Abstract
The natural course of chronic obstructive pulmonary disease (COPD) is complicated by the development of systemic consequences and co-morbidities. These may be major features in the clinical presentation of COPD, prompting increasing interest. Systemic consequences may be defined as non-pulmonary manifestations of COPD with an immediate cause-and-effect relationship, whereas co-morbidities are diseases associated with COPD. The major systemic consequences/co-morbidities now recognized are: deconditioning, exercise intolerance, skeletal muscle dysfunction, osteoporosis, metabolic impact, anxiety and depression, cardiovascular disease, and mortality. The mechanisms by which these develop are unclear. Probably many factors are involved. Two appear of paramount importance: systemic inflammation, which presents in some patients with stable disease and virtually all patients during exacerbations, and inactivity, which may be a key link to most COPD-related co-morbidities. Further studies are required to determine the role of inflammatory cells/mediators involved in systemic inflammatory processes in causing co-morbidities; the link between activity and co-morbidities; and how COPD therapy may affect activity. Both key mechanisms appear to be influenced significantly by COPD exacerbations. Importantly, although the prevalence of systemic consequences increases with increasing severity of airflow obstruction, both systemic consequences and co-morbidities are already present in the Global Initiative for Chronic Obstructive Lung Disease Stage II. This supports the concept of early intervention in chronic obstructive pulmonary disease. Although at present early intervention studies in COPD are lacking, circumstantial evidence suggests that current treatments may influence events leading to the systemic consequences and co-morbidities, and thus may affect the clinical manifestations of the disease.
| Abbreviations | ||
| ARIC | = | Atherosclerosis Risk in Communities (Study) |
| BMD | = | bone mineral density |
| COPD | = | chronic obstructive pulmonary disease |
| CRP | = | C-reactive protein |
| DALYS | = | disability-adjusted life years |
| DEXA | = | dual-energy x-ray absorptiometry |
| FEV1 | = | forced expiratory volume in 1 second |
| FVC | = | forced vital capacity |
| GOLD | = | Global Initiative for Chronic Obstructive Lung Disease® |
| HRQL | = | health-related quality of life |
| IL | = | interleukin |
| NHANES | = | National Health and Nutritional Examination Survey |
| PDE | = | phosphodiesterase |
| TLC | = | total lung capacity |
| TNF | = | tumor necrosis factor |
| TORCH | = | Towards a Revolution in COPD Health (Study) |
| UPLIFT® | = | Understanding Potential Long-term Impacts on Function with Tiotropium (Study) |
| WHO | = | World Health Organization |
INTRODUCTION
COPD is largely caused by cigarette smoking and is characterized by airflow limitation and chronic inflammation in the lungs (Citation[1]). The burden of this progressive disease is evident from mortality figures and disability-adjusted life years (DALYS). Reported as the sixth-leading cause of death in 1990, it is currently ranked fourth, and is projected to rank third by 2020 (Citation[2]). Moreover, it is the only leading cause of death with increasing prevalence (Citation[3]). Similarly, from its ranking as the 12th-leading cause of DALYS in 1990, it is projected to become the fifth by 2020.
Co-morbid conditions are frequently observed in patients with COPD. For example, COPD patients—randomly selected from 1,522 patients who enrolled in a health maintenance organization in 1997—had an average of 3.7 co-morbid conditions compared with 1.8 observed in controls (Citation[4]). The most prevalent co-morbidities or systemic consequences include: cardiovascular disease (Citation[4], Citation[5], Citation[6]), lung cancer (Citation[7]), deconditioning, exercise intolerance (Citation[8]), muscle wasting (Citation[9]), depletion of fat-free mass or obesity (Citation[10]), diabetes (Citation[11], Citation[12]), osteoporosis (Citation[13], Citation[14], Citation[15]), anemia (Citation[16], Citation[17]), and anxiety/depression (Citation[18]). These are all associated with excess morbidity and mortality (Citation[19]).
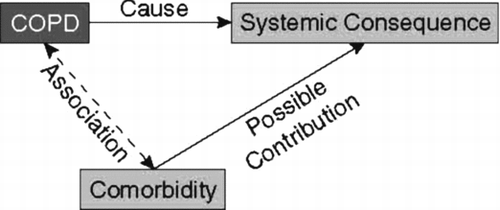
There is no consensus definition for co-morbidities that distinguishes them from systemic consequences of COPD. Intuitively, an operational definition is that the systemic consequences are direct consequences of the disease with a cause-and-effect relationship, whereas co-morbidities are diseases that occur associated with COPD, perhaps because of shared risk factors. Regardless of whether there is a causal relationship with COPD, co-morbidities may still influence the clinical course of COPD. These concepts are summarized in . In addition, systemic consequences and co-morbidities are not confined to the more advanced stages of the disease (Global Initiative for Chronic Obstructive Lung Disease[GOLD] Stage III and IV), but may also be present from the early stages of the disease (GOLD Stage I and II).
Figure 1 Interrelationships between COPD, systemic consequences, and co-morbidities.
The predominant inflammatory cells in the lung in COPD are alveolar macrophages, neutrophils, and cytotoxic T-lymphocytes (CD-8+ cells) (Citation[20]). However, COPD is increasingly recognized not to be confined to chronic inflammation of the lungs but to be associated with systemic inflammation, potentially leading to co-morbidities or systemic consequences (Citation[19], Citation[21]). This systemic inflammation may be more pronounced in some stable COPD patients than in others (Citation[22], Citation[23], Citation[24], Citation[25]). A potential major source of systemic inflammation in COPD is an exacerbation of the disease, which results in bursts of systemic inflammatory markers that are released into the systemic circulation (Citation[26], Citation[27], Citation[28], Citation[29]).
While systemic inflammation, both during exacerbations and during stable disease, is generally proposed as a contributor to the systemic consequences and co-morbidities of COPD, it may not be the only mechanism. Inactivity and deconditioning may also be implicated, and the latter has been associated with skeletal muscle dysfunction, exercise intolerance, poor health status, diabetes, osteoporosis, and cardiovascular co-morbidity (see later).
There is accumulating evidence that treatment with long-acting anticholinergics, long-acting β2-agonists and inhaled corticosteroids may affect the course of the disease by improving forced expiratory volume in 1 second (FEV1), health status, and by reducing exacerbation rate (Citation[30], Citation[31], Citation[32], Citation[33], Citation[34], Citation[35]). At present, only smoking cessation has been shown to reduce the rate of decline in FEV1 (Citation[36], Citation[37], Citation[38]). However, indirect evidence suggests that long-acting anticholinergics, long-acting β2-agonists, and inhaled corticosteroids may alleviate the progressive loss in FEV1(Citation[39], Citation[40], Citation[41]). Current hopes for conclusive evidence are with the ongoing 4-year trial assessing the effects of a long-acting anticholinergic on the rate of decline of FEV1 (Understanding Potential Long-term Impacts on Function with Tiotropium [UPLIFT®]), the results of which are expected in 2008 (Citation[42]).
This review is the result of a round-table meeting of experts held in Miami, Florida, USA, in December 2006. The purpose of the review is 4-fold: to describe the clinical course of the disease incorporating systemic consequences and co-morbidities; to briefly summarize the literature regarding the major systemic consequences and co-morbidities of COPD; to analyze the available literature on mechanisms linking COPD to systemic consequences and co-morbidities; and to examine the potential for early intervention with the treatments currently available to reduce or reverse the development of systemic consequences and co-morbidities. These goals are discussed for several systemic consequences/co-morbidities including: deconditioning, exercise intolerance and skeletal muscle dysfunction, osteoporosis, metabolic impact, anxiety and depression, and cardiovascular co-morbidity and mortality.
MECHANISMS OF SYSTEMIC CONSEQUENCES AND CO-MORBIDITIES
The link between COPD and systemic consequences and co-morbidities is poorly understood, though hypothetical mechanisms are summarized in . As already indicated, inactivity/deconditioning and systemic inflammation currently have the most supporting data.
Table 1 Potential mechanisms of systemic consequences in COPD
Inactivity is prevalent in patients with COPD and is known to be related to many systemic manifestations of COPD (see below). Inactivity is also known to be severely enhanced during and immediately after exacerbations (Citation[43], Citation[44]). We now have the appropriate tools to critically examine the relationship between inactivity and the systemic consequences and co-morbidities of COPD. Hence, the mechanism by which inactivity can mediate systemic consequences is becoming increasingly clear.
