related unsolicited Treatment-Emergent Adverse Events (TEAEs) with incidences of 2.0% to 3.0% were reported across vaccination groups. Importantly, no related Severe Adverse Events (SAEs), Adverse Events of Special Interest (AESI), or TEAEs leading to study discontinuation were reported.
4. Discussion
In both Phase 2 studies, ReCOV demonstrated significant efficacy in eliciting high levels of neutralizing antibodies (NAbs) against the SARS-CoV-2 prototype. The superiority of ReCOV was evident in both seroconversion rates (SCRs) and geometric mean titers (GMTs) of NAbs compared to the control vaccines at 14 days post-vaccination. Furthermore, robust levels of NAbs against Omicron variants were consistently higher in ReCOV groups, surpassing the INACTIVATED VACCINE group up to 6 months post-boosting in Study-1. In Study-2, ReCOV demonstrated at least non-inferiority to the BNT162b2 group at all assessed time points.
Heterologous boosting with either 20 μg or 40 μg ReCOV was well-tolerated in both studies, with no significant safety risks observed. The safety characteristics and immunogenicity features were consistent between the pilot and commercial batches of 20 μg ReCOV in Study-2, indicating a stable manufacturing process for vaccine production. Given the favorable safety profile and immunogenicity of both 20 μg and 40 μg ReCOV, the decision was made to select ReCOV at 20 μg as the candidate dose for future clinical use.
The strategy of boosting with either homologous or heterologous vaccines following the primary immunization with COVID-19 vaccines has been recommended to enhance protection against SARS-CoV-2 variants. Acknowledging the well-established advantage of a heterologous boosting strategy over homologous boosting [Citation13] we chose to use the INACTIVATED VACCINE, which holds a position in the WHO emergency use authorization (EUA) list, as the control vaccine in Study-1. This decision aimed to clearly demonstrate the superiority of ReCOV over both SARS-CoV-2 variants and the prototype, aligning with the approach taken in most heterologous boosting studies [Citation14–16].
In Study-2, we opted to use the mRNA vaccine BNT162b2 as a comparative reference. This decision was based on its endorsement by the WHO for use as a booster dose and its demonstrated substantial efficacy in mitigating the impact of the COVID-19 pandemic [Citation17–19]. In Study-2, we opted to use the mRNA vaccine BNT162b2 as a comparative reference. This decision was based on its endorsement by the WHO for use as a booster dose and its demonstrated substantial efficacy in mitigating the impact of the COVID-19 pandemic [Citation20]. However, our study results indicated that the levels of neutralizing antibodies (NAbs) induced by ReCOV were at least non-inferior, if not superior, to the well-acknowledged mRNA vaccine, BNT162b2. This consistent finding underscores the potential of heterologous boosting with ReCOV to elicit robust neutralizing activities against both Omicron variants and the prototype.
In our studies, the administration of a single booster dose of ReCOV consistently demonstrated promising cross-neutralization against Omicron subvariants. When compared with pseudovirus neutralizing antibody (NAb) levels against the prototype, only mild or moderate reductions in geometric mean titers (GMTs) for BA.4/5 and BA.2.75 were observed in Study-1, and mild reductions in GMTs for BA.4/5, BA.2.75, and BF.7 were noted in Study-2 at 14 days post-vaccination. The noteworthy finding is the persistence of significant cross-neutralizing activities, which endured for at least 6 months post-boosting, indicating the potential of ReCOV in preventing COVID-19 induced by Omicron variants.
In the exploratory analyses of Study-2, instances of immunologic escape from BQ.1.1, XBB, XBB.1.5, and CH.1.1 were observed as expected, resulting in 7.0- to 26.0-fold reductions in neutralizing antibody (NAbs) titers compared with the prototype. Despite these reductions, cross-neutralizations were still evident at 14 days post-vaccination. Notably, even the bivalent BNT162b2 vaccine (ancestral/Omicron BA.4/BA.5), widely used globally to combat Omicron-induced COVID-19, exhibited a decrease in NAbs against BQ.1.1 and XBB.1 by 15.6- and 40.7-fold, respectively, compared with the prototype [Citation21]. Boosting among populations that received 2 to 4 doses of monovalent mRNA vaccine only induced a relative vaccine efficacy of 48% against symptomatic XBB/XBB.1.5 infection [Citation22]. Therefore, alongside the development of subvariant-specific vaccines, this data strongly supports further evaluation of the role of ReCOV in preventing newly emergent variants.
