
ABSTRACT
Background
Vaccination remains the cornerstone of defense against COVID-19 globally. This study aims to assess the safety and immunogenicity profile of innovative vaccines LYB001.
Research design and methods
This was a randomized, double-blind, parallel-controlled trial, in 100 healthy Chinese adults (21 to 72 years old). Three doses of 30 or 60 µg of SARS-CoV-2 RBD-based VLP vaccine (LYB001), or the SARS-CoV-2 RBD-based protein subunit vaccine (ZF2001, control group) were administered with a 28-day interval. Differences in the incidence of adverse events (AEs) and indicators of humoral and cellular immunity among the different groups were measured.
Results
No severe adverse events were confirmed to be vaccine-related, and there was no significant difference in the rate of adverse events between the LYB001 and control group or the age subgroups (p > 0.05). The LYB001 groups had significantly higher or comparable levels of seroconversion rates, neutralization antibody, S protein-binding antibody, and cellular immunity after whole vaccination than the control group.
Conclusions
Our findings support that LYB001 developed on the VLP platform is safe and well tolerated with favorable immunogenicity for fundamental vaccination in healthy adults. Therefore, further larger-scale clinical studies are warranted.
Trial Registration
This trial was registered with ClinicalTrials.gov (NCT05552573).
1. Introduction
As of 10 March 2023, there are more than 676 million people confirmed as COVID-19 (Coronavirus Disease 2019) patients and over 6.8 million deaths have been caused by SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2) according to the Coronavirus Resource Center at Johns Hopkins University, since the end of 2019. The total administered vaccine dose has covered over 13.3 billion people worldwide [Citation1]. Although the infection rate, morbidity, and mortality of COVID-19 have been significantly reduced with mutations in the viral strain, it is imperative to implement booster vaccination or sequential immunization. Global vaccine production is limited and does not meet the global demand for enhanced vaccination or strategic stockpiles in the post-COVID-19 era.
Global epidemic control and quarantine policies are no longer implemented. However, there is still a 1.11–6.02% risk of reinfection of the public with SARS-CoV-2 or its mutated strains [Citation2]. The elusion and hesitation of the public to receive vaccination or booster doses of COVID-19 may have resulted from their mistrust due to the uncertainty of the safety and efficacy of the vaccine used at that time [Citation3]. However, the emergence of novel SARS-CoV-2 variants remains unpredictable. Vaccination remains the cornerstone of defense against COVID-19. It is urgent and necessary to explore safer and more effective vaccines with longer protection periods to combat this pandemic.
According to a WHO publication, there were 11 COVID-19 vaccines available for emergency use worldwide and 183 vaccines in clinical development as of 10 March 2023 [Citation4]. These vaccines have been developed based on different platforms, such as protein subunits, viral vectors (non-replicating), RNA, and inactivated viruses. According to current vaccination strategies, mRNA and recombinant proteins have a high efficacy of over 90% against COVID-19 [Citation5]. However, there may be higher costs and complexity in the production and storage of mRNA vaccines than recombinant vaccines. In addition, recombinant protein vaccines perform better in terms of safety and protective efficacy than inactivated vaccines widely administered in China [Citation6]. The advantages of the receptor-binding domain (RBD) for immune protection, production, and manufacturing as a COVID-19 vaccine target and virus-like particle (VLP) platform are used against SARS-CoV-2 and its variants [Citation6–8]. Owing to their high efficacy in delivering antigens to the immune system and inducing strong humoral and cellular responses, VLP technologies have achieved widespread success in the development of several vaccines, including hepatitis B virus and human papillomavirus vaccines [Citation9]. The safety and immunogenicity of the RBD-based protein subunit vaccine (ZF2001, Emergency Use Authorization/EUA in China) have been reported with high immunity and good safety from the phase 1 and 2 trials in China [Citation10]. However, scarce real-world data have been published on the RBD-VLP vaccine, and most are undergoing clinical trials because of the complex manufacturing process of the VLP vaccine [Citation6,Citation8–11]. In response to the COVID-19 pandemic, we launched a vaccine development and manufacturing program against SARS-CoV-2; LYB001 is a protein subunit vaccine targeting the RBD of the SARS-CoV-2 protein that has advanced into phase III clinical trials (Registration No. on ClinicalTrial.gov: NCT05663086, NCT05552573, NCT05683600, NCT05664932). The candidate vaccine, LYB001, was based on the VLP platform developed by Guangzhou Patronus Biotech Limited and Yantai Patronus Biotech Limited.
Here, we report the results of a clinical trial phase 1 on an RBD-based VLP vaccine in a Chinese adult population, where we investigated the safety and immunogenicity of the LBY001 vaccine on fundamental immunity in healthy, young, and older Chinese participants.
2. Methods
2.1. Ethics statement
This Clinical trial was approved by the Ethics Committee of Jiangsu Provincial Center for Disease Control and Prevention (No. JSJK2022-A029-01), validity from May 2022 to April of 2023. All the participants provided written informed consent. This study was conducted in collaboration with the Jiangsu Provincial Center for Disease Control and Prevention (Public Health Research Institute of Jiangsu Province) and was guided by the National Medical Products Administration (NMPA) and Good Clinical Practice.
2.2. Participants and enrollment
This single-center, randomized, double-blind, positive-parallel-controlled phase 1 study was conducted at Dongtai city Center for Disease Control and Prevention, Jiangsu Provincial, China. Investigators recruited the individuals publicly by issuing or posting recruitment notices approved by the Ethics committee of Jiangsu Center for Disease Control and Prevention. All participants were in good health with acceptable results on medical history, physical examination, and laboratory tests after screening. They were assigned to the younger cohort (aged 18–59 years) and the elderly cohort (aged ≥60 years) as 1:1 equally in all experiment and control subgroups. This was the first in-human trial of LYB001 that used a dose-escalation and age-sequential enrollment design. The participants were enrolled as aged 18–59 years first, and followed by those aged ≥60 years, with the low dose first, and then the high dose in each age group. The target of our research were healthy Chinese adults (aged ≥18 years) with normal body temperature (≤37.0°C), and no vaccination and infection history of any SARS-CoV-2 vaccine with a negative result for the IgG and IgM antibodies detection of wild-type SARS-CoV-2, who volunteered to participate in this clinical trial and all follow-up visits.
2.3. Exclusion criteria
Participants who met any of the following conditions were excluded from the trial. 1) Allergic to LYB001 or its excipients; 2) has a history of allergy or serious adverse reactions to other vaccines; 3) has a history of infection with SARS or MERS or a confirmed/suspected COVID-19 patient; 4) tested positive for SARS-CoV-2 nucleic acid and antibody testing (IgG; IgM); 5) had taken antipyretic, analgesic, or anti-allergic drugs within 24 h before enrollment; 6) had received vaccination within 14 days before enrollment; 7) had used blood-related products within three months before enrollment; 8) has a history of related diseases (such as infectious diseases, hepatitis B, mental disorders, immune system disorders, serious cardiovascular and cerebrovascular diseases or tumors); 8) lactating or pregnant women.