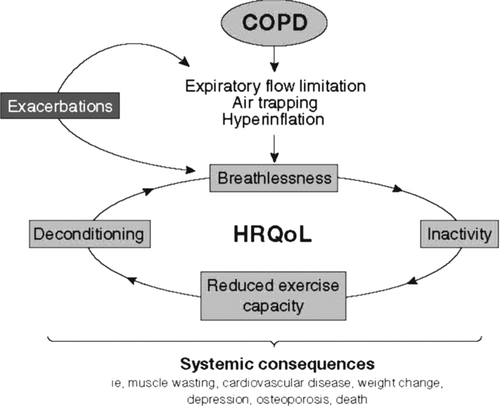
shows a schematic diagram depicting the clinical course of COPD and summarizing the extension to systemic consequences and co-morbidities (modified according to Cooper) (Citation[45]). Airflow obstruction leads to air trapping and hyperinflation, which in turn lead to dyspnea, activity limitation, and poor health-related quality of life (HRQL). Dyspnea may then cause anxiety and distress, which increase tachypnea and, thereby, enhance air trapping. Meanwhile, activity limitation leads to deconditioning, which increases ventilatory requirement and can lead to further air trapping. In addition, exacerbations contribute to the development of airflow obstruction and the progression of the disease, and also lead to worse HRQL.
Figure 2 The clinical course of COPD, showing the vicious cycle that ensues and some of the associations with systemic consequences and co-morbidities. See text for an explanation.
The mechanisms of systemic inflammation were recently reviewed by Agusti et al. (Citation[46], Citation[47]), Wouters (Citation[48]), and Sevenoaks and Stockley (Citation[21]), and we refer to these publications for a more detailed description. Systemic inflammation in COPD may result from “spill-over” of mediators, cytokines, or activated inflammatory cells from the lung into the systemic circulation. Alternatively, inflammation may arise in non-pulmonary tissues in COPD. Although not resolved (Citation[49]), considerably more evidence seems to be present in support of the former hypothesis than the latter (Citation[21], Citation[46], Citation[47], Citation[48]).
Patients with COPD have been observed, most notably during exacerbations, to have increases in systemic oxidative stress (Citation[50], Citation[51]), circulating levels of cytokines (tumor necrosis factor [TNF]-α (Citation[22], Citation[23], Citation[25]), interleukin [IL]-6 (Citation[28]), IL-8 (Citation[26], Citation[27], Citation[29])), C-reactive protein (CRP) (Citation[24]), adhesion molecules (Citation[27]), fibrinogen (Citation[24]), and circulating activated inflammatory cells, particularly neutrophils (Citation[52], Citation[53], Citation[54]). Repetitive bursts of cytokines or cells released during exacerbations most probably play a major role in producing systemic consequences of COPD. For example, the propensity of TNF-α to produce skeletal muscle dysfunction is well demonstrated (Citation[55], Citation[56]), and IL-6 in rats causes a dose-dependent, high-output cardiac failure with hemodynamics similar to those seen in septic shock (Citation[57]). These inflammatory responses may directly or indirectly add to muscle dysfunction and may also provide a mechanistic basis for the link between COPD and cardiac co-morbidity (Citation[6]).
INACTIVITY, EXERCISE TOLERANCE, DECONDITIONING, AND MUSCLES
As a larger proportion of the human population adopts a more sedentary lifestyle, it has become evident that inactivity is devastating for the human body. After many thousands of years of “survival of the fittest,” human physiology seems to cope poorly with inactivity (Citation[58]). The most studied consequences of inactivity are skeletal muscle adaptations, osteoporosis, reduced insulin sensitivity, and enhanced susceptibility to cardiovascular morbidity.
Why are COPD patients less active?
COPD patients avoid exercise and adopt a less active lifestyle. Indeed, patients become less active early in the course of the disease. Reduced walking time has been reported in patients with GOLD Stage II (Citation[59]), although this reduction was more pronounced in GOLD Stages III and IV (). Other events, such as exacerbations, further contribute to the adoption of an inactive lifestyle.
Figure 3 Reduced activity (A), prevalence of osteopenia/osteoporosis (B), depletion in fat-free mass (C), prevalence of anxiety/depression (D), cause of death (E) as a function of severity of disease (GOLD stage). Modified from (Citation[14], Citation[59], Citation[118], Citation[143], Citation[177]).
![Figure 3 Reduced activity (A), prevalence of osteopenia/osteoporosis (B), depletion in fat-free mass (C), prevalence of anxiety/depression (D), cause of death (E) as a function of severity of disease (GOLD stage). Modified from (Citation[14], Citation[59], Citation[118], Citation[143], Citation[177]).](/cms/asset/983fad05-837a-4710-ac0d-c0d28f09d9e0/icop_a_323920_uf0003_b.gif)
The profound impact of inactivity on skeletal muscle and cardiovascular function must reduce exercise capacity. However, there is a paucity of data evaluating the daily levels of physical activity of COPD patients in relation to their actual capacity for exercise (i.e., the amount to which the capacity is used on a day-to-day basis). What is known is that, once the functional exercise capacity drops below 60% of the predicted value, any further reduction in exercise tolerance is reflected in reductions in activities of daily life (Citation[59]).
It is also important to recognize that physiologic factors are not the only influence on physical activity behavior during daily life. Indeed, patients who have poor self-efficacy regarding their walking abilities also have lower physical activity levels (Citation[60]).
Which muscle abnormalities are present in COPD?
Patients with skeletal muscle weakness are likely to have excessive utilization of healthcare resources (Citation[61]). Moreover, skeletal muscle weakness increases mortality in patients with COPD (Citation[62]).
A recent literature review summarized the skeletal muscle abnormalities seen in patients with COPD (Citation[63]). Clinically, patients present with skeletal muscle weakness and impaired local skeletal muscle endurance. In 7 trials reporting both skeletal muscle strength and endurance in COPD (total n = 217) and controls (total n = 133), skeletal muscle endurance was significantly more impaired (weighted average 38% of the control values) than skeletal muscle strength (72% of control values). Indeed, skeletal muscle fatigue limited exercise tolerance in up to 50% of patients with COPD referred to a university hospital outpatient consultation (Citation[64]).
Typically, the oxidative capacity of the skeletal muscle is most disturbed. During exercise, patients prematurely reach the anaerobic threshold and, when studied during local work, it is obvious that muscle energy substrates are used up prematurely (Citation[65]). This observation is in keeping with the generally reported changes at the fiber-type level. A relatively lower proportion of oxidative (fatigue-resistant) fibers and a reduced size of all fiber types have been described. In addition, capillary:fiber ratio is significantly reduced (Citation[66]).
At the cellular level, the reduced oxidative capacity of skeletal muscle in COPD is reflected by lower activity of key enzymes in the aerobic phosphorylation pathway (e.g., lower citrate synthase and histone deacetylase activity) (Citation[67]). Furthermore, greater susceptibility of the muscle to oxidative stress has been found (Citation[68]), particularly in patients with a low body mass index (Citation[69]). Abnormalities in mitochondrial respiration further contribute to impaired skeletal muscle endurance (Citation[70]).
Finally, skeletal muscle dysfunction may contribute to exercise tolerance either directly, through the development of contractile muscle fatigue, or indirectly, through the increased ventilatory requirement during exercise.
What are the mechanisms behind abnormal muscle function?
The etiology of the skeletal muscle abnormalities in COPD remains largely unresolved; however, deconditioning is likely to play a pivotal role. Inactivity results in a tendency towards protein catabolism, which results in muscle atrophy (sarcopenia, see below). Deconditioning is typically associated with reduced oxidative capacity, fiber atrophy, and reduced skeletal muscle cross-sectional area. These changes can occur rapidly. For example, in healthy subjects, peak oxygen uptake and anaerobic threshold are reduced after only 3 days of bed rest (Citation[71]).
Systemic inflammation and excessive oxidative stress may also induce some of the changes at the cellular/molecular level. Repeated exacerbations, and the consequent increase in circulating systemic inflammatory cytokines, probably directly lead to loss in skeletal muscle function (Citation[29]). Furthermore, long-term or repeated use of oral corticosteroids may also result in steroid-induced myopathy (Citation[72]). Conversely, treatment of patients with COPD with relatively high doses of antioxidant improved skeletal muscle endurance (Citation[73]). This suggests that a poor defense against free radicals produced during dynamic exercise may contribute to impaired skeletal muscle function.
Interestingly, increased markers of oxidative stress have been convincingly shown after periods of inactivity (Citation[74]), and inactivity has also been associated with chronically increased levels of inflammatory cytokines. Speculatively, inactivity could trigger systemic inflammation, which may then lead to muscle weakness; however, this hypothesis lacks support. Indeed, most studies investigating the effect of training interventions did not show a decrease of circulating inflammatory markers with training (Citation[75]).
Aside from inactivity and inflammation, there are other factors that may influence muscle in COPD. Tissue hypoxia could further contribute to muscle weakness in hypoxemic patients or those with severely impaired capillary density. Electrolyte disturbances and general malnutrition have also been suggested to play a role in the skeletal muscle dysfunction in selected patients. Finally, hormonal factors, in particular hypogonadism, may contribute to the observed muscle weakness in a large proportion of patients (Citation[76]).
