
ABSTRACT
Some compounds of a series of novel pyrrolo-1,5-benzoxa(thia)zepine, a well-known group of tubulin targeting agents, display anti-tumor effects mainly inducing cell cycle arrest and apoptosis in several human cancer models. A member of this family, pyrrolo-1,5-benzoxazepine-15 (PBOX-15), has previously shown potent pro-apoptotic activity in a variety of human tumor cell types, with minimal toxicity toward normal blood and bone marrow cells. In this study, we evaluated the PBOX-15-mediated effects in human colorectal cancer cell (CRC) lines, DLD-1 and HT-29. The compound, used at concentrations equal to or greater than 1 μM, inhibited the proliferation of human CRC cells, inducing a significant cell cycle arrest in the G2/M phase. In DLD-1 cells, treatments prolonged over 48 h triggered a strong activation of the intrinsic apoptotic pathway as indicated by activation of caspase-9, caspase-3 and PARP cleavage. Moreover, nanomolar concentrations of PBOX-15, significantly improved the oxaliplatin and 5-fluouracil-induced anti-proliferative effects in DLD1 cell line. The observed synergistic interaction of both PBOX-15/Oxaliplatin and PBOX-15/5FU may involve activation of p38 MAPK and JNK pathway, which in turn significantly increased caspase-3 cleavage in DLD-1 cells, treated with PBOX-5/Oxaliplatin but not with PBOX-15/5FU. Moreover, PBOX-15/5FU-treated cells showed an increase in expression of the pro-apoptotic protein Bax. Taken together, these results show that PBOX-15 could represent a promising compound for the treatment of human CRC and a strong candidate for novel therapeutic options.
Abbreviations
| 5-FU | = | 5-fluorouracil |
| CB | = | cannabinoid |
| CBRs | = | cannabinoid receptors |
| CRC | = | colorectal cancer |
| FAAH | = | fatty acid amide hydrolase |
| IFNs | = | interferons |
| IL-2 | = | interleukin 2 |
| MAGL | = | monoacyl glycerol lipaseapse |
| PARP | = | Poly (ADP-ribose) polymerase |
| PBOX | = | pyrrolo-1,5-benzoxazepine |
| PBOX 15 | = | 4-acetoxy-5-(1-(naphthyl)naphtho[2,3-b]pyrrolo[2,1-d][1,4]oxazepine |
| PBR | = | peripheral-type benzodiazepine receptor. |
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide and the fourth most common cause of death in industrialized countries.Citation1 CRC develops as a consequence of aberrant crypt proliferation or progression of benign hyperplasia to benign adenoma and in most cases to adenocarcinoma.Citation2 Oxaliplatin, used in combination with 5-fluorouracil (5-FU) and leucovorin (FOLFOX) for metastatic CRC, has led to 45% response rates, with a median survival approaching 2 years,Citation3-5 and has also been found to be very effective in the adjuvant setting.Citation6 Despite these impressive accomplishments, all metastatic CRC eventually become resistant to oxaliplatin, with a median time to progression of 8 months.Citation7 Similar to oxaliplatin, 5-fluouracil resistance has also been observed in CRC patients.Citation8 Thus, new strategies to overcome chemotherapeutic resistance are under exploration and it's necessary to design new drugs for a more selective tumor therapy. Strong evidence suggests that the endogenous cannabinoid system, comprising cannabinoid receptors, endogenous ligands and enzymes for ligand biosynthesis and degradation, may provide new targets for tumor intervention,Citation9-11 and elucidate the potential role of endocannabinoids in the control of CRC growth and progression.Citation12,13
Endocannabinoid (EC) levels are strongly elevated in colorectal adenomatous polyps but slightly reduced in carcinomas.Citation14 Conversely, CB1 receptor expression in carcinoma is lower than in the healthy colon epithelium.Citation15 However, this potential protective mechanism is incomplete because of the rapid degradation ECs in vivo. Thus, a promising approach to maintain a localEC tone is to increaseEC levels by delaying their inactivation with specific inhibitors of metabolic pathway. As others reported, the inhibition of degradative enzymes (particularly, fatty acid amide hydrolase FAAH), leads to the amplification ofEC anti-tumor properties via indirect activation of CB receptors. Therefore, FAAH represents a convenient target for the development of anti-cancer drugs.
A novel series of pyrrolo-1,5-benzoxa(thia)zepine (PBOX) compounds, synthesized as high-affinity ligands to the peripheral-type benzodiazepine receptor (PBR), are implicated, among others, in cell growth and apoptosis.Citation16 In particular, PBOX-15 is a tubulin depolymerizing agent displaying a proapoptotic activity in a variety of human solid and hematological malignancies,Citation16-21 with minimal toxicity toward normal blood and bone marrow cells.Citation17 In the present study we identified PBOX-15 as a submicromolar selective inhibitor of FAAH enzyme (IC50= 0.843 μM) and we investigated the anti-tumor efficacy and mode of action of this compound on CRC cell lines. Moreover, the effect of PBOX-15-combined treatment with the chemotherapeutic agents oxaliplatin and 5-fluorouracil was investigated.
Results
PBOX-15 reduces the proliferation of FAAH-positive CRC cells
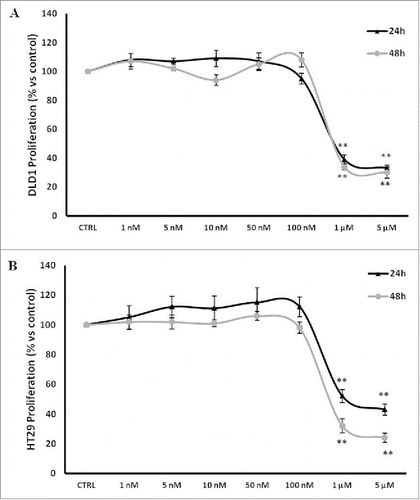
The effect of PBOX-15 on cell proliferation was specifically evaluated through BrdU incorporation analysis treating 2 common CRC cell lines, DLD-1 and HT29 cells, expressing high levels of FAAH enzyme, with increasing concentrations ranging from 1 nM to 5 µM (). PBOX-15, at the highest concentrations tested (1 µM and 5 µM) significantly inhibited cell proliferation of both DLD-1 and HT29 cell lines (). These data indicate that PBOX-15 interferes with CRC cell proliferation.
