
Abstract
Tobacco smoking is a risk factor for various diseases. The underlying cellular mechanisms are not fully characterized, but include oxidative stress, apoptosis, and necrosis. Electronic-cigarettes (e-cigarettes) have emerged as an alternative to and a possible means to reduce harm from tobacco smoking. E-cigarette vapor contains significantly lower levels of toxicants than cigarette smoke, but standardized methods to assess cellular responses to exposure are not well established. We investigated whether an in vitro model of the airway epithelium (human bronchial epithelial cells) and commercially available assays could differentiate cellular stress responses to aqueous aerosol extracts (AqE) generated from cigarette smoke and e-cigarette aerosols. After exposure to AqE concentrations of 0.063–0.500 puffs/mL, we measured the intracellular glutathione ratio (GSH:GSSG), intracellular generation of oxidant species, and activation of the nuclear factor erythroid-related factor 2 (Nrf2)-controlled antioxidant response elements (ARE) to characterize oxidative stress. Apoptotic and necrotic responses were characterized by increases in caspase 3/7 activity and reductions in viable cell protease activities. Concentration-dependent responses indicative of oxidative stress were obtained for all endpoints following exposure to cigarette smoke AqE: intracellular generation of oxidant species increased by up to 83%, GSH:GSSG reduced by 98.6% and transcriptional activation of ARE increased by up to 335%. Caspase 3/7 activity was increased by up to 37% and the viable cell population declined by up to 76%. No cellular stress responses were detected following exposure to e-cigarette AqE. The methods used were suitably sensitive to be employed for comparative studies of tobacco and nicotine products.
Introduction
Cigarette smoke is known to induce cellular oxidative stress, which is reported to be a driving factor in many smoking-related diseases (Bernhard & Wang, Citation2007; Sethi & Rochester, Citation2000; Wiseman & Halliwell, Citation1996). Oxidative stress initiates a variety of pathological processes that contribute to the development of lung cancer (Reuter et al., Citation2010), and chronic obstructive pulmonary disease (COPD). The link between smoking and COPD is well documented (Sethi & Rochester, Citation2000; Tavilani et al., Citation2012), since the majority of COPD sufferers (approximately 95%) have some form of smoking history (Barnes et al., Citation2003). The pathology of COPD is associated with ongoing and sustained bronchial oxidative stress. Oxidant exposure to the lungs is a major initiating factor of bronchial cell oxidative stress and various sources of environmental oxidant exposure exist (Ciencewicki et al., Citation2008). Cigarette smoke has been shown to be a powerful pro-oxidant, both by way of oxidation by direct-acting free radicals in the cigarette smoke and also by way of indirect generation of intracellular oxidant species resulting from smoke-toxicant-induced disruptions to cellular processes such as aerobic respiration (Faux et al., Citation2009; Gould et al., Citation2011). Furthermore, cigarette smoke irreversibly modifies the cellular antioxidant glutathione, thereby diminishing the ability of the cell to defend against oxidative damage and contributing to the chronic oxidative stress observable in COPD patients (van der Toorn et al., Citation2007).
Cells possess highly effective systems to neutralize oxidant species and repair associated oxidative damage. Many of these systems are regulated by the transcription factor nuclear factor erythroid-related factor 2 (Nrf2). Nrf2 is activated by interactions with sensors that possess the ability to detect intracellular oxidants or an increase in the oxidized form of glutathione (GSSG) (Djordjević, Citation2004; Espinosa-Diez et al., Citation2015). Cellular antioxidant mechanisms, such as the glutathione detoxification pathways, and downstream Nrf2 targets, such as haemoxygenase-1 (HO-1), c-fos, and NAD(P)H dehydrogenase quinone 1 (NQO-1), act to prevent the redox homeostatic imbalance that characterizes oxidative stress (Djordjević, Citation2004; Espinosa-Diez et al., Citation2015). Nrf2 is inactive when bound to its regulatory molecule: KEAP-1. This allows the cell to respond rapidly to the incidence of oxidative stress by dissociating Nrf2 from KEAP-1, whereby it translocates to the nucleus and acts as a promoter for the transcription of the antioxidant response element (ARE) stress response genes (Chen et al., Citation2015; Djordjević, Citation2004; Espinosa-Diez et al., Citation2015). However, when the oxidative burden or oxidative damage becomes to great to be managed by the protective antioxidant responses, cell death can occur (Kannan & Jain, Citation2000; Li et al., Citation2002).
Tobacco smoke can induce both apoptosis and necrosis in mammalian cell cultures (Vayssier et al., Citation1998), the terminal cellular response to cumulative oxidant burden (Demedts et al., Citation2006; Li et al., Citation2002; Vayssier et al., Citation1998). Apoptosis in the context of the development of COPD is be a potentially important cellular process that mediates tissue remodeling in the lung. Apoptosis allows elimination of unwanted, damaged, or infected cells and involves the activation of caspase enzymes. This protective cellular response may be induced by oxidative damage to cellular DNA and oxidative stress events, such as, mitochondrial stress or hypoxia (Demedts et al., Citation2006).
The popularity of e-cigarette consumption has increased significantly over the past decade, and potentially offers a safer substitute to conventional tobacco products (Biener & Hargraves, Citation2014; Caponnetto et al., Citation2013; Farsalinos et al., Citation2014, Citation2016; Hajek et al., Citation2014). Public Health England recently reported that e-cigarettes were 95% less harmful than tobacco products and could be a means for smokers to quit or reduce their consumption of cigarettes (Public Health England, Citation2015). This conclusion was recently supported by a report from the Royal College of Physicians (RCP), in which it was stated that “the hazard to health arising from long-term vapor inhalation from the e-cigarettes available today is unlikely to exceed 5% of the harm from smoking tobacco” (RCP, Citation2016).
The e-cigarette market is growing rapidly, with a wide variety of devices and accompanying e-liquids now available. Devices can vary from “cigilike” devices to closed systems to open-tank systems with modular units. However, the basic principle for generating e-cigarette vapor is similar across all devices. The e-cigarette vapor is produced when a carrier solution (called e-liquid and usually comprising propylene glycol and/or glycerol), which can contain nicotine, water and/or flavorings, is heated. The heat is usually generated by an electrical filament atomizer, connected to a battery within the device.
Since e-liquids contain fewer components than cigarette tobacco and do not involve combustion, they are unlikely to deliver the same breadth or quantities of harmful chemicals that are present in the smoke of conventional cigarettes. Studies to date have demonstrated that e-liquids and e-cigarette vapors contain a relatively small number of individual chemicals (Pellegrino et al., Citation2012), the levels of which are substantially lower than in cigarette smoke (Callahan-Lyon, Citation2014). These chemicals can include carbonyls, traces of tobacco-specific nitrosamines, and metals, such as cadmium, nickel, and lead, in some cases (Chen et al., Citation2015; Goniewicz et al., Citation2014). Furthermore, the levels of these compounds varies considerably between different brands of e-cigarette in the marketplace (Kosmider et al., Citation2014). We have recently reported that the levels of e-cigarette emissions from a commercially available e-cigarette (Vype closed modular device, Nicoventures, London, UK) were 92–99% lower per puff than those from a reference tobacco cigarette (University of Kentucky 3R4F) (Margham et al., Citation2016). Compounds detected included major e-liquid constituents (nicotine, propylene glycol, and glycerol), recognized impurities in pharmacopoeia-quality nicotine and eight species of thermal decomposition products of propylene glycol or glycerol. Hence, owing to the wide variety of device designs available, variation in consumer use behavior and the absence of regulated manufacturing standards to ensure product consistency, a comprehensive understanding of the health risks associated with e-cigarettes as a product category is difficult to achieve.