2.4. Randomization and masking
To balance baseline characteristics across all subgroups and increase confidence in the statistical analysis, randomization was applied in this clinical study. Based on stratified block randomization, a random list was generated by an independent statistician using SAS version 9.4. After the eligibility screening, the subject numbers were obtained from small to large and stratified by age from clinical site. Eligible participants were enrolled and assigned into two age subgroups: the younger subgroup aged 18–59 years and the elderly subgroup aged ≥60 years at a ratio of 1:1. All participants were administrated randomly in three groups in a 2:2:1 ratio to receive three-dose fundamental immunization of either 30 µg LYB001 or 60 µg LYB001, or 60 µg ZF2001.
Double blinding was used in this trial to reduce potential bias during data collection and evaluation of study endpoints. An authorized, unblinded staff member prepared the vaccines according to the participant’s assignment using the interactive response technology system, and a nurse administered the study vaccine to the participant. Unblinded staff members did not participate in the trial and were prohibited from disclosing assignment information. All other investigators, participants, laboratory staff, and sponsors were blinded throughout the trial and data analysis.
2.5. Procedure and materials
Each participant received one dose of 60 or 30 µg LYB001 as two optimal dosage selected from our preclinical studies on safety and immunogenicity, or one dose of ZF2001 by intramuscular injection into the deltoid muscle of the upper arm, with an interval of 28 days (validated on preclinical studies) on days 0, 28, and 56 for a total of three doses with acceptable time window of each timepoint as the protocol required. The vaccines used for participants in phase 1 of the clinical trial were two-dose designs (60 µg/0.5 ml/bot, 30 µg/0.5 ml/bot) of LYB001 manufactured by Yantai Patronus Biotech Limited, and a positive-control vaccine ZF2001 (0.5 ml/bot) from Anhui ZhiFei Longcom Biopharmaceutical Co., Limited. Both vaccines contained aluminum hydroxide adjuvants with the same injection procedure, based on a mechanism similar to that of Chinese hamster ovary (CHO) cells. The main component of LYB001 is RBDM (a combination of NPM-4C expressed in Escherichia coli as VLP carriers and RFD-4T expressed in CHO cells, the purified NPM-4C and RBD-4T were combined to prepare RBDM vaccine stock solution, then adsorption on aluminum hydroxide adjuvants), and the main component of ZF2001 is NCP-RBD (novel coronavirus spike glycoprotein receptor-binding region protein expressed in CHO cells) [Citation6]. The aim of vaccine LYB001 is to prevent the incidence of COVID-19.
In this trial, all participants were observed for 30 minutes, and asked to fill in their dairy card and contact card after each vaccination, and finished required measurement as the protocol for AEs recording including solicited and unsolicited AEs after each vaccination. Laboratory tests were performed before and on day 3 after each dose of vaccination as part of safety evaluation, including coagulation function, blood biochemistry, blood routine, and urine routine tests. Any change of laboratory indicators pre- and post-vaccination was reviewed for the clinical significance by the investigator, and their symptoms was judged whether it was regarded as AEs. Solicited AEs included pain, induration, swelling, rash, erythema, petechiae, and pruritus of local site adverse reactions (ARs, defined as vaccine-related); fever, drowsiness, headache, dizziness, muscle pain, cough, hypersensitivity, petechiae, fatigue, nausea, and vomiting of systemic ARs. In addition, investigators collected all SAEs and AESI from the first dose through 12 months after the full vaccination. All AEs recording material were reviewed by the principal investigator and Data and Safety Monitoring Board (DSMB) of the clinical center in a blinded manner throughout this trial.
Blood samples were collected on day 14 after the second-dose immunization, days 14 and 28, and 3, 6, and 12 months after the third-dose immunization. In this phase 1 trial, we measured serological results using a neutralization test, enzyme-linked immunosorbent assay (ELISA), and enzyme-linked immunospot (ELISPOT) assay. Humoral immunity were assessed on Day 0 (before receiving the primary dose), Day 42 (day 14 after receiving the second dose), Day 70/Day 84 (the 14th and 28th days after the third dose); cellular immunity on day 14 after the second dose and the third dose vaccination, and the anti-VLP antibodies on day 14 and 28 after the third dose vaccination. The demographic characteristics and baseline of all participants screened, enrolled, and dropped out were described for each age subgroup and dose group at different time points. For more details of the methodology used for the immunogenicity assays were showed in Supplementary Methods.
2.6. Outcomes
Our primary objectives in the phase I trial were to observe the safety of our LYB001 vaccine in healthy adults (aged 18 years and above), and secondary objectives were to evaluate the immunogenicity of our LYB001 vaccine. The incidence of solicited local reactions at the injection site and systemic adverse reactions on day 14 after each dose of vaccination and solicited adverse events (AEs) after the whole vaccination on day 28 after three-dose immunization were the primary endpoints. All solicited and unsolicited AEs were recorded during fundamental vaccination. The secondary endpoints for immunogenicity were geometric mean titers (GMTs), geometric mean fold-rises (GMFRs), seroconversion rates, and S protein-binding antibodies of IgG which were measured on days 14 and 28 after the second and third-dose injections. The seroconversion rate was defined as an increase by a factor of 4 or more in the antibody titer over the baseline. This study tends to further evaluate the immunogenicity of VLP vectors in healthy adults.
2.7. Statistical analysis
The phase I trial aimed to assess safety and tolerance and was not designed to power and analyze statistical differences in immunity. This was a small-scale trial in phase 1 with a recommended size range of 20 to 30, guided by The China Food and Drug Administration. Thus, there were 100 participants across three groups, with 40, 40, and 20 participants in the 30 µg LYB001, 60 µg LYB001, and control groups, respectively. All other investigators, participants, laboratory staff, and the sponsor remained blinded throughout the trial until all follow-up visits and reports were finished as the protocol required.
The Medical Dictionary for Regulatory Activities was applied to the medical coding of AEs and severe adverse events (SAEs), and classification statistics were based on system organ class and prothrombin time levels. Adverse events were graded according to the relevant guidelines of NMPA. Solicited and unsolicited AEs were classified according to the manufacturer’s instructions. The time of occurrence, number of doses, and severity of AE were statistically described. The measurement data were statistically described as mean, median, standard deviation (SD), maximum value, and minimum value. Count or grade data were expressed as frequencies and percentages. For spike protein IgG antibody and neutralizing antibody (nAb) titers, the geometric mean titer/concentration (GMT/GMC), geometric mean fold rise (GMFR) from baseline, and seroconversion rate (SCR) were calculated for each group at different follow-up time points.
