
Abstract
Head and neck squamous cell carcinoma (HNSCC) currently only has one FDA-approved cancer intrinsic targeted therapy, the epidermal growth factor receptor (EGFR) inhibitor cetuximab, to which only approximately 10% of tumors are sensitive. In order to extend therapy options, we subjected patient-derived HNSCC cells to small-molecule inhibitor and siRNA screens, first, to find effective combination therapies with an EGFR inhibitor, and second, to determine a potential mechanistic basis for repurposing the FDA approved agents for HNSCC. The combinations of EGFR inhibitor with anaplastic lymphoma kinase (ALK) inhibitors demonstrated synergy at the highest ratio in our cohort, 4/8 HNSCC patients' derived tumor cells, and this corresponded with an effectiveness of siRNA targeting ALK combined with the EGFR inhibitor gefitinib. Co-targeting EGFR and ALK decreased HNSCC cell number and colony formation ability and increased annexin V staining. Because ALK expression is low and ALK fusions are infrequent in HNSCC, we hypothesized that gefitinib treatment could induce ALK expression. We show that ALK expression was induced in HNSCC patient-derived cells both in 2D and 3D patient-derived cell culture models, and in patient-derived xenografts in mice. Four different ALK inhibitors, including two (ceritinib and brigatinib) FDA approved for lung cancer, were effective in combination with gefitinib. Together, we identified induction of ALK by EGFR inhibitor as a novel mechanism potentially relevant to resistance to EGFR inhibitor, a high ratio of response of HNSCC patient-derived tumor cells to a combination of ALK and EGFR inhibitors, and applicability of repurposing ALK inhibitors to HNSCC that lack ALK aberrations.
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the 6th most common cancer worldwide, affecting ∼600,000 patients per yearCitation1. Epidermal growth factor receptor (EGFR) is upregulated in up to 90% of HNSCC patients and is associated with poor survival, although its status is not associated with therapeutic responsesCitation2,Citation3. Cetuximab, a humanized monoclonal antibody targeting EGFR, has been the only FDA-approved cancer intrinsic targeted therapy for HNSCC since 2006. However, this treatment eventually fails, as patients either have intrinsic resistance to it or acquire resistance in less than 3 monthsCitation4. Given compensatory crosstalk between kinases within cancer cells, the use of combinations of other kinase inhibitors with EGFR inhibitors in HNSCC to overcome resistance to EGFR inhibitors has been investigated and suggested to be beneficial in preclinical models and under active testing in clinical trialsCitation5.
ALK is a kinase involved in various cancer types including anaplastic large-cell lymphoma, non-small-cell lung cancer (NSCLC) and neuroblastomaCitation6. Four inhibitors targeting ALK, crizotinib, alectinib, ceritinib (LDK378) and brigatinib (AP26113), have been FDA approved for the treatment of metastatic NSCLC positive for ALK fusionsCitation7,Citation8. However, the oncogenic roles of ALK and the effect of ALK inhibitors in HNSCC are less clear, due to relatively low expression and low frequency of ALK mutations or fusions in naive HNSCC tumorsCitation9. Manipulation of ALK in HNSCC has been found capable of regulating invasiveness and metastatic progression in HNSCCCitation10, and ALK is upregulated in advanced disease compared to early-stage tumorsCitation11. A recent study reported that co-targeting ALK and EGFR using TAE684 and gefitinib significantly reduces HNSCC cell proliferation in vitro and decreases tumor volumes of a cell line derived xenografts by 30%Citation11. However, whether the effectiveness of the combination of gefitinib and TAE684 was due to inhibition of EGFR and ALK was uncertain, since TAE684 has multiple targets other than ALKCitation12. More importantly, the mechanism of synergy between these two agents is unknown. Further, to better predict clinical outcome of using EGFR and ALK inhibitor combinations in treating HNSCC patients, patient-derived models are needed.
The purpose of our study was to interrogate HNSCC patient-derived epithelial tumor cells for repurposing FDA approved agents to HNSCC treatment to overcome EGFR inhibitor resistance. We used patient-derived models to examine the role of ALK in HNSCC, determine whether co-targeting ALK and EGFR could overcome EGFR resistance in HNSCC cells, and determine potential mechanisms of synergy of these agents.
Results
Inhibitor assays identified ALK and EGFR inhibitors as effective combination therapies in HNSCC patient-derived tumor cells
Given the ubiquitous role of tyrosine kinases in regulating critical cellular processes and redundant functions of kinases in cancer cells, we hypothesized that co-targeting EGFR and certain other kinase inhibitors would lead to enhanced anti-oncogenic response compared to the single-agent treatment of EGFR inhibitors. To test this hypothesis and to identify therapeutic agents that could overcome EGFR inhibitor resistance in HNSCC, we subjected patient-derived tumor cells to a small-molecule inhibitor screening assayCitation13, with or without an EGFR inhibitor, in order to identify agents that synergize with EGFR inhibitors in reducing HNSCC cell viability.
To ascertain the relevance of the inhibitor assay drug panel to HNSCC, we examined the drug target coverage of the drug panel in the context of our analysis of HNSCC somatic mutation data from the Cancer Genome Atlas (TGCA). Using a bioinformatics approach (see supplementary methods), we were able to leverage known drug-target data to discover potentially targetable HNSCC pathways. Of 224 pathways judged relevant to HNSCC in analysis of mutation enrichment from 279 TCGA HNSCC cases, 111 pathways (49.4%), which we termed “light” pathways, were targeted by the combined inhibitor panel and FDA-approved drugs based on the Cancer Targetome (an evidence-based framework of drug-target interactionsCitation14), with the remaining pathways “dark” or without current drugs targeting any members of the pathway.
In order to functionally evaluate HNSCC cell responses and their relevance to individual patients, we evaluated patient-derived tumor cells. The demographics and tumor characteristics of patients enrolled in this study include the oral and laryngeal sites predominant in TCGA HNSCC patients and alcohol and/or tobacco use in all but 1 (an HPV positive case), based on our analysis of 279 TCGA HNSCC patients (Supplementary Table S1)Citation15. Original tumor H&E staining revealed 65% (median) tumor in the specimen, and keratin and vimentin staining showed 90.5% (median) epithelial cells in the patient-derived tumor cells (data not shown).
A low dose (50 nM) of EGFR inhibitor was selected to be tested in combination with the drugs on the inhibitor assay panel. This dose is clinical achievable, and is lower than the IC50s of most HNSCC cell lines tested in the literatureCitation16; therefore it was selected as likely to allow detecting improved IC50s of combinations with the drugs on the panel and to eliminate off-target effect by a high dose of the drug.
An effective drug from the inhibitor assay for any given patient was defined as a drug that has an IC50 that is lower than 20% of the median IC50 of all the HNSCC patients tested on this panel, thus showing a degree of selectivity rather than being generally toxic to all patients’ tumor cells. A drug that was potentially synergistic to EGFR inhibitor was defined as one that decreased IC50 below 20% of the median IC50 after adding EGFR inhibitor but not as a single agent for that patient.