In order to gain a better insight of whether e-cigarette-related toxicant exposure reductions translate into reductions in biological responses and disease, further pre-clinical and clinical studies are required. In the absence of qualified biomarkers that predict the onset of smoking-related disease, research efforts have focused on using disease-relevant in vitro and animal models to assess the early biological effects associated with exposure to e-cigarette vapor (Lowe et al., Citation2015). In this study, we compared conventional cigarettes and e-cigarettes with respect to their potential to induce cellular oxidative stress in an in vitro model of the airway epithelium. This was achieved through measurements of cellular ratios of reduced glutathione (GSH) to GSSG, intracellular generation of oxidant species and the transcriptional activation of the ARE as an indirect marker of Nrf2 activity. The initiation of the apoptotic response as identified by caspase 3/7 activity and the post-exposure quantification of viable cells were included to quantify cytotoxic responses to high levels of oxidative stress.
Materials and methods
Suppliers
All chemicals were sourced from Fisher Scientific UK Ltd., Loughborough, UK, unless otherwise stated. Assay kits were purchased from Promega Ltd., Southampton, UK.
Test products
The cigarette used in this study was the 3R4F Kentucky reference cigarette. It is a US-blended king-sized product with a cellulose acetate filter and an International Organization for Standardization (ISO) tar yield of 9.4 mg/cigarette in nine puffs, and is representative of many products sold commercially around the world. The composition, construction, and mainstream smoke chemistry yields from this product have been reported previously (Roemer et al., Citation2012). Prior to analysis, cigarettes were conditioned for at least 48 h at 22 ± 1 °C and 60 ± 3% relative humidity in accordance with the ISO 3402:1999 standard on tobacco and tobacco products – atmosphere for conditioning and testing (ISO, Citation2015).
Two Vype e-cigarettes were used, a cigilike (cartomizer style product) and a closed modular product. Blended Tobacco flavored variant was chosen for this study at 36 mg/mL (cigilike) and 18 mg/mL nicotine (modular device e-liquid). The cigilike device was used at 3.7 V and the closed modular device at 4.0 V. The e-cigarette product batteries were fully charged before use and a fresh e-liquid cartridge was used to obtain each batch of aqueous aerosol extracts (AqE).
Preparation of AqE
Whole aerosols from reference 3R4F cigarettes or e-cigarettes were generated on a Borgwaldt-KC RM20H smoking machine (Borgwaldt, Richmond, VA) under the Health Canada intense (HCI) machine puffing regime (55 mL puff taken over 2 s and repeated every 30 s). Filter ventilation holes on the 3R4F cigarette were blocked. E-cigarettes were “puffed” under a modified HCI regime, in which the standard HCI bell-shaped puffing profile that was used for 3R4F was replaced with a square wave profile. In order to activate the closed modular device, the button was pressed by a puff-synchronized robot 1 s before the puff commenced and held in position for the duration of the puff. The cigilike e-cigarette operated through puff actuation rather than the pushing of a button. For this reason, the puff duration was increased to 3 s to allow sufficient time for puff actuation.
AqEs were generated by bubbling 10 × 55 mL puffs from one cigarette or e-cigarette through 20 mL DMEM/F12 medium without added supplements or serum, via an impinger. This produced a stock extract at the concentration of 0.5 puffs/mL, which was serially diluted 1:1 to produce four AqE concentrations for submerged cell culture exposure (see ). Immediately after generation, AqEs were stored in sealed, brown, smoked glass tubes at 2–8 °C until use, which was within 4 h after generation. To avoid potential cross-contamination, separate smoking machines were dedicated to either reference 3R4F tobacco products or e-cigarettes. All extractions were performed in a test atmosphere of 60 ± 5% humidity and 22 ± 2 °C (ISO, Citation2015).
Table 1. AqE exposure concentrations used during the reported studies. Neat (100%) AqE equated to 10 puffs in 20 mL capture media or 0.5 puffs/mL.
To confirm the capture of product aerosol constituents and to ensure batch-to-batch consistency between AqEs used in this study, three independent physical quality control (QC) measurements were performed. Carbon monoxide (CO) gas generated from incomplete cigarette combustion during the 3R4F AqE production was detected and quantified by a CO module incorporated in the Borgwaldt RM20H rotary smoking machine. This QC method was only used for the 3R4F-derived extracts, as CO was not detectable in the e-cigarette aerosols.
AqE nicotine concentrations were quantified by Enthalpy Analytical Inc., Durhan, NC, by gas chromatography with mass spectrometry (GC-MS) on an Agilent GC 7890 Series system (Agilent Technologies, Santa Clara, CA). Briefly a sodium hydroxide solution (2 N) was added to a 1 mL aliquot of the AqE and allowed to stand for 15 min, after which an internal standard (quinolone) and tert-butyl methyl ether was added to the mixture. This was then briefly vortexed and an aliquot of the upper organic layer was transferred to an autosampler. The nicotine content in this extract was analyzed with an Agilent CAM analytical column (30 m × 0.25 mm inner diameter; 0.25 μM film thickness) and quantified against a set of nicotine standards. The method employed a 10-point nicotine standard calibration curve in the range of 32–1600 ng/mL. The method limit of quantification was 32 ng/mL and the limit of detection was 10 ng/mL.
Tar content of the extracts was determined by optical density measurements performed with a multimode spectrophotometer at OD320 nm (Spectramax M3, Molecular Devices, Workingham, Berkshire, UK).
Quantifying carbonyl species in the AqE and whole aerosol
AqE carbonyl quantification
The quantification of acetaldehyde, acetone, acrolein, butyraldehyde, crotonaldehyde, formaldehyde, methyl ethyl ketone, and propionaldehyde by high-performance liquid chromatography (HPLC) equipped with an ultraviolet detector was conducted by Enthalpy Analytical Inc. Carbonyl quantification was performed on five independent batches of cigarette or e-cigarette AqE generated as described above, except that products were puffed on a KC Automation five-port smoking machine (Borgwaldt).
Briefly, a 1 mL aliquot of the AqE was sampled and added to 1 mL of 2,4-dinitrophenylhydrazine solution. Samples were allowed to react for 20 min before pyridine was added to quench the reaction. Samples were analyzed using the Agilent Model 1100, HPLC equipped with an ultraviolet detector operating at approximately 360 nm with an appropriate column. Method performance was monitored with QC samples, including 3R4F samples smoked into spiked cell culture media, laboratory control samples, and spiked media samples. To calculate the expected value of the spiked 3R4F AqE samples, laboratory control samples were spiked and prepared according to Enthalpy’s validated methods (AM-076).
Whole aerosol carbonyl quantification
Whole aerosol quantification of acetaldehyde, acetone, acrolein, butyraldehyde, crotonaldehyde, formaldehyde, methyl ethyl ketone, and propionaldehyde was conducted by Labstat International ULC, Kitchener, ON, Canada, using HPLC with ultraviolet detection (Health Canada method T-116) (Health Canada, Citation1999). 3R4F cigarettes and the z were puffed, respectively, under the HCI or modified HCI regimens, using a Borgwaldt-KC LM20 linear 20 port smoking machine adapted for liquid trapping into two impingers containing 25 mL of 2,4-dinitrophenylhydrazine. The resulting impinger solutions were then pooled before a 0.5 mL aliquot was added to 0.5 mL of buffer solution of sodium hydroxide and glacial acetic acid. An aliquot of the resulting solution was then analyzed with HPLC with ultraviolet detection at 380 nm, using a C18 250 mm ×4.6 mm column.
