
Abstract
Nucleus pulposus (NP) cells experience hyperosmotic stress in spinal discs; however, how these cells can survive in the hostile microenvironment remains unclear. Autophagy has been suggested to maintain cellular homeostasis under different stresses by degrading the cytoplasmic proteins and organelles. Here, we explored whether autophagy is a cellular adaptation in rat notochordal cells under hyperosmotic stress. Hyperosmotic stress was found to activate autophagy in a dose- and time-dependent manner. SQSTM1/P62 expression was decreased as the autophagy level increased. Transient Ca2+ influx from intracellular stores and extracellular space was stimulated by hyperosmotic stress. Activation of AMPK and inhibition of p70S6K were observed under hyperosmotic conditions. However, intercellular Ca2+ chelation inhibited the increase of LC3-II and partly reversed the decrease of p70S6K. Hyperosmotic stress decreased cell viability and promoted apoptosis. Inhibition of autophagy led to SQSTM1/P62 accumulation, reduced cell viability, and accelerated apoptosis in notochordal cells under this condition. These evidences suggest that autophagy induction via the Ca2+-dependent AMPK/mTOR pathway might occur as an adaptation mechanism for notochordal cells under hyperosmotic stress. Thus, activating autophagy might be a promising approach to improve viability of notochordal cells in intervertebral discs.
Abbreviations
| IDD | = | Intervertebral disc degeneration |
| NP | = | nucleus pulposus |
| ECM | = | extracellular matrix |
| MAPK | = | mitogen-activated protein kinase |
| SQSTM1 | = | Sequestosome-1 |
| mTOR | = | mammalian target of rapamycin |
| AMPK | = | AMP activated Protein Kinase |
| MDC | = | Monodansylcadaverine |
| PLC | = | phospholipase C |
| MMP | = | mitochondrial membrane potential |
| MPT | = | mitochondrial permeability transition |
| TRPV4 | = | Transient receptor potential vanilloid 4 |
| EGTA | = | ethyleneglycol-bis (2-aminoethylether)-N,N,N′, N-tetraacetic acid |
| fluo3-AM | = | fluo3 acetoximethylester |
| FITC | = | fluorescein isothiocyanate |
| PI | = | propidium iodide |
| DAPI | = | 4′, 6-diamidino-2-phenylindole |
| JC-1 | = | 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide |
| OD | = | optical density |
| BAPTA-AM | = | bis-(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid, tetra(acetoxymethyl) ester |
Introduction
Intervertebral disc degeneration (IDD) is the main cause of lower back pain, which carries a financial and health burden to the individual and to society because of its high level of morbidity. However, the mechanisms of IDD are unclear, and remain to be elucidated. Recently, IDD has been suggested to associate with a series of physiopathologic changes in nucleus pulposus (NP) cells, especially macroautophagy (hereafter autophagy).Citation1,2 Autophagy was elevated in degenerated, aged and diabetic rat discs, and autophagy could be activated by starvation in disc cells.Citation3-6 An increasing number of researches have suggested that appropriate autophagy might play a central role in IDD by regulating the adaptation of disc cells under stress.Citation7 NP cells, which are embedded in a special gelatinous tissue, are affected by various hostile stresses of their microenvironment, leading to the above changes.Citation8 Reaction and adaptation of NP cells to hypoxia, starvation, abnormal mechanics and acidic environments have been reported in multiple studies;Citation6,9-11 however, the effect of hyperosmotic stress on the cells is yet to be interpreted.
Osmolality of the extracellular matrix (ECM) around NP cells is controlled by 2 such factors: the concentration of proteoglycans and the loading on the disc.Citation8 On the one hand, owing to the high concentration of negatively charged proteoglycans controlling ionic composition in the ECM,Citation12,13 the high density of fixed charge increases the osmolality which ranges between 450 to 550 mOsm, according to Urban et al.Citation14 On the other hand, the osmolality can be upregulated when the disc undergoes extended static compression or continuous cyclic loading, which leads to the outflow of liquid and the increase in proteoglycans in NP.Citation8 Taken together, these studies show that NP cells have to contend with a hyperosmotic environment that changes dynamically due to changes in loading and matrix composition. However, how NP cells can survive in such hostile environments and what cellular adaptations they use remain unclear.
Hyperosmotic condition exerts an influence on apoptosis, metabolic activity, protein synthesis and even chromatin condensation in chondrocytes and NP cells.Citation15-17 Apoptosis is activated in porcine chondrocytes by a medium with an osmotic pressure of 600 mOsm, via the mitogen-activated protein kinase (MAPK) pathway.Citation16 However, autophagy, another type of programmed cell death, has not been reported in NP cells subjected to hyperosmotic stress. As a self protective mechanism, autophagy degrades dysfunctional cellular organelles and excess proteins by fusing with lysosomes and using the lysosomal enzymes, maintaining homeostasis within cells exposed to various stresses.Citation18,19 Microtubule-associated protein 1 light chain 3 (LC3) and Beclin-1, which participate in the autophagy process, are usually regarded as markers of autophagy,Citation20 while, Sequestosome-1 (SQSTM1)/p62 is selectively degraded by autophagy, reflecting the accumulation of protein aggregates and the degree of autophagy activation.Citation21 In order to protect cells from hostile stimulation, autophagy activation occurs in cells exposed to various types of stress. In one study, activation of autophagy inhibited the apoptotic percentage of rat annulus fibrosus cells treated with serum deprivation.Citation6 A high glucose medium promoted autophagy by inducing oxidative stress through mitochondrial damage in rat notochordal cells,Citation22 while compressive loading activated autophagy by reactive oxygen species in rat NP cells.Citation23 Given the role of autophagy in dealing with stress, we hypothesized that autophagy could be adaptively enhanced by hyperosmotic stress to address the stress. Hence, in order to demonstrate the protective role of autophagy, we considered cell viability and apoptosis regulated by hyperosmotic stress in the present study.
Recently, emerging evidence has suggested a relationship between autophagy and intercellular Ca2+, although the results are conflicting.Citation24 Several Ca2+-mobilizing agents and the endoplasmic reticulum Ca2+ ATPase inhibitor thapsigargin were shown to increase autophagy via the intercellular Ca2+-dependent mechanism.Citation25-27 Furthermore, the influx of Ca2+ was associated with AMP activated Protein Kinase (AMPK) and mammalian target of rapamycin (mTOR), which are the crucial proteins in the pathway regulating autophagy.Citation27-29 Interestingly, hyperosmotic stress could also stimulate a transient Ca2+ influx in chondrocytes and cardiomyocytes, leading to an increase in intracellular Ca2+ concentration ([Ca2+]i).Citation30,31 Therefore, we were interested to investigate whether an increase in [Ca2+]i could mediate autophagy activation exposed to hyperosmotic stress.
In this study, we set out to explore the stimulatory role of hyperosmotic stress on autophagy, and the underlying mechanism related to intercellular Ca2+, AMPK and mTOR in rat NP cells which were accurately called as notochordal cells in young rat discs. The detailed protective role of autophagy and the effect of hyperosmotic stress on cell viability and apoptosis were also investigated.
