
ABSTRACT
Intervertebral disc degeneration (IDD) is one of the main causes of lower back pain (LBP). It results from an imbalance between the degradation and synthesis of extracellular matrix (ECM) components in nucleus pulposus (NP) cells. Atorvastatin, an HMG-CoA reductase inhibitor, plays a vital role in many diseases, such as cardiovascular disease and osteoarthritis. However, the effect of atorvastatin on IDD is unclear. Herein, we demonstrated that atorvastatin affects matrix degradation induced by TNF-α and demonstrated the mechanism by which TNF-α modulates matrix metabolism in rat NP cells. Real-time PCR, western blotting and immunofluorescence staining were performed to detect the mRNA and protein expression of related genes. mRFP-GFP-LC3 adenovirus plasmid transfection and transmission electron microscopy (TEM) were used to detect cell autophagy. NLRP3 inhibitor and lentiviral vectors containing shRNA-NLRP3 were used to show the effect of NLRP3 on autophagic flux and the NF-κB signaling pathway. The results revealed that atorvastatin might suppress matrix degradation induced by TNF-α by suppressing NLRP3 inflammasome activity and inducing autophagic flux. Moreover, atorvastatin suppressed NF-κB signaling induced by TNF-α. NF-κB signaling inhibition suppressed NLRP3 inflammasome activity, and NLRP3 inhibition suppressed NF-κB signaling activation induced by TNF-α. NLRP3 inhibition or NLRP3 knockdown induced autophagic flux in the presence of TNF-α. Overall, the present study demonstrated that atorvastatin might suppress matrix degradation induced by TNF-α and further revealed the crosstalk among NLRP3 inflammasome activity, autophagy and NF-κB signaling.
1. Introduction
Lower back pain (LBP) is an extremely common symptom that occurs in all age groups and puzzles 70% of individuals in their lifetimes [Citation1,Citation2]. Sometimes, LBP can contribute to functional disability and inefficient working competence and even affect quality of life [Citation3]. Intervertebral disc degeneration (IDD) is reported to be one of the main causes of LBP, and the intervertebral disc (IVD) includes three distinct components: the inner nucleus pulposus (NP), the outer anulus fibrosus (AF) and the up and down cartilaginous endplates (EPs) [Citation4,Citation5]. As IDD proceeds, the synthesis of aggrecan and collagen type II is reduced, and the levels of inflammatory cytokines, including TNF-α, IL-1α/β, IL-6, IL-17, IL-8, and chemokines, are increased. Among these inflammatory cytokines, TNF-α and IL-1β are mostly reported to upregulate genes encoding matrix-degrading enzymes, such as matrix metalloproteinases (MMPs) and disintegrins and metalloproteinases with thrombospondin motifs (ADAMTSs) [Citation6].
Statins, HMG-CoA reductase inhibitors, prevent cardiovascular disease and stroke by altering the expression of endothelial nitric oxygen synthase and the development of cardiac hypertrophy and fibrosis and by decreasing the production of proinflammatory cytokines and reactive oxygen species [Citation7]. Atorvastatin is the most commonly used statin in the clinic and plays a pivotal role in cardiovascular disease and other inflammation-related diseases [Citation8,Citation9]. We wondered whether atorvastatin also plays a significant role in the control of IDD. Nod-like receptor pyrin domain-3 (NLRP3), caspase-1 and IL-1β are increased in degenerative human IVDs, and their expression is relevant to the grade of disc degeneration [Citation10,Citation11]. NLRP3 inflammasome activity plays a vital role in extracellular matrix (ECM) synthesis and degradation [Citation12]. Our previous results also revealed that BRD4 inhibition enhances ECM synthesis and reduces matrix-degrading enzymes by suppressing NLRP3 inflammasome activity and that nicotinamide phosphoribosyl transferase (NAMPT) controls matrix metabolism by upregulating NLRP3 inflammasome activity in rat NP cells [Citation13,Citation14].
Autophagy, a well-known form of programmed cell death, was reported to be associated with the pathological process of IDD [Citation15–17]. In monocyte and coronary artery disease, atorvastatin can inhibit NLRP3 inflammasome activity, as well as the levels of caspase-1, IL-1β, and IL-18 [Citation18,Citation19]. In addition, atorvastatin plays a protective role in the inflammatory response by inducing autophagy [Citation20]. Therefore, we hypothesized that atorvastatin controls ECM synthesis and degradation by suppressing NLRP3 inflammasome activity and inducing autophagy in rat NP cells.
In addition, the NF-κB signaling pathway is related to the occurrence of IDD [Citation21]. Our previous studies suggest that NF-κB inhibition alleviates NLRP3 inflammasome activity and promotes autophagy. Moreover, autophagy inhibition activates the NLRP3 inflammasome and upregulates ECM degradation in rat NP cells [Citation13,Citation14,Citation22]. We wondered if, in turn, NLRP3 inhibition controls autophagy and if atorvastatin regulates ECM metabolism through the NLRP3 inflammasome and autophagy feedback loop via the NF-κB signaling pathway in rat NP cells.
Therefore, in the current study, we aimed to investigate the ability of atorvastatin to control matrix metabolism and the relationships among the NLRP3 inflammasome, autophagy and the NF-κB signaling pathway in the process by which atorvastatin controls matrix metabolism in rat NP cells.
2. Material and methods
2.1 Isolation and culture of rat NP cells
The experiment was approved by the Institutional Animal Care and Use Committee, Sun Yat-sen University (Approval NO: SYSU-IACUC-2020-B0165). As described in our previous study [Citation23], SD (Sprague Dawley) rats weighing approximately 200–250 g were used. After the operation, rat NP tissues were cut into 1 mm2 pieces and then digested with filtered 0.2% pronase (Sigma-Aldrich, USA) medium and 2.5% collagenase (Sigma-Aldrich, USA) medium at 37°C for 30 min. After digestion, resuspended NP cells were cultured in 20% FBS medium containing 100 U/mL penicillin and 100 U/mL streptomycin at 37°C with 5% CO2. The culture medium was changed every three days.