The Safety Set (SS) was used for safety observations, and immunogenicity analysis was based on the Per-Protocol Set (PPS). In SS and PPS, all participants who followed the ITT principle, completed randomization and had at least one vaccination and one immunogenicity evaluation result in this trial. In this study, the missing data after immunization with humoral immunity, cellular immunity and exploratory VLP antibody levels were imputed by Last Observation Carried Forward method, and further derived to calculate the antibody positive conversion rate and other indicators. Handling of missing data for safety endpoints was not considered. Participants with missing data were reviewed by the investigator and DSMB if added to the SS or the PPS.
In this study, for categorical data, Fisher’s exact test or chi-square analysis was used to compare differences in the incidence of the above adverse events between the groups. The significance level in this study was 0.05 unless otherwise noted. For endpoints of humoral immune responses, their changes in GMTs and GMFR from baseline were tested using the t-test, chi-square test, Fisher’s exact test, and covariance analysis of the log-transformed values of titers and then back-transformed to the original scale for presentation. The differences between groups of cellular immune responses were statistically tested using the Wilcoxon rank-sum test. Confidence intervals (CI) were calculated using a two-sided 95% confidence interval. All analyses were performed using a non-parametric test via SAS software version 9.4 or higher, and figures were analyzed via the GraphPad Prism 9 software (GraphPad Software Inc., California, U.S.A.) using non-parametric tests. There were no adjustment for multiple comparisons or multiplicity in this exploratory trial.
The geometric mean and 95% confidence interval (CI) were used to calculate the GMT/GMC and GMFR of SARS-CoV-2 and the VOC-neutralizing antibody, S protein-binding antibody, and seroconversion rate before receiving the primary dose, on day 14 after receiving the second dose, and on days 14 and 28 after the three-dose vaccination. The nAb titers against the prototype SARS-CoV-2 and dominant VOCs (Omicron BA.4/BA.5) were determined using vesicular stomatitis virus (VSV)-based pseudovirus neutralizing assays and real-virus neutralizing assays. Seroconversion was defined as either a four-fold increase in post-vaccination antibody levels from seropositive (≥ cutoff value) at baseline or a seropositive conversion from seronegative (< cutoff value) at baseline. The participants in each group had negative IgG and IgM antibody test results before the primary vaccination.
The cellular immune response was detected using ELISpot assay, and it was presented as the mean of counts of spot-forming cells (SFCs) per 3 × 105 peripheral blood mononuclear cells (PBMCs) secreting interferon-γ (IFN-γ), interleukin-2 (IL-2), interleukin-4 (IL-4) when stimulated by the antigen peptide pool ex vivo. The levels of cytokines (IFN-γ, IL-2, IL-4) and their changes from baseline to day 14 after the second dose and day 14 after the entire vaccination were statistically described. The titers of anti-VLP antibodies in each group were statistically analyzed before receiving the primary dose and on days 14 and 28 after full vaccination.
3. Results
3.1. Study design and participants
A total of 149 adults were screened from 20 July 2022, to January 2023 at the Dongtai city center for disease control and prevention, Jiangsu, China. However, there were one withdrawal of consent and 48 subjects excluded who met exclusion criteria. There were 2 positive cases on hepatitis B surface antigen test, 10 abnormal cases on ECG, 1 positive case on SARS-CoV-2 nucleic acid test, 1 positive case on blood pregnancy test, 3 syphilitic cases, and 31 cases on abnormal results of laboratory tests such as transaminase, proteinuria, blood glucose and blood platelet disorder.
A total of 100 eligible participants were enrolled based on the enrollment criteria and exclusion criteria, who were assigned into two groups: the younger group (18 to 59 years old) and the elderly group (≥60 years old) and allocated 1:1 randomly in each treatment group. This was a randomized, double-blind, positive-control trial. Participants were randomized to receive three-dose fundamental immunization of LYB001 at 30 µg (aged 18–59: n = 20; aged ≥ 60: n = 20) or 60 µg (aged 18–59: n = 20; aged ≥ 60: n = 20), or three-dose ZF2001 (aged 18–59: n = 10; aged ≥ 60: n = 10) as the positive-control (See ). A sequential enrollment method was used for safety risk management. The younger cohort was enrolled first, followed by the elderly; the low-dose was administered, and the high-dose vaccination was followed in each age subgroup with a 14-day interval for safety observation. Three doses of the vaccine were administered to all participants according to the immunization schedule on days 0, 28, and 56, with an interval of 28 days for each dose.
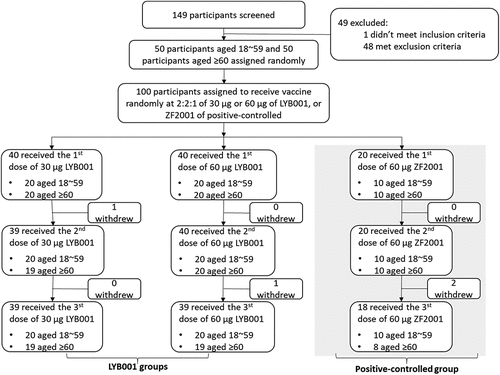
Figure 1. Study flow diagram. from 20 July 2022 to January 2023, a total of 100 eligible participants (age ≥18) out of 149 volunteers screened had been enrolled, who randomly assigned to receive 3 doses totally with LYB001 30 μg (n = 40), or LYB001 60 μg (n = 40) or vaccine ZF2001 (n = 20) at 1:1 in age subgroups (aged 18 ~ 59 years, ≥60 years). Blood samples were collected at day 14 after each vaccination, day 28 after the third dose vaccination to evaluate the post-vaccination immunogenicity. Serological samples were measured by neutralization test, ELISA and ELISPOT essay. Those were collected and assessed on the day of Day 0 (baseline), Day42 (at day 14 after the second dose vaccination), Day 70/Day 84 (the 14th day and the 28th day after the third dose vaccination) based on humoral immunity, cellular immunity and anti-VLP platform. During the second dose vaccination, 1 participant in group of vaccine LYB001 30 μg lost in follow-up visits; during the third dose vaccination, 1 in group of vaccine LYB001 60 μg, and 2 in control group were lost in follow-up visits.