Figure 1. PBOX-15 inhibits proliferation of human CRC cell lines. (A and B) CRC cell lines (1 × 104 / 0.33 cm2 plate) were incubated with increasing concentrations of PBOX-15 (from 0.001 to 5 µM) for 24 and 48 hours. Cell proliferation was evaluated by measuring BrdU incorporation during DNA synthesis through a colorimetric ELISA assay. The results are shown as the mean ± SD from triplicate cultures performed at least 3 times. (**p <0.01, ANOVA compared to the untreated control cells).
PBOX-15 induces a significant cell cycle arrest in the G2/M phase of CRC cells
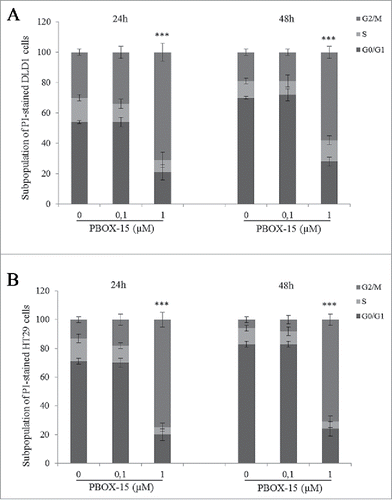
In order to investigate the effects of PBOX-15 on the cell cycle regulation in CRC cells, flow cytometry analysis was carried out. FACS analysis of PI-stained DLD-1 cells revealed that PBOX-15 (1 μM) for 24 h significantly induced a strong accumulation of the cells in the G2/M phase of the cell cycle compared to exponentially growing untreated CRC control cells. This cellular accumulation also occurred after 48 h of treatment. Accordingly, the percentage of cells in G0/G1 decreased by about 20% (). Similarly to DLD-1, also HT29 cells presented an increase in G2/M phase upon treatment with PBOX-15 (1 μM) (), showing that the modulation of the cell cycle was not cell line-specific and explaining, at least partially, the reduction in cell number after treatment as shown by BrdU assay (). As expected, treatment with PBOX-15 at a concentration of 100 nM did not change significantly the percentage distribution of the treated cells at different stages of the cycle compared to control cells treated with vehicle ().
Figure 2. PBOX-15 induces a significant cell cycle arrest in the G2/M phase of DLD-1 and HT29. The percentage of cells was determined by FACS following PtdIns incorporation. The histograms report the percentage of cells in G0/G1, S and G2/M phases after treatment of DLD1 (A) or HT29 (B) cell lines with PBOX-15 (0.1 and 1 µM) for 24 and 48 hours. Each value is the mean ± SD of 3 separate experiments performed in duplicates (***p <0.005, ANOVA).
PBOX-15 induces apoptosis in DLD-1 cells by activation of the intrinsic pathway
Since obtained data showed a good reproducibility of the PBOX-15-induced antiproliferative effect in 2 CRC cell lines and considering the similar molecular profile of DLD-1 and HT29 cells, we decided to focus our study on DLD-1 cells.
In order to analyze if the reduction in cell proliferation mediated by PBOX-15 was associated with apoptosis induction, we analyzed DLD-1 cells upon treatment with 1 μM PBOX-15. We found that at 48 h, PBOX-15 induced cellular DNA degradation as indicated by the appearance of a pattern of DNA ladders ().
Figure 3. In vitro assessment of apoptosis in DLD-1 cells. (A) DNA-fragmentation assay in DLD-1 cells.DNA was prepared from CRC untreated or treated respectively for 48 h with PBOX-15 (1 µM) and analyzed in a 1.6% agarose gel. Lane M is the 100-bp DNA ladder. (B) Annexin V-FITC/PI staining was used to corroborate the induction of apoptosis. The induction of apoptosis was determined by flow cytometric analysis of Annexin V-FITC and PtdIns-staining. Cells in the lower right quadrant indicate Annexin V-positive, early apoptotic cells. The cells in the upper right quadrant indicate Annexin V-positive/PI-positive, late apoptotic cells. (C) The histograms represents total apoptosis (% of Annexin V+ Annexin V/PtdIns positive cells) in PBOX15-treated cells compared with the untreated control.
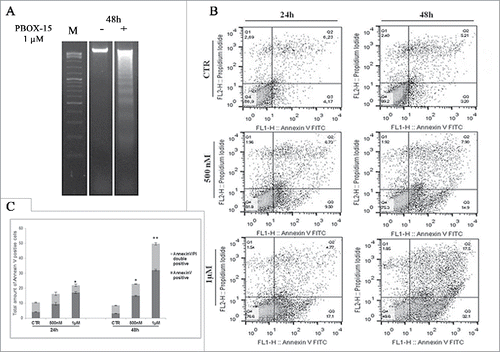
To corroborate the apoptotic fate, DLD-1 cells treated with PBOX-15 were double stained with Annexin V and PtdIns and analyzed by flow cytometry. As shown in , both Annexin V-positive (early apoptotic cells, bottom right quadrant) and Annexin V/PI-double positive cells (late apoptotic cells, top right quadrant) were significantly increased already at 500 nM at 24 h of treatment (9.27% ± 0.946 and 6.91 ± 1.179, respectively). At the highest dose of PBOX-15 (1 μM), at 48 h of treatment, we detected a significant increase of total apoptotic cells with respect to the control (50.0% ± 1.454 versus 8.46% ± 0.345).
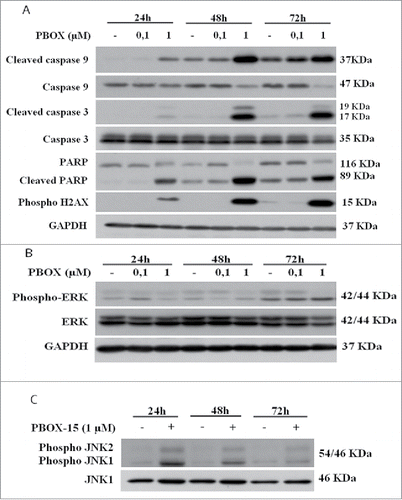
In order to dissect the molecular mechanisms underlying apoptosis induction, we examined whether PBOX-15 treatment activated caspase-dependent apoptosis in DLD-1 cells. As shown, activation of caspase-9, the initiator protease of the intrinsic apoptotic pathway also occurred after PBOX-15 treatment, indicating activation of the intrinsic pathway (). The treatment with PBOX-15 resulted in a strong cleavage and activation of the executioner caspase-3, particularly evident from 48 h of treatment. Cleavage of Poly(ADP-ribose) polymerase (PARP) also correlated with caspase-3 activation. Moreover, in accordance with DNA fragmentation, we found an increase in the phosphorylated form of histone H2AX, a nuclear marker of various types of DNA damage, starting from 24 h of treatment (). These data, taken together, indicate that PBOX-15 interferes with cell proliferation by activating the intrinsic apoptotic pathway.