To explore the effect of atorvastatin (MCE, USA) and MCC950 (MCE, USA) in the presence of TNF-α, atorvastatin or MCC950 was added 1 hour before TNF-α (50 ng/ml) stimulation of rat NP cells.
2.2 Cell counting kit‐8 (CCK-8) assay
The NP cells were seeded into 96-well plates and then incubated at 37°C with 5% CO2. When the density reached approximately 80%, the NP cells were treated with atorvastatin at different concentrations. One hour before detection, 10 μl CCK-8 was added to each well. The optical density (OD) value was measured with a microplate reader at 450 nm. The survival rate of NP cells was calculated according to the Cell Counting Kit‐8 protocol.
2.3 Total RNA isolation and RT-PCR
According to the manufacturer’s instructions, total RNA was isolated from NP cells with TRIzol Reagent (TAKARA, Dalian, China). Real-time polymerase chain reaction (real-time PCR) was performed with cDNA, forward and reverse primers, TB green (TAKARA, Dalian, China) and RNase-free water and run on a Roche Light Cycler 96 (Roche Diagnostic, Mannheim, Germany) following the manufacturer’s instructions. The data was analyzed by using 2−ΔΔCT method.
The primers were constructed by Generay, Shanghai, China, and the sequences were as follows: ADAMTS4, sense 5ʹ- GCATCCGAAACCCTGTCAACT-3ʹ and antisense 5ʹ- CAGCCATACCCAGAGCGTCAC-3ʹ; Cox-2, sense 5ʹ-TCAATGAGTACCGCAAACGC-3ʹ and antisense 5ʹ-TCAATGAGTACCGCAAACGC-5ʹ; NLRP3, sense 5ʹ-AGTGGATAGGTTTGCTGGGATA-3ʹ and antisense 5ʹ-CTGGGTGTAGCGTCTGTTGAG-3ʹ; GSDMD, sense 5ʹ-GGAGGATTTTACAGGACCAGC-3ʹ and antisense 5ʹ- GGAGGATTTTACAGGACCAGC-3ʹ; and β-actin, sense 5ʹ-TCTCTGCTCCTCCCTGTTC-3ʹ and antisense 5ʹ-ACACCGACCTTCACCATCT-3ʹ. The relative mRNA expression of the targets was normalized to the β-actin expression.
2.4 Protein extraction and western blotting
Total proteins were extracted as described previously [Citation24]. Briefly, after cell collection, the NP cells were lysed with RIPA lysis buffer (CWBIO, China) containing phosphatase inhibitor (CWBIO, China) and protease inhibitor (CWBIO, China) on ice for 20 min. The Nuclear and Cytoplasmic Extraction Kit (CWBIO, China) was used to isolate nuclear protein and cytoplasmic protein. After determining the concentration of the total proteins with BCA reagent (CWBIO, China), the proteins were mixed with 5X loading buffer (EpiZyme, Shanghai, China), heated at 98°C for 10 min, separated on sodium dodecyl sulfate-polyacrylamide gels and then transferred to PVDF membranes (Millipore, USA). The membranes were blocked with 5% bovine serum albumin (BSA) in TBST (TBS with 0.1% Tween-20) at room temperature for 1 hour and then incubated with primary antibodies in 5% BSA in TBST at 4°C overnight.
The antibodies were as follows: Cell Signaling Technology: anti-histone H3 (1:1000), anti-MMP3 (1:1000), anti-Cox2 (1:1000), anti-NLRP3 (1:1000), anti-GSDMD-NT (1:1000), anti-LC3B (1:1000), anti-P-P65 (1:1000), and anti-P65 (1:1000); Abcam: anti-MMP13 (1:1000), anti-ADAMTS4 (1:1000), and anti-P62 (1:1000); ABclonal: anti-β-actin (1:1000); and Affinity: anti-cleaved caspase-1/P20 (1:500). After incubation with primary antibodies, the membranes were washed with TBST at room temperature and then incubated with HRP goat anti-rabbit IgG (ABclonal; 1:10000) or HRP goat anti-mouse IgG (ABclonal; 1:10000) at room temperature for 1 hour. The protein bands were detected by a scanning system (Syngene G:BOX ChemiXT4, UK) with enhanced chemiluminescence (ECL) reagent. The relative nuclear protein expression was normalized to that of the internal control histone H3, while the relative cytoplasmic protein and total protein expression was normalized to that of the internal control β-actin.
2.5 Immunofluorescence staining
The NP cells were seeded into a 24-well plate. After treatment for approximately 24 hours, the medium was removed, and then the cells were washed with PBS for three times. After fixation with 4% paraformaldehyde for 30 min and permeabilization with 0.5% Triton-100 at room temperature for 20 min, the NP cells were washed with PBS for three times and blocked with 10% goat serum in PBS at room temperature for 1 hour. Later, the NP cells were incubated with primary antibodies (anti-COL2A, 1:100, Immunoway, Suzhou, China; anti-ACAN, 1:100, Affinity, OH, USA; anti-GSDMD, 1:100, ABclonal, Wuhan, China; anti-p65, Cell Signaling Technology, MA, USA) at 4°C overnight, washed with PBS, and incubated with conjugated AffiniPure goat anti-rabbit IgG (FITC or Cy3, 1:100, BOSTER Biological Technology, Wuhan, China) at room temperature for 1 hour. DAPI was used for nuclear staining at room temperature for 5 min. After the cells were washed with PBS, fluorescence was observed with an inverted fluorescence microscope (Olympus IX73, Japan).
2.6 mRFP-GFP-LC3
The mRFP-GFP-LC3 adenovirus plasmid was constructed by HanBio Technology (Shanghai, China). The NP cells were seeded into a 12-well plate. When the cell density reached almost 50%, the NP cells were transfected with mRFP-GFP-LC3 adenovirus plasmid following the manufacturer’s instructions. After 24 hours of transfection, the NP cells treated with drugs and fluorescence puncta were observed with an inverted fluorescence microscope (Olympus IX73, Japan). Yellow puncta (the combination of red and green fluorescence; mRFP+ and GFP+) indicated autophagosomes and red puncta (the extinction of GFP and the existence of RFP in the acid environment of lysosomes; mRFP+ and GFP−) corresponded to the formation of autolysosomes. At least 10 cells each group were analyzed.