3.2. Baseline information
The demographic characteristics of all participants are shown in . The mean ages of the 30 and 60 µg LYB001 groups and the control group were 44.15 (95% CI: 38.15, 50.16), 48.44 (43.62, 53.25), and 49.59 (40.76, 58.41) years old, respectively, in the younger group, while they were 65.81 (64.53, 67.10), 65.40 (63.87, 66.93), and 67.21 (64.44, 69.97) years old, respectively, in the elderly group. There were 44 males (44%) and 56 females (56%) enrolled throughout the trial, with a balanced gender distribution across all subgroups. There were no significant difference of variables at the baseline (p > 0.05, see ).
Table 1. Demographic characteristics and baseline of all participants in the phase 1 trial.
3.3. Safety and tolerance
The SS was defined as any participant who received at least one dose of a vaccine. All adverse events mostly occurred within 14 days after each vaccination as solicited AEs. The overall frequency of solicited AEs (e.g. fever, pain, etc.) did not significantly differ between 30 and 60 µg LYB001 groups and the control group within 14 days after each vaccination, at 50% (20/40), 55% (22/40), and 40% (8/20), respectively. The overall frequency of AEs was low from receiving the primary dose to day 28 after the whole vaccination for all participants in the phase 1 trial including solicited and unsolicited AEs; the AE rates were 45% (18/40), 47.5% (19/40), and 40% (8/20) for the 30 µg, 60 µg, and control groups, respectively, without a statistically significant difference (p > 0.05). Most AEs were mild or moderate (Grade ≤2) who resolved after several days without any medicine or treatment, where there were only two severe events (Grade = 3), one in the 60 µg LYB001 group (thrombocytopenia, resolved after 6 days; swelling and erythema, resolved after 12 days) and the other in the control group (swelling and erythema, resolved after 4 days). In addition, no deaths, (SAE, or suspicious and unexpected severe adverse reactions related to LYB001 occurred in the phase 1 trial. In the younger groups, the solicited adverse event rates of the 30 µg, 60 µg, and control groups were 60% (12/20), 65% (13/20), and 40% (4/10), respectively, without statistical significance, for the whole vaccination and observation period. There were mainly mild or moderate solicited AEs in the LYB001 groups and only one grade-3 event (local AE in the form of swelling and erythema, resolved after 4 days) in the control group. In the elderly group, the solicited AE rates were 40% (8/20), 45% (9/20), and 40% (4/10), respectively, of the 30 µg LYB001, 60 µg LYB001, and ZF2001 control groups without significant statistical difference (p > 0.05), with only two grade-3 cases in the 60 µg LBY001 group in the form of thrombocytopenia and swelling (resolved after 6 days), and erythema (resolved after 12 days). The common ARs that were reported were injection site pain (29 out of 100), swelling (13 out of 100), pruritus (14 out of 100), and fatigue (8 out of 100) where they could be resolved after few days without medicine or treatment. There were no significant differences between the younger and older groups of local ARs and systemic ARs (p > 0.05). There was no significant difference in the rate of clinically significant changes in blood routine, blood biochemistry, coagulation function, and urine routine between groups, which was consistent with the results of ARs. In addition, there were no AEs leading to dropout, death, SAE or Suspected and Unexpected Serious Adverse Reactions from the first dose to 28 days after the whole vaccination in this study.
On day 28 of the first or the second or the third vaccination (see ), there were no significant differences were observed on local and systemic ARs (p > 0.05) in the 60 µg LBY001, 30 µg LYB001, and positive-control groups in the younger cohort (aged 18–59) and the elderly cohort (aged ≥60).
Table 2. The solicited adverse events (AEs)* and adverse reactions (ARs)* from the first dose to the 28th day after three dose vaccination, and unsolicited adverse events based on NMPA guidelines by age and dose subgroups.
3.4. Humoral immunogenicity
The PPS included all participants who completed the three-dose vaccination without major schedule deviation/violations and who had valid immunogenicity results from baseline to day 28 after the full course of vaccination. In this phase 1 trial, we measured antibody responses based on serological RBD-binding IgG titers by ELISA. The baseline titers of the participants are shown in (Tables S1 and S2). The SARS-CoV-2 antibody responses were assessed on Day 0 (before the first vaccination), Day 42/Day 56 (14 and 28 days after the second vaccination), Day 70/Day 84 (14 and 28 days after the third vaccination) based on humoral immunity, cellular immunity, and the anti-VLP platform.
Figure 2. LYB001 induced significantly 50% live-virus and pseoduvirus-neutralizing antibody responses, compared with control group of vaccine ZF2001 by dose and age subgroups. a – d, Serum samples were collected at baseline (before prime vaccination), day 42 (at day 14 after the second dose vaccination), and day 70 and 84 (at day 14 and 28 after whole vaccination) in the LYB001 groups of 30 μg and 60 μg, and control group by the younger cohort and elderly cohort. a-b, A panel of real-virus immune responses of 50% SARS-CoV-2-neutralizing antibody GMTs measured by ELISA assay are demonstrated. c-d, A panel of pseoduvirus immune responses of 50% SARS-CoV-2-neutralizing antibody GMTs measured by ELISA assay are shown. Each point represents a serum sample, and each bar represents the mean of GMT with 95% CI by using Clopper-Pearson method. The dashed line indicates the lower limit of quantification (LLOQ). The LLOQ was 1:10 for the virus-neutralizing antibody test and 1:100 for ELISA. Neutralizing antibody responses of participants in each dose group of LYB001 were compared to the control group using analysis of covariance with log transformation. GMT as geometric mean titers; nAb as neutralizing antibody; TCID50 as 50% tissue culture infective Dose; WT as wild type strain of SARS-CoV-2; BA.4/5 as BA.4 or BA.5 of omicron strain of SARS-CoV-2; YOA as year of age; D0 as the baseline; 14d as 14 days; 28d as 28 days.
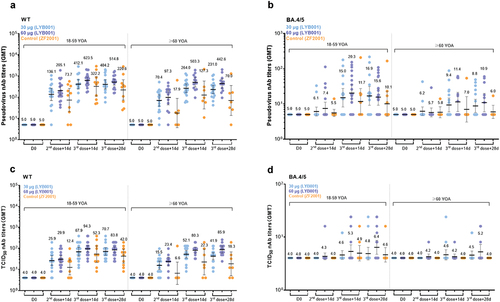
SCRs increased into 100% of the 60 µg and 30 µg LYB001 groups at Day70 of the wild-type pseudovirus platform in each age subgroup (see Table S1). At Day84, the SCRs of the LYB001 groups were higher than that of the control group (p = 0.0041) for all participants aged 60 years or above, at 100.00% (95% CI: 81.47%, 100.00%) in the 60 µg LYB001 and 30 µg LBY001 groups, and 57.14% (18.41%, 90.10%) in the control group. There was no significant difference between the 60 µg and 30 µg LYB001 groups at Day70 and D84 of the wild-type pseudovirus and live platform (see Table S1 and S2). The same trend of the wild-type live-virus testing in the elderly cohort (see Table S2) indicated that LYB001 had significantly higher SCRs than the control group at Day70 (p = 0.0019).