Figure 4. PBOX-15 affects both the apoptotic cascade and MAPKs pathway. (A and B) Lysates from DLD-1 cells, untreated (−) or treated with PBOX-15 for the indicated time points at the concentrations of 100 nM and 1 µM, were immunoblotted with anti-caspase-9, anti-caspase-3, anti-PARP (total and cleaved forms) and anti-pH2AX antibodies (panel A), and with anti-ERK or anti-pERK antibodies (panel B). To confirm equal loading the filters were stripped and reprobed with anti-GAPDH antibody. (C) Lysates from DLD-1 cells untreated (−) or treated (+) with PBOX-15 (1 µM) for the indicated incubation times, were immunoblotted with anti-JNK and anti-pJNK antibodies. Blots shown are representative of at least 3 independent experiments.
PBOX-15 modulates the phosphorylation of ERK and JNK in DLD-1 cells
To investigate the molecular mechanism by which PBOX-15 induces colon tumor cell apoptosis, we tested whether it regulates genes known to control cell death and proliferation pathways. We evaluated the extent of phosphorylation of ERK1/2 upon treatment of DLD-1 cells with PBOX-15 for increasing time points. As shown in , treatment caused a dose- and time-dependent inhibition of ERK phosphorylation. Moreover, we assessed the involvement of JNK/SAPK, another member of the MAPK family, involved in DNA damage-response and apoptosis. Phosphorylation of JNK1 and JNK2 was clearly increased at 24 h of treatment with PBOX-15 at 1 µM dose and persisted higher than the control until 72 h ().
PBOX-15 sensitizes cells to targeted colon cancer therapies
As a next step, we compared the PBOX-15 inhibitory effect on DLD-1 cell proliferation to that of 2 commercially available agents that are currently in clinical use as first-line anticancer therapeutics, oxaliplatin and 5-fluorouracil. In dose-dependent experiments, both drugs, oxaliplatin (ranging from 0.08 μM to 10 μM) and 5-FU (ranging from 3 μM to 100 μM), were unable to inhibit CRC cell proliferation when used at low concentrations for 72 h. In particular, a significant inhibition of cell proliferation was obtained with oxaliplatin 10 μM and 5-fluorouracil 50 μM or higher (fraction of cells affected/killed 15% and 13–29%, respectively) after 72 h of treatment (). Interestingly, the combined treatment of PBOX-15 with oxaliplatin (1:10 Molar ratio) () or 5-fluorouracil (1:100 Molar ratio) () inhibited DLD-1 cell proliferation more effectively than the treatment with each single agent alone. Indeed, the combination index (CI) is under 1, thus indicating synergistic interactions ().
Figure 5. Anti-proliferative effect of PBOX-15, Oxaliplatin and 5-FU in DLD-1 cells. (A) DLD-1 cells were incubated with increasing concentrations of PBOX-15 (ranging from 0.005 µM to 0.3 µM), Oxaliplatin (ranging from 0.08 µM to 10 µM) or 5-FU (ranging from 3 µM to 100 µM) for 72 hours. The dose-effect curve was performed using the CalcuSyn software and it showed the dose of the drug vs the fraction of the cells affected/killed by PBOX15, Oxaliplatin or 5-FU used alone. A representative experiment (carried out at least twice) is reported. (B) DLD-1cells were treated for 72 hours with Oxaliplatin (0.08-10 µM) or PBOX-15 in combination with Oxaliplatin (1:10 molar ratio) at the concentrations indicated in the table. (C) DLD-1 cells were treated for 72 hours with 5-FU (3-100 µM) or PBOX-15 in combination with 5-FU (1:100 molar ratio) at the concentrations indicated in the table. Cell proliferation was evaluated by measuring BrdU incorporation during DNA synthesis through a colorimetric ELISA assay. The dose-effect curve was done using the CalcuSyn software and a representative experiment (carried out at least twice) is reported.
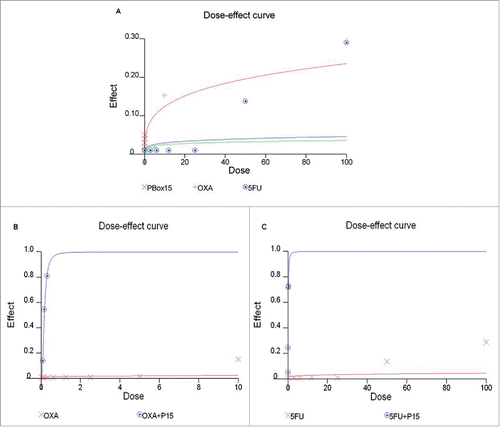
Table 1. Combination index (CI), Fraction affected (FA) and Dose Reduction Index (DRI) for PBOX-15 and OXA/5-Fluorouracil in DLD-1 cells.
Involvement of MAPK pathways in the enhancement of chemotherapy-induced apoptosis by PBOX-15
We have previously found that PBOX-15 is able to modulate oppositely ERK and JNK. Since MAPK pathways have been implicated in the modulation of chemotherapy-induced apoptosis, we evaluated whether the observed synergistic effects of PBOX-15 with Oxaliplatin and 5FU could involve MAPKs. After 48 h of combined treatment, we found a clear increase of phosphorylated form of JNK1 and JNK2, for both combination PBOX-15/Oxaliplatin and PBOX-15/5-FU combinations, compared to each single agent or to vehicle alone (). Furthermore, the combined effect also modulates the activation of p38-MAPK, as evidenced by the increase of its phosphorylated form after 72 h of treatment ().