2.7 Transmission electron microscopy (TEM)
After the culture medium was removed, the NP cells were collected into centrifuge tubes, prefixed with 2.5% glutaraldehyde for 2 hours, and then postfixed with 1% osmium tetroxide for 2 hours. Later, the NP cells were dehydrated by using graded ethanol, permeated with acetone resin, embedded in neat resin, and then cut into thin sections by ultramicrotome. Thin sections were stained with 2% uranyl acetate and lead citrate. Sections were observed with Hitachi HT-7800 transmission electron microscopy at 80kV (Hitachi, Tokyo, Japan). At least 6 cells each group were analyzed.
2.8 Knockdown of NLRP3 by shRNA
Lentiviral vectors containing shRNA-NLRP3 and shRNA-control were designed and produced by HanBio Technology Company (Shanghai, China). The sequence of shRNA-NLRP3-1 was 5ʹ-CCCTTAAGCTGGAGCTGCTCTTTGA-3ʹ, the sequence of shRNA-NLRP3-2 was 5ʹ-CAGACCTTCTGAACCGAGACGTGAA-3ʹ, and the sequence of shRNA-control was 5ʹ-TTCTCCGAACGTGTCACGTAA-3ʹ. When the cell density reached 30–50%, the NP cells were transfected with lentiviral vectors containing shRNA-NLRP3 or shRNA-control following the manufacturer’s instructions. After 24 hours of transfection, the medium containing lentivirus vectors was replaced with regular medium containing 10% FBS. Five days later, total protein was extracted from the NP cells. The transfection efficiency was measured via fluorescence intensity (Olympus IX73, Japan) and western blotting.
2.9 Statistical analysis
All data were analyzed from at least three independent experiments and expressed as the mean ± standard deviation and analyzed with GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA). The differences were analyzed by Student’s t test between two groups and by one-way ANOVA among the multiple groups. A p value<0.05 was considered to indicate statistical significance.
3. Results
3.1 Atorvastatin suppressed TNF-α-induced matrix degradation in rat NP cells
To detect the potential toxicity of atorvastatin, CCK-8 assays were performed with different concentrations of atorvastatin for 24, 48 and 72 hours. The results showed that atorvastatin significantly alleviated the proliferation of rat NP cells in a time- and concentration-dependent manner; the survival rates of cells treated with atorvastatin at 10 μM and 20 μM were above 80%. Therefore, 10 μM and 20 μM atorvastatin were used in the subsequent experiments ()).
Figure 1. Atorvastatin suppressed TNF-α-induced matrix degradation in rat NP cells. (a) The CCK-8 assay was used to detect the toxicity of atorvastatin at various concentrations and for different time points in rat NP cells. (b) Real-time PCR was used to test the effect of 20 μM atorvastatin on catabolic gene mRNA expression induced by 50 ng/mL TNF-α. (c) Western blotting analysis showed that 20 μM atorvastatin alleviated the increase in the protein expression of catabolic genes induced by TNF-α. (d, e) Immunofluorescence assays were used to observe the synthesis of collagen type II and aggrecan after 20 μM atorvastatin treatment in the presence of TNF-α (magnification: ×200, scale bar = 50 μm). The data are expressed as the mean ± standard deviation and were analyzed from three independent experiments. *p < 0.05
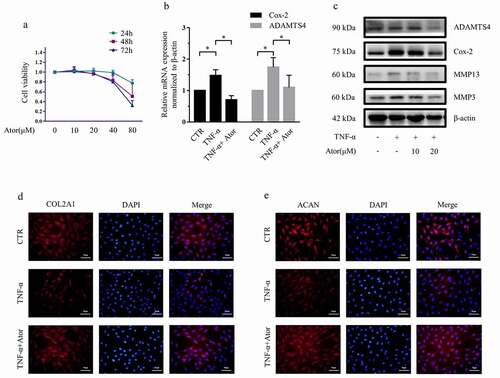
As reported previously [Citation13,Citation22], our results also revealed that TNF-α (50 ng/ml) increased the expression of matrix catabolic enzymes, including MMP3, MMP13, ADAMTS4 and Cox-2, and suppressed the expression of collagen II and aggrecan protein. However, 20 μM atorvastatin significantly attenuated the increase in ADAMTS4 and Cox-2 mRNA expression induced by TNF-α ()). Likewise, western blotting analysis indicated that 20 μM atorvastatin significantly alleviated the promotion of MMP3, MMP13, ADAMTS4 and Cox-2 protein expression ()). In addition, immunofluorescence staining demonstrated that the synthesis of collagen type II and aggrecan was reduced by TNF-α, whereas atorvastatin reversed the decreases in collagen type II and aggrecan protein expression induced by TNF-α ()).
3.2. Atorvastatin suppressed TNF-α-induced NLRP3 inflammasome activity in rat NP cells
In the process of IDD, the expression of NLRP3 is increased, and TNF-α induces NLRP3 inflammasome activity in NP cells [Citation10,Citation13]. To validate the role of atorvastatin in NLRP3 inflammasome activity, 20 μM atorvastatin was added 1 hour before TNF-α stimulation of rat NP cells. As shown in ), the mRNA expression of NLRP3 and GSDMD was increased by TNF-α stimulation, as reported by Huang et al [Citation14], while atorvastatin reduced the expression of NLRP3 and GSDMD; that is, atorvastatin significantly suppressed the effect of TNF-α.