In the titration experiment, the live virus neutralization titer of wild-type SARS-CoV-2 was defined as the reciprocal of the highest sample dilution that protected at least 50% of the cells from cytopathic effects. In the younger cohort (aged 18–59), the 50% neutralizing GMTs in the 60 µg LBY001, 30 µg LYB001, and control groups were 94.32 (67.23, 132.33), 67.86 (41.55, 110.81), and 52.33 (21.05, 130.09), respectively, at day 14 after the third dose of vaccination (Day 70). The GMFR of neutralizing antibody (nAb) titers increased of the three groups on the live-virus platform (see Table S1). In the elderly cohort (aged ≥60), the GMTs at Day 70 in the 60 µg LBY001, 30 µg LYB001, and control groups were 73.83 (51.01, 106.87), 52.14 (34.34, 79.19), and 22.27 (5.49, 90.33), respectively, in . The GMFRs at Day 70 were 18.46 (12.75, 26.72), 13.04 (8.58, 19.80), and 5.57 (1.37, 22.58) in the 60 µg LYB001, 30 µg LYB001, and control groups, respectively, compared to the baseline. The LYB001 group had significantly higher levels of GMTs and GMFRs in nAb titers than the control group (p = 0.0101 at Day84). For Omicron BA.4/5, no significant changes were observed after vaccination in the live virus assay in (see Table S1).
In the pseudovirus assay, the GMTs of nAbs increased at D42, D70, and D84 (see and Table S2) compared to that of the baseline. In the younger cohort, the GMTs of the 60 µg LYB001, 30 µg LYB001, and control groups were 623.50 (444.37, 874.85), 412.11(246.17, 689.90), and 322.19 (112.55, 922.29), respectively, at D70, respectively. The GMFRs were 124.70 (88.87, 174.97), 82.42 (49.23, 137.98), and 64.44 (22.51, 184.46) at D70 compared to the baseline in the 60 µg LYB001, 30 µg LYB001, and control groups, respectively. In the elderly cohort, the GMTs had the same trend chances from the baseline at 503.34 (304.71, 831.44), 263.96 (166.01, 419.72), and 127.33 (28.05, 578.12) at D70 in the 60 µg LYB001, 30 µg LYB001, and control groups, respectively. There were significantly higher levels of GMTs and GMFRs in nAb titers at D84 in the LYB001 groups than that of the control group (p = 0.0036) (Table S2). For Omicron BA.4/5, there were no significant changes in any age subgroup after vaccination in the pseudovirus assay in (Table S2). Given the vaccine LYB001 was designed against the prototype strain without antigen of Omicron strain. In this clinical study, the outcomes of neutralizing antibodies induced by the vaccine LYB001 were weak in , although the vaccine demonstrated strong cross-neutralizing responses against the Omicron strain in preclinical animal models [Citation12].
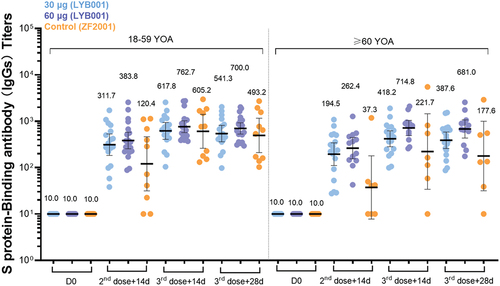
The GMCs (95% CI) for S protein-binding antibodies against SARS-CoV-2 by ELISA increased from Day42 to Day70 ( and Table S3). In the younger cohort (aged 18–59), the GMCs were 762.70 (568.53, 1023.19), 617.82 (406.70, 938.53), and 605.15 (261.05, 1402.86) in the 60 µg LYB001, 30 µg LYB001, and control groups, respectively. In the elderly cohort (aged 60 or above), the GMCs were 714.83 (491.45, 1039.74), 418.19 (281.62, 621.00), and 221.69 (33.89, 1449.99) in the above three groups, respectively. Both LYB001 groups had higher titers of SCRs and GMFRs than the control group. In addition, GMCs and GMFRs on Day84 were higher than those on Day42. All groups had 100% SCRs at Day70.
Figure 3. LYB001 induced strong S protein-binding antibody responses, compared with control group of vaccine ZF2001 by dose and age subgroups. serum samples were collected at baseline (before prime vaccination), day 42 (at day 14 after the second dose vaccination), and day 70 and 84 (at day 14 and 28 after whole vaccination) in the LYB001 groups of 30 μg and 60 μg, and control group by the younger cohort and elderly cohort. GMTs measured by ELISA assay are demonstrated. Each point represents a serum sample, and each bar represents the mean of GMT with 95% CI by using Clopper–Pearson method. The dashed line indicates the lower limit of quantification (LLOQ). The LLOQ was 1:10 for the virus-neutralizing antibody test and 1:100 for ELISA. S protein binding antibody titers of participants in each dose group of LYB001 were compared to the control group using analysis of covariance with log transformation.
3.5. Cellular immune responses
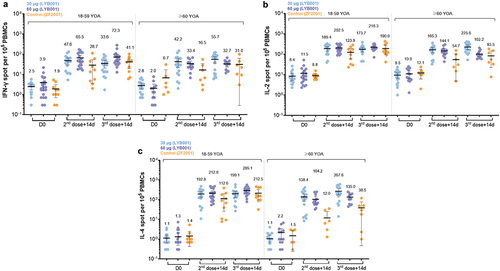
In order to observe T-cell immune responses and the symptoms of COVID-19 against SARS-CoV-2 infection and severe cases, this trial explored T-cell immune responses induced by LYB001 based on the cytokine levels of IFN-γ (Th1 type), IL-2 (Th1 type), and IL-4 (Th2 type) using the ELISPOT assay. In the younger cohort (aged 18–59), at Day42, the mean of IFN-γ levels in the 60 µg LYB001 group was 65.48 (95% CI: 42.41, 88.56) and 47.57 (28.96, 66.18) in the 30 µg LYB001 group, which were higher than that of the control group at 28.70 (3.60, 53.80), as indicated in (see Table S4). The 60 µg LYB001 groups had higher levels of IFN-γ, IL-2 (Th1 type), and IL-4 (Th2 type) than the control group at Day 42 and Day 70 (Table S4). In the elderly cohort (aged ≥60), the mean of IL-2 (Th1 type) and IL-4 in the 60 µg and 30 µg LYB001 groups were higher than that of the control group at Day 42 and Day 70 (p < 0.001, Table S4). In addition, for all participants, the mean of IFN-γ, IL-2, and IL-4 increased significantly from the baseline; furthermore, the LYB001 groups had higher levels of these molecules compared to the control group at Day 42 and Day 70 and the baseline.