Figure 6. Synergistic effect may possibly involve MAPK pathways. (A, B and C) Lysates from DLD-1 cells, untreated (−) or treated (+) with Oxaliplatin (3 μM), 5-Fluorouracil (30 μM) and PBOX-15 (300 nM) alone or in combination for 48 h (panel A) or 72 h (panels B and C) were immunoblotted with anti-JNK (total and phosphorylated forms in panel A), with anti-p38 (total and phosphorylated forms in panel B), with anti-caspase 3 (total and cleaved forms in panel C) antibodies. In B lower panel, the histograms shown represent the densitometric analyses of phospho-p38 expressed as fold change of the total p38 amount. Each value is the mean ± SD of 3 independent experiments (*p <0.05, ANOVA). (D) Lysates from DLD-1 cells, untreated or treated with 5-FU (30 μM) and PBOX-15 (300 nM) alone or in combination for 72 h, were immunoblotted with anti-Bax antibody. To confirm equal loading, the filters were stripped and reprobed with anti-GAPDH antibody. Blots shown are representative of at least 3 independent experiments with similar results.
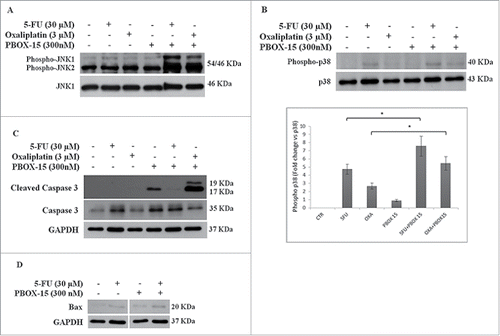
Since JNK and p38 MAPKs activate in different ways cell death processes, we analyzed the caspase-3 cleavage in DLD-1 cells treated for 72 h with PBOX-15 (300 nM) co-administered with Oxaliplatin (3 µM) or 5-FU (30 µM). The activation of caspase-3 was clearly detectable in PBOX15/Oxaliplatin-treated cells but not in DLD-1 treated with PBOX-15/5-FU in which only a very slight signal was observed (). On the other hand, at the same time point, PBOX-15/5FU-treated cells showed a slight increase of the pro-apoptotic protein Bax ().
PBOX-15 selectively inhibits FAAH enzyme activity
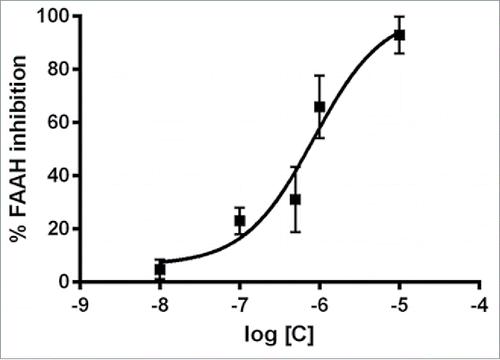
After a systematic screening of a selected number of pyrrolo-1,5-benzoxa(thia)zepine compounds, PBOX-15 (pyrrolo-1,5-benzoxazepine-15) was identified as a selective inhibitor of the FAAH enzyme. PBOX-15 was found able to inhibit FAAH with a submicromolar potency (IC50 = 847.8 nM) (). Interestingly, the compound demonstrated a high degree of selectivity.
Figure 7. PBOX-15 selectively inhibits FAAH enzyme activity. Dose–response curves of PBOX-15-mediated inhibition of FAAH activity. Data are reported as means ± SD of n = 3 experiments.
Discussion
Strong experimental evidence collected both in vitro and in vivo, proposed the endocannabinoids system as an interesting pharmacological target for the control of CRC proliferation and the inhibition of neo-angiogenic and metastatic processes, underlying the progression of CRC. These effects are obtained either by direct activation of the CB1 receptor or indirect activation through the inhibition of the main metabolic enzyme (i.e., FAAH). For this reason, the use of selective inhibitors of FAAH may represent a convenient pharmacological alternative to CB1 and CB2 receptor agonists.Citation12 The use of FAAH inhibitors rather than CB receptor agonists embraces several advantages: 1) minimize the possible psychotropic activity after systemic administration; 2) reduce the toxicity for mononuclear and bone marrow cells; 3) improve tolerance in animal models and 4) lack the recognition by proteins involved in chemoresistance (i.e., ABC transporters, P-glycoprotein, BCRP proteins).Citation24 While the efficacy of a direct activation of CBRs in tumor has been extensively described, the approach of inducing an indirect activation via the block of ligand degradation has been only seldom applied. Indeed the FAAH inhibitor N-arachidonoylserotonin increased colon endocannabinoid levels, reducing the development of precancerous lesion in the mouse colon.Citation13 Moreover the FAAH inhibitor URB597 increased the antiproliferative effects of endocannabinoids in neuroblastoma, melanoma and non-small cell lung cancer cells.Citation25-27 Therefore, the identification of a novel compound that binds and inhibits FAAH raises the obvious question of whether this molecule may interfere with proliferation of target cells.
Some compounds belonging to the pyrrolo-1,5-benzoxazepines family (PBOXs), originally developed as a novel group of tubulin targeting compounds, such as PBOX-6 and PBOX-15 have been already reported to induce a G2/M cell cycle arrest in several human cell lines.Citation17,18 Among these compounds, PBOX-6 was previously tested in CRC models using a murine colon carcinoma cell line (CT-26) and Caco-2, a human colon carcinoma derived cell line able to differentiate in culture.Citation28 In our work, during a screening program, we identified a compound belonging to the family of pyrrole-1,5-benzoxazepine, PBOX-15, as a submicromolar selective inhibitor of FAAH enzyme in rat brain membranes (IC50 = 0.843 μM).
Therefore, given the antitumor efficacy of both pyrrolo-1,5-benzoxazepines and FAAH inhibitors, we were prompted to investigate the specific anti-tumor efficacy and mode of action of PBOX-15 in FAAH-positive CRC cell lines, mainly focusing on its ability to improve the response to drugs currently used in the clinical practice for the treatment of colon cancer. The obtained results show that PBOX-15, already at 1 μM, inhibits the proliferation of human CRC cell lines in a dose- and time-dependent manner, inducing a significant cell cycle arrest in the G2/M phase with a concomitant induction of apoptosis already at 24 h from exposure to the compound. Moreover, the compound triggers the intrinsic pathway of apoptosis, as indicated by a significant occurrence of fragmentation of cellular DNA and by activation of caspase-9, caspase-3, and PARP, the enzyme responsible for DNA repair after damage.