Cell culture
Human bronchial epithelial cells (NCI-H292; American Type Culture Collection (ATCC), Teddington, Middlesex, UK) were grown in cell culture flasks. NCI-H292 cells were maintained in RPMI-1640 medium liquid, with sodium bicarbonate, without L-glutamine sterile-filtered and tested for endotoxins. The medium was supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences, Hatfield, Hertfordshire, UK), 2 mM glutamine, and 50 U/mL penicillin and 50 μg/mL streptomycin, at 37 °C in a humidified 5% carbon dioxide (CO2) incubator. All experimental cell culture (96-well) plates were seeded with 100 μL per well 1 × 105 cells/mL cell suspension and allowed to attach and grow to confluency over 72 h at 37 °C with 5% CO2 prior to experimental exposures.
The production and screening of this cell-line was conducted by Promega Corp., Madison, WI. Briefly, NCI-H292 cells were transfected with a pGL4[ARE-luc2P/Hygro] vector using FuGENE® HD at a 2:1 ratio. Positive identification of the transfected cells was achieved by antibiotic resistance to hygromycin B (150 μg/mL). A negative control consisting of non-transfected NCI-H292 cells cultured in the presence of the antibiotic confirmed the integration of the plasmid in the transfected cells. Cells were then expanded in the presence of hygromycin B (Invitrogen, Waltham, MA, Cat.# 10687-010) at 37 °C with 5% CO2.
Measurement of apoptosis and necrosis
Cell viability and apoptosis were assessed with a duplex assay in 96-well format, as per manufacturer’s protocol (Apolive-Glo™, Promega). Quantification of viable cells was by indirect observations of live-cell protease activity. The induction of apoptosis was characterized by a significant increase in caspase 3/7 activity, a known biomarker of apoptosis. Briefly, a cell permeable substrate was co-incubated with the cells, which produced a fluorescent signal upon cleavage by viable cell proteases.
A luciferase-based reaction in which a luminogenic substrate is cleaved by caspase 3/7 to produce a light signal, proportional to its activity was the basis for this assay. Fluorescence and luminescence signals were measured with a multimode plate reader. Stimulation of cells with Triton-X 100 (0.2%) or the protein kinase inhibitor Staurosporine (25 μM) allowed observations of complete cell death in the viability assay and the initiation of apoptosis via caspase 3 activation, respectively.
Assessment of oxidative stress
Glutathione ratio
The ratio of GSH to GSSG was assessed in a 96-well format assay (GSH/GSSG-Glo™, Promega), as per the manufacturer’s protocol. The GSH:GSSG ratio in NCI-H292 cells was calculated after a 4 h exposure to 100 μL of AqE for each test product, via quantification of oxidized GSSG and the total glutathione pool. Quantification of both GSH and GSSG was achieved through a GSH-dependent reaction, whereby a luciferin-NT probe was converted to luciferin by a glutathione S-transferase enzyme that had been coupled to a firefly luciferase. The luminescent signal was proportional to the amount of GSH or GSSG present. Visible light signals were measured with a multimode plate reader over a 1 s integration time. Cells were stimulated with KBrO3 (30 mM) to provide a pro-oxidant source required to lower the glutathione ratio and was the positive control for the assay.
Intracellular oxidant species generation
Increases in NCI-H292 intracellular oxidant species generation were measured with the indicator probe 2′,7′-dichlorodihydrofluorescein diacetate, which was internalized by the cell and converted to its fluorescent form 2′,7′-dichlorofluorescein by intracellular oxidant species. Briefly cells were incubated for 1 h at 37 °C with 5% CO2 with 100 μL of a 10 μM 2′,7′-dichlorofluorescein (Sigma-Aldrich, St Louis, MO) solution in DMEM/F12 supplemented with L-glutamine, without added serum. This solution was then washed off and replaced with 100 μL of the test product AqE. The experimental plates were returned to the incubator for a further 1 h, after which cells were washed again before fluorescence signals (Ex492–495 and Em517–527 nm) were measured with a multimode plate reader. a pro-oxidant source was provided by 250 μM tert-butyl hydroperoxide (Sigma-Aldrich) to generate intracellular oxidants and act as the positive control for the assay.
Activation of the antioxidant response
Transcriptional activation of the ARE by Nrf2 was determined after 6-h exposure of the stably transfected GloResponse™ H292-ARE-Luc2P cells (Promega) to the test product AqE. Luminescence signals were measured with a multimode plate reader with a 1-s integration time, directly after the addition of the ONE-Glo™ reporter substrate (Promega, Cat.# E6110) according to the manufacturer’s instructions. The isothiocyanate molecule d,l-sulforaphane (Sigma-Aldrich) is a potent inducer of Nrf2, and we used 30 μM in the H292-ARE-Luc2P reporter cell assay to confirm activation of the ARE.
Statistical analysis and calculation methods
In all cases, an analysis of variance general linear model was applied to identify statistically significant differences in assay responses between products. Dunnett’s multiple comparison tests were performed to identify significant assay responses to individual test product AqE concentrations. A value of p < 0.05 was considered as statistically significant. All assay responses were compared to the untreated (AqE capture medium only) cellular response and expressed as percentage changes in assay signal.
Results
AqE QC data
provides the product-specific values for each QC test in eight AqE productions. Cigarette smoke AqEs were confirmed as being within a defined level of variability (±2 standard deviations of the mean), based on a historical set of 40 individual QC values for each individual physical measurement (data not shown). For the e-cigarette AqE production, only the quantification of nicotine returned usable data and, therefore, this technique alone was utilized to identify batch-to-batch consistency and confirm delivery of aerosol constituents.
Table 2. QC data providing total nicotine (μg) and optical density (OD320 nm) of the AqE and the CO generated during AqE generation (10 puffs in 20 mL capture media) (LOD = ±0.01% vol).
Nicotine concentrations
The total amount of nicotine in the reference 3R4F AqE was 124.26 μg ± 16.02 μg, compared with 92.69 μg ± 18.16 μg in the Vype cigilike and 100.6 μg ± 30.84 μg in the Vype closed modular device. These values were within the pre-defined level of variability for this QC technique. Thus nicotine was reduced by 25% and 19% in the AqE from the cigilike and closed modular device, respectively, versus the 3R4F cigarette.
CO quantification
The level of CO generated during the machine puffing of the 3R4F cigarette was within the expected range for that cigarette when puffed under the HCI regime. No CO was generated during the machine puffing of the e-cigarette products because of the lack of combustion, either complete or incomplete.
Optical density (OD320 nm) of the AqE
The OD320 nm readings for the 3R4F cigarette were 0.749 ± 0.055, compared with 0.463 ± 0.023 for the cigilike AqE and 0.459 ± 0.037 for the closed modular device AqE. However, the readings for the e-cigarettes were within the OD320 nm range detected for the AqE capture medium, DMEM/F12. It was determined that the e-cigarettes did not alter optical density at that wavelength and, therefore, this method was unsuitable for assessment of e-cigarette AqE.
Carbonyl quantification in whole aerosol and AqE
The levels of individual carbonyl species that were quantified in 3R4F cigarette smoke or closed modular e-cigarette aerosol are provide in . The carbonyls that were also detected and quantified in the smoke AqE are shown in . Notably, the relative levels of individual carbonyl species in the smoke were also reflected in the AqE. The e-cigarette aerosol was found to contain four of the eight carbonyl species that were detected in the cigarette smoke, but were present at far lower levels: acetaldehyde 154 times lower, acetone 113 times lower, acrolein 26 times lower and formaldehyde 7 times lower. Specifically, e-cigarette vapors contained 10.4 μg acetaldehyde, 6.2 μg acrolein, and 12.1 μg formaldehyde per 10 puffs (55 mL), compared with 1603.9 μg acetaldehyde, 159.4 μg acrolein, and 87.9 μg formaldehyde per 10 puffs in cigarette smoke.