Figure 4. BLYB001 induced strong T cell responses in vaccinated participants compared with control group. blood specimen obtained on day 0 (baseline pre-vaccination), 42 (at day 14 in 2-dose post-vaccination) and 70 (at day 14 in whole vaccination) were stimulated overnight with overlapping peptide pools representing different portions of the wild-type sequence of SARS-CoV-2 for assessment in direct ex vivo IFN-γ ELISpot assay. Each dot represents the mean spot count from triplicate wells for one participant, after subtraction of the medium-only control by age subgroups. a-c, SARS-CoV-2 specific T cell response of IFN-γ (A), IL-2 (b), and IL-4(C) in younger (aged 18–59 years) and elderly (aged ≥60 years) participants are shown. Comparison of each cytokines between dose groups of LYB001 and control group were tested by using Kruskal-Wallis rank sum test based on geometric mean spot with 95% CIs.
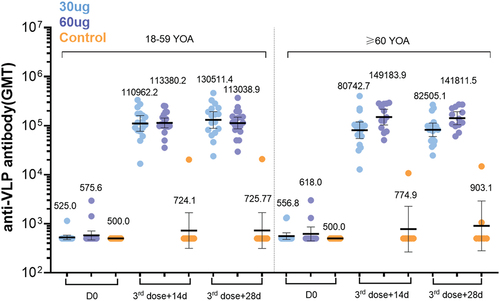
3.6. Anti-VLP antibody
These VLP nanoparticles can efficiently stimulate antigen-presenting cells, and lymphaden B and T cells for a long time and maximize the induced neutralizing antibody titer and immune memory, indicating a strong immune response. The GMTs after full vaccination increased approximately 200- to 250-fold from the baseline (see and Table S5). This approach overcomes the bottleneck of low immunogenicity caused by the small spatial structure and molecular weight of conventional recombinant protein vaccines.
Figure 5. LYB001 elicited strong anti-VLP immune responses, compared with control group of vaccine ZF2001 by dose and age subgroups. serum samples were collected at baseline (before prime vaccination), day 70 (at day 14 after whole vaccination), and day 84 (at day 28 after whole vaccination) in the LYB001 groups of 30 μg and 60 μg, and control group by the younger cohort and elderly cohort. GMTs measured by ELISA assay are demonstrated. Each point represents a serum sample, and each bar represents the mean of GMT with 95% CI by using Clopper-Pearson method. Dose groups of LYB001 were compared to the control group using analysis of variance and ANOVA SNK for pairwise comparison of post hoc test.
4. Discussion
The phase 1 clinical trial of the LYB001 vaccine has been started at 19 July 2022 and completed on 28 October 2023 in Jiangsu Province, China. This candidate vaccine for COVID-19 strongly elicited multilevel immune responses of neutralizing antibodies, S protein-binding antibodies, and T-cell immune responses based on pseudovirus assays and live-virus assays in the clinical trial.
SARS-CoV-2 causes respiratory infections mainly by converting Angiotensin-Converting Enzyme 2(ACE2) in host cells, which binds to the surface S protein of SARS-CoV-2. The S protein can induce a protective immune response, and most epitopes of neutralizing antibodies are located in the RBD of the S protein. Therefore, inhibition of the interaction of spike-RBD with ACE2 plays a prominent role in COVID-19 vaccine development and SARS-CoV-2 neutralization techniques. In the vaccine LYB001, the highly repeated display of these low immunogenic antigen proteins on VLP surface particles can be very effective in tandem with receptors on the surface of B cells and highly stimulate B cells to produce and secrete antibodies. Additionally, the particle size of the VLP in our vaccine LYB001 is between 20 and 60 nm, which is very small and can be effectively trapped by lymph nodes, so it can continuously stimulate immune cells in vivo for a long time and achieve a very high level of durable immune response. RBD-based vaccine with S protein is safer since there is no SARS-CoV-2 in vivo (viral vector vaccines) or genes are directly injected into the human body (mRNA vaccines). RBD-based COVID-19 subunit vaccine candidates developed by Anhui Zhifei Longcom, BioNTech, and Pfizer have been shown to exhibit strong immune responses and protective efficacy against COVID-19, with good safety [Citation8]. However, clinical data from healthy Chinese volunteers are limited. In this phase 1 trial, vaccination with LYB001 with the 30 or 60 μg doses and two- or three-dose schedules were well-tolerated with good safety. Generally, there was no significant difference observed between 30 μg and 60 μg LYB001 groups on immunogenicity and safety in this study.
Subunit vaccines usually require appropriate adjuvants to improve immunogenicity and efficacy, which is a key process in enhancing and prolonging immune responses. Our vaccine, LYB001, was based on genetic recombination to express its VLP vector and the RBD protein of the SARS-CoV-2 prototype strain in Escherichia coli and CHO cells, respectively, based on the highest production capacity. Then, the VLP vector and RBD protein were assembled and adsorbed on aluminum hydroxide as an adjuvant by purification with sucrose and Tris-CHI, without antibiotics and antibacterial agents. Aluminum is one of the most commonly used vaccine adjuvants among emulsions, microparticles, polysaccharides, immune-stimulating complexes, liposomes, and cytokine adjuvants as the only approved vaccine adjuvant in China. Although there are still some controversies regarding the application of aluminum in COVID-19 vaccines owing to their lower immunogenicity based on subunit vaccines [Citation13], aluminum salts have been used worldwide in vaccine formulations for a long time with good safety and immunogenicity [Citation14,Citation15], regardless of whether it is an animal model [Citation16,Citation17] or a human trial [Citation18,Citation19]. Our study showed a lower rate of adverse events and strong immune responses against wide-type strain of SARS-CoV-2, which indicates that the alum-based adjuvant is a favorable choice for LYB001 vaccine formulation with greater safety and immunogenicity in the human body. For optimize adjuvant use in vaccine, we also developed a new adjuvant, which was applied to the broad-spectrum vaccines with favorable performances of safety and immunogenicity. The immunogenicity of vaccine LYB001 against the BA.4/5 variant was not favorable in this study, which might be caused by the limitation of antigenic component (prototype strain of SARS-CoV-2 only) and immune escape from prototype. However, the immunogenicity of our VLP vaccines against the variant strains and our new adjuvant will be further confirmed by the study of our new multivalent vaccines.