Figure 2. Atorvastatin suppressed TNF-α-induced NLRP3 inflammasome activity in rat NP cells. (a) Real-time PCR was used to confirm that atorvastatin attenuated the TNF-α-induced mRNA expression of NLRP3 inflammasome-related genes. (b, c) Western blotting (b) and subsequent densitometric analyses (c) suggested that atorvastatin significantly suppressed TNF-α-induced NLRP3 inflammasome activity. (d) Immunofluorescence assays were used to observe whether TNF-α enhanced the fluorescence intensity of GSDMD, and the effect was weakened by atorvastatin (magnification: ×200, scale bar = 50 μm). The data are presented as the mean ± standard deviation and were analyzed from three independent experiments. *p < 0.05
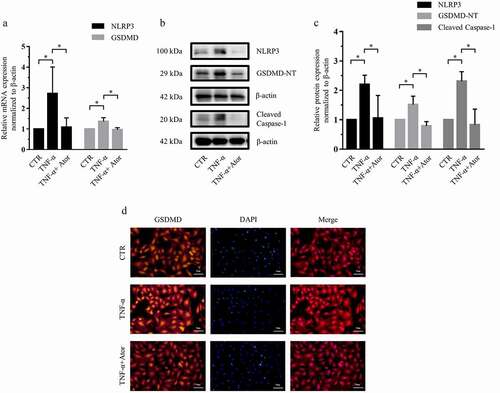
NLRP3 inflammasome activity depends on caspase-1 protein activation and GSDMD-NT formation [Citation25], so we measured the protein expression of NLRP3, cleaved caspase-1 and GSDMD-NT. Western blotting and subsequent densitometric analyses revealed that TNF-α also significantly increased NLRP3, cleaved caspase-1 and GSDMD-NT expression. However, atorvastatin significantly suppressed TNF-α-induced NLRP3, cleaved caspase-1 and GSDMD-NT protein expression by 52%, 65%, and 48%, respectively (). In addition, immunofluorescence staining further demonstrated that TNF-α-induced expression of the GSDMD protein was inhibited by atorvastatin ).
3.3 Atorvastatin promoted autophagic flux in the presence of TNF-α in rat NP cells
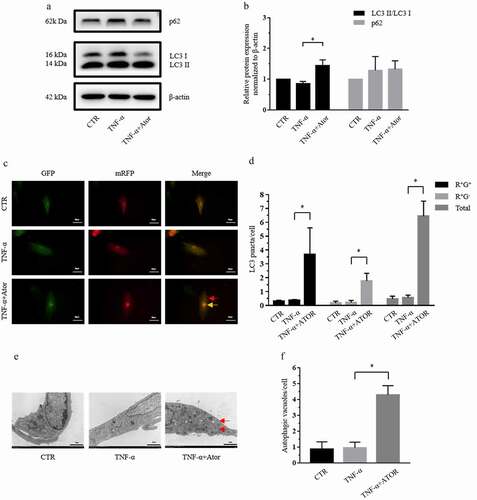
Autophagy is observed in rat NP cells and plays a protective role in the pathological process of IDD [Citation15,Citation26]. To determine the effect of atorvastatin on autophagy in the presence of TNF-α, the protein expression of LC3 II, LC3 I and P62 was measured by western blotting. Western blotting and densitometric analyses demonstrated that although TNF-α had no effect on the expression of LC3 and p62, as reported by Xu et al [Citation22], 20 μM atorvastatin significantly increased the LC3 II/LC3 I ratio in the presence of TNF-α (). The mRFP-GFP-LC3 adenovirus plasmid is commonly used to detect autophagic flux by observing fluorescence puncta, which indicate the formation of autophagy. shows that atorvastatin enhanced the formation of yellow puncta and red puncta, which promoted autophagosomes and autolysosomes in the presence of TNF-α. TEM is the gold standard technique for detecting autophagic flux. TEM demonstrated that atorvastatin increased the expression of autophagic vacuoles in NP cells in the presence of TNF-α ().
Figure 3. Atorvastatin promoted autophagy in the presence of TNF-α in rat NP cells. (a, b) Western blotting (a) and subsequent densitometric analyses (b) demonstrated that atorvastatin promoted autophagy in the presence of TNF-α. (c) Transfection of rat NP cells with the mRFP-GFP-LC3 adenovirus plasmid clarified the effect of atorvastatin on the formation of autophagosomes under TNF-α stimulation (yellow arrows indicate autophagosomes and red arrows indicated autolysosomes; magnification: ×500, scale bar = 20 μm). (d) Mean number of LC3 puncta of each cell was analyzed and plotted. (e) TEM showed that the production of autophagic vacuoles was obviously visualized in the atorvastatin treatment groups (magnification: ×5000, scale bar = 2 μm). (f) Mean number of autophagic vacuoles of each cell was analyzed and plotted. The data are presented as the mean ± standard deviation and were analyzed from at least three independent experiments. *p < 0.05
3.4 NLRP3 inhibition induced autophagic flux in rat NP cells
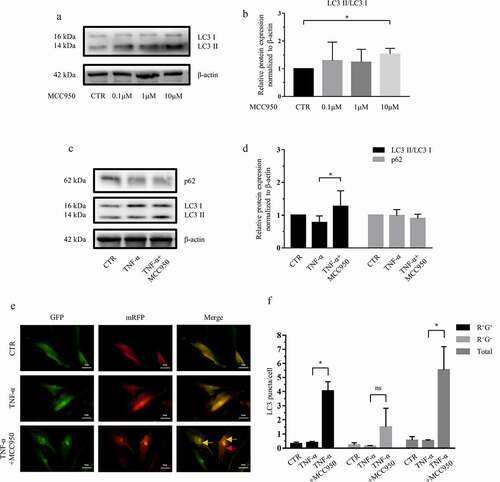
Since atorvastatin suppressed NLRP3 inflammasome activity and promoted autophagic flux, we wondered whether atorvastatin controls autophagic flux by regulating NLRP3 inflammasome activity in NP cells. To validate the effect of NLRP3 on autophagic flux, MCC950, a potent and selective NLRP3 inhibitor, was used, western blotting and subsequent densitometric analyses showed that 10 μM, not 0.1 μM or 1 μM, MCC950 significantly increased the ratio of LC3 II/LC3 I compared to that in control cells (). Moreover, in the presence of TNF-α, 10 μM MCC950 markedly increased the LC3 II/LC3 I ratio compared to that in the control cells but had no effect on the expression of p62 (). In addition, compared with the TNF-α-stimulated groups, the groups treated with 10 μM MCC950 and TNF-α exhibited more autophagic fluorescence puncta ()).