Table 3. Measurement of carbonyl-containing compounds in cigarette smoke and e-cigarette vapor.
Table 4. Measurement of carbonyl-containing compounds in AqE of cigarette smoke and e-cigarette vapor.
Bronchial epithelial cell cytotoxic responses
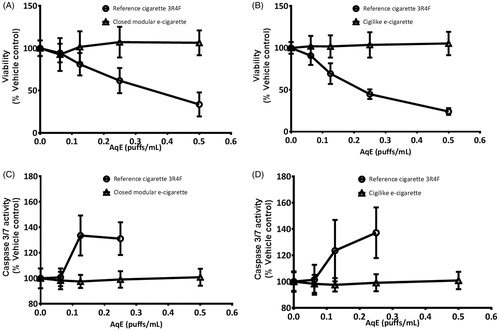
Exposure of NCI-H292 cells to 3R4F AqE-reduced cell viability across the concentration range. Significant reductions (p < .05) were observed when the 3R4F AqE exposure concentration was 0.125 puffs/mL or greater, leading to a maximum 76 ± 14% reduction in cellular viability with the neat 3R4F AqE (0.5 puffs/mL). Stimulation with the detergent Triton-X (0.2%) led to complete cytotoxicity. There was no cytotoxic effect of exposure AqE from either type of Vype e-cigarette ()). The activation of caspase 3/7 was increased in NCI-H292 cells by a maximum of 37 ± 12%, as compared to the untreated cellular response over the 4 h exposure duration. Similarly, stimulation with 25 μM staurosporine increased caspase 3/7 activity by 44 ± 26%. Significant increases (p < 0.05) in activation were measured with 3R4F AqE exposure concentrations greater than 0.125 puffs/mL (data not shown). Data for the cells exposed to the highest 3R4F AqE concentration of 0.5 puffs/mL were not included since more than 50% of the cells were non-viable at this dose. In contrast, when the NCI-H292 cells were exposed to matching concentrations of AqE from either of the Vype e-cigarette products, no apoptotic responses could be detected ()), even with undiluted (0.5 puffs/mL) e-cigarette AqEs.
Figure 1. NCI-H292 cell viability was reduced in a concentration-dependent manner in response to exposure to 3R4F AqE but not to the closed modular e-cigarette AqE (A) or cigilike AqE (B). The activation of apoptosis was increased by 3R4F AqE but not to the closed modular e-cigarette AqE (C) or cigilike AqE (D). Data are expressed as % change relative to vehicle control. Data are means of 12 replicates, repeated over a minimum of four independent experiments (±SD).
Oxidative stress
Exposure of H292 cells to 3R4F AqE induced a significant (p < 0.05) concentration-dependent lowering of the intracellular GSH:GSSG ratio. This resulted in a maximum 98 ± 0.8% lowering of the ratio with exposure to undiluted (0.5 puffs/mL) 3R4F AqE ()). Similarly, exposure to 30 mM potassium bromate induced a 97 ± 1.6% lowering of the GSH:GSSG ratio. Since measurements of total glutathione and GSSG were taken, loss to the total glutathione pool due to the cytotoxic effects shown in was accounted for.
Figure 2. Intracellular glutathione ratios were lowered in NCI-H292 cells in response to exposure to 3R4F AqE but not to the closed modular e-cigarette AqE (A) or cigilike AqE (B). Intracellular oxidant species generation was increased in H292 cells in response to exposure to 3R4F AqE but not to closed modular AqE (C) or cigilike AqE. Data are expressed as % change relative to vehicle control. Data are means of 12 replicates, repeated over a minimum of four independent experiments (±SD).
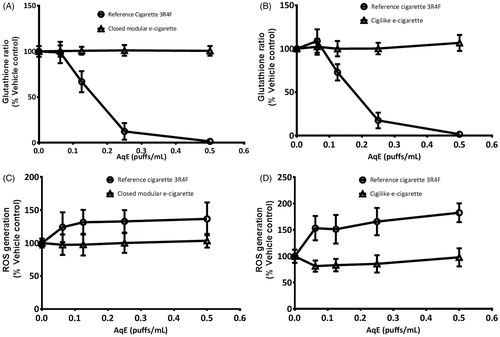
This depletion of available antioxidants occurred in addition to the significantly increased generation of intracellular oxidant species ()) that occurred after exposure to 0.063 puffs/mL 3R4F AqE. A maximum 83 ± 17% increase in intracellular oxidant species was measured after exposure to the lowest 3R4F AqE concentration, which did not significantly increase with exposure to more concentrated 3R4F AqE. Stimulation with 250 μM tert-butyl hydroperoxide induced an increase of 324 ± 159%. Taken together, these cellular responses to 3R4F AqE exposure demonstrate that NCI-H292 cells had entered into a state of oxidative stress. By contrast, cells exposed to matching concentrations of AqE from either of the Vype e-cigarette products maintained the GSH:GSSG ratio of the untreated cells, and the generation of intracellular oxidant species were not increased, even when the exposure concentration was increased to the maximum 0.5 puffs/mL. These data suggest that Vype e-cigarette AqE exposure induces negligible or no oxidative damage to NCI-H292 cells at the range of doses tested, and that any pro-oxidant species induced by exposure were not generated in sufficient quantities to overcome the cell’s internal antioxidant defense system. Thus, the cells did not enter into a state of oxidative stress and cellular redox homeostasis was maintained.
Transcription of ARE
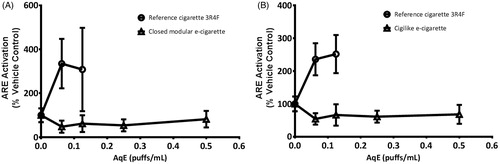
A significant activation (p < 0.05) of the ARE promoter-linked luciferase reporter function of the H292-ARE-Luc2P cell-line was induced after 3R4F AqE exposure, demonstrating a protective genetic response to oxidants. A maximum 335 ± 112% increase in ARE activation was measured after exposure to the lowest 3R4F AqE concentration of 0.063 puffs/mL ()), and this did not significantly increase with exposure to more concentrated 3R4F AqE. Data for the cells exposed to the 3R4F AqE concentrations above 0.125 puffs/mL were not included due to the extensive cytotoxic effects shown in . Similarly, stimulation with 30 μM D,L-sulforaphrane increased the activation of H292-ARE-Luc2P cell-line reporter function by 267 ± 125%. No activation of the H292-ARE-Luc2P cell-line was found after exposure to AqE from either Vype e-cigarette, even when undiluted. This observation was consistent with the previously described maintenance of the GSH:GSSG ratio in NCI-H292 cells exposed to AqE from either e-cigarette.
Figure 3. The transcriptional activation of the ARE in the GloResponse™ ARE-luc2P H292 cell-line was increased in response to exposure to combustible cigarette AqE but not to closed modular AqE (A) or cigilike AqE (B). Data are expressed as % change relative to vehicle control. Data are means of 12 replicates, repeated over a minimum of four independent experiments (±SD).
Discussion
This is the first study to demonstrate that AqE of machine-puffed e-cigarette vapors, generated under standardized conditions, do not induce oxidative stress in an in vitro model of the airway epithelium. The panel of commercially available assays used allowed the detection and measurement of acute cellular events and protective epithelial responses to oxidant exposure that can lead to oxidative stress.