With persistent infections caused by SARS-CoV-2 variants, more efficient broad-spectrum vaccines have become the focus of clinical research [Citation20]. Currently, the Mosaic-8VLP broad-spectrum nanoparticle vaccine containing eight different SARS-like β-coronavirus fragments from the California Institute of Technology in the United States is the primary frontier representative to provide broad protection against coronaviruses in animal models [Citation21]. Preclinical studies have shown that the neutralizing antibodies induced by the VLP nanoparticle vaccine have cross-neutralizing effects on other β-coronaviruses in the sarbecovirus species [Citation12,Citation22]. Domestic and international studies have reported that SARS-CoV-2 vaccines based on recombinant protein nanoparticle technology induce higher levels of neutralizing antibodies and immunogenicity than mRNA vaccines [Citation6–8]. The main component of RBDM in LYB001 has a strong binding affinity for the human ACE2 receptor, which improves the biological activity of the antigen. Moreover, compared with the RBD protein and the S-trimer, RBDM could induce stronger neutralizing antibody responses against variants of concern (VOCs) of SARS-CoV-2 in animal models, resulting from the VLP that maximized the exposure of ‘neutralizing epitopes’ of the antigen molecules that are easily recognized by the immune system [Citation23]. The VLP vector in our vaccine, LYB001, is a regular dodecahedral protein multimer with a diameter of 26 nm, which could stay in the lymph nodes for a long time to stimulate immune cells, including B cells and T cells, because of its small size. Furthermore, multiple antigen molecules displayed on the surface of the VLP vector can cross-link with B cell receptors to activate B cells and produce antibodies directly. This suggests that VLP nanoparticles are the most promising technology for achieving broad-spectrum protection against COVID-19 in the post-COVID-19 era.
Through the proprietary ‘Covalink®’ technology, the recombinant expressed SARS-CoV-2 antigens were displayed on the surface of VLP nanoparticles through protein conjugation, and a safe, stable, efficient, and high-yield recombinant SARS-CoV-2 vaccine, LYB001, against the SARS-CoV-2 prototype strain was developed, and broad-spectrum SARS-CoV-2 vaccines are currently under development. Through this ‘Covalink®’ platform, we can separate the VLP particle skeleton and antigen production, so that the modular preparation process can achieve their own optimal expression system and purification process. It has broken through the bottleneck of the industrialization of VLP nanoparticles. Several VLP vaccine candidates for SARS-CoV-2 with an AS03 adjuvant elicited strong neutralizing antibody responses in clinical trials [Citation24–26]. One study was conducted in Latin America in 2020, where the GMT was more than ten-fold higher than that of convalescent sera in 2019 [Citation24], one in South Korea [Citation25], and another in the Netherlands [Citation26]. Our SARS-CoV-2 vaccine LYB001 also had a higher GMT after three-dose primary immunization than convalescent sera at 58.3 (95% CI: 35.1, 96.8) in the live wild-type virus neutralizing assay. It might suggest that vaccine LYB001 could provide an over 84% efficacy against the wild-type of SARS-CoV-2 [Citation27].
There were some limitations on representativeness of target population to this study due to the small sample size and the restriction of the Chinese population to a single center. Serum analysis of immune responses was performed using a pseudovirus-based assay, which recommends a larger-scale study of phase III trials for real-world detection with prototype and VOC strains. In addition, the cellular immune responses elicited by RBD-dimer vaccines with aluminum hydroxide as an adjuvant were still not at a robust level after sequential vaccination. Adjuvant selection for recombinant protein vaccines must be further explored in clinical trials. All participants were required to complete a 12-month safety observation after the entire vaccination period and follow-up visits, as scheduled for the baseline in the phase 1 trial. Given an outbreak of novel coronavirus variants that infected a majority of participants in the follow-up period, the analysis of antibody persistence was not performed by consensus between the investigator and DSMB. The results of vaccine persistence over a long period and adverse events of special interest will be released in further studies. Given that phase I is an exploratory study without a statistical hypothesis or adjustment for multiplicity, the statistical power may be diminished. Based on our VLP nanoparticle technology, the favorable safety and immunogenicity profile of LYB001 was confirmed under a heterologous booster strategy following two- or three-dose inactivated CONDI-19 vaccine in a small-scale clinical trial [Citation28]. The safety, immunogenicity, and efficacy of LYB001 are favorable in a larger-scale phase III trial with lower ARs and strong immune responses induced by LYB001, and reports will be disclosed in further studies. The targets of the COVID-19 vaccine LYB001 are adults (aged 18 years and above).
In conclusion, our clinical trial demonstrated that the COVID-19 vaccine LYB001 has good safety, tolerability, and immunogenicity against SARS-CoV-2, particularly against the prototype strain in fundamental vaccination, providing the basis for the clinical application of a sequential vaccination dose and schedule. It suggests that LYB001 is a highly promising vaccine candidate against SARS-CoV-2. However, further confirmatory clinical studies on international multi-center trials are required to explore its safety, immunogenicity, and efficacy in larger populations of adults.
Declaration of interest
This research may lead to the development of products which may be licensed to Guangzhou Patronus Biotech Co., Ltd and Yantai Patronus Biotech Co., Ltd. J Jin, Y Zeng, Y Zhou, W Kang, and Z Yang are employed by Guangzhou Patronus Biotech Co. and Ltd and Yantai Patronus Biotech Co. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or material discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have received honoraria for their review work. Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose.
Author contributions
F Zhu, J Jin and H Pan initiated and designed the clinical research. Y Zeng and Z Yang contributed to the protocol and design of this study. F Zhu, H Pan, R Tang and Q Liang were responsible for supervision clinical trial and data management. Qi Liang, X Zheng and X Zang were responsible for sample collection and laboratory analysis. Y Zhou and W Kang was responsible for to the data processing and statistical analysis. W Kang, Y Zeng, Z Yang and Y Zhou drafted the manuscript. R Tang, J Jin and Y Zeng were responsible for the revision. F Zhu, J Jin and H Pan were in charge of approval. All authors reviewed and approved the final version of the manuscript. The data and decision to publish this study were reviewed and confirmed by all authors.
Supplemental Material
Download MS Word (62.9 KB)Acknowledgments
We thank all participants in our study for providing the clinical data and serum samples; and the Jiangsu Provincial Center for Disease Control and Prevention (Public Health Research institute of Jiangsu Province), for helping with clinical trial and relative work.
Data availability statement
Due to the dataset in this study is not open to the public, appropriate access to the dataset can be requested and addressed to the corresponding authors Fengcai Zhu at [email protected], or Jing Jin at [email protected]. There is no current URL for direct access to the dataset, but reasonable requests or collaboration can be made.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14760584.2024.2337051.
Additional information
Funding
References
- Coronavirus resource center at johns Hopkins university. [cited 2023 Mar 10]. Available from: https://github.com/CSSEGISandData/COVID-19
- Ma Y, Deng J, Liang W, et al. Meta-analysis of SARS-CoV-2 reinfection rate in the world. Chin J Dis Control Prev. 2023;27(2):127–135.
