
Abstract
Laterality defects include morphological anomalies with impaired left-right asymmetry induction, such as dextrocardia, situs inversus abdominis, situs inversus totalis and situs ambiguus. The different arrangement of major organs is called heterotaxy. We describe for the first time a fetus with situs viscerum inversus and azygos continuation of the inferior vena cava, due to previously unreported variants in compound heterozygosity in the CFAP53 gene, whose product is implied in cilial motility. Prenatal trio exome sequencing was performed with turn-around time during the pregnancy. The fetuses with laterality defects are suitable candidates for prenatal exome sequencing due to the emerging high diagnostic rate of this group of morphological anomalies. A timely molecular diagnosis plays a fundamental role in genetic counseling, regarding couple decisions on the ongoing pregnancy, providing recurrence risks, and in predicting possible respiratory complications due to ciliary dyskinesia.
Clinical report
A woman was referred at 12w0d of gestation for fetal laterality disorders. The non-consanguineous couple did not report family history of malformations, syndromes or suspected Mendelian disorders. Karyotype performed after amniocentesis was 46,XY. Chromosomal Microarray Analysis (CMA) detected a maternally inherited 559 kb 15q25.2 microduplication encompassing AP3B2 (MIM*602166) and HOMER (MIM*604799), without a clear pathogenic role [Citation1]. At 22w1d, fetal echocardiography showed mirror image arrangement of abdominal organs and heart, attributable to situs viscerum inversus, except for azygos continuation of the inferior vena cava (), a feature usually found in left isomerism. The heart did not show structural anomalies. Exome sequencing (ES) via Next Generation Sequencing (NGS) technologies was performed in trio on genomic DNA from amniotic fluid and parental leukocytes. Targeted enrichment was attained using the Twist Custom Panel Kit. The library was sequenced on an Illumina NovaSeq6000 platform. Fastq files were aligned to the human reference GRCh37/hg19 (Burrows-Wheeler Aligner). The BaseSpace pipeline and the TGex Knowledge Driven NGS Analysis software (LifeMap Sciences) were used for the variant calling and annotating variants, respectively. The following genes, selected using Human Phenotype Ontology searching for the term “Heterotaxy” (HP:0030853), were analyzed: ZIC3 (NM_003413), CFC1 (NM_032545.3), CIROP (NM_001354640.2), DNAH9 (NM_001372.4), FOXJ1 (NM_001454.4), ACVR2B (NM_001106.3), SMAD2 (NM_005901.6), NODAL (NM_018055.4), HAND1 (NM_004821.2), CRELD1 (NM_001031717.3), LEFTY2 (NM_003240.4), GDF1 (NM_021267), MED13L (NM_145020), PKD1L1 (NM_138295), MMP21 (NM_147191), CFAP52 (NM_145054.5), CFAP53 (NM_145020), CFAP59 (NM_181426.2), ACTG2 (NM_001615.4), CCDC32 (NM_001080792.4), CPLX1 (NM_006651.4), CTBP1 (NM_001012614.2), FANCB (NM_001018113.3), KDM3B (NM_016604.4), LETM1 (NM_012318.3), NSD2 (NM_001042424.3), PIGG (NM_001127178.3). Variants with minor allele frequency <0.01 were filtered and prioritized, and candidate variants were checked in dbSNP and GnomAD. Variants were examined for coverage and Qscore (threshold = 20) and visualized by Integrative Genome Viewer. Sanger sequencing was performed on fetal and parental DNA using the Big Terminator v1.1 Cycle Sequencing Kit and an ABI3130 GeneticAnalyzer (Applied Biosystems). NGS revealed the frameshift c.737_740del, p.(Gln246fsTer4), rs759013557 and the nonsense c.1282G > T p.(Glu428Ter), rs199505634, variants in compound heterozygosity in CFAP53 (NM_145020.4)(Cilia And Flagella Associated Protein 53, alias Coiled-Coil Domain-Containing Protein 11, CCDC11). Biallelic loss-of-function CFAP53 variants are associated with Heterotaxy Visceral 6 (MIM#614779). The first variant was inherited from the father, the second from the mother. The variants were not described in the literature. The c.737_740del variant has a frequency of 0.00000401 in the GnomAD v2.1.1 exomes database [Citation2]. It was not reported in the ClinVar database [Citation3]. It is predicted to be deleterious, with a CADDv1.6 score of 24.1 [Citation4]. The frameshift variant is expected to generate a premature stop codon, truncating the last 214 aminoacids of the protein. No functional data was available. The variant was classified as Pathogenic according to the American College of Medical Genetics and Genomics and Association for Molecular Pathology (ACMG-AMP) variant interpretation guidelines, with the PVS1, PM2-moderate and PP3-moderate criteria [Citation5].
