Abstract
Increased bronchial epithelial cell apoptosis and CD8+ T-cell numbers in the blood and airways have been reported in COPD. These cells can induce apoptosis via the granzyme-b/perforin-mediated pathway. We hypothesized that increased levels of granzyme-b/perforin would be detected in COPD, contributing to apoptosis and tissue damage. Intracellular granzyme-b/perforin were measured in blood-derived T-cells and natural killer (NK) cells from COPD subjects (30 current and 30 ex-smokers), 20 asymptomatic current-smokers and 30 never-smokers, and bronchoalveolar lavage (BAL)-derived T-cells from a cohort of these subjects using flow cytometry. Soluble granzyme-b was determined by ELISA. In blood, there was an increased percentage of T-cells expressing intracellular granzyme-b/perforin for both COPD groups but not asymptomatic smokers (versus never-smokers). Soluble granzyme-b was undetectable. In BAL, soluble granzyme-b levels and the percentage of T-cells expressing intracellular granzyme-b/perforin were increased in both COPD groups and asymptomatic smokers. There was a significant correlation between granzyme-b expression in BAL and apoptosis of bronchial epithelial cells. Most circulating NK cells expressed granzyme-b/perforin, with the median fluorescence intensity of staining increased in both COPD groups and asymptomatic smokers. Granzyme-mediated apoptosis may thus be one mechanism of lung injury in COPD. The changes that persist despite smoking cessation in COPD likely reflect pathophysiological changes in COPD as opposed to the effects of smoking per se.
BACKGROUND
Chronic obstructive pulmonary disease (COPD) arises as a result of noxious injury to the lungs, most commonly due to cigarette smoking. Currently available pharmacological therapies provide little impact on the long-term outlook for patients with COPD, and new therapeutic options are needed, based on a greater understanding of the pathogenesis of COPD, especially as distinct from the effects of smoking per se.
Increasing evidence implicates perturbations in programmed cell death (apoptosis) in COPD. In this regard, we and others have previously shown increased apoptosis of airway epithelial cells in this disease (Citation[1], Citation[2], Citation[3]). Apoptosis is an active biochemical process that progresses through ordered stages. In early apoptosis, cells lose water and shrink, with subsequent changes in mitochondrial membrane potential. Membrane alteration occurs in the next stage, with translocation of phosphatidylserine (PTS) to the outer membrane of the cell. Activation of cysteine proteases (caspases) leads to a proteolytic cascade and protein cleavage at specific aspartic acid residues. Final stages of apoptosis are characterized by dismantling of the nuclear membrane, loss of membrane pump function and fragmentation of DNA (Citation[4]). Apoptosis is associated with minimal inflammation or disruption of neighboring tissue thus efficient apoptosis and phagocytic removal of apoptotic bodies is considered to play an important role in effective repair of the injured epithelium and resolution of inflammation in the lung. However, accumulation of apoptotic material, either as a result of increased rates of apoptosis or reduced clearance of apoptotic material, may result in secondary necrosis, with the potential for inflammation of surrounding tissues (Citation[5], Citation[6]).
Several reports have shown that COPD is associated with increased numbers of cytotoxic CD8+ T-cells (Citation[7], Citation[8]), although the role of these cells in the pathogenesis of COPD is poorly understood. Cytotoxic T-cells and natural killer (NK) cells can induce apoptosis of target cells including bronchial epithelial cells via multiple mechanisms, including the granzyme-mediated pathway. Granzyme-b and perforin are stored in cytoplasmic secretory granules of these cytotoxic cells, and released into the intercellular space following adhesion to the target cell. In the presence of Ca2 +, perforin forms transmembrane pores in the target cell, facilitating the entry of granzyme-b and induction of apoptosis by activation of caspases (Citation[9]). Granzymes may be released extracellularly, particularly during cytotoxic cell degranulation (Citation[10]). It is therefore possible that levels of exogenous granzyme-b, as well as intracellular levels, may reflect cytotoxic T-cell or NK cell activity in the airways in COPD. In addition, extracellular granzyme-b has been shown to contribute to tissue destruction by degrading various extracellular proteins (Citation[11], Citation[12]) and may thus play a direct role in the disease process in COPD.
Cigarette smoking directly induces apoptosis of airway epithelial cells (Citation[13]). However, we previously demonstrated that the increased rate of apoptosis in the airways in COPD does not diminish with cessation of cigarette smoking (Citation[1]). Thus other factors that relate to perpetuation of the chronic inflammatory response must also contribute to increased apoptosis in the airways in COPD.