3R4F concentration-dependent responses to AqE demonstrated that NCI-H292 cells generated oxidant species, showed antioxidant depletion and initiated an antioxidant transcriptional response. Collectively, these effects suggest that the cells had entered into a state of oxidative stress. The activation of assay endpoints at all concentrations of 3R4F AqE confirmed that this panel was suitably sensitive to detect the deleterious pro-oxidant effects of tobacco smoke. In contrast, e-cigarette AqE showed no harmful pro-oxidant effects. This finding may indicate that assay responses were either below the limit of detection or that the chemical drivers of the assay endpoints were absent or present in insufficient concentrations in the e-cigarette AqE to elicit responses. Thus, this study demonstrated that the e-cigarettes did not induce oxidative stress or cytotoxicity under comparable experimental conditions used for cigarette smoke exposure.
Exposing bronchial epithelial cells to captured aerosols
This study utilizes a standardized aerosol extract production method that includes a defined machine puffing regime, industry recognized reference cigarettes, and QC measurements on the AqE stock solutions. The use of AqE that captures the water-soluble components of smoke are widely documented (Alexandrov et al., Citation2006; Culpitt et al., Citation2003; Facchinetti et al., Citation2007; Pryor, Citation1997; Sato et al., Citation1999; Volpi et al., Citation2011; Walters et al., Citation2005; Wirtz & Schmidt, Citation1996), but the approaches employed to generate the extracts have not been standardized. Published methods range from simple techniques, whereby smoke is drawn into a syringe and exhausted through a simple capture liquid, such as phosphate-buffered saline, to more complex systems in which rotary smoking machines puff the cigarette to standardized puffing regimes and the smoke passes through capture media in an impinger.
The 3R4F AqE exposure concentrations used were based on range-finding experiments that examined acute in vitro responses to conventional cigarettes (Taylor et al., Citation2014). The concentration range utilized was considered suitable, since concentration-dependent activation of all assay endpoints was observed. This study investigated the potential of this technique to detect in vitro epithelial responses to captured e-cigarette vapors, puff-matched to cigarette smoke, up to a maximum 0.5 puffs/mL. Future studies might investigate the potential to elicit cellular stress responses with more concentrated e-cigarette vapor extracts. However, the saturation point of individual aerosol components within AqE could be a limiting factor when attempting to achieve more concentrated extracts. Also, certain e-cigarette vapor components, such as propylene glycol, may preclude the use of highly concentrated extracts in submerged in vitro systems due to nonspecific interference with or inhibition of the assay, for instance by inducing cytotoxic effects via a change in the osmotic potential of the resulting solutions.
The development of in vitro aerosol exposure systems in which lung cells are exposed to the whole aerosol at the air–liquid interface allows exposure to the vapor phase and to particulates (Adamson & Adamson, Citation2013; Iksandar et al., Citation2016; Kilford et al., Citation2014; Scheffler et al., Citation2015a,Citationb; Schlage et al., Citation2014). In parallel with this rapidly advancing research area, much progress has been made in the production and commercialization of complex organotypic three-dimensional lung cell culture systems (Maunders et al., Citation2007; Neilson et al., Citation2015). These co-cultures of lung cells demonstrate pseudo-stratification in which mucus-producing cells afford a similar matrix and protective barrier to that found in the lungs, allowing the aerosol exposure to be physiologically closer to the in vivo situation. These techniques may be required to model the effects of repeated or chronic exposure to e-cigarette aerosols.
Confirming aerosol capture in AqE
The QC measurements that were employed in this study confirmed the capture of constituents of smoke in the 3R4F AqE. The quantification of CO produced during the machine puffing of 3R4F cigarettes confirmed consistent smoking, while the OD320 nm readings of the AqE confirmed consistent smoke capture. Chemical species that absorb light at 320 nm include the broad category of tar and, therefore, this test is a useful measurement of the relative strengths of the 3R4F. It has been widely utilized in in vitro studies of the damaging effects of tobacco smoking in which AqE was the exposure system (Culpitt et al., Citation2003; Facchinetti et al., Citation2007; Volpi et al., Citation2011; Walters et al., Citation2005) and to normalize AqE to a consistent strength by dilution (Wirtz & Schmidt, Citation1996). As the OD320 nm wavelength is not suitable for assessing e-cigarette AqE, possibly due to the lack of tar generation, another wavelength should be used. The absence of CO detected during the machine puffing confirmed that no measurable combustion, either complete or incomplete, was occurring in the e-cigarette.
The quantification of AqE nicotine provided a physical measurement that demonstrated sensitivity to both cigarette smoke and e-cigarette AqE. The lower levels of nicotine measured in the e-cigarette AqE may be due to variability or reduced capture efficiency of the e-cigarette aerosols by the media. To account for the lower nicotine levels in the e-cigarette AqEs the maximum concentration of 0.5 puffs/mL was included in all assays. The comparison of carbonyl species quantified in the AqE, whilst not demonstrating 100% transfer, provided confidence that capture efficiency during the AqE production technique was sufficient. These results also confirmed that the existing ratios of individual carbonyl species that were identified in the cigarette smoke were maintained in the AqE.
The absence of detectable epithelial cellular stress responses to the e-cigarette AqE demonstrated in this study may have result partly from the much reduced carbonyl levels and absence of CO in the e-cigarette vapor and/or tar in AqE.
The glutathione ratio as a marker for tobaccosmoke-induced oxidative stress
The use of the glutathione ratio as a marker for redox status and oxidative stress has been documented in in vitro (Djordjević, Citation2004; Espinosa-Diez et al., Citation2015; Faux et al., Citation2009; Li et al., Citation2002; Muller & Gebel, Citation1998; van der Toorn et al., Citation2007) and clinical applications (Zitka et al., Citation2012). Measurement of the ratios of GSH to GSSG in this study provide a reliable indicator of cellular health. Within healthy cells around 98% of glutathione exists in its reduced form, but when cellular redox homeostasis is disrupted, a rapid lowering of the GSH:GSSG ratio occurs.
The collective use of the key oxidative stress events measured in this study (antioxidant depletion, Nrf2 activation, and apoptosis) have been reported for an assessment of toxicity in bronchial epithelial cells following exposure to diesel exhaust particles (Li et al., Citation2002). The key events appeared to occur in sequence with exposure to increasing concentrations of diesel exhaust particles, and were associated with a concentration-dependent lowering of the GSH:GSSG ratio. Measurement of this ratio might, therefore, provide a suitable candidate marker for making observations of oxidative stress. As tobacco smoke has been reported to possess similar properties to diesel exhaust emissions in terms of particle size and toxicant profile (Invernizzi et al., Citation2004), we hypothesized that tobacco effects after smoke exposure are likely to follow a similar dose-related sequence of key events to those described by Li et al. (Citation2002). We demonstrated a 3R4F AqE concentration-dependent lowering of the ratio, which preceded apoptotic and necrotic events and occurred in parallel with activation of the ARE. Previous studies have reported that smoke-induced cellular stress responses are dependent on intracellular GSH levels. Exposure to cigarette smoke AqE that were of a similar concentration range (0.009–0.045 puffs/mL) to that employed in this study led to GSH levels decreasing in different cell-lines (Muller & Gebel, Citation1998). We also demonstrated a link between the concentration-dependent lowering of GSH levels and levels of smoke-related carbonyls, such as formaldehyde, acetaldehyde, and acrolein, in AqE.