Fourteen out of 122 drugs on the panel showed effectiveness in combination with EGFR inhibitors in the patient-derived tumor cells (Supplementary Table S2). In particular, our approach detected PI3K inhibitors PI103, BEZ235, and PP242 (as effective combinations with EGFR inhibitor, which is consistent with previous preclinical HNSCC studies in vitro and in vivo and the testing of PI3K/mTOR inhibitor combinations with EGFR inhibitors in clinical trials for HNSCCCitation17–Citation19.
Notably, 2 out of the 14 drugs effective in combination with EGFR inhibitors were ALK inhibitors. While ALK inhibitors were effective in only 1 out of 8 patients as single agents (A), with a low dose of EGFR inhibitor, 4 out of 8 patients’ tumor cells became sensitive to ALK inhibitors TAE684 and GSK1838705A (B), and only 1 out of 8 patient-derived tumor cell cultures were sensitive to EGFR inhibitors as single agents (C), suggesting synergistic effects between ALK inhibitors and EGFR inhibitors.
Table 1. ALK inhibitors synergize with EGFR inhibitors in inhibitor assays in HNSCC patient-derived tumor cells. HNSCC patient-derived tumor cells (within 0 to 2 passages) from 8 patients were screened by inhibitor assays. Patient-derived tumor cells were plated in 384-well-plates containing 122 drugs in seven serial dilutions with or without 50 nM of EGFR inhibitor. MTS cell proliferation assay was used to determine the IC50s of the drugs on the panel as single agents or in combination with EGFR inhibitors. IC50s of ALK inhibitors TAE684 and GSK1838705A as single agents (A) or in combination with 50nM gefitinib (B), as well as IC50s of three EGFR inhibitors as single agents (C) are shown. Effective drugs for an individual patient are defined as the drugs that have IC50s below 20% of median IC50.
Scale-up experiments and combination index confirmed patients’ sensitivity to different EGFR and ALK inhibitor combinations in HNSCC patient-derived tumor cells
Based upon these results from inhibitor assays, we validated responses of the patients’ tumor cells to 4 ALK inhibitors, including 2 FDA approved ALK inhibitors that are not present in the inhibitor panel. Gefitinib and TAE684 combination was reported to have efficacy in treating HNSCC cells lines and cell line derived xenograftsCitation11; therefore gefitinib was used in all follow-up validation experiments.
Since different drugs may have different pharmacokinetics in patients, IC50 of an effective drug may not be clinically achievable. Therefore, maximum plasma concentration (Cmax) of each agent (Supplementary Table S3) also was considered in determining effective drugs.
Of the 8 original cases screened, we extended validations to 6 cases, excluding a maxillary sinus SCC case (10205) that is anatomically distinct from the oral and laryngeal sites of most HNSCC, and a case lacking sufficient material (10021). We did include an HPV positive case (10159), although HPV positive HNSCC have different etiology from HPV negative cases and more effective treatment options than HPV negative (Supplementary Table S1).
Scale-up dose-response experiments confirmed synergistic effects between EGFR inhibitor gefitinib with FDA approved ALK inhibitors ceritinib and brigatinib (A and B), and with the ALK inhibitors TAE684 and GSK1838705A (C and D) that were used in inhibitor assays. IC50s as single agents and in combination with gefitinib were calculated based on dose-response-curves. In patient 10004's tumor cells, the IC50s of gefitinib and ALK inhibitors as single agents were up to 340 fold and 12 fold higher than as combinations, respectively, and the IC50s for the combination were within clinically achievable doses and statistically significantly lower than for the single agents (A-D). Similarly, in 10058, the IC50s were up to 35 fold and 12 fold higher when used as single agents than when used in combinations, respectively, and within clinically achievable doses. Therefore, 10004 and 10058 were considered as sensitive cases in follow-up studies. In contrast, in 10250, the IC50s of gefitinib and ALK inhibitors were no greater than 3.9 fold and 1.8 fold higher when used as single agents than when used in combination, respectively, and the IC50s were beyond those achievable in plasma (A and B). Therefore case 10250 was considered a relatively insensitive case.
Figure 1. Validation of EGFR and ALK inhibitor combinations in patient-derived tumor cells. Patient-derived tumor cells from patients 10004, 10054, 10058, 10139, 10159 and 10250 were treated with a dose gradient of gefitinib, ALK inhibitors including ceritinib (A), brigatinib (B), GSK1838705A (C) and TAE684 (D) or their combinations (A–D). After 72 hours, cell viability was assessed using a MTS assay and normalized to vehicle treated cells. Patient numbers are sorted based on the IC50s for each EGFR/ALK drug combination from low to high. Dotted lines in (A) and (B) indicate maxima plasma concentration in patients of the total concentration of the two drugs in patients. No information regarding maxima plasma concentration in patients for GSK1838705A (C) and TAE684 (D) was available. Data represents the mean ± SD between three independent experiments (n = 3). Combination treatment groups were compared to single agent groups by one-way ANOVA and Dunnett multiple comparison test.
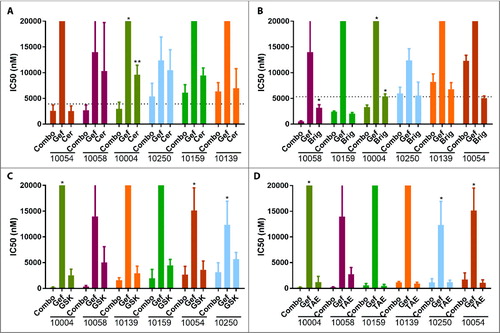
Although ceritinib and brigatinib had similar potency for ALK, with IC50 values of 0.2 nM and 0.6 nM (Supplementary Table S3)Citation20,Citation21, respectively, brigatinib had lower IC50s for all the cases as single agents compared to ceritinib (A and B). Interestingly, brigatinib is an ALK/EGFR dual inhibitorCitation21,Citation22, supporting the hypothesis that co-targeting ALK and EGFR is more effective than inhibiting either alone. Thus, our findings support potential repurposing to treat HNSCC of this newly FDA approved drug for NSCLC.
To quantify synergistic effects between gefitinib and ALK inhibitors and to differentiate between synergistic and addictive effect, the level of synergism is measured and quantified by the drug combination indexCitation23 (Supplementary Table S4). Combination index below 1 indicates synergy between the drug combination, while above 1 indicates antagonistic effects. Combination indices between gefitinib and all four ALK inhibitors tested in scale-up experiments in 10004 and 10058 were below 1, and IC50 within achievable plasma concentrations, Cmax, suggesting synergetic effects between gefitinib and these ALK inhibitors in these cases. In 10054, combination indices between gefitinib and all ALK inhibitors but brigatinib were below 1 (Supplementary Table S4), suggesting synergetic effects between gefitinib and these three ALK inhibitors in these cases. Notably, brigatinib is an ALK and EGFR dual inhibitor, which could be responsible for lack of a synergistic effect upon further addition of the EGFR inhibitor gefitinib in reducing the viability of these cells. In 10250, although combination indices between gefitinib and all ALK inhibitors but brigatinib were below 1 (Supplementary Table S4), the absolute IC50s were higher than the Cmax; therefore this case was considered relatively insensitive. Taken together, these results confirm synergistic effects between gefitinib and above ALK inhibitors.
siRNA confirmed a synergistic effect between siALK and EGFR inhibitor in HNSCC patient-derived tumor cells
While anti-cancer drugs have reported targets, nearly all, including those FDA approved drugs have additional targets besides the presumed targetCitation24. To rule out the possibility that the drug combinations' effect on reducing cell viability was due to “off-target” effects, we performed a siRNA screening assay, RAPID assaysCitation25, that target the same spectrum of targets as inhibitor assay, in the EGFR/ALK inhibitor combination sensitive and insensitive patients' patient-derived tumor cells. In RAPID assays, we exposed tumor cells to pooled siRNAs consisting of 4 different sequences of the siRNAs targeting the same gene in order to validate true targets responsible for the reduction in tumor cell viability by the drugs.