Results
Hyperosmotic stress activates autophagy in notochordal cells
To confirm the hypothesis that autophagy may be adaptively increased in rat notochordal cells exposed to hyperosmotic stress, monodansylcadaverine (MDC) staining, LysoTracker staining, and Western blotting for LC3, Beclin-1, and SQSTM1/P62 were used. LC3, Beclin-1, and SQSTM1/P62 are widely used markers for autophagy. As the osmotic pressure of the culture medium increased over 12 hours, the number of acidic granular vacuoles observed in notochordal cells stained with MDC also increased (). Because autophagic cytosolic degradation relies on the fusion of an autophagosome with a lysosome to form an autolysosome, LysoTracker Red staining, a marker of lysosomal activity, was also used to detect the autophagy process indirectly. Consistent with the MDC staining, there was an increase in punctate LysoTracker staining in notochordal cells exposed to hyperosmotic stress (). When the different figures of the same cells were merged, the MDC staining was found to co-localize to a high degree with the LysoTracker Red staining, suggesting that the increased formation of acidic vesicular organelles was reflected by both staining methods (quantitative analysis shown in Fig. S6A).
Figure 1. Typical LysoTracker Red staining, MDC staining, and Western blotting analysis of autophagy markers in notochordal cells under various osmolalities for different times. (A) After 24 hours of treatment, cells were stained with MDC and LysoTracker Red, and then observed under a fluorescence microscope. (magnification ×200). (B, C, D, F, H) Western blotting analysis of LC3-II and Beclin-1 expression in cells under increasing osmolality for 6 (B) and 12 hours (C), or under 600 mOsm for 0–24 hours (D). The optical densities of LC3-II/β-actin bands in (B and C and D) are shown in (F) and (H) respectively. (E, G, H) Western blotting analysis of SOSTM1/P62 expression in cells under increasing osmolality for 6–24 hours (E). The optical densities of SQSTM1/P62 β-actin bands in (D) and (E) are shown in (H) and (G) respectively. Values are the mean ± SD , *P < 0.05, **P < 0.01, compared with 270 mOsm or 0 hour. n = 6.
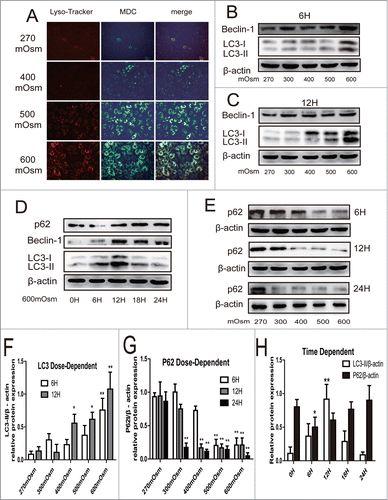
LC3-II expression was significantly increased only in the cells exposed to 600 mOsm stimulation for 6 hours (P < 0.01) compared with control cells, whereas when the treatment time was extended, a dose-dependent increase in LC3-II/β-actin expression was detected (). In addition, the effect of hyperosmotic stress on autophagy reached a peak at 12 hours, with a decline in LC3-II/β-actin at 18 and 24 hours after incubation (). A similar change in Beclin-1 was found. However, expression of SQSTM1/P62, which was selectively degraded by autophagy through binding to LC3 decreased as the osmotic pressure increased in the medium, showing the opposite tendency to that of LC3-II and Beclin-1 (). Interestingly, when the incubation was sustained for 24 hours, SQSTM1/P62 was significantly degraded even in the cells exposed to 300 mOsm compared with those exposed to 270 mOsm (P < 0.01), suggesting a consistently increased autophagy flux.
To corroborate these findings, we also detected autophagy flux by treating cells with rapamycin and Bafilomycin A1. Western blotting and immunochemistry were used to analyze LC3-II expression. In cells exposed to 500 and 600 mOsm, the distinct puncta of LC3 protein were distributed in the cytoplasm, which was consistent with the finding in cells treated with the classic autophagy activator rapamycin (). By contrast, fewer cells with LC3-positive puncta were observed in the 270 mOsm group (). In addition, cells were transfected with LC3B-GFP and observed under confocal microscope (see supplemental materials and methods). Similar trend of LC3b-GFP puncta number was found in transfected cells under different osmotic mediums (Fig. S1). By analyzing LC3-II expression using Western blotting, both rapamycin-treated and 600 mOsm-treated cells were found to have significantly higher expression of LC3-II/β-actin compared with control cells (P < 0.01) (). In addition, an increase in autophagic flux was demonstrated with Bafilomycin A1 treatment, which acted as a inhibitor of H+-ATPase and impeded the maturation of autophagosomes, leading to the decrease in the conversion of LC3-II to LC3-I. Compared with the cells treated with 600 mOsm alone, co-treatment with hyperosmotic pressure and Bafilomycin A1 significantly increased LC3-II/ β-actin (P < 0.01) ().
Figure 2. Autophagy analyzed by immunofluorescence and Western blotting analysis for LC3, as well as transmission electron microscopy to detect autophagosomes. Representative images are shown. (A, D) After 12 hours of treatment with 270, 500, or 600 mOsm or 10 μM rapamycin, LC3 expression was analyzed by immunofluorescence under confocal microscopy (magnification ×400). The percentage of LC-3-positive cells is shown in (D). (B, E) Under the same treatment as above, LC3 expression was detected by Western blotting. The optical densities ofthe LC3-II/β-actin bands are shown in (E). (C, F) Cells were treated with 600 mOsm or Bafilomycin A1, or co-treatment with both for 12 hours, and LC3 expression was detected by Western blotting. The optical densities of theLC3-II/β-actin bands are shown in (F). Values are the mean ± SD , *P < 0.05, **P < 0.01. n = 6. (G) Notochordal cells were treated with 270, 500 or 600 mOsm for 24 hours, and transmission electron microscopy was used to detect autophagosomes andautophagolysosomes. White star, mitochondria; white arrow: autophagosome; white triangle, autophagolysosome.
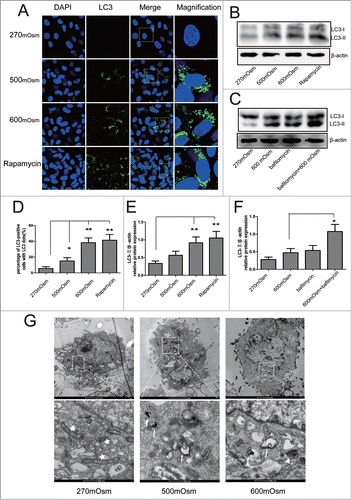
Autophagosomes and autophagolysosomes were observed by TEM, which is a standard method to check autophagy activation. Compared with the cells treated with 270 mOsm, the 500 and 600 mOsm-treated cells exhibited more autophagosomes and autophagolysosomes in cytoplasm ().
Involvement of intracellular Ca2+ level in the autophagy activation
Because it has been reported that there is an influence of Ca2+ influx on autophagy in the acidic environment,Citation24 we speculated that autophagy activation in notochordal cells exposed to hyperosmotic stress might also require the involvement of intracellular Ca2+ concentration ([Ca2+]i). As shown in , exposure of notochordal cells to 600 mOsm medium led to a transient increase of [Ca2+]i at about 200 seconds after stimulation, accompanied by different ranges of oscillations.
Figure 3. Involvement of [Ca2+]i levels in hyperosmotic stress-induced autophagy. (A, B) Cells preloaded with fluo3-AM were stimulated by 600 mOsm medium and examined under a confocal microscope. Serialfluorescent images were captured every 20 seconds. Some representative images are shown in (A)(magnification ×600). Relative fluorescence values were calculated as ΔF/F0 to represent the relative [Ca2+]i levels. To detect the Ca2+ pathway, cells were pre-incubated with 100 mM BAPTA-AM, 1 mM EGTA or 10 μM U73122, following fluo3-AM fluorescence analysis (B). (C, D) Western blotting analysis for LC3 in cells treated with 270 mOsm, 600 mOsm, 270+ BAPTA-AM, or 600 mOsm+ BAPTA-AM. Values are mean ± SD , **P < 0.01. n = 6.