Lu and colleaguesCitation29 demonstrate that phosphorylated form of histone H2AX is essential for Caspase-Activated DNase (CAD)-mediated DNA fragmentation and that cJun NH2-terminal kinase (JNK), a member of the stress-activated MAP kinase group, can phosphorylate its non-canonical substrate H2AX (Ser139), in vitro. Our results show that the dose- and time-dependent increase of phospho-H2AX corresponds to a transient increase in the phosphorylated form of JNK, after PBOX-15 exposure. Furthermore, the compound used at 1 μM reduced ERK phosphorylation, starting from 24 h of treatment. Accordingly with a lack of effect on CRC proliferation at the concentration of 100 nM, PBOX-15 does not alter the levels of activated ERK. These results suggest that the effect of PBOX-15 is mediated, at least in part, via MAPK pathways. At the moment we are not able to dissect what percentage of PBOX-15-induced inhibition of FAAH is able to take part in the antiproliferative observed effects.
Indeed, PBOX-15 has been reported to induce G2/M arrest and apoptosis in HeLa cells where no expression of FAAH was found.Citation18 Wild-type HeLa cells suggests that FAAH activity is not an absolute requirement for cell growth control and, as a consequence, the anti-proliferative effects could be ascribed, only in part, to FAAH inhibition. However, the obtained results demonstrate that PBOX-15 is able to significantly modulate cellular pathways involved in the proliferation and progression of CRC.
Intriguingly, several reports have shown that in leukemia and in multiple myeloma PBOX-15 has a reduced, if not absent, toxicity in normal cells compared to transformed ones.Citation19,30,31 Therefore, it seemed interesting to assess whether, at doses lower than those used in the assessment of cell proliferation, PBOX-15 is able to improve the sensitivity to treatment with oxaliplatin or with 5-FU. Oxaliplatin in combination with 5-Fluorouracil and leucovorin (FOLFOX) has been approved for metastatic CRC therapy. Despite clinical success, patients who initially respond to the treatment may subsequently become refractory,Citation5 and thus the attention has been directed toward alternative strategies, including the use of combined therapies, able to contrast resistance occurrence and to decrease the effective doses and, consequently, the adverse effects of the drugs used in combination. Therefore, we tested the antiproliferative activity of PBOX-15 in combination with the cytotoxic agents, oxaliplatin and 5-FU. Interestingly, when used at low doses in combination with oxaliplatin (5 μM) or 5-FU (50 μM), PBOX-15 significantly enhanced the antiproliferative effects of these chemotherapeutics. Moreover, combination index (CI) obtained for the combined use of PBOX-15 with both Oxaliplatin and 5-FU shows a strong synergistic effect (CI<1.0) of the drugs used in combination and a good DRI for both chemotherapic compounds ().
Targeting MAPKs pathway is one of the best strategies in the control of tumor progression. The role of stress-activated MAPKs such as p38 and JNK in cancer cells outcome seems to vary in a tumor-dependent manner, but their modulation may be implicated in response to anticancer drugs.Citation32,33 It has been reported that in HCT-116 CRC cell line, p38 MAPK plays an important role in oxaliplatin-induced apoptosis.Citation34 Here we found that the combination of sub-micromolar doses of PBOX-15 (300 nM) and Oxaliplatin cooperates to induce apoptosis possibly through activation of p38 and JNK MAPKs, as evidenced by the cleavage of caspase 3. For PBOX-15/5-FU combination, we also found the activation of both p38 and JNK, but we are not able to demonstrate the activation of caspase-dependent cell death. de la Cruz-Morcillo et al reported that p38 MAPK inhibition is associated, in p53-dependent manner, to 5-FU resistance. Moreover, p38 is associated with autophagic response to 5-FU.Citation35 Other authors have demonstrated that resistance to long-term 5-FU exposure, was dependent not only on p53 status, but also on the levels of anti- and pro-apoptotic protein, such as Bax.Citation36 Our results show that combined treatment PBOX-15/5-FU is able to increase Bax levels, but the cleavage of caspase 3 was less than in the treatment with PBOX-15 alone.
Recently, it has been shown that PBOX-15 acts as autophagic inducer in Caco2 cells and the inhibition of late stage autophagy by bafilomycin-A1, a vacuolar H + ATPase inhibitor, does not increase PBOX-15-induced cell death.Citation28 On the other hand, unlike DLD-1, Caco2 cell in vitro differentiation dramatically modify FAAH expression and the sensitivity to cannabinoids.Citation14 We speculate that the different genetic profiles of the 2 cell lines, could at least partly explain the percentage of apoptosis induced by PBOX-15 in DLD-1, higher than that observed in Caco2 cells.
Thus, further studies are required to clarify if the synergistic effect observed with PBOX-15/5-FU combined treatment may involve caspase-independent cell death mechanisms or whether this combination may trigger an autophagic response following long-term treatment. Even if we cannot conclude that the observed effect on cell proliferation are mediated, at least in part, through a direct involvement of FAAH, our data show that the pyrrolo-1,5-benzoxazepine PBOX-15 has anti-proliferative and pro-apoptotic activity in an in vitro model of human CRC FAAH positive cells.
Although further studies are needed to fully elucidate the molecular mechanisms triggered by PBOX-15 and the specific consequences of FAAH inhibition, this compound can synergize with the chemotherapeutics currently used for this type of cancer, Oxaliplatin and 5-FU. These results suggest that rational combination strategies should be further investigated in the preclinical setting using PBOX-15 to potentiate the effects of FOLFOX-based chemotherapy.
Materials and methods
Chemicals
The 4-acetoxy-5-(1-(naphthyl))naphtho[2,3-b]pyrrolo[2,1-d][1,4] oxazepine (PBOX-15) was synthesized as previously describedCitation16 and dissolved in dimethylsulfoxide (DMSO). The chemotherapeutic agents used were oxaliplatin, kindly provided by Sanofi-Aventis Research, and 5- Fluorouracil (Sigma Aldrich).
FAAH activity assay
The effect of different concentrations of compounds (from 0.5 to 10 μM) and vehicle (DMSO) on the hydrolysis of anandamide was measured by incubating the membrane fractions from rat brain (70 μg/sample) in Tris–HCl 50 mM, pH 9 for 30 min at 37°C in presence of [14C]-anandamide properly diluted with unlabeled anandamide. After incubation, the amount of [14C]-ethanolamine produced enzymatically was measured by scintillation counting of the aqueous phase after extraction of the incubation mixture with 2 volume-equivalents of chloroform/methanol (1:1 V/V). Data are expressed as the concentration exerting 50% inhibition of AEA hydrolysis (IC50), calculated with Prism 6.0 software (GraphPads®). Data are reported as means ± SD of n = 3 experiments.