We thus hypothesized that increased granzyme-b/perforin would be detected in the airways in COPD, contributing to increased apoptosis of bronchial epithelial cells and associated tissue damage. In the current study, we assessed soluble and intracellular granzyme-b and perforin in BAL from well-characterized current and ex-smoking COPD subjects, asymptomatic smokers and never-smokers.
Although the link between T-cells in the peripheral blood and airways is still a controversial area, our previous investigations have shown that several of the changes noted in the airways in COPD are reflected in the peripheral blood (Citation[14]), supporting the view that COPD is a systemic disease. Based on these findings, we also investigated granzyme-b and perforin in peripheral blood-derived CD8 T-cells and CD56+ NK cells.
METHODS
Subject population
COPD volunteers were specifically recruited for the study and informed consent obtained. There was no exacerbation of COPD for 6 weeks prior to involvement in the study. Exclusion criteria included very severe COPD disease (FEV1 < 1.4L) other co-existing lung disease and age greater than 75 years. Ethics approval was obtained from the Royal Adelaide Hospital. The diagnosis of COPD was established using the GOLD criteria with clinical correlation (Mild COPD: FEV1/FVC < 70% but FEV1 ≥ 80% predicted; Moderate COPD FEV1 50% ≤ 80% predicted, Severe COPD FEV1 < 30% predicted) (Citation[15]). 30 of the COPD subjects were ex-smokers (at least 1 year) and 30 were current smokers. A further 20 current smokers of at least 10 pack-years with no evidence of COPD or other lung disease were also recruited (hereinafter referred to as “asymptomatic smokers”). Specimens were also obtained from 30 never-smokers (). These were healthy, recruited volunteers with no history of airways disease. Patients underwent spirometry and chest X-ray as part of their routine clinical assessment.
Table 1 Demographic characteristics of the population studied and BAL leucocyte counts
Bronchoscopy procedure
BAL was obtained via flexible bronchoscopy, as we have previously described (Citation[16]). Briefly, a 50 ml aliquot of sterile normal saline (at room temperature) was instilled into the airway with a syringe then aspirated using low suction. To obtain sufficient T-cells for analysis, 2 further aliquots of saline were instilled and aspirated in the same way. The first aliquot was processed for microbiological testing. For each collection from an individual patient the aspirated BAL specimens 2 and 3 were pooled, kept on ice and processed within 1 hour of collection (the first aliquot was not included due to airway mucus contamination). For a cohort of subjects, bronchial brushings were obtained by positioning the tip of the bronchoscope in a subsegmental airway, then gently advancing a standard cytology brush (Fuginon Inc., Wayne, NJ, USA) into 4 to 6 medium-sized airways. Airway epithelial cells (AEC) were obtained with several gentle passages of the brush into each airway so as to avoid bleeding. Cells were deposited by washing the brush in 10 ml RPMI 1640 media (Gibco, BRL, Germany), in a 10 ml conical polypropylene tubes (Johns Professional Products, Sydney, Australia), and kept on ice. Cells were processed within 1h of collection. Particular attention was given to use a minimal amount of Xylocaine (100 mg) in the airways in view of the adverse effect it has on epithelial cell viability.
Preparation of ex vivo samples
BAL cells were washed in RPMI 1640 (Gibco, BRL, Germany), and re-suspended in RPMI 1640 media supplemented with 10% fetal calf serum (Gibco) and 1% weight per volume penicillin/streptomycin (Gibco) (hereinafter referred to as “culture medium”) at a concentration of 3 × 106/ml. Total cell counts in BAL were performed using a modified Neubauer haemocytometer. Cytospin preparations were made using 200 μl of sample (82 × g for 10 minutes; Shandon Cytospin III, Shandon, Cheshire, UK) and stained by Giemsa technique carried out using an automated staining machine (Shandon Veristat Southern Products, Astmore, UK). BAL differential cell counts were performed using flow cytometry as previously reported (Citation[17]). Venous blood was collected into tubes containing 10 U/ml preservative free sodium heparin (DBL, Sydney, Australia). Brushing-derived epithelial cells were pelleted by centrifuging at 500 × g for 5 minutes, supernatant was discarded, and cells re-suspended to a cell count of 4 × 105 cells/ml with RPMI 1640 media.