Various carbonyls have also been detected within AqE of cigarette smoke (Horinouchi et al., Citation2016; Noya et al., Citation2013), and pro-oxidant species, such as hydroxyl radicals, hydrogen peroxide and peroxynitrite, and have been reported to be formed within tobacco smoke AqE (Cosgrove et al., Citation1985; Muller et al., Citation1997). However, Muller & Gebel (Citation1998) reported that it was the carbonyl species in the extracts alone that were responsible for the GSH depletion observed in the different cell-lines studied. Only when GSH was depleted by smoke-derived carbonyls or when a capture medium was spiked with comparable levels of carbonyls (6 mM total carbonyls) did oxidative stress occur, as identified by the induction of stress response genes such as c-fos or HO-1 (Muller & Gebel, Citation1998). Thus, pro-oxidant species alone do not induce cellular oxidative stress, but require depletion of the GSH pool by carbonyls. Furthermore, it has been reported that aldehydes directly and irreversibly react with GSH, forming non-reducible GSH derivatives (van der Toorn et al., Citation2007). In this study, GC-MS of primary or secondary lung epithelial cell lysates following exposure to tobacco smoke identified GSH plus acrolein and GSH plus crotonaldehyde adducts, which suggests a mechanism for the impaired antioxidant capacity observed in COPD patients (Kluchová et al., Citation2007; Tavilani et al., Citation2012). This mechanism for smoke-induced depletion of the total glutathione pool is independent to the oxidation of GSH to GSSG, which may be reversed by enzymes under the control of Nrf2.
Apoptosis, a terminal response to oxidative stress
The absence of bronchial epithelial cell cytotoxicity observed in response to the e-cigarette AqE indicates that those species responsible for inducing cell death in cigarette smoke, which are likely to result from the process of tobacco combustion were either absent or not present in sufficient levels to induce cytotoxic effects or apoptosis. Apoptosis is a tightly regulated mechanism of programmed cell death that allows for the elimination of unwanted, damaged, or infected cells. In the context of the development of COPD, apoptosis has been found to be a potentially important cellular process in mediating tissue remodeling in the lung (Demedts et al., Citation2006). This process also has relevance for emphysema. Airway epithelial cell apoptosis can be induced in a number of ways, for example by chemically induced mitochondrial stress or hypoxia (Kannan & Jain, Citation2000) and by epithelial-derived cytokine release (Demedts et al., Citation2006). A cumulative oxidant burden leading to oxidative stress can initiate apoptosis and necrosis, and has been shown to involve mitochondrial stress (Espinosa-Diez et al., Citation2015; Li et al., Citation2002). Tobacco smoke has been demonstrated to induce both apoptosis and necrosis in mammalian cell cultures (Vayssier et al., Citation1998). Studies have reported mechanisms for the activation of apoptosis in which pivotal roles are described for reactive oxygen species and oxidative stress resulting signal transduction pathways, activated due to mitochondrial dysfunction, These pathways involve the activation of pro-caspases, leading to caspase-dependent initiation of apoptosis (Kannan & Jain, Citation2000). Therefore, the caspase 3/7 activity marker of apoptosis utilized in this study would likely cover both apoptosis induced by epithelial-derived cytokines and from mitochondrial stress.
The relative safety of electronic cigarette usage
Consumers concerned about the health consequences of tobacco smoking have sought alternatives to combustible nicotine delivery and the rapidly developing market of e-cigarettes may provide a consumer-acceptable alternative. Since no combustion occurs during the vaping of an e-cigarette and the e-liquids themselves contain relatively few individual chemicals (Callahan-Lyon, Citation2014; Cheng, Citation2014; Goniewicz et al., Citation2014; Margham et al., Citation2016), they are unlikely to deliver the concentrations of harmful species present within tobacco smoke. The chemical analysis of e-cigarette vapors has identified potentially damaging chemicals such as free radical species (Goel et al., Citation2015), carbonyls, for example acrolein, formaldehyde and acetaldehyde (Bekki et al., Citation2014; Jensen et al., Citation2015; Kosmider et al., Citation2014), polyaromatic hydrocarbons, traces of carcinogenic nitrosamines, and some toxic metals such as cadmium, nickel, and lead (Cheng, Citation2014; Goniewicz et al., Citation2014; Kosmider et al., Citation2014). These compounds are generally reported to exist at substantially lower levels than in mainstream cigarette smoke or environmental tobacco smoke (Burstyn, Citation2014). However, researchers have described relatively high yields of carbonyls being generated by e-cigarettes during the e-liquid vaporization process (Bekki et al., Citation2014; Farsalinos et al., Citation2015; Jensen et al., Citation2015; Kosmider et al., Citation2014). Publications describing high carbonyl generation can include analysis of vapors produced where little or no e-liquid remains in the device (Jensen et al., Citation2015; Kosmider et al., Citation2014). This has been termed “dry puffing” and results in higher than normal vaping temperatures, particularly in high-voltage devices, leading to what consumers describe as “a hot and unpleasant taste”. Studies have shown that e-cigarette consumers respond to this sensation by discontinuing their vaping session until the device is replenished with e-liquid (Farsalinos et al., Citation2015). Studies that include analysis of vapors produced with the contribution of dry puffing have been criticized by the American Council on Science and Health as being potentially unrealistic to consumer e-cigarette usage (ACSH, Citation2015). In stark contrast, reported levels of aldehydes within e-cigarette vapors, generated when sufficient e-liquid was available to the device were considerably lower than in cigarettes (Farsalinos et al., Citation2015): the levels of aldehydes per 10 puffs quantified under “normal vaping” conditions were typically 4.5 μg for acetaldehyde, 1.0 μg for acrolein, and 11.3 μg for formaldehyde, compared with 1240.3 μg for acetaldehyde, 120.4 μg for acrolein, and 74.0 μg for formaldehyde detected in cigarette smoke in the same study. This pattern was consistent with the carbonyl chemistries observed in this study.
In support of the reduced carbonyl levels shown in chemical analysis of e-cigarette vapors, in vitro studies have demonstrated reduced or negative adverse cellular responses to e-cigarette vapors when compared with tobacco smoke (Azzopardi et al., Citation2016; Farsalinos et al., Citation2013; Iksandar et al., Citation2016; Misra et al., Citation2014; Romagna et al., Citation2013; Thorne et al., Citation2016). These findings were supported by reports from the RCP (2016) and Public Health England (2015), which indicated that e-cigarettes are around 95% less harmful to health than normal cigarettes. Certain commercially available e-cigarettes have been shown to generate oxidant species during the vaporization of the e-liquids (Lerner et al., Citation2015), which contributes to a cytotoxic response to e-cigarette vapor. Cytotoxic effects in vitro caused by e-liquid vapor have been reported in myocardial cells (Farsalinos et al., Citation2013), human embryonic stem cells, mouse neural stem cells, human pulmonary fibroblasts (Bahla et al., Citation2012) and human lung epithelial cells (Scheffler et al., Citation2015a). In contrast to our data, another study by Scheffler et al. (Citation2015b) reported reduced cell viability following exposure to e-cigarette vapor. However, this study utilized a whole aerosol exposure system in which human bronchial epithelial cells were exposed to 200 puffs of e-cigarette vapor over a period of 30 min. The contrasting data could potentially be explained by differences between the two studies in terms of exposure regime, different e-cigarettes used and differences in the total constituent dose delivered to the cell cultures. Further studies are required to investigate this in more depth.
In certain studies, e-cigarette vapors induced an inflammatory response, as characterized by secretion of interleukins 6 and 8 from H292 cells, which was suggested in a subsequent analysis to be due to particular e-liquids flavors (Lerner et al., Citation2015). Nevertheless, the overwhelming majority of studies to date have reported e-liquids and e-cigarette aerosols to be markedly less toxic than tobacco smoke or smoke extracts. In our own studies, we have seen 70–95% reductions in cell cytotoxic effects with H292 cells (Azzopardi et al., Citation2016) and found no mutagenic effects with Ames in vitro genotoxicity testing (Thorne et al., Citation2016).