Poor exercise tolerance, muscle weakness, and dyspnea have profound effects on the systemic consequences of COPD. Dyspnea is often perceived by patients as a distressing sensation that should be avoided (Citation[77]). Dyspnea is a consequence of increased ventilation and dynamic hyperinflation, but its perception is further shaped by patient experience of previous episodes (Citation[78]). Hence, inactivity may be a “learned behavior” to avoid dyspneic stimuli that are perceived as distressing. A vicious cycle ensues in which deconditioning reinforces dyspnea, causing further deconditioning (). This progression ultimately results in profoundly impaired exercise tolerance, which is relatively poorly related to the lung function impairment (Citation[9]).
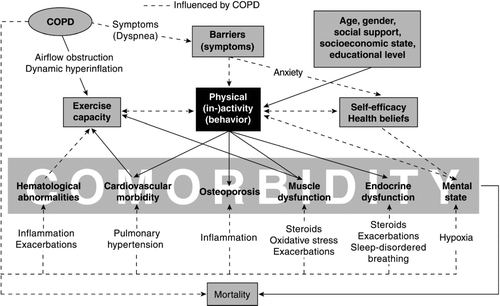
The interrelationships between COPD, activity levels, other causative factors, and systemic consequences and co-morbidities are summarized in . Among other consequences, inactivity leads to enhanced osteoporosis. The relation between inactivity and loss of bone mineral density (Citation[79]) may further be aggravated by long-term exposure to cigarette smoke (Citation[80]) or hypogonadism (Citation[81]), which are associated with reduced bone mineral density in men and women. Inactivity may also alter mood. For example, 1 study found a significant relation between deterioration in dyspnea and psychologic morbidity; however, a clear relation was not found between deterioration in mood and change in peak oxygen consumption over time (Citation[82]).
Figure 4 Interrelationships between activity and co-morbidities in COPD. See text for an explanation.
How can treatment improve skeletal muscle function and exercise tolerance?
Exercise capacity in many patients with COPD is limited as ventilation during exercise approaches the maximum ventilatory capacity. Increase in minute ventilation is accompanied by dynamic hyperinflation, whereby flow limitation causes more air to be trapped in the lung with each breath. The unfavorable effects of dynamic hyperinflation on ventilatory mechanics and the sensation of dyspnea cannot be underestimated. There is evidence that, when operational lung volumes increase to a certain level, the sensation of dyspnea intensifies (Citation[83]). Furthermore, termination of exercise can be anticipated when the end-inspiratory lung volume approaches within 500 ml of total lung capacity (Citation[45], Citation[84]). Therefore, essentially 3 strategies may be used to improve exercise capacity: increase in ventilatory capacity; reduction of operational lung volumes; and reduction in ventilatory requirement at a given exercise level.
In the mild-to-severe patient, increased ventilatory capacity and reduced operational lung volumes are primarily achieved by optimal bronchodilator therapy. Bronchodilators improve the flow-volume relationship during exercise. Consequently, the operational lung volume is reduced, which results in reductions in the work of breathing and dynamic hyperinflation at isowork in concert with a reduced sensation of dyspnea. However, a sub-group of patients exist for whom bronchodilators provide little benefit (Citation[85]). This sub-group has been reported to be limited predominantly by skeletal muscle fatigue (see above).
The second strategy that may enhance exercise performance is to increase ventilatory capacity. In patients suffering from deconditioning, exercise training may result in a rightward shift of the “anaerobic threshold” and, as an immediate metabolic consequence, the ventilatory requirement for a given level of exercise is reduced. An additional benefit of exercise training is that the skeletal muscle becomes less fatigable (Citation[86]); hence, patients can exercise for longer. In addition, enhanced mechanical efficiency during exercise ensures less oxygen consumption and ventilation at iso-work. Last, a more efficient breathing (i.e., slower with larger tidal volume) pattern allows a similar alveolar ventilation with less ventilation (Citation[87], Citation[88]).
An alternative strategy to reduce ventilatory requirement is oxygen supplementation, which improves exercise performance by reducing hypoxic ventilatory drive, thus reducing respiratory rate and ameliorating dynamic hyperinflation (Citation[89]). Even in non-hypoxemic patients, oxygen supplementation reduces ventilatory drive, without compromising oxygen delivery to the working skeletal muscle. Walking aids used by more severe patients during walking exercise is a second strategy that may enhance ventilatory capacity, presumably due to more effective recruitment of shoulder girdle and rib cage muscles (Citation[90]). Last, in very severe patients, surgical strategies (lung volume reduction surgery, transplantation) can enhance ventilatory capacity and increase exercise performance (Citation[91]).
Combining strategies that enhance ventilatory capacity with interventions that reduce the ventilatory requirement can be very successful, as they act through complementary mechanisms. At least two such combinations deserve particular attention. A recent study investigating the combination of the bronchodilator tiotropium, in combination with exercise training, showed that both interventions could be combined successfully (Citation[92]). The effects on exercise endurance and on symptoms were greater when strategies were combined, and effects were maintained after the training intervention was discontinued. Interestingly, the effects of the combination of tiotropium and exercise training on the transitional dyspnea index were far greater compared with the effects typically found after bronchodilation therapy or rehabilitation alone.
Second, bronchodilator therapy may be combined with oxygen supplementation. Complementary effects of both interventions have been reported in laboratory-based exercise tests (Citation[93]). In clinical practice, however, it is difficult to prescribe ambulatory oxygen therapy, especially in patients who are non-hypoxemic. Furthermore, oxygen therapy is prone to non-compliance and its overall effects outside the laboratory remain uncertain, particularly in the non-hypoxemic patient (Citation[94]).
OSTEOPOROSIS
Osteoporosis is a disorder most prevalent in post-menopausal women of Northern European descent who, consequently, have high rates of fractures (Citation[95]). However, the frequency of osteoporotic fractures is also high in Asian and other populations and is likely to increase further as life expectancy increases (Citation[96]). The overall prevalence of vertebral deformity was 12% in a multinational study of over 15,000 men and women aged 50 to 79 years (Citation[97]). The prevalence increased in both sexes with age, but the rate was more rapid in women. The probability that a 50-year-old person would have a hip fracture in his or her lifetime was 5–6% for a White male and 14% for a White female. The risk for African Americans at age 50 is approximately half that of non-Hispanic Whites (Citation[98]).
Osteoporosis, like COPD, has both a clinical definition and a physiologic definition. The clinical definition of osteoporosis is a “a disease characterized by low bone mass and microarchitectural deterioration of bone tissue, leading to enhanced bone fragility and a consequent increase in fracture risk” (Citation[99]). The World Health Organization (WHO) has established physiologic definitions of osteoporosis and osteopenia based on bone mineral density (BMD).
BMD is usually assessed by dual-energy X-ray absorptiometry (DEXA). The technique is simple to use, accurate, and precise, and requires radiation levels approximately 90% less than a standard chest radiograph. The unit of measurement for BMD with the use of DEXA is area density (g/cm2); however, DEXA results are usually reported as either Z-scores (compared with age- and sex-matched controls) or T-scores (compared with young adult sex-matched controls) (Citation[100]).
The WHO has established that osteopenia is a BMD of 1 to 2.5 standard deviations below the mean for young adults, and that osteoporosis is a BMD less than 2.5 standard deviations below the mean for young adults (Citation[100]). These definitions are arbitrary and controversial, but the risk of fractures has been shown to increase as the T-score decreases (Citation[101]).
What is the relationship between COPD and osteoporosis?
Osteoporosis is remarkably common among women with COPD. One of the largest studies to examine associations between airflow obstruction and BMD was the third National Health and Nutritional Examination Survey (NHANES III; 13). Sin and colleagues used the NHANES III data to demonstrate that airflow obstruction is independently associated with reduced bone mineral density, and in doing so, highlighted the magnitude of the problem among women with COPD (Citation[14]). The analysis was limited to the 9502 non-Hispanic White participants aged 20 years and older and severity of airflow obstruction (i.e., FEV1/forced vital capacity [FVC] ratio < 0.7) was stratified using the GOLD classification (Stage I, mild: FEV1 > 80% predicted; Stage II, moderate: FEV1 50–79%; Stage III and IV, severe and very severe: FEV1 < 50%). Osteopenia and osteoporosis were defined using the WHO criteria.
The prevalence of either osteopenia or osteoporosis among women with mild airflow obstruction was slightly higher than expected in a population-based cohort of this age. Prevalence increased as the severity of airflow obstruction increased, with approximately 33% of women with severe COPD having osteoporosis, and almost all of the rest having osteopenia (). Notable was that even in GOLD Stage II, the prevalence was significantly enhanced compared with the overall population. Hence, an increased prevalence of both osteopenia and osteoporosis is already present in the early stages of the disease.
In comparison, among men with severe airflow obstruction, the prevalence of osteoporosis was 11% and the prevalence of osteopenia was 60%. Although men were at less risk than women, this was still approximately 3 times higher than expected.
Patients with COPD tend to have other risk factors for osteoporosis, including use of oral corticosteroids, inactivity (see above), malnutrition, active smoking, and hypogonadism. However, even after adjustments for these confounding factors, men and women with moderate and severe airflow obstruction still had an elevated risk of developing osteoporosis (Citation[14]).