Blood differential cell counts were performed using a CELL DYN 4000 (Abbott Diagnostics, Sydney, Australia). Blood films were stained by Giemsa technique and white cell differential counts checked by morphological assessment microscopically. For BAL and peripheral blood, the percentages of CD3 and CD8 and CD3-CD56+ lymphocytes were calculated using flow cytometry. Absolute numbers were calculated from total leucocyte counts.
Determination of apoptosis of bronchial epithelial cells
To allow correlation between apoptosis of bronchial epithelial cells and of the percentage of airway T-cells expressing granzyme-b, bronchial brushings were obtained from a cohort of 20 subjects (5 in each group of never-smokers, asymptomatic current smokers and current and ex-smoking COPD subjects). Bronchial epithelial cells were identified and staining with 7-amino-actinomycin D (7-AAD) was used to quantify apoptosis as we have previously described (Citation[1]).
Reagents
The following monoclonal antibodies (Mabs) and immunological reagents were employed: CD3[PC5] and CD56 [PC5] (Immunotech/Coulter, Marseille, France), CD3 and CD8 [FITC] (BD Biosciences, San Jose, CA, USA), granzyme-b [PE] (Serotec, Oxford, UK) and perforin [PE] (BD). A control IgG [PE] Mab, FacsPerm and FACSlyse were obtained from BD Biosciences; 7-AAD was purchased from Sigma (Sydney, Australia).
Intracellular staining with Mabs to granzyme-b and perforin
Measurement of intracellular granzyme-b and perforin was performed in peripheral blood from all subjects. Analysis of BAL was performed on 14 of the never-smokers, 30 of the COPD subjects (15 current and 15 ex-smokers) and 10 asymptomatic smokers. Intracellular granzyme-b and perforin was measured on CD3+ and CD3+CD8+ T-cells and CD56+CD3-natural killer (NK) cells using flow cytometric methods as previously reported (Citation[18]). Briefly, red blood cells were lysed, cells washed then stained with CD8 and CD3 or CD3 and CD56 for 30 minutes prior to washing. Cell membranes were permeabilized with FACSPerm (BD) then cells washed and stained with Mabs to granzyme-b or perforin for a further 30 min. Washed cells were analyzed using a FACSCalibur flow cytometer (BD) using Cell Quest software (BD) as shown in .
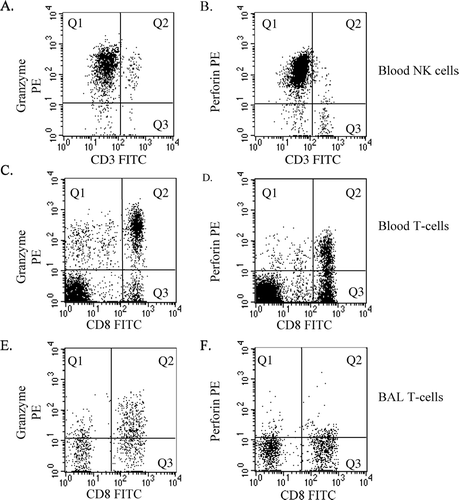
Figure 1 Representative flow cytometric dot plots of granzyme-b and perforin expression by NK cells and T-cells. (A,B) Peripheral blood-derived NK cells were identified in R1 as staining brightly with CD56PC5 (not shown). Subsequent analysis was carried out on cells from R1. NK cells expressing (A) granzyme-b or (B) perforin were identified in quadrant 1 (Q1) by negative staining with CD3 (ie. excluding NKT cells) and bright staining with Mabs to granzyme-b or perforin. Data was expressed as a percentage of total CD3 negative NK cells. (C,D) Peripheral blood-derived T-cells were identified in R1 as staining brightly with CD3PC5 (not shown). Subsequent analysis was carried out on cells from R1. T-cells expressing granzyme-b or perforin were identified by bright staining with Mabs to (C) granzyme-b or (D) perforin (Q1 +Q2). Data was expressed as a percentage of total CD3 T-cells and as a percentage of CD8+ T-cells (Q2/Q2+Q3). (E,F) BAL-derived T-cells. Analysis of BAL was performed as described for blood.
Soluble granzyme-b levels by ELISA
ELISA determination of granzyme levels was performed on plasma from all subjects and BAL from 24 of the never-smokers, 23 of the COPD subjects (10 current and 13 ex-smokers) and 11 asymptomatic smokers, using specific enzyme-linked immunosorbent assay (ELISA) (Diaclone, Besancon, France) according to manufacturer's instructions. Samples of plasma and BAL were stored at −70°C and tested on the same day.