Figure 4. NLRP3 inhibition induced autophagy in rat NP cells. (a, b) Western blotting (a) and subsequent densitometric analyses (b) revealed the effect of various concentrations of the NLRP3 inhibitor MCC950 on autophagy in rat NP cells. (c, d) Western blotting (c) and subsequent densitometric analyses (d) demonstrated that MCC950 treatment enhanced autophagy in the presence of TNF-α. (e) Transfection with the mRFP-GFP-LC3 adenovirus plasmid further confirmed that MCC950 elevated the production of autophagosomes in the presence of TNF-α (yellow arrows indicate autophagosomes and red arrows indicated autolysosomes; magnification: ×500, scale bar = 20 μm). (f) Mean number of LC3 puncta of each cell was analyzed and plotted. The data are presented as the mean ± standard deviation and were analyzed from three independent experiments. *p < 0.05
3.5 NLRP3 knockdown promoted autophagy in rat NP cells
To further verify the effect of NLRP3 inhibition, NLRP3 knockdown was performed by using lentiviral vectors containing shRNA-NLRP3. As demonstrate, although both vectors were effectively transfected, knockdown was more effective with sequence-1 (expression significantly decreased by approximately 60%), and shNLRP3-1 was used for the subsequent experiments. To further test the effect of shRNA-NLRP3 on matrix metabolism, as reported by Zhao et al [Citation12], the expression of catabolic genes, such as Cox-2, MMP3 and MMP13, was detected, and western blotting and subsequent densitometric analysis also demonstrated that NLRP3 knockdown markedly suppressed the protein expression of Cox-2, MMP3 and MMP13 (by 67%, 65% and 78%, respectively) in the presence of TNF-α ()).
Figure 5. NLRP3 knockdown promoted autophagy in rat NP cells. (a) Rat NP cells were transfected with lentiviral vectors containing shRNA-NLRP3 and shRNA-negative control, and the transfection efficiency was evaluated via fluorescence intensity; magnification: ×100, scale bar = 100 μm. (b, c) Western blotting (b) and subsequent densitometric analyses (c) verified which shRNA-NLRP3 sequence was effective in the presence of TNF-α. (d, e) Western blotting (d) and subsequent densitometric analyses (e) showed that NLRP3 knockdown dramatically suppressed the expression of catabolic genes in the presence of TNF-α in rat NP cells. (f, g) Western blotting (f) and subsequent densitometric analyses (g) demonstrated the role of NLRP3 knockdown on the LC3 II/LC3 I ratio and P62 expression level under TNF-α stimulation in rat NP cells. The data are presented as the mean ± standard deviation and were analyzed from three independent experiments. *p < 0.05
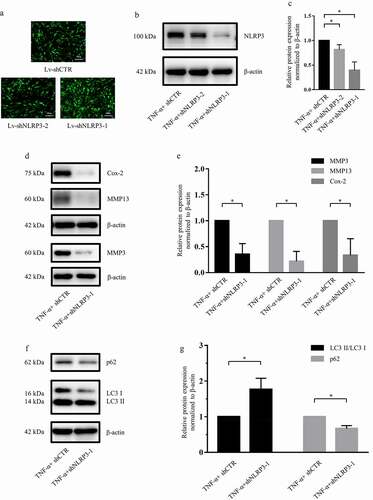
In addition, similar to the NLRP3 inhibition studies, western blotting and subsequent densitometric analyses showed that NLRP3 knockdown significantly downregulated the expression of P62 (to 67%) and markedly increased the LC3 II/LC3 I ratio (to 177%) in the presence of TNF-α ()).
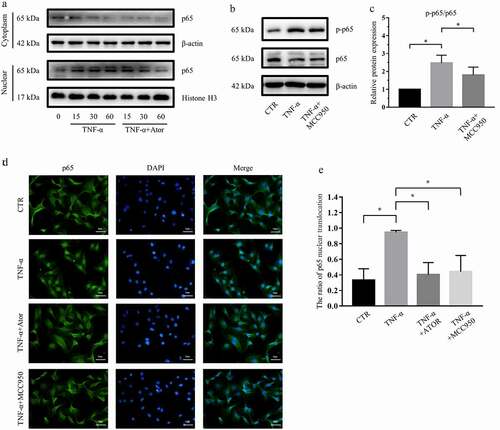
3.6 Atorvastatin and NLRP3 inhibition suppressed the activity of NF-κB signaling in rat NP cells
NF-κB signaling plays a critical role in inflammation, autophagy and IDD [Citation27]. To explore the effect of NF-κB signaling in the process of atorvastatin modulation on the NLRP3 inflammasome and autophagy, we detected p65 protein expression after atorvastatin and MCC950 stimulation via western blotting. ) demonstrates the expression of p65 protein in the nucleus and cytoplasm of NP cells and reveals that TNF-α increased the nuclear translocation of p65 and that atorvastatin reversed the effect of TNF-α. That is, atorvastatin suppressed the nuclear translocation of p65 induced by TNF-α. In addition, western blotting and subsequent densitometric analyses showed that TNF-α markedly increased the p65 phosphorylation/p65 ratio, as reported by Chen et al [Citation28], while MCC950 significantly attenuated the increase in the p65 phosphorylation/p65 ratio induced by TNF-α (). Moreover, immunofluorescence staining revealed that TNF-α promoted the nuclear translocation of p65, but this effect was restrained by atorvastatin or MCC950 ()).
Figure 6. Atorvastatin and NLRP3 inhibition suppressed the activity of NF-κB signaling in rat NP cells. (a) Western blotting analysis revealed that atorvastatin inhibited the nuclear translocation of p65 induced by TNF-α. (b, c) Western blotting (b) and subsequent densitometric analyses (c) suggested that NLRP3 inhibition by MCC950 suppressed TNF-α-mediated p65 phosphorylation and p-p65/p65 ratios. (d) Immunofluorescence staining showed that the nuclear translocation of p65 induced by TNF-α was repressed by atorvastatin and MCC950 in rat NP cells (magnification: ×500, scale bar = 20 μm). (e) The ratio of cells with p65 located in the nucleus was analyzed and plotted. The data are presented as the mean ± standard deviation and were analyzed from three independent experiments. *p < 0.05
4. Discussion
In the present study, for the first time, we found that atorvastatin could suppress matrix degradation induced by TNF-α in rat NP cells. Moreover, atorvastatin decreased NLRP3 inflammasome activity and induced autophagy, and NLRP3 inhibition increased autophagic flux. In addition, we found that NF-κB signaling plays a pivotal role in NLRP3 inflammasome activity and autophagy controlled by atorvastatin and autophagic flux modulated by NLRP3 in rat NP cells.