What are the mechanisms for osteoporosis in COPD?
The effects of the loss of vertebral height on total lung capacity (TLC) and the associated impairment in ventilatory mechanics may contribute to the association between osteoporosis and COPD. In a study of 74 women with COPD, Leech and colleagues demonstrated that lung function and vital capacity incrementally declined as the number of thoracic vertebral fractures increased (Citation[102]). Women with 3 or more fractures had an average loss of 25% of predicted TLC, and 32% of FVC. In another study, the degree of kyphosis was directly related to the loss of FVC and FEV1 (Citation[103]). Kyphosis caused by osteoporosis caused limitation in rib mobility and inspiratory muscle function (Citation[104]).
Systemic inflammation in COPD could also be a contributing factor to osteoporosis. COPD has been associated with increased circulating levels of IL-6 and TNF-α, which are potent promoters of osteoclast generation and activity. Interestingly, polymorphism in TNF-α has been suggested to explain some of the variation in genetic susceptibility to osteoporosis seen among older women in the general population (Citation[105]).
Can treatment have an impact on osteoporosis?
All women with COPD meet the criteria for referral for DEXA testing, according to guidelines for the prevention and treatment of osteoporosis. Men with COPD who have had weight loss or evidence of osteoporotic fractures should also be tested (Citation[106]). Prevention and early treatment for osteoporosis are indicated, though specific studies on treatment of osteoporosis in COPD are unavailable. Whenever possible, the use of systemic and high-dose inhaled steroids in the treatment of COPD patients should be minimized. It is presently unknown whether maintaining optimal lung function from early in the disease affects bone disease, though enhanced activity levels resulting from better lung function may contribute to the prevention of osteoporosis. Smoking cessation is always advisable, but may be particularly important because of the adverse effects of smoking on bone density (Citation[80]).
METABOLIC IMPACT
In previous decades, body habitus was considered an important manifestation of two classical phenotypes of COPD: the “pink puffer” (the emphysematous type) and the “blue bloater” (the bronchitic type) (Citation[107]). A low body weight and weight loss were negatively associated with survival in COPD as early as the1960s (Citation[108]). At the time, however, weight loss was generally considered as a terminal phase of the disease and, hence, inevitable and irreversible. A significant portion of COPD patients (27–47%) have a history of significant weight loss exceeding 5–10% over 1 year (Citation[109], Citation[110], Citation[111]).
What is the metabolic impact of COPD?
Low body weight is usually determined from the ideal body weight as derived from average statistics for height, frame size, and sex, or the ratio of body weight divided by height squared. The prevalence of a low body weight was investigated in 3 large cohorts of stable COPD patients in the early 1990s (Citation[112], Citation[111], Citation[114]). These studies suggest that being underweight is particularly associated with a loss of diffusing capacity for carbon monoxide (Citation[115]).
Assessment of nutritional status by extrapolation from body weight is limited as body weight provides no qualitative information on body composition. Hence, to allow recognition of different patterns of tissue depletion, the body needs to be partitioned into at least two compartments: fat mass and lean body mass. Indeed, cachexia, sarcopenia, muscle atrophy, and semi-starvation are all terms used to indicate changes in body composition. Cachexia is usually defined by a disproportional loss of fat-free mass. Sarcopenia also refers to the loss of lean body mass, but this is usually in the absence of weight loss and is related to aging. This pattern is also seen in patients with muscle atrophy, which is related to inactivity. Semi-starvation is the loss of fat mass and is caused by energy imbalance.
Loss in lean body mass is a prevalent finding even in mild COPD patients (Citation[114], Citation[116], Citation[117], Citation[118]). In the majority of patients, depletion in muscle mass is associated with a loss in body mass; in a sub-group of patients, the loss in muscle mass is masked by an increase in fat mass. Depletion of muscle mass is particularly prevalent in emphysematous patients, largely related to a reduction in trunk fat-free mass, while extremity fat-free mass is not significantly different between emphysematous and non-emphysematous patients (Citation[10], Citation[119]). shows the increasing prevalence of depletion of fat-free mass with each increasing GOLD stage. In the earlier stages, depletion of fat-free mass is present in 20% of the patients, indicating that this may occur even in the early stages of the disease.
Recent data suggest that a metabolic syndrome is common in patients with COPD (Citation[12]). Although the finding may not be all that surprising, since metabolic syndrome has been associated with inactivity, systematic studies in COPD are lacking. Metabolic syndrome represents a cluster of risk factors for developing diabetes mellitus, non-fatal and fatal cardiovascular disease, as well an increased risk of mortality from all causes. Considering these increased risks, assessment for features of metabolic syndrome seems prudent as part of the management of COPD.
What are the mechanisms of the metabolic changes in COPD?
Weight loss, and particularly loss of fat mass, occurs if energy expenditure exceeds dietary energy sources. Total dietary energy expenditure is usually divided into 3 components: resting energy expenditure, comprising sleeping metabolic rate and the energy cost of arousal; thermogenesis induced by food intake; and thermogenesis induced by physical activity. Based on the assumption that resting energy expenditure is the major component of total energy expenditure in sedentary persons, several studies have measured resting energy expenditure in COPD patients.
In one study, resting energy expenditure was elevated in 25% of COPD patients, after adjustment for the metabolically active lean body mass (Citation[120]). Furthermore, whereas the amount of lean body mass explained up to 84% of the individual variation in resting energy expenditure in healthy subjects, it explained only 43% in patients with COPD (Citation[120]). Thus, other factors, such as the work of breathing, drug therapy, and systemic inflammation, may explain the inter-subject variability in resting energy expenditure of patients with COPD.
A likely cause of the increased resting metabolic rate in patients with COPD is increased respiratory muscle work. The energy cost of ventilation is higher in patients with advanced disease than in healthy controls of comparable age and gender. Contrary to this, no reduction in resting energy expenditure was observed in a study with nasal intermittent positive-pressure ventilation to eliminate diaphragmatic and intercostal activity, despite baseline hypermetabolic state (Citation[121]). In contrast, systemic inflammation appears to be related to augmented metabolic state (Citation[23], Citation[25]), which suggests this may be the mechanistic link between COPD and metabolic syndrome.
From a metabolic point of view, total energy expenditure in normal, daily living conditions is more relevant than the resting component alone. Using the doubly-labeled water technique to measure total energy expenditure, patients with COPD had significantly higher total energy expenditure, particularly the non-resting component of total energy expenditure, than did healthy subjects (Citation[122]). Furthermore, no significant difference in daily living total energy expenditure was found between clinically stable patients with COPD with an elevated resting energy expenditure and those with a normal metabolic state (Citation[123]). To account for these findings, it is hypothesized that part of the increased oxygen consumption during exercise is related to inefficient ventilation in the presence of increased ventilatory demands, especially under conditions of dynamic hyperinflation.
To better understand the possible course of body compositional changes and weight loss in COPD patients, energy balance has been evaluated during acute phases of the disease. A negative energy balance has been reported during the first days after an exacerbation-related hospital admission and this has been related to a drop in energy intake (Citation[124]). During recovery, the reduction in breathlessness is significantly related to the changes in resting energy expenditure. Also noteworthy is that these changes during acute exacerbations are significantly related to changes in leptin homeostasis (Citation[125]).
Muscle wasting is also a consequence of an imbalance between protein synthesis and protein breakdown. Several studies report associations between various circulating markers of inflammation and the loss of fat-free mass (Citation[22], Citation[23], Citation[24], Citation[25]). However, data providing genuine insight into the pathobiology of cachexia associated with chronic low-grade inflammation are still scarce. Based on cancer models, activation of the ubiquitin proteasome pathway is suggested as a mechanism leading to cachexia. However, the slow progression of muscle depletion in COPD suggests deregulation of muscle homeostasis at levels other than protein energy metabolism, e.g., the maintenance of functional skeletal muscle mass by muscle fiber apoptosis and regeneration.
Does treatment have an impact on the metabolic consequences?
Based on the contemporary understanding of insights into the pathophysiology of COPD, current treatments seem to have limited effect on the metabolic imbalances seen in COPD patients. Maintaining optimal lung function may contribute to a reduction in activity-related energy expenditure, though objective data are lacking.
PSYCHOSOCIAL IMPACT
Anxiety and depression are significant clinical problems in patients with COPD. The prevalence of clinical anxiety and mood disorders in patients with COPD is significantly higher than in the age-matched population. Estimates for specific anxiety disorders range from a 3- to 10-fold increase in COPD. The highest rates were found for panic disorder, which may occur in as many as one-third of COPD patients () (Citation[126], Citation[127], Citation[128]). Anxiety symptoms are very common in COPD patients (Citation[129], Citation[130], Citation[131], Citation[132], Citation[133], Citation[134]), exceeding that for patients with other chronic medical conditions such as heart failure and cancer (Citation[135]). Clinically significant depression symptoms are seen in almost half of patients with COPD, and about 20% of patients will meet psychiatric criteria for major depressive or dysthymic disorder (Citation[136], Citation[137]).