Statistical analysis
The Kruskall–Wallis and Mann–Whitney U-tests were applied to analyze the non-normally distributed data. Correlation between apoptosis of bronchial epithelial cells and intracellular levels of granzyme-b in BAL-derived T-cell was performed using Spearman's rank correlation. Analysis was performed using SPSS software. p values < 0.05 were considered significant.
RESULTS
Identification and differential counting of BAL-and peripheral blood derived lymphocytes
Flow cytometry was applied for differential counting of BAL, as previously described (Citation[17]). It is noteworthy that the vast majority of subjects had no significant bacterial growth in their BAL (occasional “oral flora”). Thus significant clinical infection was excluded. There was no significant difference in absolute leucocyte counts in BAL from ex-smoking COPD subjects compared to never-smokers (). However, absolute leucocyte numbers were significantly increased in both current smoking COPD subjects and asymptomatic smokers (). To account for these differences, all cell numbers were thereafter calculated as absolute numbers relative to BAL leucocyte counts. For BAL, absolute numbers of lymphocytes were not significantly different between COPD and never-smokers (current smoking COPD subjects vs never-smokers (). CD8+ T-cell numbers were higher in BAL from current- and ex-smoking COPD subjects and asymptomatic smokers compared to never-smokers (). There were no differences in NK numbers in BAL from any groups (data not shown).
For peripheral blood the number of lymphocytes ranged from 0.8–5.4 × 109/l and was not significantly higher in blood from COPD patients than from never-smokers. Similarly, absolute numbers of NK cells were not significantly different between COPD and never-smokers [range 0.01–0.25 × 109/l]. The absolute number of CD8+ T-cells was significantly higher in peripheral blood from current and ex-smoking COPD subjects compared to never-smokers (current smoking COPD subjects median 0.005 [range 0.001–0.117]; ex-smoking COPD subjects median 0.008 [range 0.002–0.100] vs never-smokers median 0.003 [range 0.0002–0.012 × 109/l]. There were no significant changes in any lymphocyte subsets in the peripheral blood from asymptomatic smokers without COPD.
Intracellular staining of granzyme-b and perforin
The analysis approach is depicted in . For both current and ex-smoking COPD subjects, the percentage of both blood and BAL-derived CD3+ and CD8+ T-cells expressing intracellular granzyme-b and perforin were significantly higher than controls ( and ). Both granzyme-b and perforin were highly expressed in peripheral blood-derived CD56+CD3- NK cells with a significant increase in the median fluorescence intensity (MFI) of staining in both COPD groups (granzyme-b: current smoking COPD subjects median MFI 220 [range 51–649] p = 0.01; ex-smoking COPD subjects median MFI 191 [range 95–487] p = 0.02; vs never-smokers median MFI 155 [range 45–340]; perforin: current smoking COPD subjects median MFI 199 [range 38–380] p = 0.001; ex-smoking COPD subjects median MFI 158 [range 108–280] p = 0.03 vs never-smokers median MFI 125 [range 39–323]). Granzyme-b or perforin could not be accurately measured in NK cells from the BAL due to small NK numbers in BAL.
Figure 2 Intracellular expression of granzyme-b and perforin in peripheral blood derived T-cells. Expression of granzyme-b and perforin was quantified on A. CD3+ T-cells and B. CD8+ T-cells by flow cytometry. Note significantly increased expression of granzyme-b and perforin in current- (COPD current smokers, n = 30) and ex- (COPD ex-smokers, n = 30) smokers with COPD but no changes in asymptomatic smokers (Smokers, n = 18) compared to never-smokers (n = 30). *significant increase (p ≤ 0.05) in granzyme-b compared to control; **(p ≤ 0.05) significant increase in perforin compared to never-smokers. Box plots shows median, quartiles, and ranges.