A healthy intervertebral disc requires the dynamic balance of anabolic and catabolic factors, and IDD is caused by a reduction in the synthesis of aggrecan and collagen type II and an increase in catabolic factors [Citation29]. TNF-α and IL-1β are important catabolic factors in IDD that upregulate the expression of matrix catabolic enzymes such as MMP3, MMP13, ADAMTS4, and ADAMTS5 [Citation30–32]. Atorvastatin, an HMG-CoA reductase inhibitor, not only has advantages in cardiovascular disease treatment but also has an anti-inflammatory effect in inflammation-related diseases, such as osteoarthritis [Citation33]. In rat cartilage explants, atorvastatin reduced IL-1β-mediated TNF-α and MMP13 expression, and in chondrocytes, atorvastatin decreased MMP13 mRNA expression and increased COL2A and aggrecan expression [Citation9,Citation34]. In vivo, atorvastatin can postpone the development of osteoarthritis induced by hypercholesterolemia [Citation35]. Herein, our results showed that atorvastatin inhibited TNF-α-induced matrix degradation in NP cells. This suggests that atorvastatin plays a protective role in rat NP cells.
The inflammasome is a polymeric protein complex that activates pyroptosis, a programmed cell death method that differs from apoptosis and necroptosis, by binding to caspase-1 [Citation36]. It was reported that the expression of NLRP3, caspase-1 and IL-1β are increased in degenerative human IVDs and that their expression is associated with the grade of disc degeneration [Citation10,Citation11]. Our previous study also revealed that the NLRP3 inflammasome plays a critical role in the effect of BRD4 and NAMPT in rat NP cells [Citation13,Citation14]. Zhao et al [Citation12] reported that NLRP3 inhibition and knockdown relieve the downregulation of ECM components and the upregulation of matrix-degrading enzymes induced by lactate. Herein, we also demonstrated that knockdown of NLRP3 decreased the expression of matrix catabolic enzymes in the presence of TNF-α. In addition, in vitro and clinical studies have revealed that atorvastatin can be used to treat cardiovascular disease via inhibition of the NLRP3 inflammasome [Citation18,Citation19]. In the current study, we also explored the anti-inflammatory effect of atorvastatin in rat NP cells. Overall, atorvastatin exerts an anti-inflammatory effect that is dependent on NLRP3 inflammasome activity alleviation in rat NP cells.
Autophagy plays an important role in many kinds of diseases and is relevant to the pathological process of IDD, and autophagy stimulation can reduce its catabolic effect in rat NP cells [Citation15,Citation22,Citation37]. In cardiovascular disease, atorvastatin plays a protective role by activating autophagy [Citation20,Citation38]. Thus, we wondered whether atorvastatin has a similar effect in IDD. Our results suggested that autophagy-related protein LC3 II/LC3 I ratios were upregulated by atorvastatin in the presence of TNF-α. Furthermore, the imaging data showed that atorvastatin promoted the formation of autophagosomes and autolysosomes under TNF-α stimulation. This suggests that atorvastatin plays a protective role by enhancing autophagic flux in rat NP cells.
In addition, it was reported that there is crosstalk between autophagy and the NLRP3 inflammasome [Citation39]. Autophagy can negatively regulate the NLRP3 inflammasome, and in turn, quite a few studies have demonstrated that suppressing the NLRP3 inflammasome can control autophagy. Under pathological conditions, combined treatment with MCC950 and metformin has a protective role in dextran sodium sulfate (DSS)-mediated colitis by inducing autophagy [Citation40]. Herein, we revealed that NLRP3 inhibition by MCC950 and NLRP3 knockdown promoted autophagy in the presence or absence of TNF-α stimuli. This further verified the mechanism by which MCC950 or advanced glycation end products enhance ECM synthesis by controlling NLRP3 inflammasome activity in rat IDD models or human NP cells [Citation11,Citation41].
Activation of autophagy by rapamycin, a well-known inducer that increases autophagy activity, can restrain the formation of NLRP3 inflammasome, while autophagy inhibition with chloroquine, an autophagic flux inhibitor, can exacerbate the production of the NLRP3 inflammasome in the liver [Citation42,Citation43]. Our previous data also proved that autophagy inhibition with bafilomycin A1 can increase NLRP3 inflammasome formation in the presence of TNF-α and JQ1 in rat NP cells [Citation13]. Therefore, we concluded that atorvastatin may control matrix metabolism through the NLRP3 inflammasome and autophagy loop in NP cells.
The NF-κB signaling pathway contributes to the pathogenesis of IDD [Citation21,Citation44]. In cardiovascular disease and cardiomyopathy, atorvastatin exerts an anti-inflammasome effect via the inhibition of the NF-κB signaling pathway [Citation19,Citation45]. In C28I2 chondrocytes, atorvastatin reduces the nuclear translocation of NF-κB [Citation40]. Similarly, herein, we revealed that atorvastatin could alleviate the nuclear translocation of P65 induced by TNF-α in rat NP cells. It was reported that NF-κB inhibition could induce autophagy in NP cells [Citation13]. Therefore, in the presence of TNF-α, atorvastatin induces autophagic flux by suppressing NF-κB signaling.