What is the psychosocial impact of COPD?
Anxiety and depression have a significant effect on the course of COPD and impact of the disease on patients. Anxiety in patients with COPD is associated with decreased HRQL, greater disability, and impaired functional status (Citation[138], Citation[139]), even after controlling for lung function, dyspnea, and the presence of other chronic diseases (Citation[131], Citation[140]). Anxiety is also a significant predictor of the frequency of hospitalizations for acute exacerbations of COPD (Citation[141]). Likewise, depression with COPD is associated with poorer treatment adherence, decreased HRQL, greater disability and increased mortality (Citation[131], Citation[138], Citation[139], Citation[142]).
Gender differences in anxiety/depression in COPD
As in the general population, anxiety and depression accompanying COPD are more common in women than in men. Women are 1.5–2 times more likely than men to have problems with anxiety or depression (Citation[143]). A number of factors may lead to this gender difference. First, women appear to be impacted more by COPD than are men. For the same levels of COPD severity, women report more dyspnea and worse HRQL than men (Citation[144], Citation[145]). An age interaction with gender is also evident on depression. Younger women with COPD report significantly higher depression than older women, whereas age is only modestly associated with depression reports for men. A similar interaction between age and gender on depression was also reported in healthy subjects (Citation[146]). Gender differences in rumination tendencies are one well-studied phenomenon that might explain this interaction. Rumination is associated with increased risk for depression. Women tend to ruminate more than men and, among women, rumination is inversely related to age (Citation[147]).
What are the mechanisms of anxiety/depression in COPD?
The cause of anxiety and depression in patients with COPD is probably multifactoral and varies between patients. As with other co-morbidities, anxiety and depression can be viewed as a systemic consequence of COPD. Several pathologic systemic processes could contribute to the development of anxiety and depression in both direct and indirect ways.
The effects of chronic inflammatory processes potentially explain the increased rates of depression and anxiety in patients with COPD. Depressed patients without COPD or other chronic medical illnesses show elevated circulating levels of pro-inflammatory cytokines including IL-1, IL-6, and TNF-α (Citation[148], Citation[149]). In addition, administration of cytokines to healthy individuals produces symptoms of depression, including fatigue, hypersomnia, irritability, decreased appetite, and cognitive deficits (Citation[150]). Furthermore, cytokines may induce neurendocrine and neurotransmitter changes reminiscent of those found in depression (Citation[151]).
Hypoxemia may also contribute to the development of anxiety and depression. Chronic hypoxia may lead to structural brain changes that cause neurocognitive deficits and impairment in mood regulation. Even mild hypoxia produces alterations in neurotransmitter systems essential to cognitive-emotional functioning (Citation[152]). These impairments in brain function may result directly in psychiatric symptoms, but may also impair the individual's ability to adapt and cope with challenge, including the stress associated with managing a chronic illness. Repeated experiences with hypoxia may also sensitize the brain circuitry involved in the control of fear responses to overreact either to subsequent episodes of hypoxia and hypercapnia due to COPD or to sensations previously associated with these states (e.g., dyspnea) (Citation[153]).
In addition, patients may become more vigilant to respiratory sensations and react to these sensations with increased anxiety and physiologic arousal. This may produce an escalating cycle of increasing anxiety that leads to a panic attack (Citation[154]). Panic disorder is common in patients with COPD, with approximately one-third of COPD patients reporting having panic attacks (Citation[127], Citation[155]), and approximately 8% meeting full psychiatric criteria for the disorder (Citation[126]). Fearful thoughts and beliefs about respiratory symptoms can increase risk for anxiety and panic attacks. The common development of these frightening thoughts is understandable considering COPD can be associated with near-death episodes, need for ventilatory support, and other illness experiences.
Hyperventilation could be another contributor to COPD-related anxiety. In COPD, increased ventilation may be partly adaptive in counteracting hypercapnic states caused by airway obstruction. However, excessive ventilation could lead to counterproductive effects, such as airway narrowing and increased respiratory distress.
Living with a chronic illness like COPD may lead to feelings of frustration, helplessness, and hopelessness in patients as they struggle with the impact of COPD on their physical, psychologic, and social functioning. COPD patients who report worse HRQL as a result of having COPD are more likely to have problems with anxiety and depression (Citation[131]). Poorer general health, decreased physical functioning, impaired social functioning, and frequency of exacerbations have all been shown to contribute to worse HRQL in COPD patients, as well as to anxiety and depression (Citation[134]).
In some cases, COPD may not be the primary cause of increased rates of anxiety and depression. The onset of anxiety or depression long precedes the onset of COPD in at least a subset of patients. Cigarette smoking rates are higher among those with anxiety and depression (Citation[156], Citation[157]). Thus, as a result, pre-morbid anxiety and depression may be overrepresented in COPD. However, it is still unclear whether common factors influence the development of anxiety, depression, smoking, and COPD. Indeed, it is unknown whether anxiety and depression lead to smoking or whether the converse is true. More complex potential mechanisms include the possibility that anxiety and depression, in the presence of smoking, may increase the risk for developing COPD. Finally, pre-existing vulnerabilities to anxiety or depression may also lead to other clinical disorders in the presence of the consequent physiologic and psychologic stress of COPD.
Does COPD treatment impact on anxiety/depression?
Given the strong relationship between COPD, anxiety, and depression, it is surprising that few studies have directly examined the impact of COPD treatments on these important psychiatric outcomes. Studies with pharmacologic treatment have largely excluded specific assessments of anxiety or depression from their outcome measures. However, several studies have used HRQL instruments, which typically include emotional or psychosocial functioning components (Citation[30], Citation[31], Citation[158], Citation[159]). In as much as these measures can be taken as a proxy for anxiety and depression status, studies generally support the hypothesis that pharmacologic treatments that improve or stabilize physiologic functioning in patients with COPD also positively impact anxiety and depression symptoms. Studies of non-pharmacologic treatments for COPD have more often included direct measures of anxiety and depression. In this regard, pulmonary rehabilitation (Citation[160], Citation[161]), long-term oxygen therapy (Citation[162]), and COPD self-management programs (Citation[163]) have all demonstrated significant and specific benefits for anxiety and depression in COPD patients.
The mechanisms by which treatments for COPD reduce anxiety and depression have not been well studied. Changes in hypothalamo-pituitary-adrenal system functioning have been implicated as a mediator of the beneficial effect of exercise on clinical depression in patients without COPD (Citation[164], Citation[165]) and may also partly explain some of the benefits observed in COPD patients following pulmonary rehabilitation. Pulmonary rehabilitation (Citation[166]), long-term oxygen therapy (Citation[89]), and self-management programs (Citation[167]) induce decreases in dyspnea in COPD patients. These reductions in dyspnea potentially increase tolerance of activity and trigger fewer panic attacks. Oxygen therapy may additionally partially restore neurocognitive functioning, leading to improvements in coping capacity.
Treatment programs may also increase self-confidence in the patient's ability to manage their disease more effectively, leading to decreased feelings of frustration, helplessness, and hopelessness, and improvements in anxiety and depression. Central cholinergic hyperactivity has been implicated in some biologic models of depression (Citation[168]), and may be affected by medications with anticholinergic effects, though the commonly used topical anticholinergic bronchodilators are not believed to have significant effects on the central nervous system.
CARDIOVASCULAR CO-MORBIDITY
Cardiovascular disease is a major co-morbid condition in patients with COPD (Citation[4], Citation[169]). “Cardiovascular disease” is a term that encompasses several diverse disease processes with different etiologies relating to COPD in different ways. These include pulmonary vascular disease, congestive heart failure, coronary artery disease, peripheral vascular disease, and occasionally may also include biologic markers of disease, such as lipid abnormalities and inflammatory markers.
What is the impact of COPD on cardiovascular function and cardiovascular disease?
The relationship between COPD and the components of cardiovascular disease is complex. For example, it is possible that, in some cases, the presence of COPD leads to the development of cardiovascular disease; in other cases, a shared risk factor (i.e., smoking) may result in development of both COPD and cardiovascular disease; and, in further cases, cardiovascular disease might result in abnormalities in the respiratory system (e.g., cardiac asthma).
In general, people with more severe lung function impairment are more likely to develop adverse outcomes. In an analysis of data from NHANES, Sin et al. demonstrated that cardiovascular mortality was associated with reduced lung function at all levels of lung function, even with small decrements within the “normal range.” This relationship was also noted among non-smokers in a meta-analysis of published studies (Citation[6]).
What are the mechanisms of this association?
While the precise mechanisms for these extra-pulmonary manifestations of COPD are unknown, spillover of inflammatory cells from the lung into the systemic circulation, tissue hypoxemia, and inactivity have been suggested. These may be related through neurohumoral activation (Citation[170]) or oxidative stress and activation of TNF-α (Citation[21]). Alternatively, the associations may be independently related to the causes of COPD, such as smoking or other factors. Since intervention studies investigating the link between COPD and cardiovascular disease have largely had mortality as an endpoint, these are specifically discussed below. Other factors that may contribute to this association include inactivity/deconditioning, hypoxemia, pulmonary hypertension, etc.