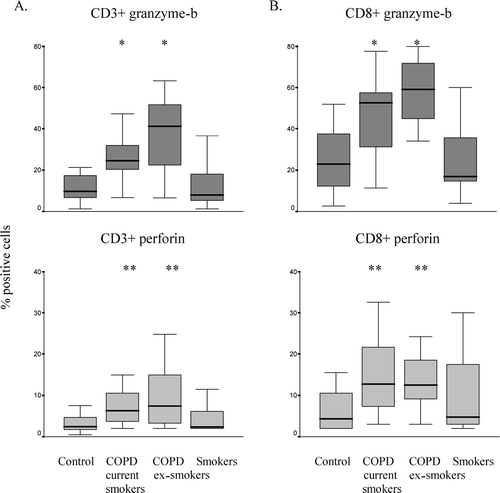
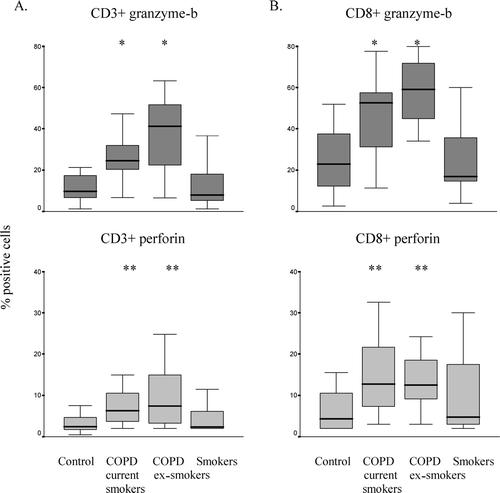
Figure 3 Intracellular expression of granzyme-b and perforin in BAL derived T-cells. Expression of granzyme-b and perforin was quantified on A. CD3+ T-cells and B. CD8+ T-cells by flow cytometry. Note significantly increased expression of granzyme-b and perforin in current- (COPD current smokers, n = 15) and ex- (COPD ex-smokers, n = 15) smokers with COPD and asymptomatic smokers (Smokers, n = 10) compared to never-smokers (n = 12). *significant increase (p ≤ 0.05) in granzyme-b compared to never-smokers; **(p ≤ 0.05) significant increase in perforin compared to never-smokers. Box plots shows median, quartiles, and ranges.
Effects of smoking on intracellular granzyme-b and perforin
There were no significant differences in intracellular granzyme-b and perforin levels in blood from the group of asymptomatic smokers versus never-smokers (). However, increased granzyme-b and perforin were noted in the BAL from this group (). There was a trend for an increase in the MFI of granzyme-b staining in blood-derived NK cells from asymptomatic smokers (median 183 [range 68–378] vs never-smokers median MFI 155 [range 45–340] p = 0.08) and a significant increase in the MFI of perforin staining in NK cells from this group (median 196 [range 94–355] vs never-smokers median MFI 125 [range 39–323] p = 0.01).
Soluble granzyme-b levels measured by ELISA
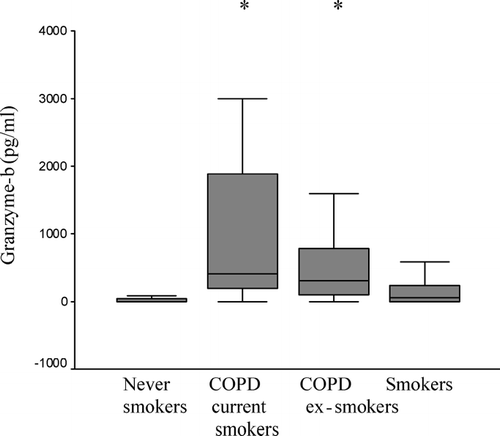
Soluble granzyme-b was undetectable in plasma. Granzyme-b in BAL was significantly increased in both current and ex-smoking COPD subjects versus never-smokers. There was a non-significant trend for increased granzyme-b in BAL from asymptomatic smokers without COPD ().
Figure 4 Soluble granzyme-b levels in BAL. Granzyme-b levels were measured by ELISA in BAL. Note significantly increased expression of granzyme-b in current- (COPD current smokers, n = 10), ex- (COPD ex-smokers, n = 13) smokers with COPD but no changes in asymptomatic smokers (Smokers, n = 11) compared to never-smokers (n = 24). *significant increase (p ≤ 0.05) in granzyme-b compared to never-smokers. Box plot shows median, quartiles, and ranges.