Several limitations should be considered in the current study. First, the sample sizes in both Phase 2 studies were limited due to the nature of Phase 2 trials, and conclusions regarding safety and immunogenicity should be confirmed in larger-scale studies for boosting vaccination with ReCOV. In Study-2, the neutralizing antibody (NAbs) assessments against newly emergent variants were conducted on a subset of subjects as an exploratory analysis, and studies with a larger sample size are necessary to validate the current observations. Second, the continuous variation in SARS-CoV-2 and limitations in testing methods make it challenging to comprehensively cover neutralization assessments for all emerging strains [Citation23]. Third, efficacy in preventing COVID-19 was not addressed in the two studies; this aspect is under investigation in the Phase III trial for ReCOV (NCT05398848). Fourth, the unavailability of new-generation COVID-19 vaccines at the time of study design led to the use of prototype-based first-generation vaccines as active controls. Fifth, the investigational vaccine, ReCOV, is derived from the Wuhan strain but demonstrated strong cross-protection against all tested variants. Sixth, the evaluation of immunity focused on humoral responses, and cellular responses, which play a crucial role in decreasing the severity of infection, were not assessed. Subvariant-specific vaccines, supported by structure-based design and potent adjuvants, are currently under development to further enhance neutralization activities and effectiveness against emerging variants [Citation24].
5. Conclusions
Heterologous boosting with ReCOV is well tolerated with good safety profile, and could elicit strong, broad, and persistent cross-neutralization against multiple SARS-CoV-2 variants. The significant advantages compared with those induced by inactivated vaccine or BNT162b2 could strongly support the further clinical investigation in preventing COVID-19. Subvariant specific vaccines on account of the structure-based design are under development and may contribute to cope with the emerging variants.
Declaration of interests
Jia-Ping Yu, Wen-Rong Yao, Zijing Yue, Ying Ma, Chen Mo, Qing-Shuang Wang, Ren-Du Wen, Zheng Yao, Jian-Hui Zhang, Kun-Xue Hong, Yong Liu are employees of Jiangsu Recbio Technology Co., Ltd. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author contributions
Suad Hannawi and Abundio Balgos are the principal investigators of Study-1 and Study-2, respectively; Alaa Abuquta, Linda Safeldin, Aala Hassan, and Ahmad Alamadi are sub-investigators of Study-1, Louie Tirador, Aniuli May Jaen, and Ralph Elvi Villalobos are sub-investigators of Study-2. Zijing Yue, Ying Ma, and Chen Mo are medical leads for both studies, and Chen Mo drafted of the manuscript. Qing-Shuang Wang and Ren-Du Wen are the project leads for two studies. Zheng Yao is the statistician taking charge of data statistical analysis for both studies. Jian-Hui Zhang, Jing-Xin Li, and Kun-Xue Hong contributed to critical review and revising of the manuscript. Jian-Hui Zhang, Yong Liu, and Jing-Xin Li contributed to study supervision. Jia-Ping Yu and Wen-Rong Yao contributed to the design of the investigational vaccine. All authors reviewed and approved the final manuscript.
Data sharing statement
Data Sharing Statement will be available with the full text of this article upon publication.