An effective siRNA was defined as a siRNA reduced cell viability below mean viability of the whole panel minus two standard deviations (SD) and statistically significantly different from the non-specific siRNA controls.
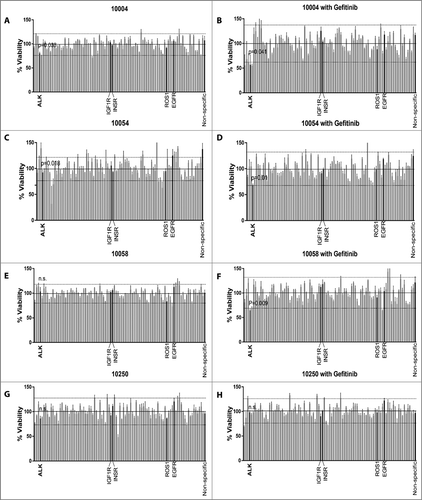
We transfected HNSCC patient-derived tumor cells from ALK inhibitor sensitive patients (10004, 10054 and 10058) and a relatively insensitive patient (10250) with a panel of siRNAs targeting the entire tyrosine kinase gene family in addition to NRAS and KRAS (93 genes total)Citation25. siRNA against ALK alone did not reduce relative cell viability significantly in any of the patients’ tumor cells. However, after adding 50 nM gefitinib to the siRNA panel, siRNA targeting ALK reduced the relative cell viability below the mean-2SD in tumor cells derived from patients 10004, 10054 and 10058 (A–F) but did not reduce viability in 10250 (G and H), consistent with the effects using EGFR and ALK inhibitor combinations. In addition, based on the results of these siRNA screens, potential alternative targets, such as IGF-1R, insulin receptor (InsR) and ROS1, which are common targets of ALK inhibitors, including TAE684, GSK1838705A, ceritinib and brigatinib, but with lower affinity than ALK (Supplementary Table S3)Citation12,Citation20,Citation26–Citation28, were ruled out as true targets of the inhibitor combinations. These data indicate that the cell viability reduction by the ALK inhibitors in combination with gefitinib was likely to be specific to ALK, not an “off-target” effect.
Figure 2. Synergy between siALK and gefitinib in HNSCC patient-derived tumor cells. Patient-derived tumor cells from patients indicated were transfected with siRNA pools individually targeting each member of the receptor tyrosine kinome in addition to NRAS and KRAS co-treated with vehicle (A, C, E, and G) or with 50nM gefitinib (B, D, F, and H). Cell viability was calculated by normalizing absorbance at 490 nM (as determined by the MTS assay) to the median plate value after 96 hours of treatment. Dotted lines indicate mean ± 2SD. Effective siRNAs are defined as those that inhibit cell viability 2SD below the mean-of all siRNAs and were statistically different from the non-specific siRNA controls. Error bar represents the mean ± SEM, each containing three replicates (n = 3).
Co-targeting EGFR and ALK decreased HNSCC patient's tumor cell number, colony formation ability and increased annexin V staining
To further determine the effects of EGFR and ALK inhibition in HNSCC patient-derived tumor cells, cell number, colony formation ability and annexin V staining were evaluated after treatment with EGFR/ALK inhibitor or siRNAs.
In cases 10004 and 10054, cell numbers by nucleus staining were decreased by gefitinib and ceritinib combination compared to single agents (A; Supplementary Fig. S1A), consistent with the cell viability reduction and low IC50 (below Cmax), suggesting EGFR and ALK were not just important in cell metabolism. In 10250, although gefitinib and ceritinib combination significantly decreased cell number compared to vehicle control (Supplementary Fig. S1B), the absolute IC50s (A and B) were still higher than the Cmax, therefore this case was considered insensitive.
Figure 3. Co-targeting EGFR and ALK decreases HNSCC patient's tumor cell number, cell colony formation ability and increases cell apoptosis in patient 10004 derived tumor cells. A, Patient 10004 derived tumor cells were treated with 1 μM gefitinib, 600 nM ceritinib or their combination for 72h, cells were fixed and stained by DAPI and cell number was counted. B, Patient 10004 derived tumor cells were transfected with siRNA pools targeting ALK, EGFR or their combination for 96 hours; 500 cells were plated for each group after treatment for colony formation assays, colony numbers were determined by crystal violet staining after 12 days. C, Patient 10004 derived tumor cells were treated with 1 μM gefitinib, 600 nM ceritinib, or the combination of 500 nM gefitinib and 300 nM ceritinib for 72 hours. Cells were stained by annexin V and PI, and flow cytometry was performed to determine the ratio of annexin V positive and PI negative cells for each group. Data represents the mean ± SD, each containing three replicates (n = 3). A one-way ANOVA was performed for statistical analysis.
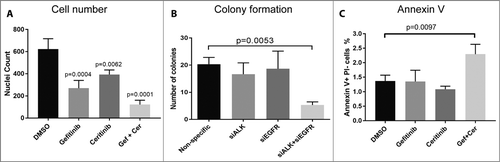
To determine the effects of co-targeting EGFR and ALK on cell reproductive ability, colony formation assay was performed in the most sensitive case, 10004, to determine the ability of single cells to grow into colonies. Colony formation ability in tumor cells derived from 10004 was statistically significantly reduced when EGFR and ALK were simultaneously co-targeted by siRNAs compared to single siRNAs (B), suggesting that co-targeting EGFR and ALK could impair cancer cell's reproductive ability. In addition, the effect of EGFR/ALK inhibitor combination on cell apoptosis, annexin V and PI staining was evaluated in these tumor cells. Gefitinib and ceritinib combination statistically significantly increased annexin V positive and PI negative cell population compared to single agents in these tumor cells, suggesting that co-targeting EGFR and ALK could increase cell apoptosis (C).
The synergy between ALK inhibitor and gefitinib is associated with phosphorylation and/or expression of ALK after gefitinib treatment in patient tumor-derived tumor cells
RNAseq analysis of our HNSCC patients’ native tumors and patient-derived tumor cells without any treatment showed low expression levels of ALK mRNA (data not shown). Therefore, based upon the idea that compensating pathways are activated after monotherapy treatment of anti-cancer drugs, we hypothesized that synergy between EGFR and ALK inhibitors was due to induction of ALK by EGFR inhibitor, as a mechanism to explain why ALK inhibitors were not effective as single agents but showed significant effects when combined with an EGFR inhibitor. We tested this hypothesis by determining ALK phosphorylation and expression levels in patient-derived tumor cells after gefitinib treatment.