In addition, our previous data confirmed that inhibition and silencing of NF-κB can suppress NLRP3 inflammasome activity [Citation13,Citation14]. Moreover, several studies have demonstrated that in other kinds of cells, such as Kupffer cells, NLRP3 inflammasome activity regulates the NF-κB signaling pathway, and the effect can be alleviated by MCC950 via blockage of the NF-κB signaling pathway [Citation46,Citation47]. Likewise, the present data revealed that NLRP3 inhibition by MCC950 reduced the phosphorylation of p65 and delayed the nuclear translocation of p65 induced by TNF-α. Thus, these data show that atorvastatin can directly regulate TNF-α-mediated NF-κB activity and indirectly modulate NF-κB signaling by controlling NLRP3 inflammasome activity, which further illuminates the interplay between the NLRP3 inflammasome and the NF-κB signaling pathway in rat NP cells.
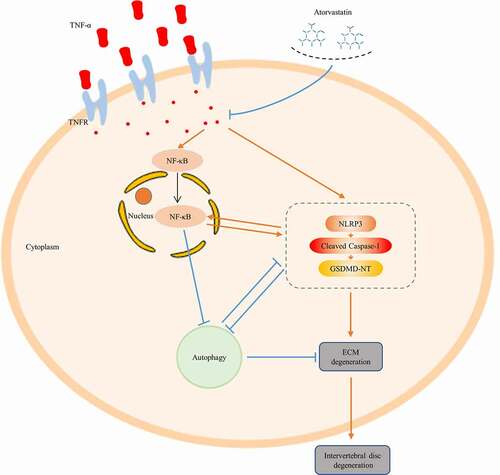
In summary, our research puts forward a new standpoint regarding the potential treatment of IDD, as we found that atorvastatin might control ECM synthesis by alleviating NLRP3 inflammasome activity and promoting autophagy in the presence of TNF-α. The findings of this and our previous study also further clarify the crosstalk among the NLRP3 inflammasome, autophagy and the NF-κB signaling pathway in rat NP cells (). Nevertheless, the effect of atorvastatin on IDD needs to be tested in rat models.
Figure 7. Schematic graph. Atorvastatin inhibits matrix degradation induced by TNF-α, and atorvastatin suppresses NF-κB signaling, while NF-κB signaling inhibition suppresses NLRP3 inflammasome activity and induces autophagy in rat NP cells. NLRP3 inhibition suppresses TNF-α-induced NF-κB signaling activation and induces autophagy. The present study further demonstrates the crosstalk among NLRP3 inflammasome activity, autophagy and NF-κB signaling
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81572197, 81702225 and 82002776); the Natural Science Foundation of Guangdong Province, China (2018A0303130349, 2020A151511538 and 2020A1515010060); the Guangdong Basic and Applied Basic Research Foundation (2021A1515010345); the Medical Scientific Research Foundation of Guangdong Province, China (B2019094); and the Foundation of the Guangzhou Health Bureau (20181A011092).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hartvigsen J, Hancock MJ, Kongsted A, et al. What low back pain is and why we need to pay attention. Lancet. 2018;391(10137):2356–2367.
- Frymoyer JW, Cats-Baril WL. An overview of the incidences and costs of low back pain. Orthop Clin North Am. 1991;22(2):263–271.
- Vlaeyen J, Maher C, Wiech K, et al. Low back pain. Nat Rev Dis Primers. 2018;4:52.
- Livshits G, Popham M, Malkin I, et al. Lumbar disc degeneration and genetic factors are the main risk factors for low back pain in women: the UK Twin Spine Study. Ann Rheum Dis. 2011;70(10):1740–1745.
- Cheung KM. The relationship between disc degeneration, low back pain, and human pain genetics. Spine J. 2010;10(11):958–960.
- Risbud M, Shapiro I. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol. 2014;10(1):44–56.
- Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. 2017;120(1):229–243.
- Arca M. Atorvastatin efficacy in the prevention of cardiovascular events in patients with diabetes mellitus and/or metabolic syndrome. Drugs. 2007;67(1):43–54.
- Pathak N, Lingaraju M, Balaganur V, et al. Anti-inflammatory and chondroprotective effects of atorvastatin in a cartilage explant model of osteoarthritis. Inflamm Res. 2015;64(3–4):161–169.
- Chen Z, Jin S, Wang M, et al. Enhanced NLRP3, caspase-1, and IL- 1β levels in degenerate human intervertebral disc and their association with the grades of disc degeneration. Anat Rec (Hoboken). 2015;298(4):720–726.
- Song Y, Wang Y, Zhang Y, et al. Advanced glycation end products regulate anabolic and catabolic activities via NLRP3-inflammasome activation in human nucleus pulposus cells. J Cell Mol Med. 2017;21(7):1373–1387.
- Zhao K, An R, Xiang Q, et al. Acid-sensing ion channels regulate nucleus pulposus cell inflammation and pyroptosis via the NLRP3 inflammasome in intervertebral disc degeneration. Cell Prolif. 2021;54(1):e12941.
- Hong J, Li S, Markova D, et al. Bromodomain-containing protein 4 inhibition alleviates matrix degradation by enhancing autophagy and suppressing NLRP3 inflammasome activity in NP cells. J Cell Physiol. 2020;235(7–8):5736–5749.
- Huang Y, Peng Y, Sun J, et al. Nicotinamide phosphoribosyl transferase controls NLRP3 inflammasome activity through MAPK and NF-κB signaling in nucleus pulposus cells, as suppressed by melatonin. Inflammation. 2020;43(3):796–809.
- Ye W, Xu K, Huang D, et al. Age-related increases of macroautophagy and chaperone-mediated autophagy in rat nucleus pulposus. Connect Tissue Res. 2011;52(6):472–478.
- Lan T, Shiyu H, Shen Z, et al. New insights into the interplay between miRNAs and autophagy in the aging of intervertebral discs. Ageing Res Rev. 2021;65:101227.
- Ye W, Zhu W, Xu K, et al. Increased macroautophagy in the pathological process of intervertebral disc degeneration in rats. Connect Tissue Res. 2013;54(1):22–28.
- Satoh M, Tabuchi T, Itoh T, et al. NLRP3 inflammasome activation in coronary artery disease: results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin Sci (Lond). 2014;126(3):233–241.
- Kong F, Ye B, Lin L, et al. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF-κB signaling in PMA-stimulated THP-1 monocytes. Biomed Pharmacother. 2016;82:167–172.