Apoptosis of bronchial epithelial cells and correlation with BAL T-cell granzyme-b
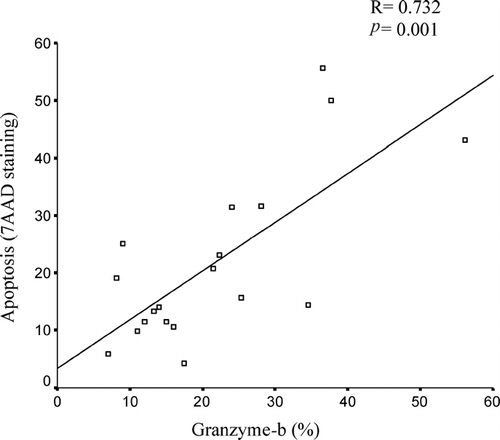
We investigated the correlation between BAL T-cell granzyme-b and bronchial epithelial cell apoptosis. Consistent with our previous study, the percentage of apoptotic cells was significantly increased in COPD subjects compared to never smokers (COPD 25.4% ± SD12.9% vs never-smokers 12.5% ± SD 3.5%, p = 0.02). There was good correlation between intracellular granzyme-b in BAL-derived T-cells and apoptosis of brushing-derived bronchial epithelial cells (R = 0.732, p = 0.000) ().
Figure 5 Correlation between T-cell granzyme-b levels in BAL and bronchial epithelial apoptosis. Apoptosis was measured in bronchial brushing derived epithelial cells by 7AAD staining. Note good correlation between intracellular granzyme-b in BAL-derived T-cells and apoptosis of bronchial epithelial cells.
DISCUSSION
Intracellular granzyme-b and perforin were significantly increased in selected BAL and peripheral blood T-cell populations from subjects with COPD. Levels of these cytotoxic mediators were not affected by smoking status among those with COPD. To our knowledge, this is the first comprehensive report of granzyme-b and perforin in cytotoxic T-cells obtained from the peripheral blood and lungs of patients with COPD. A few studies have reported increased levels of granzyme-b in other chronic inflammatory lung diseases including septic acute respiratory distress syndrome (Citation[19]) and atopic asthma (Citation[20]). Importantly, these studies suggested that the granzyme-b/perforin apoptotic pathway might contribute to the lung injury noted in these diseases. Consistent with our findings, another study reported increased intracellular perforin in sputum CD8+ lymphocytes from current -smoking COPD subjects (Citation[21]).
Our studies extend the previous observations that CD8+ T-cell numbers are increased both in the peripheral blood and airway in COPD (Citation[7], Citation[8]). The precise mechanisms for cytotoxic T-cell damage in COPD has not been fully elucidated, in particular the cytotoxic T-cell mediated cytolysis of airway epithelial cells. We now show that the granzyme-b/perforin pathway is highly upregulated in CD8+ T-cells in both peripheral blood and airways in COPD. Importantly, a significant correlation was also found between intracellular granzyme-b in BAL and bronchial epithelial apoptosis. The increased rate of apoptosis of bronchial epithelial cells COPD subjects in the present study is consistent with our previous findings (Citation[1]). It has been convincingly demonstrated that uncleared apoptotic material may then undergo secondary necrosis (Citation[6]), with the potential for inflammation of surrounding tissues (Citation[5], Citation[6]). Taken together, these findings suggest that upregulation of the granzyme apoptotic pathway may at least partially explain the prevalence of chronic inflammation and defective repair processes that are the hallmark of COPD pathogenesis.
In the present study, increased granzyme and perforin was noted for both current and ex-smoking COPD subjects. Cigarette smoking has been shown to induce apoptosis of airway epithelial cells (Citation[13]). However, consistent with our current findings for granzyme-b and perforin, we previously found that cessation of smoking did not prevent the increased rates of apoptosis in COPD (Citation[1]). This suggests that factors other than cigarette smoke must play a role in lung injury and chronic inflammation once the COPD disease is established. Our current findings suggest that the granzyme-b/perforin cytotoxic pathway may be one factor that relates to perpetuation of the chronic inflammatory response in COPD by contributing to increased apoptosis in the airways. Interestingly, there were no changes in the median expression of intracellular granzyme-b or perforin in peripheral blood from asymptomatic smokers, although several individual smokers demonstrated very high levels of these mediators, raising the possibility that these markers may serve as early markers for developing airways disease.