![Figure 3. Involvement of [Ca2+]i levels in hyperosmotic stress-induced autophagy. (A, B) Cells preloaded with fluo3-AM were stimulated by 600 mOsm medium and examined under a confocal microscope. Serialfluorescent images were captured every 20 seconds. Some representative images are shown in (A)(magnification ×600). Relative fluorescence values were calculated as ΔF/F0 to represent the relative [Ca2+]i levels. To detect the Ca2+ pathway, cells were pre-incubated with 100 mM BAPTA-AM, 1 mM EGTA or 10 μM U73122, following fluo3-AM fluorescence analysis (B). (C, D) Western blotting analysis for LC3 in cells treated with 270 mOsm, 600 mOsm, 270+ BAPTA-AM, or 600 mOsm+ BAPTA-AM. Values are mean ± SD , **P < 0.01. n = 6.](/cms/asset/c9a60690-9b97-4728-b221-6ca0b79e6daa/kccy_a_1004946_f0003_oc.gif)
To further elucidate the pathways involved in the increase of Ca2+, bis-(o-aminophenoxy)ethane-N,N,N',N′-tetraacetic acid, tetra(acetoxymethyl) ester (BAPTA-AM, cell-permeable cytosolic Ca2+ chelator), ethyleneglycol-bis (2-aminoethylether)-N,N,N′,N-tetraacetic acid (EGTA, extracellular Ca2+ chelator) and U73122 (phospholipase C (PLC) inhibitor) were incubated with cells prior to hyperosmotic stimulation. Both EGTA and U73122 abolished the intracellular Ca2+ change elicited by hyperosmotic stress, suggesting that both intracellular Ca2+ release and extracellular Ca2+ influx participate in the accumulation of [Ca2+]i (). BAPTA-AM also suppressed the rise in [Ca2+]i (). Interestingly, the hyperosmotic stress-induced increase of LC3-II/β-actin was significantly (P < 0.01) reversed by BAPTA-AM (), suggesting that there is an association of [Ca2+ ]i with autophagy activation in cells exposed to hyperosmotic stress.
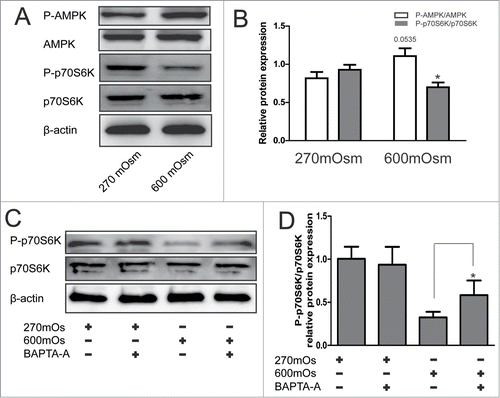
Hyperosmotic stress regulates autophagy by the Ca2+ -dependent AMPK-mTOR pathway
The downstream signaling pathways following Ca2+ influx were explored by analyzing the expression of p-AMPK, AMPK, p70S6K, and P-p70S6K. P70S6K is a downstream target of mTOR, representing the activity of the mTOR pathway. Compared with the 270 mOsm-treated cells, the cells exposed to hyperosmotic stress for 12 hours showed significantly higher expression of p-AMPK/AMPK (p = 0.0535) and significantly lower expression of P-p70S6K/p70S6K (P < 0.05) (), implying the activation of AMPK pathway and inhibition of mTOR pathway under hyperosmotic stress. Moreover, it was found that BAPTA-AM reversed the inhibition of p70S6K regulated by hyperosmotic stress, demonstrating the regulatory role of [Ca2+]i upstream of AMPK and mTOR (P < 0.05) ().
Figure 4. Activation of the AMPK/mTOR pathway detected by Western blotting analysis under hyperosmotic conditions. Typical bands are shown. (A, B) Cells were treated with 270 or 600 mOsm for 12 hours and analyzed by Western blotting analysis. The optical densities ofP-p70S6K/ p70S6K and P-AMPK/AMPK bands in (A) are shown in (B). Values are mean ± SD . *P < 0.05 versus 270 mOsm respectively. n = 6. (C, D) Notochordal cells were treated with 270 mOsmor with 270+600 mOsm. The optical densities ofthe LC3-II/β-actin bands are shown in (D). Values aremean ± SD , *P < 0.05. n = 6.
Silencing of ATG5 inhibited autophagy and induced SQSTM1/P62 accumulation
To further corroborate the protective role of autophagy under hyperosmotic stress, we inhibited autophagy by using RNAi against ATG5, which is a key protein of autophagosome formation. First, reduction of ATG5 protein and mRNA in notochordal cells was validated by Western blotting and Real-time PCR (; Fig. S2C and D). Silencing of ATG5 attenuated the increase of LC3-II expression treated with 500 and 600 mOsm medium for 24 hours(), indicating functional inhibition of autophagy. However, siRNA-treated cells which were exposed to hyperosmotic stress for 24 hours showed higher SQSTM1/p62 expression than the 600 mOsm-treated cells and control cells (). A similar result was observed using immunofluorescence for SQSTM1/p62 (), suggesting that SQSTM1/p62 accumulated in autophagy-deficient cells exposed to hyperosmotic stress (quantitative analysis shown in ).
Figure 5. Inhibition of autophagy by ATG5 siRNA. Typical images and bands are shown. (A) Western blotting analysis for ATG5 in cells after transfection with ATG5 or control siRNA. (B, C) Western blotting analysis for LC3 in cells treated with normal medium or hyperosmotic medium combined with ATG5 siRNA or control siRNA. (D, E) Western blotting and immunofluorescence analysis for SQSTM1/P62 in cells treated with normal medium or hyperosmotic medium combined with ATG5 siRNA or control siRNA. (magnification × 400).
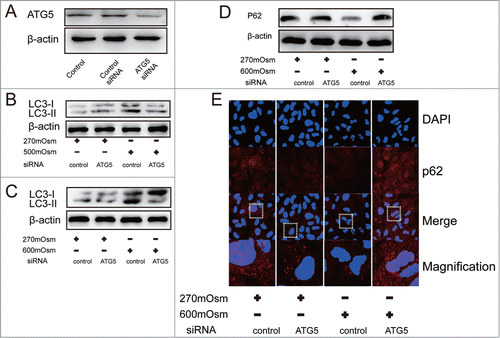
Figure 6. Apoptosis under hyperosmotic stress and inhibition of autophagy exacerbates apoptosis in notochordal cells. Typical images are shown. (A) Cell viability was analyzed by CCK-8 assay. *P < 0.05, **P < 0.01, vs. 270 mOsm for the same durations. (B, C) Following treatment with the indicated media for 24 hours, notochordal cells were stained with DAPI (B) or co-stained with Annexin-V and PI (C), followed by observation by fluorescence microscopy or flow cytometry to analyze the percentage of apoptotic cells. (D) Notochordal cells were treated with 500 or 600 mOsm, or co-treated with 500 or 600 mOsm plus ATG5 siRNA transfection for 24 hours. Apoptotic incidence (E) was calculated according to the flow cytometry results (D). Cell viability was analyzed by CCK-8 assay. (G) *P < 0.05, **P < 0.01 versus 270 mOsm for the same durations.