Cell culture
Human colon DLD-1 (Interlab Cell Line Collection ICLC no. HTL 95011) and HT29 (ICLC no. HTL 99026) cells were, respectively, grown in RPMI-1640 and McCoy′s 5A (Lonza) medium supplemented with 2 mM L-glutamine, 10% fetal bovine serum (Invitrogen) and 100 U/ml penicillin, 100 mg/ml streptomycin (P/S; Euroclone) at 37°C in a humidified 5% CO2 air atmosphere.
Western blotting
CRC cells were washed twice in ice-cold phosphate-buffered saline (PBS) and lysed in buffer A (50 mM Tris–HCl pH8.0 buffer containing 150 mM NaCl, 1% Nonidet P-40, 2 mg/ml aprotinin, 1 mg/ml pepstatin, 2 mg/ml leupeptin, 1 mM Na3VO4). Protein concentration was determined by the Bradford assay using bovine serum albumin as standard. Cell lysates were subjected 10-12 % SDS-PAGE. Gels were electroblotted into nitrocellulose membranes (Millipore Co.) and filters were probed with the indicated primary antibodies: anti-caspase 3 (#9662, Cell Signaling), anti-cleaved caspase-3 (#9664, Cell Signaling) anti-caspase 9 (#9502, Cell Signaling), anti-cleaved caspase-9 (#7237, Cell Signaling), anti-PARP (#9542, Cell Signaling), anti-cleaved PARP (#5625, Cell Signaling), anti-ERK1/2 (#4695, Cell Signaling), anti-phospho ERK1/2 (#4370, Cell Signaling), anti-Bax (#5023, Cell Signaling), anti-phospho p38 (#4511, Cell Signaling), anti-p38 (#9212, Cell Signaling), anti-phospho JNK (#4668, Cell Signaling) anti-phospho H2AX (#9718, Cell Signaling) and anti-GAPDH (#2118, Cell Signaling), anti-JNK1 (ab10664, Abcam). Proteins were visualized with peroxidase-conjugated secondary antibodies using the enhanced chemiluminescence system (Amersham-Pharmacia Biosciences LTD).
Cell proliferation ELISA
Cell proliferation was evaluated, in vitro, by measuring BrdU incorporation during DNA synthesis through a colorimetric ELISA kit (Roche Diagnostics GmbH). In brief, DLD-1 and HT29 were seeded in triplicate into 96-well plates. Cells (1 × 104 /0.33 cm2 plate) were treated with the drugs (ranging from 1 nM to 5 μM) and incubated for 24 or 48 h. According to the protocol provided by the manufacturer, at the end of treatment, cells were first fixed with a denaturation solution for 30 min. and then incubated with anti-Bromodeoxyuridine (BrdU) antibody peroxidase conjugated solution (anti-BrdU-POD) for about 1 h followed by incubation with substrate solution for 20 minutes. The colorimetric reaction was measured through a microplate reader (Multiskan™ GO Microplate Spectrophotometer, Thermo Scientific) at 370 nm. The blank was performed in each experimental setup. The absorbance value of the blank was subtracted from other experimental values and cell proliferation was expressed as the percentage of absorbance values ± SD of treated samples to untreated controls.
Cell cycle analysis
To assess cell cycle profiles, CRC cells treated with vehicle or PBOX-15 for 24 and 48 h, (2.5 × 104 /6 cm plate) were collected, fixed in 70% ethanol and kept at −20°C overnight. Propidium iodide (PtdIns; 50 µg/ml) in ice-cold phosphate-buffered saline (PBS) was added to the cells for 15 min at room temperature. The cells were acquired by a FACS-Calibur flow cytometer (BD Biosciences, San Jose, CA, USA). The analysis was performed with ModFit LT v3.2 (Verity Software House, Inc.).
DNA fragmentation analysis
DLD-1 cells were cultured as previously described for cell cycle analysis and treated with PBOX-15 (1 μM) for 48 h. Adherent and floating CRC cells were harvested, washed once with ice-cold PBS and incubated with lysis buffer (5 mM Tris–HCl pH 8.0, 10 mM EDTA, 0.5% Triton X-100) on ice for 20 minutes. Then, cell lysates were clarified by centrifugation at 12,000 rpm for 20 min. The obtained supernatant was treated with RNase A (1 mg/ml) at 37°C for 60 min, and Proteinase K (8 mg/ml) at 65°C for 15 min and subsequent incubation at 37°C for 12 h. The total DNA, extracted by purification with phenol/chloroform/isoamyl alcohol and ethanol precipitation, was dissolved in 20 μl TE buffer (10 mM Tris–HCl—1 mM EDTA, pH 8,0), analyzed by 1.8% agarose gel electrophoresis. Approximately 20 μl DNA was loaded, stained by ethidium bromide and visualized under UV light.
Annexin V/Propidium iodide staining
Adherent cells were harvested by trypsinization and washed once with warm PBS. Cells were double stained with FITC-conjugated Annexin V (BioLegend) in a binding buffer (10 mM HEPES/NaOH, pH 7; 140 mM NaCl; 2.5 mM CaCl2) for 20 min at room temperature and then with PI for additional 15 min in the dark. The cells were analyzed on a FACS-Calibur flow cytometer (BDIS, Becton Dickinson). FITC and propidium iodide emissions were detected in the FL-1 and FL-2 channels, respectively. For each sample, at least 20,000 cells were recorded in list mode on logarithmic scales. Subsequent analysis was performed with FlowJo®software (BDIS).
Drug combination analysis
The combination index (CI) was calculated by the CalcuSyn software based on the Chou-Talalay equation,Citation22 which takes into account both potency (Dm or IC50) and shape of the dose-effect curve. Briefly, CI<1, CI = 1, and CI>1 indicate synergism, additive effect and antagonism, respectively. Dose reduction index (DRI) representing the measure of how much the dose of each drug in a combination may be reduced at a given effect level compared with the doses of each drug alone.Citation23 To explore the relative contribution of each agent to the anti-proliferative effect, combinations with 1:10 PBOX-15/Oxaliplatin and 1:100 PBOX-15/5-Fluorouracil molar ratios, were tested in DLD-1 cell line. Assessment of drug interaction was performed calculating the Combinatorial Index (CI). CI/fractional effect curves represent the CI vs. the fraction of cells affected/killed by oxaliplatin or 5-Fluorouracil and PBOX-15 in combination.