- Lazarus JV, Wyka K, White TM, et al. A survey of COVID-19 vaccine acceptance across 23 countries in 2022. Nat Med. 2023;29(2):366–375. doi: 10.1038/s41591-022-02185-4
- COVID-19 vaccine tracker and landscape. [cited 2023 Mar 10]. Available from: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines
- Li M, Wang H, Tian L, et al. COVID-19 vaccine development: milestones, lessons and prospects. Signal Transduct Target Ther. 2022;7(1):146. doi: 10.1038/s41392-022-00996-y
- Dai L, Gao L, Tao L, et al. Efficacy and safety of the RBD-dimer–based COVID-19 vaccine ZF2001 in adults. N Engl J Med. 2022;386(22):2097–2111. doi: 10.1056/NEJMoa2202261
- Kleanthous H, Silverman JM, Makar KW, et al. Scientific rationale for developing potent RBD-based vaccines targeting COVID-19. NPJ Vaccin. 2021;6(1):128. doi: 10.1038/s41541-021-00393-6
- Zhao X, Zheng A, Li D, et al. Neutralisation of ZF2001-elicited antisera to SARS-CoV-2 variants. Lancet Microbe. 2021;2(10):e494. doi: 10.1016/S2666-5247(21)00217-2
- Brisse M, Vrba SM, Kirk N, et al. Emerging concepts and technologies in vaccine development. Front Immunol. 2020;11:583077. doi: 10.3389/fimmu.2020.583077
- Yang S, Li Y, Dai L, et al. Safety and immunogenicity of a recombinant tandem-repeat dimeric RBD-based protein subunit vaccine (ZF2001) against COVID-19 in adults: two randomised, double-blind, placebo-controlled, phase 1 and 2 trials. Lancet Infect Dis. 2021;21(8):1107–1119. doi: 10.1016/S1473-3099(21)00127-4
- Zhao X, Zhang R, Qiao S, et al. Omicron SARS-CoV-2 neutralization from inactivated and ZF2001 vaccines. N Engl J Med. 2022;387(3):277–280. doi: 10.1056/NEJMc2206900
- Ma X, Zou F, Yu F, et al. Nanoparticle vaccines based on the receptor binding domain (RBD) and heptad repeat (HR) of SARS-CoV-2 elicit robust protective immune responses. Immunity. 2020;53(6):1315–1330.e9. doi: 10.1016/j.immuni.2020.11.015
- Shi J, Zhao Y, Peng M, et al. Screening of efficient adjuvants for the RBD-based subunit vaccine of SARS-CoV-2. Vaccines. 2023;11(4):713. doi: 10.3390/vaccines11040713
- Firdaus FZ, Skwarczynski M, Toth I. Developments in vaccine adjuvants. Methods Mol Biol. 2022;2412:145–178.
- Kim E, Attia Z, Woodfint RM, et al. Inhibition of elastase enhances the adjuvanticity of alum and promotes anti-SARS-CoV-2 systemic and mucosal immunity. Proc Natl Acad Sci USA. 2021;118(34):e2102435118. doi: 10.1073/pnas.2102435118
- Pino M, Abid T, Pereira Ribeiro S, et al. A yeast expressed RBD-based SARS-CoV-2 vaccine formulated with 3M-052-alum adjuvant promotes protective efficacy in non-human primates. Sci Immunol. 2021;6(61):eabh3634. doi: 10.1126/sciimmunol.abh3634
- Routhu NK, Cheedarla N, Bollimpelli VS, et al. SARS-CoV-2 RBD trimer protein adjuvanted with alum-3M-052 protects from SARS-CoV-2 infection and immune pathology in the lung. Nat Commun. 2021;12(1):3587. doi: 10.1038/s41467-021-23942-y
- Guo W, Duan K, Zhang Y, et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18 years or older: a randomized, double-blind, placebo-controlled, phase 1/2 trial. EClinicalmedicine. 2021;38:101010. doi: 10.1016/j.eclinm.2021.101010
- Bravo L, Smolenov I, Han HH, et al. Efficacy of the adjuvanted subunit protein COVID-19 vaccine, SCB-2019: a phase 2 and 3 multicentre, double-blind, randomised, placebo-controlled trial. Lancet. 2022;399(10323):461–472. doi: 10.1016/S0140-6736(22)00055-1
- Zhou J, Liu Z, Zhang G, et al. Development of variant‐proof severe acute respiratory syndrome coronavirus 2, pan‐sarbecovirus, and pan‐β‐coronavirus vaccines. J Med Virol. 2023;95(1):e28172. doi: 10.1002/jmv.28172
- Cohen AA, van Doremalen N, Greaney AJ, et al. Mosaic RBD nanoparticles protect against challenge by diverse sarbecoviruses in animal models. Science. 2022;377(6606):eabq0839. doi: 10.1126/science.abq0839
- Cohen AA, Gnanapragasam PNP, Lee YE, et al. Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science. 2021;371(6530):735–741. doi: 10.1126/science.abf6840
- Li Y, Zhang Y, Zhou Y, et al. An RBD virus-like particle vaccine for SARS-CoV-2 induces cross-variant antibody responses in mice and macaques. Signal Transduct Target Ther. 2023;8(1):173. doi: 10.1038/s41392-023-01425-4
- Ward BJ, Gobeil P, Séguin A, et al. Phase 1 randomized trial of a plant-derived virus-like particle vaccine for COVID-19. Nat Med. 2021;27(6):1071–1078. doi: 10.1038/s41591-021-01370-1
- Song JY, Choi WS, Heo JY, et al. Safety and immunogenicity of a SARS-CoV-2 recombinant protein nanoparticle vaccine (GBP510) adjuvanted with AS03: a randomised, placebo-controlled, observer-blinded phase 1/2 trial. EClinicalmedicine. 2022;51:101569.
- Smit MJ, Sander AF, Ariaans MBPA, et al. First-in-human use of a modular capsid virus-like vaccine platform: an open-label, non-randomised, phase 1 clinical trial of the SARS-CoV-2 vaccine ABNCoV2. Lancet Microbe. 2023;4(3):e140–e148. doi: 10.1016/S2666-5247(22)00337-8
- Khoury DS, Cromer D, Reynaldi A, et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat Med. 2021;27(7):1205–1211. doi: 10.1038/s41591-021-01377-8
- Yong X, Liu J, Zeng Y, et al. Safety and immunogenicity of a heterologous booster with an RBD virus-like particle vaccine following two- or three-dose inactivated COVID-19 vaccine. Hum Vaccin Immunother. 2023;19(3):2267869. doi: 10.1080/21645515.2023.2267869