Although COPD is primarily a disease that affects the airway, it is being increasingly recognized as a systemic disease (Citation[22], Citation[23]). Lymphocytes have been shown to traffic from the bloodstream to the bronchoalveolar space and vice versa (Citation[24]). We now show that intracellular stores of granzyme-b and perforin are upregulated in both BAL-derived T-cells and blood-derived T-cells and NK cells in both current- and ex-smokers with COPD, further confirming the systemic nature of the disease. In addition, both percentage and absolute numbers of CD8+ cytotoxic T-cells were increased in both peripheral blood and BAL in COPD. Such systemic inflammatory effects could be a factor in the cachexia sometimes noted with severe disease. One could also speculate that the T-cells might be primed with granzyme-b and perforin before they enter the lungs
Not all granzymes enter the target cell; some may also leak out into surrounding biological fluids, after their release from cytoplasmic granules of the cytotoxic T-cell. Interestingly, the present study found a significant increase in soluble granzyme-b in the BAL but not in the peripheral blood in COPD. This finding was in contrast to intracellular granzyme-b that was increased in both blood and BAL. The normal lung is protected from the detrimental effects of proteases by serine protease inhibitors including α 1-antitrypsin, secretory leucocyte protease inhibitor and elafin (Citation[25]) that may play a role in inhibition of granzyme activity in the lung. It is thus possible that granzyme activity may be poorly controlled by these protease inhibitors in COPD. Consistent with this, a study by Tremblay et al showed that granzyme activity in the inflamed lung in hypersensitivity pneumonitis was not controlled by the main endogenous proteinase inhibitors (Citation[26]). Alternatively, the levels of free enzyme may indicate the persistent presence of cells that are expressing the two proteins, and that, given the right reactivity, could conceivably contribute to apoptosis in the lung and contribute to the destructive changes noted in emphysema.
In conclusion, our data suggests that the granzyme-b/perforin pathway is upregulated in current and ex-smoking COPD subjects but relatively unaffected by smoking per se, raising the possibility that identification of these proteins may serve as a useful diagnostic marker for early onset of the disease. Our results also suggest that granzyme-mediated apoptosis may be one mechanism of lung injury in COPD. The increased granzyme-b that persists despite smoking cessation in COPD may reflect the ongoing pathology of COPD rather than the effects of smoking per se.
SH conceived of the study, participated in its design, participated in experiments, and drafted the manuscript; GH and JN carried out the experiments. MH and PR participated in the design and coordination of the study and were responsible for obtaining patient samples. All authors helped to draft the manuscript and read and approved the final manuscript.
The authors thank the staff of Thoracic Medicine, Royal Adelaide Hospital, for their help in obtaining patient samples. The work was supported by NHMRC Project Grant, NHMRC Clinical Career Development Award, Allen & Hanburys Respiratory Research Fellowship and Royal Adelaide Hospital Clinical Project Grant.
REFERENCES
- Hodge S, Hodge G, Holmes M, Reynolds P N. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J 2005; 25: 447–454, [INFOTRIEVE], [CSA]
- Kasahara Y, Tuder R M, Cool C D, Lynch D A, Flores S C, Voelkel N F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 2001; 163(3 Pt 1)737–744, [INFOTRIEVE], [CSA]
- Majo J, Ghezzo H, Cosio M G. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J 2001; 17(5)946–953, [INFOTRIEVE], [CROSSREF], [CSA]
- Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz M A, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry 1992; 13: 795–808, [INFOTRIEVE], [CROSSREF], [CSA]
- Hart S P, Haslett C, Dransfield I. Recognition of apoptotic cells by phagocytes. Experientia 1996; 52: 950–956, [INFOTRIEVE], [CROSSREF], [CSA]
- Knapp S, Leemans J C, Florquin S, Branger J, Maris N A, Pater J, van Rooijen N, van der Poll T. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med 2003; 167: 171–179, [INFOTRIEVE], [CROSSREF], [CSA]