Although potentially harmful chemicals within e-cigarette aerosols may be present at low levels, early disease-relevant biological effects could still occur following more intensive consumer use. A need exists, therefore, for suitable in vitro approaches that are capable of both determining biological responses in direct comparison to conventional and e-cigarette aerosols (e.g. dose matching by puff or by nicotine delivery), and also to accommodate exposures of e-cigarette constituents to provide information which could be relevant to e-cigarette use patterns in consumers (e.g. greater puff volumes and puff frequency). These investigations will contribute to the available evidence and help to inform regulatory decision making regarding the efficacy of e-cigarette use as a safer alternative to tobacco smoking.
Conclusions
The exposure of NCI-H292 human bronchial epithelial cells to Vype e-cigarette AqE did not induce significant responses in the in vitro assays of oxidative stress or cytotoxicity at concentrations up to and including 0.5 puffs/mL. Therefore, the AqEs either do not contain the chemical drivers of the individual assays or they are present at insufficient concentrations to be detected with these in vitro assays. However, we saw sensitivity to detect NCI-H292 cell responses to low exposure concentrations of cigarette smoke AqE (0.063 puffs/mL). Indeed, this finding highlights the scale of difference in potency between the two product categories. The stress-related responses to e-cigarettes and conventional cigarettes could be clearly differentiated and, therefore, this approach is suitably sensitive to be employed for future assessments or comparative studies of novel tobacco and nicotine delivery products supported by other pre-clinical and clinical studies. In the meantime, we have demonstrated that under comparable experimental conditions, when compared with conventional cigarettes, e-cigarettes do not activate cellular stress responses in an in vitro model of the airway epithelium.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Funding
The authors are employees of British American Tobacco. All experimental work were funded by Nicoventures Ltd., UK, is a wholly-owned subsidiary of British American Tobacco.
References
- Alexandrov K, Rojas M, Rolando C. (2006). DNA damage by benzo(a)pyrene in human cells is increased by cigarette smoke and decreased by a filter containing rosemary extract, which lowers rree radicals. Cancer Res 66:11938–45.
- American Council on Science and Health (ACSH). (2015). E-cigarette study gets big headlines, but what does it mean? [Internet]. New York (NY): ACSH. Available from: http://acsh.org/news/2015/01/22/poorly-done-e-cig-vapor-study-gets-big-headlines-means-nothing/ [last accessed 8 Jul 2016].
- Azzopardi D, Patel K, Jaunky T, et al. (2016). Electronic cigarette aerosol induces significantly less cytotoxicity than tobacco smoke. Toxicol Meth Mech; in press.
- Bahla V, Lin S, Xu N, et al. (2012). Comparison of electronic cigarette refill fluid cytotoxicity using embryonic and adult models. Reprod Toxicol 34:529–53.
- Barnes P, Shapiro SD, Pauwels RA. (2003). Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J 22:672–88.
- Bekki K, Uchiyama S, Ohta K, et al. (2014). Carbonyl compounds generated from electronic cigarettes. Int J Environ Res Public Health 11:11192–200.
- Bernhard D, Wang XL. (2007). Smoking, oxidative stress and cardiovascular diseases-do anti-oxidative therapies fail? Curr Med Chem 14:1703–12.
- Biener L, Hargraves JL. (2014). A longitudinal study of electronic cigarette use among a population-based sample of adult smokers: association with smoking cessation and motivation to quit. Nicotine Tob Res 17:127–33.
- Burstyn I. (2014). Peering through the mist: systematic review of what the chemistry of contaminants in electronic cigarettes tells us about health risks. BMC Public Health 14:18.
- Callahan-Lyon P. (2014). Electonic cigarettes: human health effects. Tob Cont 23:ii36–40.
- Caponnetto P, Campagna D, Cibella F, et al. (2013). EffiCiency and safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One 8:e66317.
- Chen B, Liu Y, Chen Y, et al. (2015). The role of Nrf2 in oxidative stress-induced endothelial injuries. J Endocrinol 225:R83–99.
- Cheng T. (2014). Chemical evaluation of e-cigarettes. Tob Cont 23:ii11–17.
- Ciencewicki J, Trivedi S, Kleeberger SR. (2008). Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol 122:456–70.
- Cosgrove JP, Borish ET, Church DF, et al. (1985). The metal mediated formation of hydroxyl radical by aqueous extracts of cigarette tar. Biochem Biophys Res Commun 132:390–6.
- Culpitt S, Rogers DF, Fenwick PS, et al. (2003). Inhibition by red wine extract, resveratrol, of cytokine release by alveolar macrophages in COPD. Thorax 58:942–6.
- Demedts IK, Demoor T, Bracke KR, et al. (2006). Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res 7:53.
- Djordjević VB. (2004). Free radicals in cell biology. Int Rev Cytol 237:57–89.
- Espinosa-Diez C, Miguel V, Mennerich D, et al. (2015). Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol 6:183–97.
- Facchinetti F, Amadei F, Geppetti P, et al. (2007). Alpha,beta-unsaturated aldehydes in cigarette smoke release inflammatory mediators from human macrophages. Am J Respir Cell Mol Biol 37:617–23.
- Farsalinos KE, Romagna G, Allifranchini E, et al. (2013). Comparison of the cytotoxic potential of cigarette smoke and electronic cigarette vapor extract on cultured myocardial cells. Int J Environ Res Public Health 10:5146–62.
- Farsalinos KE, Romagna G, Tsiapras D, et al. (2014). Characteristics, perceived side-effects and benefits of electronic cigarette use: a worldwide survey of more than 19,000 consumers. Int J Environ Res Public Health 11:4356–73.
- Farsalinos KE, Voudris V, Poulas K. (2015). E-cigarettes generate high levels of aldehydes only in ‘dry puff’ conditions. Addiction 110:1352–6.
- Farsalinos K, Cibella F, Caponnetto P, et al. (2016). Effect of continuous smoking reduction and abstinence on blood pressure and heart rate in smokers switching to electronic cigarettes. Intern Emerg Med 11:85–94.
- Faux S, Tai T, Thorne D, et al. (2009). The role of oxidative stress in the biological responses of lung epithelial cells to cigarette smoke. Biomarkers 14:90–6.
- Goel R, Durand E, Trushin N, et al. (2015). Highly reactive free radicals in electronic cigarette aerosols. Chem Res Toxicol 28:1675–57.
- Goniewicz ML, Knysak J, Gawron M, et al. (2014). Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob Cont 23:133–9.
- Gould NS, Min E, Gauthier S, et al. (2011). Lung glutathione adaptive responses to cigarette smoke exposure. Respir Res 12:133.
- Hajek P, Etter JF, Benowitz N, et al. (2014). Electronic cigarettes: review of use, content, safety, effects on smokers and potential for harm and benefit. Addiction 109:1801–10.
- Health Canada. (1999). Official method T-116. “Determination of 1,3-butadiene, isoprene, acrylonitrile, benzene and toluene in mainstream tobacco smoke”. Ottawa: Health Canada.
- Horinouchi T, Higashi T, Mazaki Y, et al. (2016). Carbonyl compounds in the gas phase of cigarette mainstream smoke and their pharmacological properties. Biol Pharm Bull 39:909–14.
- Iksandar AR, Gonzalez-Suarez I, Majeed S, et al. (2016). A framework for in vitro systems toxicology assessment of e-liquids. Toxicol Mech Meth 26:1–24.
- International Organization for Standardization. (2015). ISO 3402:1999. Tobacco and tobacco products – atmosphere for conditioning and testing. Geneva: International Organization for Standardization.