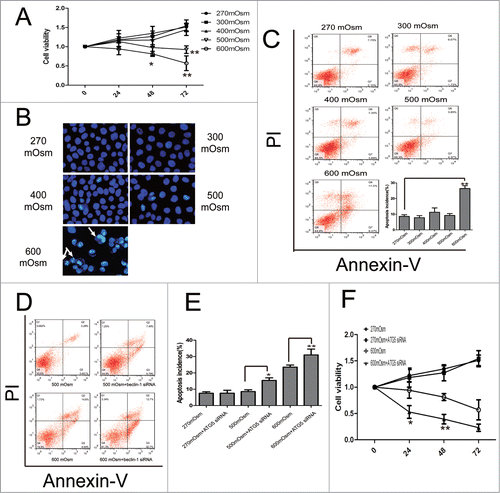
Inhibition of autophagy exacerbates apoptosis under hyperosmotic condition
Normal notochordal cells and autophagy-deficient cells were exposed to various osmotic pressures for 24 hours. Cell viability and apoptosis were determined by Cell Counting Kit-8 (CCK-8), 4′,6-diamidino-2-phenylindole (DAPI), and Annexin-V/ propidium iodide (PtdIns) staining. Unexpectedly, we found that osmotic pressure exerted no effect on cell viability, with the exception of the 600 mOsm medium which reduced the optical density (OD) value after 24 hours incubation (). Similar results were also observed under phase-contrast microscopy (Fig. S3). However, cell viability in the 500 and 600 mOsm groups declined significantly as the incubation time was extended to 72 hours (P < 0.05, ).
With regard to apoptosis, the percentage of notochordal cells with brightly stained condensed nuclei was significantly upregulated in 600 mOsm-treated cells after DAPI staining, although the difference between the other groups was similar (). Double-staining with Annexin V and PI showed that only the 600 mOsm medium significantly increased the percentage of apoptotic cells, which rose from 8.120 ± 1.103 in the 270 mOsm group to 26.40 ± 1.835 in the 600 mOsm group (P < 0.01) ().
At 48 hours after transfection with ATG5 siRNA, cells were exposed to 500 mOsm and 600 mOsm medium for 24 hours. As shown in , there was no influence of ATG5 siRNA on apoptosis or cell viability in the 270 mOsm medium. However, autophagy-deficient cells had a significantly higher percentage of apoptotic cells than did wild-type cells exposed to the same hyperosmotic stress (P < 0.05) (). In terms of cell viability, silencing of ATG5 significantly exacerbated cell death induced by hyperosmotic stress at 24 and 48 hours after incubation (P < 0.05), although this difference disappeared when the incubation time was extended to 72 hours ().
Finally, we investigated the mechanism involved in the association between autophagy and apoptosis. Bax and cytochrome c was increased in hyperosmotic stress-treated cells, however Bcl-2 was decreased (Fig. S4A). The mitochondrial membrane potential (MMP) was evaluated by 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) staining. MMP was inhibited by hyperosmotic stress, as evidenced by the decrease in the ratio of red to green fluorescence (JC-1 aggregates to JC-1 monomers) (Fig. S4B). Consistent with the widely-reported role of Ca2+ in the change in mitochondrial polarization state,Citation32 BAPTA-A was able to partly reverse the decrease in MMP, demonstrating the involvement of Ca2+ in hyperosmotic stress-induced apoptosis. However, silencing of ATG5 enhanced green fluorescence in cells subjected to hyperosmotic stress (Fig. S5), suggesting that mitochondria might be the target of autophagy in the process of cell protection.
Discussion
Hyperosmotic stress is a well-recognized microenvironmental factor contributing to IDD,Citation33 therefore, the current research into the response and adaption of notochordal cells under such stimulation might provide some clues to therapy for IDD. The present study first demonstrated a dose- and time-dependent increase in autophagy level in rat notochordal cells exposed to hyperosmotic pressure. This stress provoked a similar level of autophagy in notochordal cells to that induced by rapamycin, and an increase in autophagic flux was demonstrated by the increase in LC-3 II level after co-treatment with Bafilmycin A1 and 600 mOsm medium. Secondly, we found that intracellular Ca2+, AMPK, and mTOR participated in autophagy activation under hyperosmotic pressure. As indicated by previous studies,Citation15,16 hyperosmotic stress activates apoptosis, which might be mediated by intracellular Ca2+-induced mitochondrial dysfunction. Finally, inhibition of autophagy by targeted silencing of ATG5 exacerbated apoptosis, reduced cell viability, and led to SQSTM1/P62 protein accumulation in notochordal cells. All of this evidence indicated that autophagy might be a cellular adaptation for notochordal cells under hyperosmotic condition.
Because autophagy is a mechanism controlling cellular homeostasis by self-digestion using lysosome enzymes, an increasing number of papers have shown a protective role of autophagy under stress.Citation34 In osteoarthritis, autophagy is regarded as the principal mechanism responsible for cartilage and cellular homeostasis, although its negative and positive effect on osteoarthritis both have been reported.Citation35,36 Appropriate autophagy activation by rapamycin can even retard the progression of osteoarthritis in rats.Citation37 Notochordal cells and chondrocytes, both long-lived post-mitotic cells, have to rely on autophagy when confronted by stress during aging and degeneration.Citation38 Consequently, autophagy was elevated in the NP of aged rats.Citation4 However, duo to the dural roles of autophagy in cell survival and death, it is so important and yet so difficult to control the degree of autophagy in notochordal cells, especially in vivo. In mice, autophagy activated by daily intra-peritoneal injections of rapamycin at 1 mg/kg body weight/dose was reported to protect osteoarthritis.Citation37 Whereas, how to activate autophagy and the appropriate level of autophagic activation in discs were not described in the recent papers. In order to accurately measure and control autophagic level in vivo, transgenic mice with green fluorescent protein fused to LC3 (LC3-GFP) were generated and might provide more convenience and assistance to researchers.Citation39
In our study, hyperosmotic pressure, a major stress in disc tissue, increased the autophagy level in notochordal cells analyzed by several different methods, namely MDC and LysoTracker Red staining, TEM assessment, and Western blotting for LC3, Beclin-1, and SQSTM1/P62. Importantly, autophagy was activated as early as 6 hours after exposure to 600 mOsm, with the highest level at 12 hours and a gradual decrease at 18 and 24 hours. However, apoptosis was activated after 24 hours of exposure, and the percentage of apoptotic cells increased continuously as the exposure time increased. The different activation times suggested that early exposure might promote autophagy to adapt to stress; however, when the stress is prolonged and becomes more severe, apoptosis is activated, leading to irreversible cell death. Sasaki et al.Citation40 also suggested that autophagy can be increased in osteoarthritis during the initial degenerative phase, whereas autophagy declined as the cartilage degeneration became severe.