Statistical analysis
Statistical differences between the treatments and the control were evaluated by one-way analysis of variance (ANOVA). A P value less than 0.05 was considered statistically significant.
Note
A previous, related manuscript was retracted after publication in the European Journal of Pharmacology (retraction doi: 10.1016/j.ejphar.2014.01.024). Data included in the present manuscript published by Cancer Biology & Therapy are original, produced through experiments re-performed by the authors and include new results obtained in order to evaluate different pathways not investigated previously. The authors can provide the raw data of each experiment, as well as any material required, upon request. Table 1 is identical in both the present and retracted manuscripts because it presents the result of the pharmacological combinations evaluated by CalcuSyn software. This specific result was not among the reasons for the prior retraction since it contains sound data, from both a procedural and a statistics point of view. The authors are ready to provide any raw data used in the computational analysis.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was partially supported by National Interest Research Projects (PRIN 2008, Grant “Design, synthesis and antitumor efficacy of new modulators of the endocannabinoid system,” DM n. 1407, December 4, 2008prot.20088SPEFN) and by Associazione Educazione e Ricerca Medica Salernitana (ERMES).
M.C. P. was supported by fellowships from FIRC (Italian Foundation for Cancer Research).
References
- Haggar FA, Boushey RP. Colorectal cancerepidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg 2009; 22:191-7; PMID:21037809; http://dx.doi.org/10.1055/s-0029-1242458
- Wada-Hiraike O, Warner M, Gustafsson JA. New developments in oestrogen signalling in colonic epithelium. Biochem Soc Trans 2006; 34(Pt 6):1114-6; PMID:17073763; http://dx.doi.org/10.1042/BST0341114
- Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med 2005; 352:476-87; http://dx.doi.org/10.1056/NEJMra040958
- Alberts SR, Horvath WL, Sternfeld WC, Goldberg RM, Mahoney MR, Dakhil SR, Levitt R, Rowland K, Nair S, Sargent DJ, et al. Oxaliplatin, fluorouracil, and leucovorin for patients with unresectable liver-only metastases from colorectal cancer: a North Central Cancer Treatment Group Phase II Study. J Cli Oncol 2005; 23: 9243-9; http://dx.doi.org/10.1200/JCO.2005.07.740
- Cassidy J, Tabernero J, Twelves C, Brunet R, Butts C, Conroy T, Debraud F, Figer A, Grossmann J, Sawada N, et al. XELOX (capecitabine plus oxaliplatin): active first-line therapy for patients with metastatic colorectal cancer. J Clin Oncol 2004; 22:2084-91; http://dx.doi.org/10.1200/JCO.2004.11.069
- André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, Topham C, Zaninelli M, Clingan P, Bridgewater J, et al. Multicenter International Study of Oxaliplatin/5-Fluorouracil/Leucovorin in the Adjuvant Treatment of Colon Cancer (MOSAIC) Investigators. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med 2004; 350:2343-51; http://dx.doi.org/10.1056/NEJMoa032709
- Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, Alberts SR. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2004; 22:23-30; http://dx.doi.org/10.1200/JCO.2004.09.046
- Zhang N, Yin Y, Xu SJ, Chen WS. 5-Fluorouracil: mechanisms of resistance and reversal strategies. Molecules 2008; 13:1551-69; PMID:18794772; http://dx.doi.org/10.3390/molecules13081551
- Malfitano AM, Ciaglia E, Gangemi G, Gazzerro P, Laezza C, Bifulco M. Update on the endocannabinoid system as an anticancer target. Expert Opin Ther Targets 2011; 15:297-308; http://dx.doi.org/10.1517/14728222.2011.553606
- Pisanti S, Picardi P, D'Alessandro A, Laezza C, Bifulco M. The endocannabinoid signaling system in cancer.Trends Pharmacol Sci 2013; 34:273-82; PMID:23602129; http://dx.doi.org/10.1016/j.tips.2013.03.003
- Guida M, Ligresti A, De Filippis D, D'Amico A, Petrosino S, Cipriano M, Bifulco G, Simonetti S, Orlando P, Insabato L, et al. The levels of the endocannabinoid receptor CB2 and its ligand 2-arachidonoylglycerol are elevated in endometrial carcinoma. Endocrinology 2010; 151:921-8; PMID:20133454; http://dx.doi.org/10.1210/en.2009-0883
- Proto MC, Gazzerro P, Di Croce L, Santoro A, Malfitano AM, Pisanti S, Laezza C, Bifulco M. Interaction of endocannabinoid system and steroid hormones in the control of colon cancer cell growth. J Cell Physiol 2012; 227:250-8; PMID:21412772; http://dx.doi.org/10.1002/jcp.22727
- Izzo AA, Aviello G, Petrosino S, Orlando P, Marsicano G, Lutz B, Borrelli F, Capasso R, Nigam S, Capasso F, et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J MolMed (Berl) 2008; 86:89-98; PMID:17823781
- Ligresti A, Bisogno T, Matias I, De Petrocellis L, Cascio MG, Cosenza V, D'argenio G, Scaglione G, Bifulco M, Sorrentini I, et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology 2003; 125:677-87; PMID:12949714; http://dx.doi.org/10.1016/S0016-5085(03)00881-3
- Notarnicola M, Messa C, Orlando A, Bifulco M, Laezza C, Gazzerro P, Caruso MG. Estrogenic induction of cannabinoid CB1 receptor in human colon cancer cell lines. Scand J Gastroenterol 2008; 43:66-72; PMID:18938775; http://dx.doi.org/10.1080/00365520701559011
- Campiani G, Nacci V, Fiorini I, De Filippis MP, Garofalo A, Ciani SM, Greco G, Novellino E, Williams DC, Zisterer DM, et al. Synthesis, biological activity, and SARs of pyrrolobenzoxazepine derivatives, a new class of specific “peripheral-type” benzodiazepine receptor ligands. J Med Chem 1996; 39:3435-50; PMID:8784441; http://dx.doi.org/10.1021/jm960251b