Supplemental Material
Download Zip (1.6 MB)Acknowledgments
For Study-1, we are grateful for all investigators at AI Kuwait Hospital, Dubai UAE who contributed to the site work of the trial. For Study-2, we are grateful for all investigators at The Health Centrum, Roxas City, St. Paul’s Hospital, Iloilo City, The Medical City Iloilo, University of the Philippines, Philippine General Hospital who contributed to the site work of the trial. For both studies, we are grateful for all investigators at the central laboratory G42 laboratory LLC, the United Arab Emirates, and investigators at central laboratory Gobond Testing Technology (Beijing) Co., Ltd., China, who performed laboratory tests of the trials, respectively. We thank all the subjects in two clinical studies for their dedication to the trials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14760584.2024.2334423
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Pérez-Then E, Lucas C, Monteiro VS, et al. Neutralizing antibodies against the SARS-CoV-2 delta and omicron variants following heterologous CoronaVac plus BNT162b2 booster vaccination. Nature Med. 2022;28(3):481–485. doi: 10.1038/s41591-022-01705-6
- World Health Organization. Summary status of COVID-19 vaccines within WHO EUL-PQ evaluation process. 2024 Jan 25. https://www.american-club.com/files/files/WHO_COVID-19_EUL_23-March-2021.pdf
- World Health Organization. WHO roadmap on uses of COVID-19 vaccines in the context of omicron and high population immunity: an approach to optimize the global impact of COVID-19 vaccines at a time when omicron and its sub-lineages are the dominant circulating variants of concern, based on public health goals, evolving epidemiology, and increasing population-level immunity, irst issued 20 October 2020, updated: 13 November 2020, updated: 16 July 2021, updated: 21 January 2022, updated: 30 March 2023, latest update: 10 November 2023[M]//WHO roadmap on uses of COVID-19 vaccines in the context of omicron and high population immunity: an approach to optimize the global impact of COVID-19 vaccines at a time when omicron and its sub-lineages are the dominant circulating variants of concern, based on public health goals, evolving epidemiology, and increasing population-level immunity, first issued 20 October 2020, updated: 13 November 2020, updated: 16 July 2021, updated: 21 January 2022, updated: 30 March 2023, latest update: 10 November 2023. 2023.
- Luvira V, Pitisuttithum P. Effect of homologous or heterologous vaccine booster over two initial doses of inactivated COVID-19 vaccine. Expert Rev Vaccines. 2024;23(1):283–293. doi: 10.1080/14760584.2024.2320861. just-accepted.
- Zuo F, Abolhassani H, Du L, et al. Heterologous immunization with inactivated vaccine followed by mRNA-booster elicits strong immunity against SARS-CoV-2 omicron variant. Nat Commun. 2022;13(1):2670. doi: 10.1038/s41467-022-30340-5
- World Health Organization. WHO SAGE roadmap on uses of COVID-19 vaccines in the context of OMICRON and substantial population immunity: an approach to optimize the global impact of COVID-19 vaccines at a time when omicron and its sub-lineages are the dominant circulating variants of concern, based on public health goals, evolving epidemiology, and increasing population-level immunity, first issued 20 October 2020, updated: 13 November 2020, updated: 16 July 2021, update: 21 January 2022. 2023 Mar 30.
- Zhang L, Cao L, Gao XS, et al. A proof of concept for neutralizing antibody-guided vaccine design against SARS-CoV-2. Natl Sci Rev. 2021;8(8):nwab053. doi: 10.1093/nsr/nwab053
- Yu J, Yao W, Hu Y, et al. A trimeric NTD and RBD SARS-CoV-2 subunit vaccine induced protective immunity in CAG-hACE2 transgenic mice and rhesus macaques. bioRxiv. 2021-11