Generally, the autophagic pathways include mTOR-dependent and mTOR-independent pathways; however, the upstream molecules of mTOR have recently been investigated extensively.Citation41 Intracellular Ca2+, a second messenger, has dual roles in regulating autophagy and apoptosis, and its level can be increased via Ca2+ influx from intracellular stores or the extracellular space.Citation24 Our previous study showed that [Ca2+]i transiently released from Ca2+ intracellular stores contributed to endplate chondrocyte apoptosis in an acidic environment.Citation42 In the present study, EGTA, BAPTA-AM, and U73122 all inhibited the transient Ca2+ influx, implying that both the intracellular and extracellular pathways are involved in the increase of [Ca2+]i in notochordal cells exposed to hyperosmotic stress. We also found that BAPTA-AM, a rapid intracellular Ca2+ buffer, inhibited LC3-II expression, indicating that [Ca2+]i mediates autophagy activation. Corroborating these results, BAPTA-AM had no effect on basal autophagy level, although other studies have reported that it has inhibitory and stimulatory effects on autophagy.Citation43,44
AMPK, an important metabolic sensor, is comprised of α, β, and γ subunits, and is crucial to regulate cell response to metabolic stresses.Citation45 To regulate energy metabolism under stress, including hyperosmotic conditions and starvation,Citation46 AMPK is activated and then inhibits mTOR, leading to autophagy activation. Inhibition of mTOR was shown to change mRNA translation and cell metabolism.Citation46 Consistent with previous research, we found that hyperosmotic stress activated AMPK and inhibited p70S6K. BAPTA-AM could partly attenuate the decrease in phosphorylation of p70S6K, suggesting that intracellular Ca2+ is involved in the activation of AMPK/mTOR pathway.
Induction of apoptosis in response to hyperosmotic stress has been found in a number of different cells, including chondrocytes and ovary cells.Citation16,47 Apoptosis contributes to the development of IDD, which could be activated via the death receptor, the mitochondrial, and the endoplasmic reticulum pathway by various types of stimulations.Citation48 Mitochondrial dysfunction, including the decrease of MMP and mitochondrial permeability transition (MPT), as well as the release of cytochrome C, triggers caspase activation, leading to apoptosis.Citation49 In the present study, we found that 600 mOsm increased Bax and cytochrome c, and decreased Bcl-2 and MMP in notochordal cells, suggesting that the mitochondrial pathway might be involved in hyperosmotic stress-induced apoptosis. Furthermore, chelation of intracellular Ca2+ by BAPTA-AM partly attenuated the decrease in MMP, demonstrating that Ca2+ might mediate the mitochondrial dysfunction. To the best of our knowledge, this is the first report demonstrating that the mitochondrial pathway is involved in apoptosis induced by hyperosmotic stress in notochordal cells. In addition, we showed that hyperosmotic stress could enhance apoptosis and autophagy simultaneously, although their activation time was different.
To reveal the relationship between autophagy and apoptosis, autophagy was inhibited by knocking down ATG5 using RNAi technology. ATG5-deficient cells showed decreased LC3 II and increased SQSTM1/P62 expression, demonstrating that the autophagy process was directly blocked and that SQSTM1/P62 was accumulating in notochordal cells under hyperosmotic stress without autophagic degradation. Moreover, autophagy-deficient cells showed reduced cell viability and an increased percentage of apoptotic cells, suggesting that appropriate autophagy protected cells under hyperosmotic stress. How can autophagy regulate cell viability and apoptosis? Autophagy maintains mitochondrial integrity and even allows engulfment of impaired mitochondria,Citation50 which might be a target for apoptosis regulation by autophagy in such an environment. In our research, we found that autophagy-deficiency worsened the decrease of MMP induced by hyperosmotic stress, partly confirming this hypothesis.
Hyperosmotic stress activated a series of changes in notochordal cells, but what is the receptor mediating these alterations? Transient receptor potential vanilloid 4 (TRPV4) was indicated as a mechanosensor responsible for the effect of hyperosmotic stress in chondrocytes.Citation51,52 Importantly, knocking out this channel abolished the transient [Ca2+]i change, suggesting that TRPV4 mediation of the change in intracellular Ca2+ might be the first step of this series of changes. Therefore, further research is required to explain whether TRPV4 mediates autophagy activation in notochordal cells, which might be a therapeutic target to treat IDD.
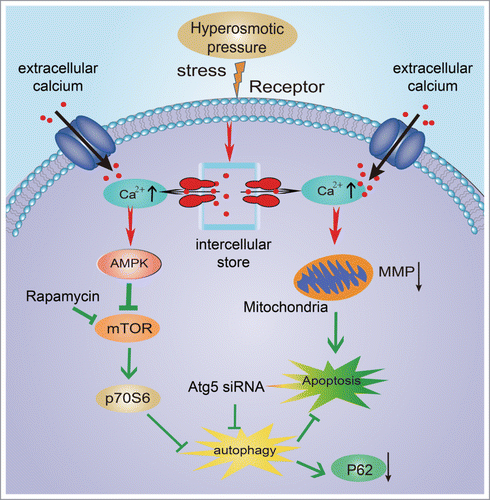
In conclusion, our studies revealed that hyperosmotic stress could induce autophagy in a dose- and time-dependent manner. Transient [Ca2+]i influx was stimulated by hyperosmotic conditions, and inhibition of [Ca2+]i abolished the increase in LC3-II under hyperosmotic conditions. Hyperosmotic stress activated AMPK and inhibited p70S6K, but chelation of intracellular Ca2+ partly attenuated the decrease in p70S6K. Notochordal cells under hyperosmotic stress showed apoptosis and a decline in cell viability. Inhibition of autophagy was achieved by targeted knockdown of ATG5, which led to accumulation of SQSTM1/P62, increased apoptosis, and decreased cell viability, suggesting a protective role of autophagy under such an environment. Based on previous findings, we propose a mechanism by which hyperosmotic stress induces autophagy and apoptosis (). Because notochordal cell death in a hyperosmotic environment contributes to the development of IDD, it is reasonable to infer that autophagy activation in vivo might retard the progress of IDD, which will be investigated in our future research. In addition, receptors of hyperosmotic stress, which might be a promising target of IDD treatment, will also be investigated.
Figure 7. Schematic diagram of the mechanism involved in the protective role of autophagy under hyperosmotic stress.
Materials and Methods
The experimental animal protocol was approved by the Animal Care and Use Committee of Fudan University.
Reagents and antibodies
The reagents and drugs, type II collagenases, MDC, NaCl, rapamycin, EGTA, Bafilomycin A1, BAPTA-AM, and U-73122 hydrate were purchased from Sigma Aldrich (St. Louis, MO, USA). The protein extraction and measurement reagents, Lyso-Tracker Red, fluo3 acetoximethylester (fluo3-AM), JC-1, DAPI, and Annexin V-fluorescein isothiocyanate (FITC)/ PtdIns were obtained from Beyotime (Shanghai, China). Pluronic F-127 was bought from Biotium (Hayward, CA, USA). The primary antibodies against Beclin-1, SQSTM1/P62, β-actin, AMPK and p-AMPK were bought from Abcam (Cambridge, UK), and antibodies of microtubule-associated protein 1 light chain 3 B (LC3B), p70S6K, p-p70S6K and ATG5 were purchased from CST (Massachusetts, USA). All cell culture reagents were acquired from Hyclone (Utah, USA).
Notochordal cell culture
Forty Sprague-Dawley (SD) rats from department of laboratory animal science, which were 3 months old and about 250 g in weight, were used. As described in our previous paper,Citation53 the gel-like NP tissue was separated from the lumbar disc under a surgical microscope. To avoid cell infection, SD rats were euthanized by asphyxiation, and immersed in 75% alcohol for 10 minutes before this operation. The tissue was digested with 0.1% collagenase II for 4 hours, and then filtered through a 70μm commercial filter. Notochordal cells were cultured with high glucose Dulbecco modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells at passage 2 were used for the following investigation.