What effect can maintaining optimal lung function and impacting the clinical course of COPD have on cardiovascular function and cardiovascular disease?
It is unknown whether maintaining optimal lung function will ultimately improve cardiovascular outcomes, and this is an area that merits additional study. It is likely, however, that any intervention that increases the capacity for physical activity (assuming of course that the patient becomes more physically active) would result in improved cardiovascular outcomes. However, the effects of bronchodilator treatment on activity levels have, hitherto, not been studied. Smoking cessation is undoubtedly beneficial, probably as is avoidance of other respiratory irritants. Similarly, one would hope that decreasing exacerbations would also result in better cardiovascular outcomes.
MORTALITY
During 2000, COPD was responsible for over 119,000 deaths in the United States (Citation[171]). During the period 1980–2000, the most substantial change in the COPD death rate was seen among women, increasing from 20.1/100,000 in 1980 to 56.7/100,000 in 2000, compared with the more modest increase in death rate among men, from 73.0/100,000 in 1980 to 82.6/100,000 in 2000. In 2000, for the first time, the number of women dying from COPD surpassed the number of men dying from COPD (59,936 versus 59,118) (Citation[171]).
What are the causes of death in COPD?
One of the limitations of the U.S. mortality files is that many decedents with COPD have their death attributed to another cause (Citation[172]). For example, in 1998 only 45.4% of the 233,610 decedents with COPD mentioned anywhere on their death certificates had this coded as the underlying cause of death. Other research has shown that people with COPD listed anywhere on their death certificate frequently had severe COPD (Citation[173]). Progressive respiratory failure accounts for approximately one-third of the COPD-related mortality, hence, factors other than progression of lung disease would be expected to play a substantial role in COPD-related mortality (Citation[174], Citation[175]).
A long-standing history of smoking tobacco in COPD patients may increase the risk for developing co-morbidities, such as cardiovascular disease and cancer. In a nationally representative sample of 47 million hospitalizations from 1979 to 2001, hospital discharges with a diagnosis of COPD were more likely to have been hospitalized with pneumonia, hypertension, heart failure, ischemic heart disease, pulmonary vascular disease, thoracic malignancies, and ventilatory failure, when compared with age-adjusted discharges without COPD. Furthermore, having a diagnosis of COPD was associated with higher age-adjusted in-hospital mortality for pneumonia, hypertension, heart failure, ventilatory failure, and thoracic malignancies, when compared with hospital discharges with these co-morbidities without COPD. These results suggest that the burden of disease associated with COPD is largely under-estimated, since having a diagnosis of COPD is associated with an increased risk for hospitalization and in-hospital mortality from other common diagnoses (Citation[176]).
One method of determining the causes of death associated with COPD is to follow populations prospectively for several years. In an analysis of data from the Atherosclerosis Risk in Communities (ARIC) study, subjects with severe or moderate COPD had a higher mortality than subjects with normal lung function (42.9 and 18.1 deaths per 1,000 person years vs. 5.4 deaths per 1,000 person years, respectively) (Citation[177]). Among people with severe COPD (GOLD Stage III or IV), 32% of the deaths were due to respiratory causes, whereas 24% were due to lung cancer and 13% were cardiac related (). Among people with moderate COPD (GOLD Stage II), only 4% of the deaths were respiratory related, 25% were due to lung cancer and 28% were cardiac related. These data are similar to data from other prospective research (Citation[178]), and suggest that a substantial proportion of the early mortality among COPD patients, particularly those with mild or moderate disease, is related to cardiac disease, lung cancer, and other causes.
What have intervention studies taught us about the causes of death in COPD?
Increasing life expectancy and improving the HRQL in patients with COPD is a goal towards which clinicians strive. Achieving this in most patients, however, has not been possible and, in the context of randomized controlled trials, is difficult to demonstrate. Even though landmark trials, such as Towards a Revolution in COPD Health (TORCH) (Citation[179]) and UPLIFT® (Citation[42]), have enrolled large numbers of patients, a persistent problem is that sicker people are more likely to drop out of studies, thereby potentially biasing the results towards not finding an effect of interventions.
Nevertheless, two interventions have clearly been shown to improve survival in COPD patients. The first is smoking cessation. People who stop smoking, even in middle age, have a survival benefit (Citation[180]). The Lung Health Study clearly demonstrated that current smokers who were able to stop smoking had less lung function decline and improved survival (Citation[37], Citation[38]). It is not clear, however, whether the observed mortality benefit is related to fewer COPD mortalities or fewer of the other causes of mortality associated with smoking (heart disease, cancers, etc).
The second intervention known to improve survival is long-term oxygen therapy. Studies from 30 years ago have demonstrated that patients with hypoxemia have improved survival if they stay on oxygen therapy for more than 16 hours each day (Citation[181], Citation[182]). With the advancement in medicine over the past 30 years, however, questions are raised whether this mortality benefit continues to exist and whether groups other than those with resting hypoxemia can benefit from oxygen therapy.
Beyond these two interventions, data for delaying mortality in COPD are sparse, but there are some promising areas of investigation. Data from TORCH suggest that the combination of fluticasone and salmeterol may provide a degree of long-term mortality benefit, though the difference compared with placebo was not statistically significant (Citation[179]). Patients who have reversibility and are adherent to therapy may have better survival (Citation[183]). COPD exacerbations are associated with mortality, so one might expect treatments that reduce exacerbation rate to decrease mortality (Citation[142], Citation[184]). Similarly, respiratory symptoms and inactivity also predict mortality, so interventions that decrease these symptoms and increase a patient's activity levels could be expected to decrease mortality (Citation[177], Citation[185]).
Would early treatment of COPD impact mortality?
A hallmark of medical interventions is that earlier treatment of disease, before complications develop, results in better outcomes than later treatment of disease. This is certainly the case in the treatment of chronic diseases such as diabetes and renal failure, and in the treatment of malignant disease such as breast cancer. Thus, it is probable that earlier detection and intervention in COPD would result in better outcomes, though this has not been proven.
A substantial proportion of COPD in the United States is undiagnosed, particularly in its earlier stages (Citation[13]). As noted, the only proven intervention in early COPD is smoking cessation. However, there is currently no evidence from randomized trials that knowledge of impaired lung function results in higher rates of smoking cessation (Citation[186]), though a recent non-randomized trial from Poland suggests that this may occur (Citation[187]). Results from TORCH and UPLIFT®, may point to other interventions in addition to smoking cessation for early-stage COPD (Citation[188]). Some evidence in support of these interventions is available. A nested case-control study, for example, has suggested that exposure to low (100 μ g/day) but not high (300 μ g/day) doses of inhaled steroids resulted in a decreased risk of acute myocardial infarction (Citation[189]).
Currently, interventions in COPD are primarily aimed at reducing patient symptoms and improving their HRQL. Some patients with poor lung function are able to maintain active lifestyles, whereas many others do not. The hope is that, if patients are assisted in maintaining active and healthy lifestyles, despite their disease, they will have both a longer life and a higher HRQL. Since, average activity levels decrease at all levels of COPD, and since cardiovascular disease is the major cause of death in mild and moderate COPD, any intervention that increases activity levels over the long term would probably result in improved survival.
SYSTEMIC EFFECTS OF EXACERBATIONS
Although much has been learnt about the systemic effects of COPD, the information regarding systemic effects of exacerbations is relatively limited. Studies to measure the systemic effects of exacerbations should ideally be of a longitudinal design, where patients are assessed not only in the stable state prior to the exacerbation, but also during the exacerbation, and following resolution of the exacerbation. Such a design would possibly allow separation of the systemic effects of exacerbation from the systemic effects of COPD itself, as well as to determine the time-frame of resolution of these systemic effects. An easier approach is to perform cross-sectional studies of systemic effects at the time of exacerbations or to compare systemic effects between patients who experience frequent exacerbations and those that have infrequent exacerbations.
What are the mechanisms for systemic effects of exacerbations?
Both the local and systemic inflammation associated with COPD increase with exacerbations. Papi et al. showed that blood neutrophil counts are increased during exacerbations and that the extent of this increase correlates with the decrement in FEV1 with the exacerbation (Citation[190]). Oudijk et al. demonstrated that blood neutrophils are primed during exacerbations, and that this priming phenotype correlates with the amount of oxidative stress in the airways and the severity of the exacerbation as measured by the Borg score (Citation[191]). Several mediators and markers of inflammation have been reported to be increased with exacerbations. These include serum CRP, IL-6, IL-8, myeloid progenitor inhibitory factor-1, pulmonary and activation-regulated chemokine, adipocyte-specific serum protein-30, soluble intercellular adhesion molecule-1, serum amyloid-A, and pro-calcitonin. It is noteworthy that inflammation and oxidative stress are intimately related. Hence, it is not surprising that markers of oxidative stress and reduced antioxidant status were reported to be elevated in patients suffering from acute exacerbations (Citation[51], Citation[192]).