- Invernizzi G, Ruprecht A, Mazza R, et al. (2004). Particulate matter from tobacco versus diesel car exhaust: an educational perspective. Tob Cont 13:219–21.
- Jensen RP, Luo W, Pankow JF, et al. (2015). Hidden formaldehyde in e-cigarette aerosols. N Engl J Med 372:392–4.
- Kannan K, Jain SK. (2000). Oxidative stress and apoptosis. Pathophysiology 7:153–63.
- Kilford J, Thorne D, Payne R, et al. (2014). A method for assessment of the genotoxicity of mainstream cigarette smoke by use of the bacterial reverse-mutation assay and an aerosol-based exposure system. Mutat Res Genet Toxicol Environ Mutagen 769:20–8.
- Kluchová Z, Petrásová D, Joppa P, et al. (2007). The association between oxidative stress and obstructive lung impairment in patients with COPD. Physiol Res Acad Sci Bohemoslov 56:51–6.
- Kosmider L, Sobczak A, Fik M, et al. (2014). Carbonyl compounds in electronic cigarette vapors: effects of nicotine solvent and battery output voltage. Nicotine Tob Res 16:1319–26.
- Lerner CA, Sundar IK, Yao H, et al. (2015). Vapors produced by electronic cigarettes and e-juices with flavorings induce toxicity, oxidative stress, and inflammatory response in lung epithelial cells and in mouse lung. PLoS One 10:e0116732.
- Li N, Kim S, Wang M, et al. (2002). Use of a stratified oxidative stress model to study the biological effects of ambient concentrated and diesel exhaust particulate matter. Inhal Toxicol 14:459–86.
- Lowe F, Fearon IM, Camacho O, et al. (2015). A framework for the assessment of reduced-risk tobacco and nicotine products. 69th Tobacco Science Research Conference (TSRC), 20–23 September 2015, Naples, FL.
- Margham J, McAdam K, Forster M, et al. (2016). Chemical composition of an e-cigarette aerosol: a quantitative comparison with cigarette smoke. Chem Res Toxicol. doi: 10.1021/acs.chemrestox.6b00188.
- Maunders H, Patwardhan S, Phillips J, et al. (2007). Human bronchial epithelial cell transcriptome: gene expression changes following acute exposure to whole cigarette smoke in vitro. Am J Physiol Lung Cell Mol Physiol 292:L1248–56.
- Misra M, Leverette RD, Cooper BT, et al. (2014). Comparative in vitro toxicity profile of electronic and tobacco cigarettes, smokeless tobacco and nicotine replacement therapy products: e-liquids, extracts and collected aerosols. Int J Environ Res Public Health 11:11325–47.
- Muller T, Haussmann HJ, Schepers G. (1997). Evidence for peroxynitrite as an oxidative stress-inducing compound of aqueous cigarette smoke fractions. Carcinogenesis 18:295–301.
- Muller T, Gebel S. (1998). The cellular stress response induced by aqueous extracts of cigarette smoke is critically dependent on the intracellular glutathione concentration. Carcinogenesis 19:797–801.
- Neilson L, Mankus C, Thorne D, et al. (2015). Development of an in vitro cytotoxicity model for aerosol exposure using 3D reconstructed human airway tissue: application for assessment of e-cigarette aerosol. Toxicol in Vitro 29:1952–62.
- Noya Y, Seki K, Asano H, et al. (2013). Identification of stable cytotoxic factors in the gas phase extract of cigarette smoke and pharmacological characterization of their cytotoxicity. Toxicology 314:1–10.
- Pellegrino RM, Tinghino B, Mangiaracina G, et al. (2012). Electronic cigarettes: an evaluation of exposure to chemicals and fine particulate matter (PM). Ann Ig 24:279–88.
- Pryor WA. (1997). Cigarette smoke radicals and the role of free radicals in chemical carcinogenicity. Environ Health Persp 105:875–82.
- Public Health England. (2015). E-cigarettes: an evidence update. London: Public Health England.
- Reuter S, Gupta SC, Chaturvedi MM, et al. (2010). Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49:1603–16.
- Roemer E, Shramke H, Weiler H, et al. (2012). Mainstream smoke chemistry and in vitro and in vivo toxicity of the reference cigarettes 3R4F and 2R4F. Beiträge zur Tabakforschung International/Contrib Tob Res 25:316–35.
- Romagna G, Allifranchini E, Bocchietto E, et al. (2013). Cytotoxicity evaluation of e-cigarette vapor extract on cultured mammalian fibroblasts (clearstream-life): comparison with tobacco cigarette smoke extract. Inhal Toxicol 25:354–61.
- Royal College of Physicians. (2016). Nicotine without smoke: tobacco harm reduction. London: RCP.
- Sato E, Koyama S, Takamizawa A, et al. (1999). Smoke extract stimulates lung fibroblasts to release neutrophil and monocyte chemotactic activities. Am J Physiol 277:L1149–57.
- Scheffler S, Dieken H, Krischenowski O, et al. (2015a). Cytotoxic evaluation of e-liquid aerosol using different lung-derived cell models. Int J Environ Res Public Health 12:12466–74.
- Scheffler S, Dieken H, Krischenowski O, et al. (2015b). Evaluation of e-cigarette liquid vapor and mainstream cigarette smoke after direct exposure of primary Human bronchial epithelial cells. Int J Environ Res Public Health 12:3915–25.
- Schlage WK, Iskandar AR, Kostadinova R, et al. (2014). In vitro systems toxicology approach to investigate the effects of repeated cigarette smoke exposure on human buccal and gingival organotypic epithelial tissue cultures. Toxicol Mech Meth 24:470–87.
- Sethi JM, Rochester CL. (2000). Smoking and chronic obstructive pulmonary disease. Clin Chest Med 21:67–86. viii.
- Tavilani H, Nadi E, Karimi J, et al. (2012). Oxidative stress in COPD patients, smokers, and nonsmokers. Respir Care 57:2090–4.
- Taylor M, Vella L, Carr T. (2014). Assessment of an in vitro model of lung epithelial cell stress responses. Free Rad Biol Med 75:S51.
- Thorne D, Adamson J. (2013). A review of in vitro cigarette smoke exposure systems. Exp Toxicol Pathol 65:1183–93.
- Thorne D, et al. (2016). The mutagenic assessment of electronic-cigarettes and tobacco smoke using the Ames assay in strains TA98 and TA100. Mutat Res, in press.
- van der Toorn M, Smit-de Vries MP, Slebos DJ, et al. (2007). Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 293:L1156–62.
- Vayssier M, Banzet N, François D, et al. (1998). Tobacco smoke induces both apoptosis and necrosis in mammalian cells: differential effects of HSP70. Am J Physiol 275:L771–9.
- Volpi G, Facchinetti F, Moretto N, et al. (2011). Cigarette smoke and α,β-unsaturated aldehydes elicit VEGF release through the p38 MAPK pathway in human airway smooth muscle cells and lung fibroblasts. Br J Pharmacol 163:649–61.
- Walters MJ, Paul-Clark MJ, McMaster SK, et al. (2005). Cigarette smoke activates human monocytes by an oxidant-AP-1 signaling pathway: implications for steroid resistance. Mol Pharmacol 68:1343–53.
- Wirtz HR, Schmidt M. (1996). Acute influence of cigarette smoke on secretion of pulmonary surfactant in rat alveolar type II cells in culture. Eur Respir J 9:24–32.
- Wiseman H, Halliwell B. (1996). Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J 313:17–29.
- Zitka O, Skalickova S, Gumulec J, et al. (2012). Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett 4:1247–53.