Experimental design
The osmotic pressure of DMEM obtained from Hyclone was 270 mOsm (control medium), and it was modified to give 300, 400, 500, and 600 mOsm DMEM by adding NaCl to the medium, which was measured with a Vapro vapor pressure osmometer (model 5500, Wescor, USA). For all experiments, the pH of the medium was maintained at 7.4 ± 0 .2 to exclude the influence of acid. The dose-dependent and time-dependent effects of osmotic stress on autophagy and apoptosis were assessed. In order to detect autophagy flux, 10 μM rapamycin and 0.1 μM Bafilomycin A1 were used to treat cells 1 hour prior to osmotic stimulation. When exploring the involvement of intracellular Ca2+, BAPTA-AM, EGTA and U73122 were utilized to chelate the cytosolic Ca2+, to chelate the extracellular Ca2+ and to inhibit phospholipase C (PLC) respectively. They were added into medium 1 hour prior to osmotic stimulation. Before all the above stimulations, the cells were cultured with 1% FBS DMEM for 12 hours. All the following tests were performed at least 6 times.
Cell counting assay
Notochordal cells were placed in 96-well plates and treated with increasing osmolalities for 24 hours. Then these cells were incubated with 10 μl CCK-8 reagent (Dojindo, Tokyo, Japan) for 1 hour in a culture chamber. Absorbance of the cells exposed to hyperosmotic stress was normalized to the absorbance of cells exposed to 270 mOsm, which was measured at 450 nm.
Autophagy related gene 5 (Atg5) siRNA for inhibition of autophagy
Atg5, a key protein for autophagosome formation, is the common target to inhibit autophagy by the technology of RNA interfering.Citation40 The sequences of small interfering RNAs (siRNAs) are shown in Table S1. These base sequences were designed previouslyCitation54 and synthesized by GenePharma (Shanghai, China). We used lipofection to deliver 100 nmol/L ATG5 siRNA into notochordal cells using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. In order to analysis the transfection efficiency, cells transfected by control siRNA labled with a reporter dye FAM (6-carboxy-fluorescein) were observed under fluorescence microscope (Fig. S2A and B). 48 hours after transfection, the expression of ATG5 protein and ATG5 mRNA were determined, and notochordal cells were treated with different stimuli as described in experimental design. In order to confirm the success of silencing, notochordal cells were analyzed by Western blotting and Real-time PCR. The primers of ATG5 are shown in Table S2.
Monodansylcadaverine (MDC) staining
MDC staining has been widely reported to label autophagic vacuoles in vitro.Citation55 Following the relevant stimulation, notochordal cells were incubated with 0.05 mM MDC without paraformaldehyde fixation in 24-well plates for 30 minutes at 37°C. These cells were washed with phosphate buffer solution (PBS) 3 times and observed using a fluorescence microscope with a UV filter to detect autophagic vacuoles.
LysoTracker Red staining for lysosomal activity
Following each treatment, these same 24-well plates were rinsed 3 times with fresh medium, then these cells were incubated with 66nM Lyso-Tracker Red for 30 minutes at 37°C, washed with PBS, and observed under fluorescence microscopy with a green excitation filter. In order to detect the co-staining of MDC and LysoTracker, notochordal cells were stained with MDC for 15 minutes first, followed by a co-staining with a fresh supply of MDC plus LysoTracker Red for another 30 minutes. The same cells were loaded with MDC and Lyso Tracker Red simultaneously.
Western blotting
Cold PBS was used to stop each treatment. Total protein was extracted using RIPA buffer and 1 mM phenylmethanesulfonylfluoride (PMSF), followed by measurement of protein concentration using a BCA Protein Assay Kit (Beyotime, Shanghai, China). An aliquot (30 mg) was separated by SDS-PAGE and transferred to a PVDF membrane. The membrane was blocked with 5% non-fat milk, and then incubated with the primary antibodies overnight at 4°C. All of the primary antibodies were diluted to 1:1000 with the exception of β-actin, which was diluted to 1:2000. The corresponding HRP-conjugated secondary antibodies combined with the primary antibodies, and enhanced chemiluminescence (ECL plus kit; Beyotime, Shanghai, China) plus was used to detect protein expression with an ECL detection system (PerkinElmer, USA). AlphaEaseFC 4.0 software was used to analyze protein band intensity.
Immunofluorescence
To detect LC3 and SQSTM1/P62 expression in notochordal cells by confocal microscopy, cells were cultured in cover glass-bottom dishes. Notochordal cells were fixed with 4% paraformaldehyde for 10 minutes. For SQSTM1/P62, notochordal cells were incubated with 0.2% Triton X-100 for 15 min, and for LC3, ice-cold 100% methanol was used to treat cells for 10 min at –20°C. Cells were blocked with 5% bovine serum albumin, then incubated with primary antibodies (SQSTM1/P62 and LC3, diluted to 1:100 and 1:50, respectively), then with FITC- conjugated second antibodies. These Cells were labeled with DAPI for 5 minutes. Finally, cells were observed under an inverted confocal microscope (Nikon Japan) .
Transmission electron microscopy
Cells in the culture flasks were fixed with 2.5% glutaraldehyde overnight. The cells were post-fixed with 2% osmium tetroxide, block-stained with 2% uranyl acetate, dehydrated in an acetone series, and then embedded in Epon 812. Using semi-thin sections stained with Toluidine Blue as reference, the cells were cut into ultra-thin sections and observed under a transmission electron microscope (TEM) (Hitachi, Japan).
Measurement of intracellular Ca2+ level
Notochordal cells were loaded with 4 mM fluo3-AM to detect the change of intracellular Ca2+ under an inverted confocal laser scanning microscopy (Nikon Japan), as described previously.Citation42 Cells cultured in cover glass-bottom dishes were incubated with 4 mM fluo3-AM and 0.02% Pluronic F-127 for 45 minutes, and rinsed 3 times with PBS. Confocal microscopy with excitation and emission wavelengths of 488 nm and 525 nm, respectively, was used to detect fluorescence intensity. By sketching the contour of the whole cell, the fluorescence signal and image was captured every 20 seconds, and this procedure continued for 1,200 seconds. At about 20 seconds, the 600 mOsm medium (with or without 100 mM BAPTA-AM, 1 mM EGTA and 10 μM U73122) was immediately injected into the dishes with a pipettor. The signal of at least 20 different cells in the dishes was acquired each time. The fluorescence signal was analyzed by the LSM 5 Image software. F0 was the basal fluorescence signal, and ΔF was the difference between the experimental and basal signals. The equation ΔF /F0 was used to express the relative level of intracellular calcium.
DAPI staining
Notochordal cells were fixed with 4% paraformaldehyde for 15 minutes at 4°C and rinsed 3 times with PBS, followed by incubation with DAPI staining solution for 5 minutes. The nucleus changes were observed under a fluorescence microscope with a UV filter.
Flow cytometry analysis
An Annexin V-FITC/PI staining kit was used to detect the percentage of apoptotic cells, according to the manufacturer's protocol. Notochordal cells were digested with 0.25% Trypsin and centrifuged (1000g for 5 minutes), followed by incubation with Annexin V-FITC for 10 minutes, and then PtdIns for 10 minutes at 20–25°C. Stained cells were quantified by flow cytometry (FACSCaliber; Becton Dickinson, Heidelberg, Germany).