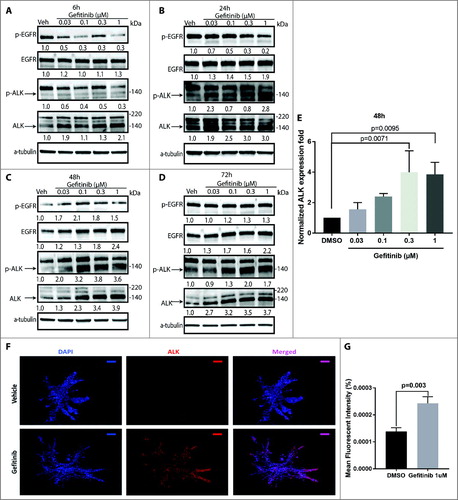
In 10004, a case that was the most sensitive to EGFR/ALK inhibitor combination, on average a 4-fold increase in total level of ALK protein compared to vehicle controls was detected by western blotting in patient 10004's cells 48 hours after 0.03 μM gefitinib treatment, with increases in phosphorylation of ALK up to 3.8 fold (A–E). Notably, EGFR phosphorylation was inhibited early (at 6 hours) as expected and restored after 48 hours gefitinib treatment.
Figure 4. ALK protein and phosphorylation levels increase after gefitinib treatment in HNSCC patient-derived tumor cells. Patient-derived tumor cells from patient 10004 were treated with gefitinib at 0.03-1 μM or vehicle for 6 hours (A), 24 hours (B), 48 hours (C) and 72 hours (D). Levels of total and phospho-EGFR and ALK, as well as alpha-tubulin, were assessed by immunoblot analysis. Total and phospho-ALK bands are indicated by arrows. E, Quantification of ALK protein expression after 48 hours of gefitinib treatment. Data represents the mean ± SD, each containing three independent experiments (n = 3). F, Immunofluorescent staining of ALK protein (red) and the nucleus (blue) on 10004 patient tumor cell derived spheroids treated with gefitinib 1 μM or vehicle. Scale bar = 100µm G, Data represents the mean fluorescent intensity of ALK normalized to the volume of the gefitinib treated (n = 9) and vehicle treated (n = 5) spheroids.
Patient 10058's tumor cells were also sensitive to EGFR/ALK inhibitor combinations, although increases in ALK protein level were small, up to 1.3 fold when treated with gefitinib at a clinical achievable dose 1 μM (Supplementary Fig. S2B). Notably, 10058's tumor cells had a higher basal level of total ALK expression compared to 10004 (Supplementary Fig. S3). This is a possible explanation for the initial sensitivity of 10058 to GSK1838705A and TAE684 in the inhibitor assay (), due to less reliance upon induction of ALK for ALK inhibitor sensitivity in this case.
Although 10054's sensitivity to gefitinib and four ALK inhibitors in scale-up studies was variable (), RAPID assay () and cell nuclear staining (Supplementary Fig. S1) showed sensitivity to ALK inhibition in combination with gefitinib, and induction of ALK protein after gefitinib treatment up to 2.1 fold was detected (Supplementary Fig. S2A).
In contrast, ALK protein was not induced above vehicle control in patient 10250's tumor cells when treated with gefitinib at a clinical achievable dose 1 μM (Supplementary Fig. S2C), the case that was relatively resistant to the EGFR/ALK inhibitor combinations as shown in .
Of note, a previously reportedCitation29–Citation31 but less investigated 140 kDa ALK form, rather than the 220 kDa form, was induced by gefitinib in these HNSCC patient-derived tumor cells (A–E; Supplementary Fig. S2), warranting further investigation of distinct oncogenic roles of ALK forms in HNSCC and other cancers.
Taken together, the level of ALK induction or the basal level of ALK correlated with the responses to gefitinib/ALK inhibitor and gefitinib/siALK combination. In the most sensitive case 10004, we observed the highest level of ALK induction after gefitinib treatment (average 4 fold, up to 5.6 fold); in another sensitive case 10058, although only up to 1.7 fold induction was detected, there was a higher basal level of ALK expression in naïve tumor cells compared to 10004 (Supplementary Fig. S3); in an intermediate responder 10054, moderate level of ALK was induced after treatment (up to 2.1 fold); while in a resistant case 10250, no more than 1.1 fold induction of ALK was observed by gefitinib treatment lower than 1 μM, a clinically achievable dose. These results suggest that ALK induction after gefitinib treatment and high basal level of ALK may be responsible for sensitivity to gefitinib and ALK inhibitor combinations in these tumor cells.
ALK expression was induced after gefitinib treatment in patient tumor-derived spheroids and xenografts
To evaluate the mechanism of EGFR inhibitor resistance under culture conditions considered more relevant to tissue than 2D culture, a patient tumor cell-derived spheroid model using the most sensitive case 10004 was generated. ALK protein was determined by immunofluorescent staining in gefitinib or vehicle-treated spheroids after treatment for 72 hours. Consistent with 2D culture, ALK protein levels were induced in spheroids treated with 1μM gefitinib compared to vehicle controls (F and G). These results support ALK induction and activation after EGFR inhibition as a mechanism underlying the observed synergistic effect of EGFR and ALK inhibitors in combination.
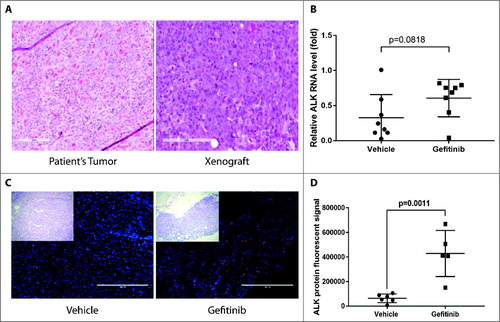
In order to evaluate whether gefitinib treatment induced ALK expression in patient-derived xenografts in mice, patient-derived tumor cells from case 10004 were subcutaneously injected into NSG mice, and upon tumor growth to 500 mm3, mice were treated with 100 mg/kg gefitinib or vehicle daily for 48 hours. Morphology of xenograft tumors and the original patient tumor were evaluated by H&E staining and similar differentiation status were found in xenograft tumors and the original tumor (A). Levels of ALK RNA and protein were assessed by qRT-PCR and immunofluorescent staining, respectively. ALK RNA expression levels tended to increase after 48 hours of gefitinib treatment (p = 0.0818) (B). Immunofluorescent staining showed statistically significant induction of ALK protein in 10004 patient-derived xenograft tumors treated with gefitinib compared to vehicle-treated controls (C and D).
Figure 5. ALK RNA and protein levels increase after gefitinib treatment in HNSCC patient-derived xenografts. Patient-derived tumor cells from patient 10004 were subcutaneously injected into NSG mice. Mice were treated with 100 mg/kg gefitinib or vehicle for 48 hours. A, Morphology of a xenograft tumor and the original patient tumor were shown. B, Quantification of ALK RNA expression after 48 hours of gefitinib treatment. Data represents the mean ± SD (n = 8). C, Immunofluorescent staining of ALK protein (red) and the nucleus (blue) on 10004 patient-derived xenografts treated with gefitinib or vehicle. Scale bar = 400µm D, Data represents the integrated density (positive area x mean fluorescent intensity) of ALK staining in gefitinib treated and vehicle treated mice (n = 5-6).
Thus, expression of ALK after gefitinib treatment was induced in patient tumor-derived spheroids and xenografts, uncovering a potential patient-specific mechanism underlying synergy between ALK inhibitor and gefitinib in HNSCC.