More important is whether this heightened state of systemic inflammation has any clinical consequences, especially among the classic systemic consequences of COPD. One example noted above, is that Creutzberg et al. showed that dietary intake declines during an exacerbation and that the resting energy expenditure increases (Citation[125]), which may explain the weight loss and nutritional depletion seen in patients with COPD. Moreover, they were able to show an inverse correlation between the dietary intake/resting energy expenditure with serum leptin and soluble TNF receptor-55, suggesting that systemic inflammation mediates this energy imbalance.
Systemic inflammation in exacerbations may also contribute to the muscle weakness in patients with COPD. Skeletal muscle weakness in these patients is undoubtedly multi-factorial; however, peripheral muscle force, as measured by quadriceps peak torque, was diminished during hospitalized exacerbations of COPD and correlated with increases in serum IL-8 (Citation[29]). The increased cardiovascular deaths in COPD patients may also be related, at least in part, to exacerbations. It is possible that exacerbations increase the risk of acute ischemic heart disease through increased systemic inflammation or by inducing a pro-coagulant state, in addition to profound inactivity, hypoxemia, pulmonary vascular disease, etc.
In all the above studies, all exacerbations were regarded to be similar in their systemic consequences. However, exacerbations are of varied etiology and the etiology can influence the inflammatory process associated with exacerbations and, likely, their systemic effects. A recent study has examined whether elevations in serum CRP were associated with etiology of exacerbations (Citation[193]). Exacerbations that were bacterial, as identified by acquisition of new strains of bacterial pathogens, were clearly associated with much larger increases in serum CRP from baseline. Furthermore, these increases in serum CRP could extend beyond clinical resolution and were associated with early recurrence of an exacerbation. Hence, the systemic consequences of exacerbations may persist longer than the clinical symptoms.
Though the focus in many studies has been on systemic inflammation, non-inflammatory mechanisms could also contribute to the systemic effects of exacerbations. Walking time and time spent outside are known to be reduced during exacerbations (Citation[43], Citation[44]). However, these studies did not directly correlate these changes with decrements in peripheral muscle force. Nevertheless, the inactivity and bed rest associated with exacerbations could contribute to the enhanced bone loss in COPD.
Can treatment of exacerbations reduce these systemic effects?
Adequate treatment of exacerbations may restrict the systemic effects. Alternatively, the adverse effects of treatment could actually worsen the systemic effects of COPD. For example, use of systemic corticosteroids on a long-term basis worsens the skeletal muscle weakness and bone loss associated with COPD. Decramer et al. determined that, in a group of patients hospitalized for COPD exacerbations, respiratory and skeletal muscle strength was associated with average daily dose of corticosteroids (Citation[72]). Much of this steroid use was in the form of pulse therapy for exacerbations.
A decrease in the frequency of exacerbations of COPD is the optimal prevention of systemic effects due to exacerbations; however, the preventive therapies used should not worsen the systemic effects on their own. The influence of current preventative therapies on systemic effects of COPD is largely unknown. Unfortunately, studies that have examined the prevention of exacerbations with bronchodilators did not measure systemic effects of COPD to determine if these were improved in line with the reduction of exacerbations. Data with inhaled steroids are unclear, with triamcinolone being associated with a decrement in bone density (Citation[194]), while such an effect was not seen with fluticasone (Citation[195]).
IMPACT OF TREATMENT ON SYSTEMIC CONSEQUENCES OF COPD
COPD is a disease of progressive airway inflammation resulting in partially reversible airflow obstruction (Citation[196]). As a result of the airflow obstruction, and in some cases co-existent emphysema, COPD patients develop air trapping and hyperinflation. As noted, this in turn leads to other more complex pathophysiology with a main consequence being activity limitation (Citation[45]). More severe COPD is characterized by exacerbations that occur with increasing frequency (Citation[197]). Exacerbations worsen the pathophysiology but also, themselves, are associated with increased healthcare utilization, worsening HRQL, and increased mortality (Citation[198]).
Understanding the interrelationships between airway inflammation, pathophysiology, systemic inflammation and exacerbations reveals 4 essential mechanisms by which effective treatment could benefit patients with COPD. Firstly, effective treatment could reduce the pathophysiologic consequences of airway inflammation (without necessarily suppressing inflammation itself) and thus improve patient-reported outcomes. Secondly, effective treatment could directly suppress airway inflammation, thereby reducing systemic inflammation, and systemic consequences.
Thirdly, effective treatment could prevent exacerbations (through hitherto unknown mechanisms), thus reducing airway inflammation, systemic inflammation, and systemic consequences. Finally, effective treatment could be directed at the systemic consequences themselves rather than at the underlying COPD (e.g., exercise prescription, anabolic supplementation, antidepressants, etc).
Impact of treatment on pathophysiology
The role of various classes of bronchodilator drugs has been exhaustively studied in COPD (). Guidelines ascribe roles for anticholinergic, β -adrenergic agonists, and theophylline (Citation[199]). However, in terms of clinical decision-making, it has been recommended to prescribe short-acting bronchodilators for mild, intermittent symptoms, and to introduce long-acting bronchodilators as maintenance therapy for patients with persistent symptoms, including activity limitation (Citation[199]). However, this regimen may not be ideal. Mild, intermittent symptoms are likely to be the result of dynamic hyperinflation induced by exercise. More regular therapy with bronchodilators to provide 24-hour bronchodilation may improve overall activity and prevent symptoms associated with dynamic hyperinflation (Citation[200]).
Figure 5 Schematic representation of the primary and potential effects of a bronchodilator on the systemic consequences (A) and co-morbidities (B) of COPD.
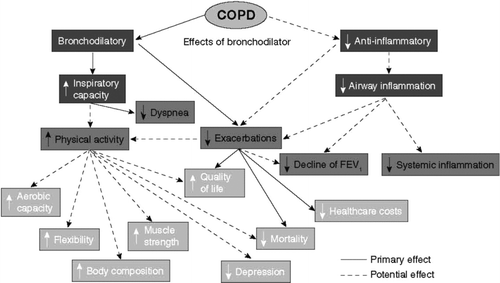
Bronchodilators relax airway smooth muscle, but also reduce static hyperinflation, an effect that has been demonstrated for theophylline (Citation[201]), albuterol (Citation[202], Citation[203]), salmeterol (Citation[83]), ipratropium (Citation[204]), and tiotropium (Citation[205]). Several of these studies also demonstrated amelioration of dynamic hyperinflation; notably with salmeterol (Citation[83]), ipratropium, (Citation[204]) and tiotropium (Citation[206], Citation[207]). It is reasonable to conclude that, depending on the bronchodilator responsiveness of the individual COPD patient, the more effective the bronchodilator, the greater the impact will be on hyperinflation (Citation[45]).
Other therapeutic or experimental interventions also reduce static and/or dynamic hyperinflation. These include supplemental oxygen (Citation[89]), exercise training (Citation[88]), helium-oxygen breathing (Citation[208], Citation[209]), and bronchoscopic lung volume reduction surgery (Citation[210]). Supplemental oxygen seems to have a substantial effect on dynamic but not static hyperinflation by reducing respiratory rate (Citation[89]). The effects with other non-bronchodilator therapies are noticeably less than with bronchodilators (Citation[45]).
Impact of treatment on inflammation
There have been multiple endeavors to discover a drug with direct anti-inflammatory effects in the airway, but convincing evidence remains scarce. Selective β2-adrenoceptor-agonists have yielded disappointing results. The cholinergic pathway, however, may have a previously unrecognized important role in mediating inflammation in the lung. Koyama et al. have shown that acetylcholine, acting via the muscarinic receptor, stimulates bronchial epithelial cells to release neutrophil and monocyte chemotactic activity (Citation[211]). Furthermore, acetylcholine-stimulated alveolar macrophages induce neutrophil chemotaxis and this response is blocked by 4-diphenylacetoxy-N-methylpiperidine methobromide (an M3 muscarinic antagonist) but not by pirenzipine (an M1 antagonist) or gallamine (an M2 antagonist) (Citation[212]). Thus, it is interesting that tiotropium also suppressed acetylcholine induced macrophage migrationin vitro (Citation[213]). Using a smoking mouse model, Fukuchi et al. showed that nebulized tiotropium reduced the accumulation of neutrophils and macrophages in bronchoalveolar lavage fluid in a dose-dependent manner (Citation[214]).