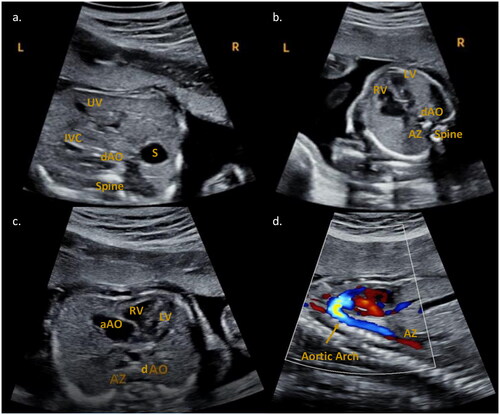
Figure 1. Fetal echocardiography performed at 22w1d. (a) Abdominal situs view. The stomach (S) is localized on the right side of the fetus. In front of and slightly to the left of the spine is the descending aorta (dAO). The umbilical vein (UV) and the inferior vena cava (IVC) are present anteriorly and to the left. (b) Four chamber view. The dAO and the azygos vein (AZ, further to the left) are anterior to the spine. RV: right ventricle; LV: left ventricle. (c) Transverse view. RV: right ventricle; LV: left ventricle; aAO: ascending aorta. The dAO and the AZ (to the left) are anterior to the spine. (d) Sagittal aortic arch and azygos vein view. Inferior to the flow of the aortic arch a red flow is visible, attributable to the azygos continuation of the inferior vena cava.
The c.1282G > T variant has a frequency of 0.000272 in the GnomAD v2.1.1 exomes database [Citation2]. It is reported as Variant of Uncertain Significance (VUS) in the ClinVar database, with no information on the zygosity status and phenotype of the identified carrier [Citation3]. It is predicted to be highly deleterious, with a CADDv1.6 score of 40 [4]. The truncating variant is expected to disrupt the last 87 amino acids of the 514-aminoacid CFAP53 protein. No functional data was available. The variant was classified as Likely Pathogenic according to the ACMG-AMP guidelines, with the PM3-moderate, PM2-moderate and PP3-moderate criteria [Citation5]. Given the distal position of the stop codon on the transcript, and the absence of genotype-phenotype correlations for variants disrupting the distal portion of the CFAP53 protein, we did not apply the PVS1 criterion. Sanger sequencing confirmed fetal variants, with paternal segregation for p.Gln246fsTer4 and maternal segregation for p.Glu428Ter. After birth, the newborn did not show respiratory involvement. Magnetic Nuclear Resonance imaging and echocardiography confirmed the prenatal findings. We describe for the first time a fetus with situs inversus and azygos continuation of the inferior vena cava due to novel compound heterozygous variants in CFAP53, focusing on the importance a molecular diagnosis of a condition not associated with respiratory involvement for decision making on the ongoing pregnancies.
Discussion and review of the literature
Vertebrate morphogenesis occurs along three orthogonal axes. While anteroposterior and dorsoventral defects usually result in conditions so severe they are non-vital, laterality defects include a broad spectrum of anomalies in left-right asymmetry patterns. They can result in dextrocardia, situs inversus abdominis, situs inversus totalis (SIT), situs ambiguous (heterotaxy), left or right isomerism. CFAP53 is a cytoplasmic and cilial protein containing coiled-coil domains expressed in ciliated cells in different tissues (http://www.proteinatlas.org [Citation6]), involved in primary cilia beating and laterality induction [Citation6–9]. Few cases of CFAP53-related disorders are reported, with different phenotypes of the laterality defect spectrum, all caused by homozygous variants in inbred families [Citation10,Citation11]. Perles described a 14-year-old with complete unbalanced atrioventricular canal defect, hypoplastic left ventricle and single atrium, double outflow right ventricle, transposition of great arteries with severe pulmonary valve stenosis, right aortic arch, abnormal systemic venous return and total anomalous pulmonary venous drainage, consistent with right isomerism [Citation11]. He also showed midline liver and inverted stomach and spleen. The asymptomatic brother was detected with situs inversus totalis. Both siblings presented the homozygous splice-site mutation c.1213 + 1 G > A in CFAP53 [11]. A patient with situs inversus totalis and sinusitis was detected with the nonsense homozygous c.121C > T, p.Arg41Ter variant in CFAP53. This group investigated CFAP53 expression and functioning, identifying the protein localization in both spinal canal cilia and in the base of Kupffer’s vesicle (left-right asymmetry inductor in zebrafish), playing a pivotal role in the rotational movement pattern. They speculated that CFAP53 can guide the transport of the outer dynein arms proteins to the axoneme through the base of the cilia, also mediating protein-protein interactions [Citation10]. In a further report two families presenting with laterality defects were described [Citation12]. In the first one, an individual showed dextrocardia and asplenia, while their sibling had situs inversus. ES identified the homozygous c.472A > G p.(Arg158Gly) missense variant in CFAP53. In the second family, an individual presented dextrocardia and a biallelic p.(Leu101_Glu125del) in-frame deletion was found [Citation12]. Noel [Citation12] observed in zebrafish the cfap53 expression in the midline, without cilial defects in the spinal cord. Other experiments verified that cilia in Kupffer’s vesicles were significantly reduced (8–10 somite stage embryos) [Citation13]. This suggests that Cfap53 represents a centriolar satellite component, regulating cilia assembly and function and left-right asymmetry in zebrafish.