Discussion
The current study identifies EGFR and ALK inhibitor combinations as effective combination therapies in EGFR inhibitor-resistant HNSCC patient-derived tumor cells. Four different ALK inhibitors, including two that are FDA-approved, ceritinib and brigatinib, showed synergistic effects with gefitinib in patient-derived tumor cells, suggesting a potential benefit of using ALK inhibitors in combination with an EGFR inhibitor for treating HNSCC patients.
Importantly, we found a correlation between the responsiveness to gefitinib and ALK inhibitor combinations and the level of ALK induction or the basal level in ALK wild-type HNSCC patients. ALK has been implicated in the development and progression of many malignancies, including anaplastic large-cell lymphoma, NSCLC, and neuroblastoma. ALK fusions, mutations and amplifications are the most common alterations in ALK in cancersCitation6,Citation9, and ALK fusions and other rearrangement have been used as a patient selection criteria for ALK inhibitors in NSCLC patientsCitation32,Citation33. Furthermore, wild-type expression of ALK was also strongly correlated with poor prognosis in cancer patientsCitation34. ALK alterations are not commonly detected (4% according to TCGA ciBioPortalCitation9 analysis) in HNSCC, and no expression level changes or mutations in ALK were detectable by RNAseq and whole exome sequencing analysis in our patients’ tumors. The functional screens using viable patient-derived tumor cells treated with gefitinib uncovered a mechanism of EGFR inhibitor resistance through ALK induction that was missed by genomic and transcriptomic analysis in naive tumors alone. The effectiveness of ALK inhibitors plus gefitinib in patient-derived cells was associated with strong induction of total ALK protein, induction of phosphorylated ALK and/or higher basal levels of ALK protein. Further, our findings indicate efficacy of EGFR and ALK inhibitors in tumor cells without ALK fusions or mutations.
Although ALK protein expression is relatively low in naive HNSCC patient tumors (The Human Protein AtlasCitation35), in late-stage HNSCC, ALK promoter is hypomethylated and there is increased ALK activity in late-stage human HNSCC tumors and invasive OSCC cell linesCitation11, suggesting plastic regulation of ALK expression in HNSCC. Given compensatory cross-talk between kinases, it is plausible that ALK upregulated in cancers after EGFR inhibitor treatment is responsible for the broader sensitivity seen in response to this combination. Support for this view comes from reports of different mechanisms of EGFR inhibitor resistance in various cancers including upregulation of parallel pathways such as MET, aurora kinase A and HER336–38 and heterodimerization and transactivation of other RTKs such as MET, ERBB2, and IGF-1RCitation37,Citation39. Nuclear EGFR has been shown to function as a co-transcription factor alongside STAT3, E2F1 and STAT5 to enhance the transcription of aurora kinase A and c-MycCitation40–Citation44. Studies have shown that EGFR inhibitors including gefitinib and cetuximab could induce EGFR translocation into the nucleusCitation45,Citation46. Nuclear EGFR can contribute to acquired resistance to gefitinib and cetuximab by elevating expression of proteins including breast cancer resistance protein (BCRP/ABCG2)Citation47 and by upregulation of cyclin D1 and B-mybCitation48, respectively. ALK RNA expression levels tended to be increased after 48 hours of gefitinib treatment (B), suggesting ALK upregulation, at least partially, was at RNA level. Whether ALK is induced through nuclear EGFR as a co-transcription factor has yet to be determined. However, RNA stability, post-transcriptional, translational and post-translational regulation may also play roles in ALK induction in these tumors.
ALK is expressed as the 220 kDa full-length transmembrane receptor, and a shorter form of 140 kDa could result from the extracellular proteolytic cleavage of the full-length receptorCitation49. A significant correlation was reported between 140 kDa ALK protein levels and ALK inhibitor TAE684 response in wild-type neuroblastoma cell linesCitation50, suggesting potential kinase dependence of these cancer cells on 140 kDa ALK protein and the benefit of targeting ALK in ALK wild-type cancers. Our finding that 140 kDa ALK protein expression and phosphorylation was induced after EGFR inhibitor treatment in ALK wild-type HNSCC provides evidence warranting further study of the oncogenic functions of this form in HNSCC and other cancers and possible specific targeting strategies in these diseases.
The use of patient-derived cells to functionally evaluate HNSCC cell sensitivity to drugs and siRNAs may provide critical guidance for precision medicine clinical trials in the future. The results of inhibitor and RAPID assays can be available in 4 to 8 weeks, well within the two-year timeframe of recurrence of most HNSCCCitation51. Thus, providing functional information for stratification to match individual patients to effective drugs in clinical trials is feasible. Future development of HNSCC-specific inhibitor and siRNA panels based on highly altered pathways in big data sets such as TCGA HNSCC cohort could expand drugs and siRNAs targeting “dark” pathways that are not represented in the current inhibitor assay panels or FDA approved drugs. This promises to be beneficial in identifying more effective agents and combination therapies for HNSCC and also applicable to other cancer types.
Our in vitro dose-response studies demonstrated that the anti-EGFR mAb cetuximab also reduced cell viability when combined with the ALK inhibitor ceritinib better than ceritinib alone, but this was not statistically significantly different from cetuximab alone (data not shown). However, discrepancies between in vitro and in vivo efficacy of cetuximab have been reportedCitation52. Thus, ALK inhibitors could enhance the efficacy of cetuximab in HNSCC in vivo, although this remains to be determined.
Overall, our study reports evidence for ALK induction and activation after EGFR inhibitor treatment in HNSCC, suggesting a novel mechanism for and strategy to address EGFR inhibitor resistance using FDA approved ALK inhibitors. These findings provide a rationale to test EGFR and ALK inhibitor combinations in clinical trials and further investigate this phenomenon in other cancer types where EGFR inhibitors are relevant.
Materials and methods
Collection of patient samples and cell culture
Clinical samples were obtained from patients treated at Oregon Health &Science University upon informed consent under approval IRB00010071 by the Oregon Health & Science University Institutional Review Board (Portland, OR). Primary human HNSCC cell lines were developed from excised tumors, using IRB-approved collection techniques. Minced tumors were placed in collagen-I coated (Gibco Coating Matrix Kit, Hyclone, R-011-K) Petri dishes and/or 490 cm2 roller bottles using cell culture media developed by James Rheinwald of the Harvard Skin Disease Research Center, Boston, MACitation53–Citation55. DMEM/F12 Media (Gibco, 11320082), supplemented with 5% BCS (Hyclone, SH3007203,), antibiotic/antimitotic (Gibco, 15240112), 1.8 × 10−4 M adenine (Sigma, A2786), 0.4 µg/mL hydrocortisone (Sigma, H0888), 1 × 10−10 M cholera enterotoxin (Sigma, C8052), 2 × 10−11 M triiodothyronine (Sigma, T6397), 5 µg/mL insulin (Sigma, I9278) and 10 µg/mL epidermal growth factor (Gibco, PHG0311), was used for cell maintenance. Differential (1–3 minute) trypsinization (0.25% with EDTA, Gibco, 25200114) was used as needed to remove fibroblasts. Cells used for experiments were within passage 5.