- Saetta M, Di Stefano A, Turato G, Facchini F M, Corbino L, Mapp C E, Maestrelli P, Ciaccia A, Fabbri L M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 157: 822–826, [INFOTRIEVE], [CSA]
- de Jong J W, van der Belt-Gritter B, Koeter G H, Postma D S. Peripheral blood lymphocyte cell subsets in subjects with chronic obstructive pulmonary disease: association with smoking, IgE and lung function. Respir Med 1997; 91: 67–76, [INFOTRIEVE], [CROSSREF], [CSA]
- Darmon A J, Nicholson D W, Bleackley R C. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 1995; 377: 446–448, [INFOTRIEVE], [CROSSREF], [CSA]
- Takayama H, Sitkovsky M V. Antigen receptor-regulated exocytosis in cytotoxic T lymphocytes. J Exp Med 1987; 166: 725–743, [INFOTRIEVE], [CROSSREF], [CSA]
- Simon T, Opelz G, Wiesel M, Ott R C, Susal C. Serial peripheral blood perforin and granzyme B gene expression measurements for prediction of acute rejection in kidney graft recipients. Am J Transplant 2003; 3: 1121–1127, [INFOTRIEVE], [CROSSREF], [CSA]
- Froelich C J, Zhang X, Turbov J, Hudig D, Winkler U, Hanna W L. Human granzyme B degrades aggrecan proteoglycan in matrix synthesized by chondrocytes. J Immunol 1993; 151: 7161–7171, [INFOTRIEVE], [CSA]
- D'Agostini F, Balansky R M, Izzotti A, Lubet R A, Kelloff G J, De Flora S. Modulation of apoptosis by cigarette smoke and cancer chemopreventive agents in the respiratory tract of rats. Carcinogenesis 2001; 22: 375–380, [INFOTRIEVE], [CROSSREF], [CSA]
- Hodge S J, Hodge G L, Reynolds P N, Scicchitano R, Holmes M. Increased production of TGF-beta and apoptosis of T lymphocytes isolated from peripheral blood in COPD. Am J Physiol Lung Cell Mol Physiol 2003; 285: L492–L499, [INFOTRIEVE], [CSA]
- Fabbri L M, Hurd S S. GOLD Scientific Committee. Global strategy for the diagnosis, management, and prevention of COPD 2003. Update Eur Respir J 2003; 22: 1–2, [CROSSREF], [CSA]
- Hodge S, Hodge G, Scicchitano R, Reynolds P N, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol 2003; 81: 289–296, [INFOTRIEVE], [CROSSREF], [CSA]
- Hodge S J, Hodge G L, Holmes M, Reynolds P N. Flow cytometric characterization of cell populations in bronchoalveolar lavage and bronchial brushings from patients with chronic obstructive pulmonary disease. Cytometry B Clin Cytom 2004; 61: 27–34, [INFOTRIEVE], [CROSSREF], [CSA]
- Hodge S, Hodge G, Flower R, Han P. Surface activation markers of T lymphocytes: role in the detection of infection in neonates. Clin Exp Immunol 1998; 113: 33–38, [INFOTRIEVE], [CROSSREF], [CSA]
- Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H. Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med 2000; 161: 237–243, [INFOTRIEVE], [CSA]
- Bratke K, Bottcher B, Leeder K, Schmidt S, Kupper M, Virchow J C, Jr., Lutterman W. Increase in granzyme B+ lymphocytes and soluble granzyme B in bronchoalveolar lavage of allergen challenged patients with atopic asthma. Clin Exp Immunol 2004; 136: 542–548, [INFOTRIEVE], [CROSSREF], [CSA]
- Chrysofakis G, Tzanakis N, Kyriakoy D, Tsoumakidou M, Tsiligianni I, Klimathianaki M, Siafakas N M. Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest 2004; 125: 71–76, [INFOTRIEVE], [CROSSREF], [CSA]
- Wouters E F. Chronic obstructive pulmonary disease. 5: systemic effects of COPD. Thorax 2002; 57: 1067–1070, [INFOTRIEVE], [CROSSREF], [CSA]
- Oudijk E J, Lammers J W, Koenderman L. Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J Suppl 2003; 46: 5s–13s, [INFOTRIEVE], [CSA]
- Lehmann C, Wilkening A, Leiber D, Markus A, Krug N, Pabst R, Tschernig T. Lymphocytes in the bronchoalveolar space reenter the lung tissue by means of the alveolar epithelium, migrate to regional lymph nodes, and subsequently rejoin the systemic immune system. Anat Rec 2001; 264: 229–236, [INFOTRIEVE], [CROSSREF], [CSA]
- Tremblay G M, Sallenave J M, Israel-Assayag E, Cormier Y, Gauldie J. Elafin/elastase-specific inhibitor in bronchoalveolar lavage of normal subjects and farmer's lung. Am J Respir Crit Care Med 1996; 154: 1092–1098, [INFOTRIEVE], [CSA]
- Tremblay G M, Wolbink A M, Cormier Y, Hack C E. Granzyme activity in the inflamed lung is not controlled by endogenous serine proteinase inhibitors. J Immunol 2000; 165: 3966–3969, [INFOTRIEVE], [CSA]