We report the first prenatal case of laterality defects due to CFAP53, describing a compound heterozygosity for the first time, with the extremely uncommon finding of azygos continuation of the inferior vena cava in situs inversus. Indeed, sonography detected mirror image arrangement of abdominal organs and heart, as found in situs viscerum inversus, but unexpectedly this picture was associated with azygos continuation of the inferior vena cava, anatomic characteristic usually found in left isomerism. The ES diagnostic rate in this group of anomalies, usually with autosomal recessive inheritance pattern, appears to be very high [Citation14]. According to recent publications, ES or genome sequencing should be offered to patients with familial history of heterotaxy and to syndromic individuals with lower priority for the others [Citation15]. As we have seen in the present case, a timely molecular diagnosis plays a fundamental role in genetic counseling, regarding ongoing pregnancies and recurrence risks, and in preventing respiratory complications due to ciliary dyskinesia. Prenatal diagnosis in cases with laterality defects is essential to discriminate isolated from possibly complicated forms. In particular, CFAP53 has not been associated with respiratory involvement and its detection in a fetus with laterality defects can guide the couple’s decision on the prosecution of pregnancy.
Ethical approval
The study conforms to the ethical guidelines of the 1975 Declaration of Helsinki. Informed consent was obtained from the couple.
Acknowledgements
We thank Dr. E. Maggi for the very early ultrasonographic suspicion and for referring the patient.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Monkam CY, Kemeny S, Miret A, et al. A case which further refines the critical region for 15q25.2 microduplication phenotypes. Acta Neurol Belg. 2016;116(4):683–685.
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans [published correction appears in nature [published correction appears in nature. 2021;597(7874):E3-E4]. Nature. 2020;581(7809):434–443.
- Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D1067.
- Rentzsch P, Schubach M, Shendure J, et al. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021;13(1):31.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424.
- Levin M, Johnson RL, Stern CD, et al. A molecular pathway determining left right asymmetry in chick embryogenesis. Cell. 1995;82(5):803–814.
- Uhlén M, Fagerberg L, Hallström BM, et al. Proteomics. Tissue based map of the human proteome. Science. 2015;347(6220):1260419.
- Pagán-Westphal SM, Tabin CJ. The transfer of left-right positional information during chick embryogenesis. Cell. 1998;93(1):25–35.
- Psychoyos D, Stern CD. Restoration of the organizer after radical ablation of Hensen’s node and the anterior primitive streak in the chick embryo. Development. 1996;122(10):3263–3273.
- Narasimhan V, Hjeij R, Vij S, et al. Mutations in CCDC11, which encodes a coiled-coil containing ciliary protein, causes situs inversus due to dysmotility of monocilia in the left-right organizer. Hum Mutat. 2015;36(3):307–318.
- Perles Z, Cinnamon Y, Ta-Shma A, et al. A human laterality disorder associated with recessive CCDC11 mutation. J Med Genet. 2012;49(6):386–390.
- Noël ES, Momenah TS, Al-Dagriri K, et al. A zebrafish loss-of-Function model for human CFAP53 mutations reveals its specific role in laterality organ function. Hum Mutat. 2016;37(2):194–200.
- Silva E, Betleja E, John E, et al. Ccdc11 is a novel centriolar satellite protein essential for ciliogenesis and establishment of left-right asymmetry. Mol Biol Cell. 2016;27(1):48–63.
- Bolkier Y, Barel O, Marek-Yagel D, et al. Whole-exome sequencing reveals a monogenic cause in 56% of individuals with laterality disorders and associated congenital heart defects. J Med Genet. 2022;59(7):691–696.
- Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Europace. 2022;24(8):1307–1367.