Inhibitor assay
HNSCC patient-derived cells were examined for sensitivity against a panel of 122 small-molecule inhibitors as previously describedCitation13. Briefly, 6–8 × 103 cells per well were treated with a threefold interval dilution series totaling 8 concentrations of each drug (including no-drug control) in 384-well plates. After 72 hours, relative cell viability was determined using a tetrazolium-based MTS assay (Promega, PR-63581), and IC50 values were determined from the dose-response curves. A final concentration of 50 nM of one of three EGFR inhibitors used in clinical trials for HNSCC or other cancers was used in combination with the inhibitors on the panels to identify synergistic agents (lapatinib (Selleck, S1028) in patient 10004's and 10021's cells, erlotinib (Selleck, S1023) in patient 10054's and 10058's cells, and gefitinib (Selleck, S1025) in patient 10139's, 10159's, 10205's and 10250's cells).
Scale-up inhibitor validation studies
Patient-derived tumor cells were distributed in 96-well plates with dilution series totaling 10 concentrations of each drug. Cells were plated at a density of 8 × 103 cells/well and treated with the following inhibitors or combination of inhibitors for 72 hours: gefitinib, TAE684, GSK1838705A, ceritinib and brigatinib. For the drug combinations, gefitinib and ALK inhibitors were used at a ratio of 2:1. All inhibitors were purchased from Selleck Chemicals. All conditions were plated in triplicates. Cell viability was measured using MTS assay, and absorbance (490 nm) was read at 1 to 4 hours after adding reagent using a BioTek Synergy 2 plate reader. MTS absorbance values of inhibitor-treated wells were normalized to those of untreated cells. IC50 values were determined by CalcuSyn (BioSoft).
RAPID assay
The RNAi-assisted protein target identification (RAPID) assay has been previously described Citation25. All siRNAs were SMARTpool: siGENOME siRNA pools (GE Dharmacon).
Quantification of annexin V/propidium iodide staining
Cells were stained using annexin V Apoptosis Detection Kit APC (eBioscience, 88–8007) according to the manufacturer's protocol. Cells were treated with 1 μM gefitinib, 600 nM ceritinib, 500 nM gefitinib and 300 nM ceritinib, or vehicle control (DMSO) for 72h prior to analysis. Cells were re-suspended in annexin V binding buffer containing APC conjugated annexin V, followed by addition of propidium iodide (PI). Subsequently, cells were analyzed by flow cytometry, using a Canto II. Data were analyzed using FlowJo_v10. Unstained cells as well as annexin V, and PI single stained cells were used as negative controls.
Colony formation assay
After exposure to the pooled siRNAs for 12 days, cells were washed with PBS, then fixed and stained with a mixture of 6.0% glutaraldehyde (Sigma, 340855-25ml) and 0.5% crystal violet (Sigma, C0775-25G) for 30 minutes. Plates were rinsed with tap water and dried in normal air at room temperature (20 °C). Colonies were counted manually.
Nucleated cell number analysis
Patient-derived tumor cells in triplicate wells were treated with either 1 uM gefitinib, 600 nM ceritinib, the combination of 1 uM gefitinib+ 600 nM ceritinib or vehicle for 48 hours. After treatment, cells were fixed using 4% paraformaldehyde (Electron Microscopy Science, 15710), blocked using 5% goat serum (Abcam, AB138478) and 0.3% Triton X-100 (Sigma, T8787), and stained with Hoechst (Thermo Fisher, 62249). After subsequent washing with PBS and 0.5% Tween20 (Sigma, P7949), coverslips were mounted using prolong diamond antifade (Fisher, P36961). Images were taken using an EVOS FL microscope (Thermo Fisher) with a 10x objective. Five images were taken per coverslip, resulting in 15 images for each treatment group. Nuclei were counted using Image J56.
Immunoblotting
Patient-derived tumor cells were treated with cell lysis buffer (Cell Signaling Technologies, 9803S) with complete mini protease inhibitor mixture tablets (Roche, 11836153001), and PhosSTOP Phosphatase Inhibitor Cocktail Tablets (Roche, 4906845001). Lysates were spun at 8,000 xg for 10 minutes at 4°C to pellet cell debris, mixed 3:1 with 4x Laemmli Sample Buffer (Bio-Rad, 1610747) with β-ME, and heated at 95°C for 5 minutes. Lysates were run on 4% to 15% Criterion TGX Precast Midi Protein Gel (Bio-Rad, 5671083), transferred to a polyvinylidene difluoride membrane (Bio-Rad, 1704157), and blocked for 1 hour in TBS-T with 5% BSA. Blots were probed overnight at 4°C with anti-ALK rabbit antibody (1:300, 3333), Phospho-ALK (Tyr1604) Antibody (1:300, 3341), EGF Receptor (D38B1) XP® Rabbit mAb (1:1000, 4267), Phospho-EGF Receptor (Tyr1068) (1H12) Mouse mAb (1:1000, 2236) or α-Tubulin (DM1A) Mouse mAb (1:1000, 3873), followed by anti-rabbit or anti-mouse IgG HRP conjugated secondary antibodies. All primary antibodies are from Cell Signaling. Blots were developed using Clarity™ or Clarity Max™ Western ECL Substrate (Bio-Rad, 1705060 and 1705062) and imaged using a Bio-Rad ChemiDoc touch MP Imaging System. Optimal exposure time was automatically determined by ChemiDoc touch. The SK-N-SH cell line which expresses both 220 kDa and 140 kDa ALK was used as an ALK-positive control to confirm molecular weight in all western blot experiments for ALK and p-ALK detection.
Patient-tumor-cell-derived spheroids formation, treatment, and immunofluorescent staining
Patient-derived tumor cells were grown into spheroids using a hanging drop method as previously describedCitation57. Spheroids were embedded in Collagen I (Corning, 354236) and were cultured for four days. 1 μM gefitinib diluted in DMEM/F12 (Gibco, 11330057) supplemented with 5% bovine calf serum (Hyclone, SH3007203) or vehicle was applied to the cells for 72 hours. The experimental procedure for embedding and staining was as previously reported with minor modificationsCitation58. Spheroids were fixed in 4% paraformaldehyde and 1% Triton X 100 (Sigma, T8787), and then washed with PBS and 10mM Glycine (Bio-Rad, 1610717). Spheroids were dehydrated in an ascending series of methanol, then rehydrated before blocking overnight at 4°C with 3% normal goat serum (Abcam, AB138478) in PBS. ALK antibody (1:100, Cell Signaling, 3633) was diluted in 3% normal goat serum and 0.1% Triton X 100 in PBS and incubated for two nights at 4°C. Alexa Goat Anti-Mouse Texas Red antibody (Invitrogen, T2767) and Hoechst (Thermo Fisher, 62249) was applied at 1:1000 and 1:2000 dilution overnight at 4°C. Cells were imaged using a Nikon/Yokogawa CSU-W1 Spinning Disk Confocal. Replicate spheroids of each treatment condition were completed and imaged. Mean fluorescent intensity was quantified using Fiji (ImageJ, NIH, Bethesda, MD) at five separate levels in the z stack, consistent across all images and was normalized to the volume of the spheroid, calculated using Bitplane Imaris (Oxford Instruments).