Statistical Analysis
All experiments were carried out at least 6 times. Data were expressed as mean ± SD . The SPSS statistical software program (version 15; SPSS Inc., Chicago, IL, USA) was used to perform the statistical analyses. The differences between the groups were evaluated by one-way analysis of variance (ANOVA) and Tukey test to explore the differences between the 2 groups. The differences were considered to be statistically significant at an error level of P < 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1004946_Supplementary_Materials.zip
Download Zip (9 MB)Funding
The study was supported by Natural Science Foundation of China (81372002), Natural Science Foundation of China (31170925), Natural Science Foundation of China (81401820), “Technology Innovation Action Plan” Key Project of Shanghai Science and Technology Commission (12411951300 and12JC1402600).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- Ding F, Shao ZW, Xiong LM. Cell death in intervertebral disc degeneration. Apoptosis 2013; 18:777-85; PMID:23512131; http://dx.doi.org/10.1007/s10495-013-0839-1
- Chen JW, Ni BB, Li B, Yang YH, Jiang SD, Jiang LS. The responses of autophagy and apoptosis to oxidative stress in nucleus pulposus cells: implications for disc degeneration. Cell Physiol Biochem 2014; 34:1175-89; PMID:25277442; http://dx.doi.org/10.1159/000366330
- Ye W, Zhu W, Xu K, Liang A, Peng Y, Huang D, Li C. Increased macroautophagy in the pathological process of intervertebral disc degeneration in rats. Connect Tissue Res 2012; 54:22-8; PMID:22835012; http://dx.doi.org/10.3109/03008207.2012.715702
- Ye W, Xu K, Huang D, Liang A, Peng Y, Zhu W, Li C. Age-related increases of macroautophagy and chaperone-mediated autophagy in rat nucleus pulposus. Connect Tissue Res 2011; 52:472-8; PMID:21591930; http://dx.doi.org/10.3109/03008207.2011.564336
- Jiang L, Zhang X, Zheng X, Ru A, Ni X, Wu Y, Tian N, Huang Y, Xue E, Wang X, et al. Apoptosis, senescence, and autophagy in rat nucleus pulposus cells: Implications for diabetic intervertebral disc degeneration. J Orthop Res 2013; 31:692-702; PMID:23238821; http://dx.doi.org/10.1002/jor.22289
- Shen C, Yan J, Jiang LS, Dai LY. Autophagy in rat annulus fibrosus cells: evidence and possible implications. Arthritis Res Ther 2011; 13:R132; PMID:21846367; http://dx.doi.org/10.1186/ar3443
- Jiang L, Yuan F, Yin X, Dong J. Responses and adaptations of intervertebral disc cells to microenvironmental stress: a possible central role of autophagy in the adaptive mechanism. Connect Tissue Res 2014; 55:311-21; PMID:25000273; http://dx.doi.org/10.3109/03008207.2014.942419
- Urban JP. The role of the physicochemical environment in determining disc cell behaviour. Biochem Soc Trans 2002; 30:858-64; PMID:12440933; http://dx.doi.org/10.1042/BST0300858
- Shoukry M, Li J, Pei M. Reconstruction of an in vitro niche for the transition from intervertebral disc development to nucleus pulposus regeneration. Stem Cells Dev 2013; 22:1162-76; PMID:23259403; http://dx.doi.org/10.1089/scd.2012.0597
- Johnson WE, Stephan S, Roberts S. The influence of serum, glucose and oxygen on intervertebral disc cell growth in vitro: implications for degenerative disc disease. Arthritis Res Ther 2008; 10:R46; PMID:18433481; http://dx.doi.org/10.1186/ar2405
- Xu HG, Li ZR, Wang H, Liu P, Xiang SN, Wang CD, Zhang XL. Intermittent cyclic mechanical tension-induced down-regulation of ectonucleotide pyrophosphatase phosphodiesterase 1 gene expression is mainly dependent on TGF-beta1 in end-plate chondrocytes. Orthopaedic Surgery 2013; 5:40-5; PMID:23420746; http://dx.doi.org/10.1111/os.12028
- Ishihara H, Warensjo K, Roberts S, Urban JP. Proteoglycan synthesis in the intervertebral disk nucleus: the role of extracellular osmolality. Am J Physiol 1997; 272:C1499-506; PMID:9176140
- Urban JP, Maroudas A. Swelling of the intervertebral disc in vitro. Connect Tissue Res 1981; 9:1-10; PMID:6456121; http://dx.doi.org/10.3109/03008208109160234
- JP U, A M. The measurement of fixed charged density in the intervertebral disc Biochim Biophys Acta 1979; 586:166-78; http://dx.doi.org/10.1016/0304-4165(79)90415-X
- Takeno K, Kobayashi S, Negoro K, Uchida K, Miyazaki T, Yayama T, Shimada S, Baba H. Physical limitations to tissue engineering of intervertebral disc cells: effect of extracellular osmotic change on glycosaminoglycan production and cell metabolism. Laboratory investigation. J Neurosurg Spine 2007; 7:637-44; PMID:18074689; http://dx.doi.org/10.3171/SPI-07/12/637
- Racz B, Reglodi D, Fodor B, Gasz B, Lubics A, Gallyas F, Jr., Roth E, Borsiczky B. Hyperosmotic stress-induced apoptotic signaling pathways in chondrocytes. Bone 2007; 40:1536-43; PMID:17392049; http://dx.doi.org/10.1016/j.bone.2007.02.011
- Irianto J, Swift J, Martins RP, McPhail GD, Knight MM, Discher DE, Lee DA. Osmotic challenge drives rapid and reversible chromatin condensation in chondrocytes. Biophys J 2013; 104:759-69; PMID:23442954; http://dx.doi.org/10.1016/j.bpj.2013.01.006
- Shapiro IM, Layfield R, Lotz M, Settembre C, Whitehouse C. Boning up on autophagy: the role of autophagy in skeletal biology. Autophagy 2014; 10:7-19; PMID:24225636; http://dx.doi.org/10.4161/auto.26679
- Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol cells 2013; 36:7-16; PMID:23708729; http://dx.doi.org/10.1007/s10059-013-0140-8
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Chen C, Deng M, Sun Q, Loughran P, Billiar TR, Scott MJ. Lipopolysaccharide stimulates p62-dependent autophagy-like aggregate clearance in hepatocytes. Biomed Res Int 2014; 2014:267350; PMID:24683544
- Park EY, Park JB. High glucose-induced oxidative stress promotes autophagy through mitochondrial damage in rat notochordal cells. Int Orthop 2013; 37:2507-14; PMID:23907350; http://dx.doi.org/10.1007/s00264-013-2037-8
- Ma KG, Shao ZW, Yang SH, Wang J, Wang BC, Xiong LM, Wu Q, Chen SF. Autophagy is activated in compression-induced cell degeneration and is mediated by reactive oxygen species in nucleus pulposus cells exposed to compression. Osteoarthritis Cartilage 2013; 21:2030-8; PMID:24120490; http://dx.doi.org/10.1016/j.joca.2013.10.002
- Decuypere JP, Bultynck G, Parys JB. A dual role for Ca(2+) in autophagy regulation. Cell Calcium 2011; 50:242-50; PMID:21571367; http://dx.doi.org/10.1016/j.ceca.2011.04.001