- Thomas SJ, Moreira ED Jr, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine through 6 months. N Engl J Med. 2021;385(19):1761–1773. doi: 10.1056/NEJMoa2110345
- Polack FP, Thomas SJ, Kitchin N, et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N Engl J Med. 2020;383(27):2603–2615. doi: 10.1056/NEJMoa2034577
- Barbara L, Miriam S. Safety platform for emergency vaccines-D2.3 priority list of adverse events of special interest: COVID-19. 2020. https://www.bing.com/ck/a?!&&p=5e2cd235b068b450JmltdHM9MTcxMTMyNDgwMCZpZ3VpZD0xNGExOTM5Yy1lOTQzLTY3ZDAtM2ZkZC04NzhmZTgyNTY2YWQmaW5zaWQ9NTE5MA&ptn=3&ver=2&hsh=3&fclid=14a1939c-e943-67d0-3fdd-878fe82566ad&psq=SPEAC+D2.3&u=a1aHR0cHM6Ly96ZW5vZG8ub3JnL3JlY29yZHMvNjY5NzI5Mi9maWxlcy9TUEVBQ19EMi4xX1YyLjBfQ09WSUQtMTlfMjAyMDA1MjVfcHVibGljLnBkZg&ntb=1
- China National Medical Product Administration. Guidelines for adverse event classification standards for clinical trials of preventive vaccines. 2019. https://www.nmpa.gov.cn/xxgk/ggtg/ypggtg/ypqtggtg/20191231111901460.html
- Deng J, Ma Y, Liu Q, et al. Comparison of the effectiveness and safety of heterologous booster doses with homologous booster doses for SARS-CoV-2 vaccines: a systematic review and meta-analysis. Int J Environ Res Public Health. 2022;19(17):10752. doi: 10.3390/ijerph191710752
- Roa CC Jr, de Los Reyes MRA, Plennevaux E, et al. Superior boosting of neutralizing titers against omicron SARS-CoV-2 variants by heterologous SCB-2019 vaccine vs. A homologous booster in CoronaVac-primed adults. J Infect Dis. 2023;228(9):1253–1262. doi: 10.1093/infdis/jiad262
- Zhao X, Zhang R, Qiao S, et al. Omicron SARS-CoV-2 neutralization from inactivated and ZF2001 vaccines. N Engl J Med. 2022;387(3):277–280. doi: 10.1056/NEJMc2206900
- Kaabi NA, Yang YK, Liang Y, et al. Safety and immunogenicity of a broad-spectrum mosaic vaccine as a booster dose against SARS-CoV-2 omicron and other circulating variants. medRxiv. 2022;2022.09. 05.22279589.
- Cheng SM, Mok CKP, Leung YW, et al. Neutralizing antibodies against the SARS-CoV-2 omicron variant BA.1 following homologous and heterologous CoronaVac or BNT162b2 vaccination. Nature Med. 2022;28(3):486–489. doi: 10.1038/s41591-022-01704-7
- Wan EYF, Mok AHY, Yan VKC, et al. Effectiveness of BNT162b2 and CoronaVac vaccinations against SARS-CoV-2 omicron infection in people aged 60 years or above: a case-control study. J Travel Med. 2022;29(8):1–12. doi: 10.1093/jtm/taac119
- Ranzani OT, Hitchings MD, de Melo RL, et al. Effectiveness of an inactivated COVID-19 vaccine with homologous and heterologous boosters against omicron in Brazil. Nat Commun. 2022;13(1):5536. doi: 10.1038/s41467-022-33169-0
- Chi WY, Li YD, Huang HC, et al. COVID-19 vaccine update: vaccine effectiveness, SARS-CoV-2 variants, boosters, adverse effects, and immune correlates of protection. J Biomed Sci. 2022;29(1):1–27. doi: 10.1186/s12929-022-00853-8
- Zou J, Kurhade C, Patel S, et al. Neutralization of BA.4-BA.5, BA.4.6, BA.2.75.2, BQ.1.1, and XBB.1 with bivalent vaccine. N Engl J Med. 2023;388(9):854–857. doi: 10.1056/NEJMc2214916
- Link-Gelles R, Ciesla AA, Roper LE, et al. Early estimates of bivalent mRNA booster dose vaccine effectiveness in preventing symptomatic SARS-CoV-2 infection attributable to omicron BA.5 and XBB/XBB.1.5 related sublineages among immunocompetent adults – increasing community access to testing program, US, Dec 2022 – Jan 2023. Morbidity Mortality Weekly Rep. 2023;72(5):119.
- Carabelli AM, Peacock TP, Thorne LG, et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nature Rev Microbiol. 2023;21(3):162–177. doi: 10.1038/s41579-022-00841-7
- Tregoning JS, Flight KE, Higham SL, et al. Progress of the COVID-19 vaccine effort: viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat Rev Immunol. 2021;21(10):626–636. doi: 10.1038/s41577-021-00592-1