- Liu D, Cui W, Liu B, et al. Atorvastatin protects vascular smooth muscle cells from TGF-β1-stimulated calcification by inducing autophagy via suppression of the β-catenin pathway. Cell Physiol Biochem. 2014;33(1):129–141.
- Sun Z, Zhao S, Liu C, et al. Effects of nuclear factor kappa B signaling pathway in human intervertebral disc degeneration. Spine (Phila Pa 1976). 2015;40(4):224–232.
- Xu K, Chen W, Wang X, et al. Autophagy attenuates the catabolic effect during inflammatory conditions in nucleus pulposus cells, as sustained by NF-κB and JNK inhibition. Int J Mol Med. 2015;36(3):661–668.
- Sun J, Hong J, Sun S, et al. Transcription factor 7-like 2 controls matrix degradation through nuclear factor κB signaling and is repressed by microRNA-155 in nucleus pulposus cells. Biomed Pharmacother. 2018;108:646–655.
- Zhou J, Sun J, Markova D, et al. MicroRNA-145 overexpression attenuates apoptosis and increases matrix synthesis in nucleus pulposus cells. Life Sci. 2019;22:274–283.
- Swanson K, Deng M, Ting J. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–489.
- Zhang S, Yang W, Wang C, et al. Autophagy: a double-edged sword in intervertebral disk degeneration. Clin Chim Acta. 2016;457:27–35.
- Yi W, Wen Y, Tan F, et al. Impact of NF-κB pathway on the apoptosis-inflammation-autophagy crosstalk in human degenerative nucleus pulposus cells. Aging (Albany NY). 2019;11:7294–7306.
- Chen L, Xie Z, Liu L, et al. Nuclear factor-kappa B-dependent X-box binding protein 1 signalling promotes the proliferation of nucleus pulposus cells under tumour necrosis factor alpha stimulation. Cell Prolif. 2019;52:e12542.
- Kadow T, Sowa G, Vo N, et al. Molecular basis of intervertebral disc degeneration and herniations: what are the important translational questions. Clin Orthop Relat Res. 2015;473(6):1903–1912.
- Wang Y, Che M, Xin J, et al. The role of IL-1β and TNF-α in intervertebral disc degeneration. Biomed Pharmacother. 2020;131:110660.
- Wang J, Huang C, Lin Z, et al. Polydatin suppresses nucleus pulposus cell senescence, promotes matrix homeostasis and attenuates intervertebral disc degeneration in rats. J Cell Mol Med. 2018;22:5720–5731.
- Chen J, Xuan J, Gu Y, et al. Celastrol reduces IL-1β induced matrix catabolism, oxidative stress and inflammation in human nucleus pulposus cells and attenuates rat intervertebral disc degeneration in vivo. Biomed Pharmacother. 2017;91:208–219.
- Hosseinzadeh A, Bahrampour Juybari K, Kamarul T, et al. Protective effects of atorvastatin on high glucose-induced oxidative stress and mitochondrial apoptotic signaling pathways in cultured chondrocytes. J Physiol Biochem. 2019;75(2):153–162.
- Simopoulou T, Malizos KN, Poultsides L, et al. Protective effect of atorvastatin in cultured osteoarthritic chondrocytes. J Orthop Res. 2010;28(1):110–115.
- Gierman L, Kühnast S, Koudijs A, et al. Osteoarthritis development is induced by increased dietary cholesterol and can be inhibited by atorvastatin in APOE*3Leiden.CETP mice–a translational model for atherosclerosis. Ann Rheum Dis. 2014;73(5):921–927.
- Xue Y, Enosi Tuipulotu D, Tan W, et al. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019;40(11):1035–1052.
- Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383:1564–1576.
- Peng S, Xu L, Che X, et al. Atorvastatin inhibits inflammatory response, attenuates lipid deposition, and improves the stability of vulnerable atherosclerotic plaques by modulating autophagy. Front Pharmacol. 2018;9:438.
- Cao Z, Wang Y, Long Z, et al. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim Biophys Sin. 2019;51:1087–1095.
- Saber S, El-Kade E. Novel complementary coloprotective effects of metformin and MCC950 by modulating HSP90/NLRP3 interaction and inducing autophagy in rats. Inflammopharmacology. 2020. DOI:https://doi.org/10.1007/s10787-020-00730-6
- He D, Zhou M, Bai Z, et al. Propionibacterium acnes induces intervertebral disc degeneration by promoting nucleus pulposus cell pyroptosis via NLRP3-dependent pathway. Biochem Biophys Res Commun. 2020;526(3):772–779.
- Shan S, Shen Z, Zhang C, et al. Mitophagy protects against acetaminophen-induced acute liver injury in mice through inhibiting NLRP3 inflammasome activation. Biochem Pharmacol. 2019;169:113643.
- Weng Z, Xu C, Zhang X, et al. Autophagy mediates perfluorooctanoic acid-induced lipid metabolism disorder and NLRP3 inflammasome activation in hepatocytes. Environ Pollut. 2020;267:115655.
- Nasto L, Seo H, Robinson A, et al. ISSLS prize winner: inhibition of NF-κB activity ameliorates age-associated disc degeneration in a mouse model of accelerated aging. Spine (Phila Pa 1976). 2012;37:1819–1825.
- Ren X, Zuo G, Wu W, et al. Atorvastatin alleviates experimental diabetic cardiomyopathy by regulating the GSK-3β-PP2Ac-NF-κB signaling axis. PLoS One. 2016;11(11):e0166740.
- Dolunay A, Senol SP, Temiz-Resitoglu M, et al. Inhibition of NLRP3 inflammasome prevents LPS-induced inflammatory hyperalgesia in mice: contribution of NF-κB, caspase-1/11, ASC, NOX, and NOS Isoforms. Inflammation. 2017;40(2):366–386.
- Zhang W, Fang Z, Liu W. NLRP3 inflammasome activation from Kupffer cells is involved in liver fibrosis of Schistosoma japonicum-infected mice via NF-κB. Parasit Vectors. 2019;12(1):29.