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol Cell 2007; 25:193-205; PMID:17244528; http://dx.doi.org/10.1016/j.molcel.2006.12.009
- Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci 2008; 65:3640-52; PMID:18850067; http://dx.doi.org/10.1007/s00018-008-8383-9
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-β peptide metabolism. J Biol Chem 2010; 285:9100-13; PMID:20080969; http://dx.doi.org/10.1074/jbc.M109.060061
- Law BY, Wang M, Ma DL, Al-Mousa F, Michelangeli F, Cheng SH, Ng MH, To KF, Mok AY, Ko RY, et al. Alisol B, a novel inhibitor of the sarcoplasmic/endoplasmic reticulum Ca(2+) ATPase pump, induces autophagy, endoplasmic reticulum stress, and apoptosis. Mol Cancer Ther 2010; 9:718-30; PMID:20197400; http://dx.doi.org/10.1158/1535-7163.MCT-09-0700
- Zhang J, Chiu J, Zhang H, Qi T, Tang Q, Ma K, Lu H, Li G. Autophagic cell death induced by resveratrol depends on the Ca(2+)/AMPK/mTOR pathway in A549 cells. Biochem Pharmacol 2013; 86:317-28; PMID:23680031; http://dx.doi.org/10.1016/j.bcp.2013.05.003
- Chiong M, Parra V, Eisner V, Ibarra C, Maldonado C, Criollo A, Bravo R, Quiroga C, Contreras A, Vicencio JM, et al. Parallel activation of Ca(2+)-induced survival and death pathways in cardiomyocytes by sorbitol-induced hyperosmotic stress. Apoptosis 2010; 15:887-903; PMID:20454859; http://dx.doi.org/10.1007/s10495-010-0505-9
- Erickson GR, Alexopoulos LG, Guilak F. Hyper-osmotic stress induces volume change and calcium transients in chondrocytes by transmembrane, phospholipid, and G-protein pathways. J Biomech 2001; 34:1527-35; PMID:11716854; http://dx.doi.org/10.1016/S0021-9290(01)00156-7
- Ip SW, Weng YS, Lin SY, Mei D, Tang NY, Su CC, Chung JG. The role of Ca+2 on rhein-induced apoptosis in human cervical cancer Ca Ski cells. Anticancer Res 2007; 27:379-89; PMID:17352257
- Wognum S, Huyghe JM, Baaijens FP. Influence of osmotic pressure changes on the opening of existing cracks in 2 intervertebral disc models. Spine (Phila Pa 1976) 2006; 31:1783-8; PMID:16845351; http://dx.doi.org/10.1097/01.brs.0000227267.42924.bb
- Martinez-Borra J, Lopez-Larrea C. Autophagy and self-defense. Adv Exp Med Biol 2012; 738:169-84; PMID:22399380; http://dx.doi.org/10.1007/978-1-4614-1680-7_11
- Lotz MK, Carames B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat Rev Rheumatol 2011; 7:579-87; PMID:21808292
- Chang J, Wang W, Zhang H, Hu Y, Wang M, Yin Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int J Mol Med 2013; 32:1311-8; PMID:24126970
- Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis 2011; 71:575-81; PMID:22084394; http://dx.doi.org/10.1136/annrheumdis-2011-200557
- Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT. Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 2010; 12:503-35; PMID:19650712; http://dx.doi.org/10.1089/ars.2009.2598
- Carames B, Kiosses WB, Akasaki Y, Brinson DC, Eap W, Koziol J, Lotz MK. Glucosamine activates autophagy in vitro and in vivo. Arthritis Rheum 2013; 65:1843-52; PMID:23606170; http://dx.doi.org/10.1002/art.37977
- Sasaki H, Takayama K, Matsushita T, Ishida K, Kubo S, Matsumoto T, Fujita N, Oka S, Kurosaka M, Kuroda R. Autophagy modulates osteoarthritis-related gene expressions in human chondrocytes. Arthritis Rheum 2011; 64:1920-8; PMID:22147463; http://dx.doi.org/10.1002/art.34323
- Esclatine A, Chaumorcel M, Codogno P. Macroautophagy signaling and regulation. Curr Top Microbiol Immunol 2009; 335:33-70; PMID:19802559
- Yuan FL, Wang HR, Yuan W, Zhao MD, Cao L, Duan PG, Jiang YQ, Li XL, Dong J. Ovarian cancer G protein-coupled receptor 1 is involved in acid-induced apoptosis of endplate chondrocytes in intervertebral discs. J Bone Miner Res 2014; 29:67-77; PMID:23821474; http://dx.doi.org/10.1002/jbmr.2030
- Gordon PB, Holen I, Fosse M, Rotnes JS, Seglen PO. Dependence of hepatocytic autophagy on intracellularly sequestered calcium. J Biol Chem 1993; 268:26107-12; PMID:8253727
- Khan MT, Joseph SK. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J Biol Chem 2010; 285:16912-20; PMID:20308071; http://dx.doi.org/10.1074/jbc.M110.114207
- Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, et al. Mammalian AMP-activated protein kinase subfamily. J Biol Chem 1996; 271:611-4; PMID:8557660; http://dx.doi.org/10.1074/jbc.271.2.611
- Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F, Carling D, Hardie DG, Baldwin SA. Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 2002; 115:2433-42; PMID:12006627
- Han YK, Kim YG, Kim JY, Lee GM. Hyperosmotic stress induces autophagy and apoptosis in recombinant Chinese hamster ovary cell culture. Biotechnol Bioeng 2010; 105:1187-92; PMID:20014438
- Zhao CQ, Jiang LS, Dai LY. Programmed cell death in intervertebral disc degeneration. Apoptosis 2006; 11:2079-88; PMID:17051327; http://dx.doi.org/10.1007/s10495-006-0290-7
- Ravindran J, Gupta N, Agrawal M, Bala Bhaskar AS, Lakshmana Rao PV. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 2011; 16:145-61; PMID:21082355; http://dx.doi.org/10.1007/s10495-010-0554-0
- Ryter SW, Choi AM. Regulation of autophagy in oxygen-dependent cellular stress. Curr Pharm Des 2013; 19:2747-56; PMID:23092322; http://dx.doi.org/10.2174/1381612811319150010
- Phan MN, Leddy HA, Votta BJ, Kumar S, Levy DS, Lipshutz DB, Lee SH, Liedtke W, Guilak F. Functional characterization of TRPV4 as an osmotically sensitive ion channel in porcine articular chondrocytes. Arthritis Rheum 2009; 60:3028-37; PMID:19790068; http://dx.doi.org/10.1002/art.24799
- Clark AL, Votta BJ, Kumar S, Liedtke W, Guilak F. Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: age- and sex-dependent progression of osteoarthritis in Trpv4-deficient mice. Arthritis Rheum 2010; 62:2973-83; PMID:20583100; http://dx.doi.org/10.1002/art.27624
- Wu W, Zhang X, Hu X, Wang X, Sun L, Zheng X, Jiang L, Ni X, Xu C, Tian N, et al. Lactate down-regulates matrix systhesis and promotes apoptosis and autophagy in rat nucleus pulposus cells. J Orthop Res 2013; 32:253-61; PMID:24307209; http://dx.doi.org/10.1002/jor.22503
- Yu KY, Wang YP, Wang LH, Jian Y, Zhao XD, Chen JW, Murao K, Zhu W, Dong L, Wang GQ, et al. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Res Cardiol 2014; 109:416; PMID:24847907; http://dx.doi.org/10.1007/s00395-014-0416-y
- Yin J, Ye AJ, Tan KS. Autophagy is involved in starvation response and cell death in Blastocystis. Microbiology 2010; 156:665-77; PMID:19910412; http://dx.doi.org/10.1099/mic.0.033944-0