- Mulligan JM, Greene LM, Cloonan S, Mc Gee MM, Onnis V, Campiani G, Fattorusso C, Lawler M, Williams DC, Zisterer DM. Identification of tubulin as the molecular target of proapoptotic pyrrolo-1,5-benzoxazepines. MolPharmacol 2006; 70:60-70
- Greene LM, Campiani G, Lawler M, Williams DC, Zisterer DM. BubR1 is required for a sustained mitotic spindle checkpoint arrest in human cancer cells treated with tubulin-targeting pyrrolo-1,5-benzoxazepines. Mol Pharmacol 2008; 73:419-30
- McElligott AM, Maginn EN, Greene LM, McGuckin S, Hayat A, Browne PV, Butini S, Campiani G, Catherwood MA, Vandenberghe E, et al. The novel tubulin-targeting agent pyrrolo-1,5-benzoxazepine-15 induces apoptosis in poor prognostic subgroups of chronic lymphocytic leukemia. Cancer Res 2009; 69:8366-75; PMID:19826055; http://dx.doi.org/10.1158/0008-5472.CAN-09-0131
- Nathwani SM, Butler S, Fayne D, McGovern NN, Sarkadi B, Meegan MJ, Lloyd DG, Campiani G, Lawler M, Williams DC, et al. Novel microtubule-targeting agents, pyrrolo-1,5-benzoxazepines, induce apoptosis in multi-drug-resistant cancer cells. Cancer Chemother Pharmacol 2010; 66:585-96
- Bright SA, McElligott AM, O'Connell JW, O'Connor L, Carroll P, Campiani G, Deininger MW, Conneally E, Lawler M, Williams DC, et al. Novel pyrrolo-1,5-benzoxazepine compounds display significant activity against resistant chronic myeloid leukaemia cells in vitro, in ex vivo patient samples and in vivo. Br J Cancer 2010; 102:1474-82; PMID:20407438; http://dx.doi.org/10.1038/sj.bjc.6605670
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27-55; PMID:6382953; http://dx.doi.org/10.1016/0065-2571(84)90007-4
- Takahashi N, Li W, Banerjee D, Guan Y, Wada-Takahashi Y, Brennan MF, Chou TC, Scotto KW, Bertino JR. Sequence-dependent synergistic cytotoxicity of ecteinascidin-743 and paclitaxel in human breast cancer cell lines in vitro and in vivo. Cancer Res 2002; 62:6909-15; PMID:12460906
- Bifulco M, Di Marzo V. Targeting the endocannabinoid system in cancer therapy: a call for further research. Nat Med 2002; 8:547-50; PMID:12042794; http://dx.doi.org/10.1038/nm0602-547
- Hamtiaux L, Hansoulle L, Dauguet N, Muccioli GG, Gallez B, Lambert DM. Increasing antiproliferative properties of endocannabinoids in N1E-115 neuroblastoma cells through inhibition of their metabolism. PLoS One 2011; 6:e26823; http://dx.doi.org/10.1371/journal.pone.0026823
- Hamtiaux L, Masquelier J, Muccioli GG, Bouzin C, Feron O, Gallez B, Lambert DM. The association of N-palmitoylethanolamine with the FAAH inhibitor URB597 impairs melanoma growth through a supra-additive action. BMC Cancer 2012; 12:92; PMID:22429826; http://dx.doi.org/10.1186/1471-2407-12-92
- Ravi J, Sneh A, Shilo K, Nasser MW, Ganju RK. FAAH inhibition enhances anandamide mediated anti-tumorigenic effects in non-small cell lung cancer by downregulating the EGF/EGFR pathway. Oncotarget 2014; 5:2475-86; PMID:24811863; http://dx.doi.org/10.18632/oncotarget.1723
- Greene LM, Nolan DP, Regan-Komito D, Campiani G, Williams DC, Zisterer DM. Inhibition of late-stage autophagy synergistically enhances pyrrolo-1,5-benzoxazepine-6-induced apoptotic cell death in human colon cancer cells. Int J Oncol 2013; 43:927-35; PMID:23799546
- Lu C, Zhu F, Cho YY, Tang F, Zykova T, Ma WY, Bode AM, Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell 2006; 23:121-32; PMID:16818236; http://dx.doi.org/10.1016/j.molcel.2006.05.023
- Lysaght J, Verma NK, Maginn EN, Ryan JM, Campiani G, Zisterer DM, Williams DC, Browne PV, Lawler MP, McElligott AM. The microtubule targeting agent PBOX-15 inhibits integrin-mediated cell adhesion and induces apoptosis in acute lymphoblastic leukaemia cells. Int J Oncol 2013; 42:239-46; PMID:23135704
- Maginn EN, Browne PV, Hayden P, Vandenberghe E, MacDonagh B, Evans P, Goodyer M, Tewari P, Campiani G, Butini S, et al. PBOX-15, a novel microtubule targeting agent, induces apoptosis, upregulates death receptors, and potentiates TRAIL-mediated apoptosis in multiple myeloma cells. Br J Cancer 2011; 104:281-89; PMID:21179037; http://dx.doi.org/10.1038/sj.bjc.6606035
- Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, Pan H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett 2014; 344:174-79; PMID:24333738; http://dx.doi.org/10.1016/j.canlet.2013.11.019
- Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene 2004; 23:2838-49; PMID:15077147; http://dx.doi.org/10.1038/sj.onc.1207556
- Dey H, Liu ZR. Phosphorylation of p68 RNA helicase by p38 MAP kinase contributes to colon cancer cells apoptosis induced by oxaliplatin. BMC Cell Biol 2012; 13:27; PMID:23110695; http://dx.doi.org/10.1186/1471-2121-13-27
- de la Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-González L, Melgar-Rojas P, Galán-Moya EM, García-Gil E, García-Cano J, Sánchez-Prieto R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene 2012; 31:1073-85; PMID:21841826; http://dx.doi.org/10.1038/onc.2011.321
- Violette S, Poulain L, Dussaulx E, Pepin D, Faussat AM, Chambaz J, Lacorte JM, Staedel C, Lesuffleur T. Resistance of colon cancer cells to long-term 5-fluorouracil exposure is correlated to the relative level of Bcl-2 and Bcl-X(L) in addition to Bax and p53 status. Int J Cancer 2002; 98:498-504; PMID:11920608; http://dx.doi.org/10.1002/ijc.10146