Xenograft mouse model for ALK induction after Gefitinib treatment
NOD SCID GAMMA (NSG) mice (8-16 weeks old) were used for this study. Mice were divided into 2 groups (n = 8 mice per group), 1) vehicle control; 2) 100 mg/kg gefitinib. Patient-derived tumor epithelial cells (2 × 106 in 0.1 ml) from 10004 were inoculated subcutaneously into the right flank of all mice. Treatments were initiated when tumors reached 500 mm2. Body weights and tumor dimensions were determined 3 times a week, and tumor volumes were calculated from measurements of 3 diameters of individual tumors based on the following formula: tumor volume (mm3) = 1/2(length × width Citation2). Treatment with gefitinib or vehicle control (dimethyl sulfoxide 5% and corn oil 95%) administered daily by oral gavage for 2 days. The volume of liquid for oral gavage was 0.1 ml / 10 g. Mice were sacrificed and the tumors were harvested 2 days after the last gefitinib treatment. All studies were performed according to guidelines approved by OHSU Institutional Animal Care and Use Committee.
Statistical analyses
For the scale-up IC50 experiments, nucleated cell count assay, colony formation assay, annexin V/PI staining assay and western blot quantification, a one-way ANOVA test with Dunnett's multiple comparisons were carried out for each treatment condition compared with monotherapy treated groups, vehicle-treated cells or appropriate controls. For spheroid staining, ALK RNA expression, xenograft ALK protein staining experiments, student's t-tests were performed for treatment groups compared with vehicle groups. Combination indices were calculated using CalcuSyn (Biosoft), whereby data points for combinations with upper confidence limits below 1 are considered synergistic Citation23.
Grant Support
This study was supported by NIH grants R01 CA192405, R21 CA180576 and Knight Cancer Institute P30 CA069533.
Disclosure of Potential Conflicts of Interest
Xiaoming Ouyang, Ashley Barling, Aletha Lesch, Gabrielle Choonoo, Christina Zheng, Sophia Jeng, Toni M. West, Daniel Clayburgh and Molly Kulesz-Martin declare no potential conflicts of interest.
Jeffrey W. Tyner: Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Takeda and Leap Oncology.
Sara A. Courtneidge: Crown Biosciences.
Shannon K. McWeeney: Aptose.
2018CBT11285-s05.docx
Download MS Word (2 MB)Acknowledgments
We are grateful to all the patients who contributed their tumors and pathologists Dr. Rosemary Makar and Dr. Terry K. Morgan. We thank Dr. Monika A. Davare for providing ceritinib and brigatinib for initial testing and SK-N-SH cell line as an ALK-positive control. We thank Dr. Zhiping Wang, Dr. Yuangang Liu, Dr. Shinji Iizuka, Rachel De La Torre, Aurelie Snyder and Yang Wang for their technical advice and assistance and Clara Stemwedel for editorial assistance.
References
- Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 2010; 127:2893–917.
- Cassell A, Grandis JR. Investigational EGFR-targeted therapies in HNSCC. Expert Opin Investig Drugs 2010; 19:709–22.
- Ang KK, Berkey BA, Tu X, Zhang H-Z, Katz R, Hammond EH, Fu KK, Milas L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res 2002; 62:7350–6.
- Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013; 19:1389–400.
- Aung KL, Siu LL. Genomically personalized therapy in head and neck cancer. Cancers Head Neck 2016; 1:2.
- Mologni L. Inhibitors of the anaplastic lymphoma kinase. Expert Opin Investig Drugs 2012; 21:985–94.
- Blackhall F, Cappuzzo F. Crizotinib: from discovery to accelerated development to front-line treatment. Ann Oncol Off J Eur Soc Med Oncol 2016; 27 Suppl 3:iii35–iii41.
- Shaw AT, Engelman JA. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med 2014; 370:2537–9.
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov 2012; 2:401–4.
- Huang T-T, Gonzales CB, Gu F, Hsu Y-T, Jadhav RR, Wang C-M, Redding SW, Tseng C-E, Lee C-C, Thompson IM, et al. Epigenetic deregulation of the anaplastic lymphoma kinase gene modulates mesenchymal characteristics of oral squamous cell carcinomas. Carcinogenesis 2013; 34:1717–27.
- Gonzales CB, De La Chapa JJ, Saikumar P, Singha PK, Dybdal-Hargreaves NF, Chavez J, Horning AM, Parra J, Kirma NB. Co-targeting ALK and EGFR parallel signaling in oral squamous cell carcinoma. Oral Oncol 2016; 59:12–9.
- Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci 2007; 104:270–5.
- Tyner JW, Yang WF, Bankhead A 3rd, Fan G, Fletcher LB, Bryant J, Glover JM, Chang BH, Spurgeon SE, Fleming WH, et al. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res 2013; 73:285–96.
- Blucher AS, Choonoo G, Kulesz-Martin M, Wu G, McWeeney SK. Evidence-Based Precision Oncology with the Cancer Targetome. Trends Pharmacol Sci 2017;
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015; 517:576–82.
- Kondo N, Ishiguro Y, Kimura M, Sano D, Fujita K, Sakakibara A, Taguchi T, Toth G, Matsuda H, Tsukuda M. Antitumor effect of gefitinib on head and neck squamous cell carcinoma enhanced by trastuzumab. Oncol Rep 2008; 20:373–8.
- Wang Z, Martin D, Molinolo AA, Patel V, Iglesias-Bartolome R, Sol Degese M, Vitale-Cross L, Chen Q, Gutkind JS. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst 2014; 106.
- De Felice F, Guerrero Urbano T. New drug development in head and neck squamous cell carcinoma: The PI3-K inhibitors. Oral Oncol 2017; 67:119–23.
- Jimeno A, Shirai K, Choi M, Laskin J, Kochenderfer M, Spira A, Cline-Burkhardt V, Winquist E, Hausman D, Walker L, et al. A randomized, phase 2 trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase Inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann Oncol 2014;:mdu574.
- Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, et al. Synthesis, structure-activity relationships, and in vivo efficacy of the novel potent and selective anaplastic lymphoma kinase (ALK) inhibitor 5-chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulfonyl)phenyl)pyrimidine-2,4-diamine (LDK378) currently in phase 1 and phase 2 clinical trials. J Med Chem 2013; 56:5675–90.
- Huang W-S, Liu S, Zou D, Thomas M, Wang Y, Zhou T, Romero J, Kohlmann A, Li F, Qi J, et al. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J Med Chem 2016; 59:4948–64.
- Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, Fujita N, Katayama R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat Commun 2017; 8:14768.
- Chou T-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res 2010; 70:440–6.
- Law V, Knox C, Djoumbou Y, Jewison T, Guo AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, et al. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res 2014; 42:D1091–1097.
- Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, Willis SG, Erickson H, Kovacsovics T, O'Hare T, Heinrich MC, et al. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc Natl Acad Sci U A 2009; 106:8695–700.
- Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, Atkins C, Dumble M, Yang J, Anderson K, et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Mol Cancer Ther 2009; 8:2811–20.
- Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci 2011; 108:7535–40.
- Davare MA, Vellore NA, Wagner JP, Eide CA, Goodman JR, Drilon A, Deininger MW, O'Hare T, Druker BJ. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci U S A 2015; 112:E5381–90.
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene 1997; 14:439–49.
- Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene 1997; 14:2175–88.
- Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, Créminon C, Vigny M. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J Biol Chem 2005; 280:26039–48.
- Shaw AT, Kim D-W, Nakagawa K, Seto T, Crinó L, Ahn M-J, De Pas T, Besse B, Solomon BJ, Blackhall F, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013; 368:2385–94.
- Kwak EL, Bang Y-J, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou S-HI, Dezube BJ, Jänne PA, Costa DB, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363:1693–703.
- Passoni L, Longo L, Collini P, Coluccia AML, Bozzi F, Podda M, Gregorio A, Gambini C, Garaventa A, Pistoia V, et al. Mutation-independent anaplastic lymphoma kinase overexpression in poor prognosis neuroblastoma patients. Cancer Res 2009; 69:7338–46.
- Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 2010; 28:1248–50.
- Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors—impact on future treatment strategies. Nat Rev Clin Oncol 2010; 7:493–507.
- Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P, Kulmala J, Pulkkinen J, Grenman R, Elenius K. Signaling via ErbB2 and ErbB3 Associates with Resistance and Epidermal Growth Factor Receptor (EGFR) Amplification with Sensitivity to EGFR Inhibitor Gefitinib in Head and Neck Squamous Cell Carcinoma Cells. Clin Cancer Res 2006; 12:4103–11.
- Hoellein A, Pickhard A, von Keitz F, Schoeffmann S, Piontek G, Rudelius M, Baumgart A, Wagenpfeil S, Peschel C, Dechow T, et al. Aurora kinase inhibition overcomes cetuximab resistance in squamous cell cancer of the head and neck. Oncotarget 2011; 2:599–609.
- Jameson MJ, Beckler AD, Taniguchi LE, Allak A, VanWagner LB, Lee NG, Thomsen WC, Hubbard MA, Thomas CY. Activation of the Insulin-like Growth Factor-1 Receptor Induces Resistance to Epidermal Growth Factor Receptor Antagonism in Head and Neck Squamous Carcinoma Cells. Mol Cancer Ther 2011; 10:2124–34.
- Hanada N, Lo H-W, Day C-P, Pan Y, Nakajima Y, Hung M-C. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog 2006; 45:10–7.
- Hung L-Y, Tseng JT, Lee Y-C, Xia W, Wang Y-N, Wu M-L, Chuang Y-H, Lai C-H, Chang W-C. Nuclear epidermal growth factor receptor (EGFR) interacts with signal transducer and activator of transcription 5 (STAT5) in activating Aurora-A gene expression. Nucleic Acids Res 2008; 36:4337–51.
- Jaganathan S, Yue P, Paladino DC, Bogdanovic J, Huo Q, Turkson J. A functional nuclear epidermal growth factor receptor, SRC and Stat3 heteromeric complex in pancreatic cancer cells. PloS One 2011; 6:e19605.
- Lo H-W, Hsu S-C, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih J-Y, Hung M-C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005; 7:575–89.
- Lo H-W, Cao X, Zhu H, Ali-Osman F. Cyclooxygenase-2 is a novel transcriptional target of the nuclear EGFR-STAT3 and EGFRvIII-STAT3 signaling axes. Mol Cancer Res MCR 2010; 8:232–45.
- Liao H-J, Carpenter G. Cetuximab/C225-induced intracellular trafficking of epidermal growth factor receptor. Cancer Res 2009; 69:6179–83.
- Tan X, Lambert PF, Rapraeger AC, Anderson RA. Stress-Induced EGFR Trafficking: Mechanisms, Functions, and Therapeutic Implications. Trends Cell Biol 2016; 26:352–66.
- Huang W-C, Chen Y-J, Li L-Y, Wei Y-L, Hsu S-C, Tsai S-L, Chiu P-C, Huang W-P, Wang Y-N, Chen C-H, et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem 2011; 286:20558–68.
- Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009; 28:3801–13.
- Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Carvalho NB, Bureau J, Créminon C, Vigny M. Activation and Inhibition of Anaplastic Lymphoma Kinase Receptor Tyrosine Kinase by Monoclonal Antibodies and Absence of Agonist Activity of Pleiotrophin [Internet]. [cited 2017 May 31]; Available from: http://www.jbc.org
- Duijkers FAM, Gaal J, Meijerink JPP, Admiraal P, Pieters R, de Krijger RR, van Noesel MM. Anaplastic lymphoma kinase (ALK) inhibitor response in neuroblastoma is highly correlated with ALK mutation status, ALK mRNA and protein levels. Cell Oncol Dordr 2011; 34:409–17.
- Leemans CR, Tiwari R, Nauta JJ, van der Waal I, Snow GB. Recurrence at the primary site in head and neck cancer and the significance of neck lymph node metastases as a prognostic factor. Cancer 1994; 73:187–90.
- Dassonville O, Bozec A, Fischel JL, Milano G. EGFR targeting therapies: Monoclonal antibodies versus tyrosine kinase inhibitors: Similarities and differences. Crit Rev Oncol Hematol 2007; 62:53–61.
- Rheinwald JG, O'connell TM. Intermediate Filament Proteins as Distinguishing Markers of Cell Type and Differentiated State in Cultured Human Urinary Tract Epithelia. Ann N Y Acad Sci 1985; 455:259–267.
- Mesosecrin: a secreted glycoprotein produced in abundance by human mesothelial, endothelial, and kidney epithelial cells in culture. J Cell Biol 1987; 104:263–75.
- Kulesz-Martin MF, McLimans WF. Steady-state cultures of human skin. Exp Cell Biol 1982; 50:195–206.
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676–82.
- Foty R. A simple hanging drop cell culture protocol for generation of 3D spheroids. J Vis Exp JoVE 2011;
- Weiswald L-B, Guinebretière J-M, Richon S, Bellet D, Saubaméa B, Dangles-Marie V. In situ protein expression in tumour spheres: development of an immunostaining protocol for confocal microscopy. BMC Cancer 2010; 10:106.
- Cooper MR, Chim H, Chan H, Durand C. Ceritinib: A New Tyrosine Kinase Inhibitor for Non-Small-Cell Lung Cancer. Ann Pharmacother 2015; 49:107–12.
- Khozin S, Blumenthal GM, Zhang L, Tang S, Brower M, Fox E, Helms W, Leong R, Song P, Pan Y, et al. FDA Approval: Ceritinib for the Treatment of Metastatic Anaplastic Lymphoma Kinase–Positive Non–Small Cell Lung Cancer. Clin Cancer Res 2015; 21:2436–9.
- Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA drug approval summary: gefitinib (ZD1839) (Iressa) tablets. The Oncologist 2003; 8:303–6.
- Brehmer D, Greff Z, Godl K, Blencke S, Kurtenbach A, Weber M, Müller S, Klebl B, Cotten M, Kéri G, et al. Cellular Targets of Gefitinib. Cancer Res 2005; 65:379–82.
- McKillop D, Partridge EA, Kemp JV, Spence MP, Kendrew J, Barnett S, Wood PG, Giles PB, Patterson AB, Bichat F, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther 2005; 4:641–9.
- Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. ZD 1839 (Iressa): An Orally Active Inhibitor of Epidermal Growth Factor Signaling with Potential for Cancer Therapy. Cancer Res 2002; 62:5749–54.