Phosphodiesterase (PDE) inhibitors are a family of enzymes involved in the degradation of cyclic adenosine monophosphate, which is a modulator of inflammation. Selective PDE4 inhibitors possess anti-inflammatory activity in vitro (Citation[215]). Cilomilast reduces CD8+ T cells in the airway mucosa of patients with COPD (Citation[216]). Roflumilast has been shown to reduce acute inflammation induced by tobacco smoke in a smoking mouse model (Citation[217]). Whether these PDE4 inhibitors will find a role in the clinical management of COPD is yet to be established. Other anti-inflammatory drugs currently under investigation include TNF-α antagonists, peroxisome proliferator-activated receptor-γ agonists, transforming growth factor-β inhibitors, matrix metalloproteinase inhibitors and leukotriene B4 antagonists (Citation[218]).
Evidence of benefit from earlier improvement of lung function
Although there is currently no direct evidence of a benefit from maintaining optimal lung function with maintenance therapy earlier in the course of COPD, there is considerable circumstantial evidence to support such a treatment strategy. By inference from clinical experience and certain published studies, we can deduce that, as pulmonary function declines, many of the accompanying systemic consequences worsen in parallel. Foglio et al. showed that the decline in FEV1 is accompanied by progressively worsening hyperinflation (Citation[219]). Pitta et al. have shown that physical activity declines with worsening disease severity, particularly in the transition from GOLD Stage II to III (Citation[59]). A lack of adequate physical activity predictably leads to deconditioning and an accelerated rate of decline in aerobic performance, as has been reported in a cohort of COPD patients in Japan (Citation[220]). Activity limitation, therefore, plays a central role in the development of systemic consequences and is likely to influence multiple co-morbidities.
Maintenance of physical activity is also vitally important in other diseases as well as apparently healthy older adults. Myers, et al. have used a simple exercise intensity scale, the Veterans Specific Activity Questionnaire, to assess habitual activity levels in a variety of patients with chronic diseases (Citation[221]). Their study showed that the level of physical activity independently predicted mortality in patients with hypertension, diabetes mellitus, and obesity, as well as apparently healthy smokers and those with COPD. Other investigators have shown that daily energy expenditure through physical activity was associated with a lower risk of mortality in healthy older adults (Citation[222]). In this study of 302 high-functioning, community-dwelling older adults (aged 70–82), followed on average for 6.15 years, the absolute risk of death was 12.1% in the highest tertile vs. 24.7% in the lowest tertile for energy expenditure and activity level. These effects changed little after adjustment for smoking status, educational level, and prevalent health conditions.
Further important circumstantial evidence is provided in the study that assessed the effect of maintenance treatment with a long-acting bronchodilator (tiotropium) on the improvement in exercise endurance gained from participation in a pulmonary rehabilitation program (Citation[92]). After 8 weeks of structured exercise training with target work rates, patients randomized to receive tiotropium had an average endurance time for a sub-maximal constant work rate exercise test on a treadmill ergometer that was 5.35 minutes longer than patients receiving only a short-acting β2-adrenergic-agonist (albuterol). Perhaps more impressive was the difference in endurance time after a further 12 weeks of follow-up, during which the patients received no further supervised exercise training. The patients taking tiotropium maintained their exercise endurance whereas those taking placebo in addition to albuterol experienced a gradual decline in exercise endurance. Although the evidence is indirect, these observations likely indicate that prescription of the long-acting bronchodilator enabled the patients to maintain a higher level of physical activity and thus preserve their exercise endurance.
Considering that exacerbations increase in frequency as pulmonary function declines and begin to manifest themselves when FEV1falls below 50% of predicted, we can deduce that maintenance of optimal lung function should forestall the development of exacerbations. Indeed, several inhaled therapies have been shown to reduce the frequency of exacerbations including tiotropium, long-acting β2-adrenergic-agonists and inhaled corticosteroids (Citation[35], Citation[223], Citation[224]). A compelling hypothesis to explain this effect is that maintenance of airway patency improves clearance of secretions and thus impacting bacterial colonization and averting exacerbations. Furthermore, if exacerbations truly accelerate the decline in FEV1 (Citation[225]), reduction in exacerbations should have a favorable effect on disease progression.
CONCLUSIONS
The systemic consequences and co-morbidities of COPD have great clinical impact on the natural course of COPD, on patient-reported outcomes and on mortality. Future clinical studies, including clinical trials with new drugs, will need to include patients with systemic consequences and co-morbidities rather than excluding them systematically, as was hitherto the case. As a consequence of the exclusion of patients with co-morbidities, trials have often been performed on selected populations that are not representative for the COPD population as a whole. Clinical guidelines, based largely on the results of these trials, are not likely to address adequately these co-morbidities. While co-morbidities increase with increasing disease severity, major COPD-related systemic consequences and co-morbidities, such as inactivity, osteoporosis, metabolic impact, cardiovascular disease, and enhanced mortality, are present in GOLD Stage II and, hence, in mild disease. In these patients, the co-morbidities are likely the major clinical feature. Thus, future research needs to characterize the early stages of COPD and the response to treatment of both the lung disease and the systemic manifestations.
The authors acknowledge the financial support of Boehringer Ingelheim (Ingelheim, Germany) and Pfizer Inc. (New York, USA) in the organization of the Round Table meeting held on 9–10 December 2006 from which this manuscript was developed.
REFERENCES
- Pauwels R A, Buist A S, Calverley P M, Jenkins C R, Hurd S S, GOLD Scientific Committee. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop Summary. Am J Respir Crit Care Med 2001; 163: 1256–1276
- Murray C JL, Lopez A D. Alternative projections of mortality and disability by cause 1990–2020: Global burden of disease study. Lancet 1997; 349: 1498–1504
- Lopez A D, Shibuya K, Rao C, Mathers C D, Hansell A L, Held L S, Schmid V, Buist S. Chronic obstructive pulmonary disease: current burden and future projections. Eur Respir J 2006; 27: 397–412
- Mapel D W, Hurley J S, Frost F J, Petersen H V, Picchi M A, Coultas D B. Health care utilization in chronic obstructive pulmonary disease. A case-control study in a health maintenance organization. Arch Intern Med 2000; 160: 2653–2658
- Sorlie P D, Kannel W B, O'Connor G. Mortality associated with respiratory function and symptoms in advanced age. The Framingham Study. Am Rev Respir Dis 1989; 140: 379–384
- Sin D D, Wu L, Man S F. The relationship between reduced lung function and cardiovascular mortality: a population-based study and a systematic review of the literature. Chest 2005; 127: 1952–1959
- Mannino D M, Aguayo S M, Petty T L, Redd S C. Low lung function and incident lung cancer in the United States: data From the First National Health and Nutrition Examination Survey follow-up. Arch Intern Med 2003; 163: 1475–1480
- O'Donnell D E, Revill S M, Webb K A. Dynamic hyperinflation and exercise intolerance in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164: 770–777
- Gosselink R, Troosters T, Decramer M. Peripheral muscle weakness contributes to exercise limitation in COPD. Am J Respir Crit Care Med 1996; 153: 976–980
- Engelen M P, Schols A M, Lamers R J, Wouters E F. Different patterns of chronic tissue wasting among patients with chronic obstructive pulmonary disease. Clin Nutr 1999; 18: 275–280
- Antonelli Incalzi R, Fuso L, De Rosa M, Forestiere F, Rapiti E, Nardecchia B, Pistelli R. Co-morbidity contributes to predict mortality of patients with chronic obstructive pulmonary disease. Eur Respir J 1997; 10: 2794–2800
- Marquis K, Maltais F, Duguay V, Bezeau A M, LeBlanc P, Jobin J, Poirier P. The metabolic syndrome in patients with chronic obstructive pulmonary disease. J Cardiopulm Rehabil 2005; 25: 226–232
- Mannino D M, Gagnon R C, Petty T L, Lydick E. Obstructive lung disease and low lung function in adults in the United States: data from the National Health and Nutrition Examination Survey, 1988-1994. Arch Intern Med 2000; 160: 1683–1689
- Sin D D, Man J P, Man S F. The risk of osteoporosis in Caucasian men and women with obstructive airways disease. Am J Med 2003; 114: 10–14
- Bolton C E, Ionescu A A, Shiels K M, Pettit R J, Edwards P H, Stone M D, Nixon L S, Evans W D, Griffiths T L, Shale D J. Associated loss of fat-free mass and bone mineral density in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2004; 170: 1286–1293
- Similowski T, Agusti A, MacNee W, Schönhofer B. The potential impact of anaemia of chronic disease in COPD. Eur Respir J 2006; 27: 390–396
- Cote C, Zilberberg M D, Mody S H, Dordelly L J, Celli B. Haemoglobin level and its clinical impact in a cohort of patients with COPD. Eur Respir J 2007; 29: 923–929
- Wagena E J, Arrindell W A, Wouters E F, van Schayck C P. Are patients with COPD psychologically distressed?. Eur Respir J 2005; 26: 242–248
- Sin D D, Anthonisen N R, Soriano J B, Agusti A G. Mortality in COPD: role of comorbidities. Eur Respir J 2006; 28: 1245–1257
- Barnes P J. Chronic obstructive pulmonary disease (review article). N Engl J Med 2000; 343: 269–280
- Sevenoaks M J, Stockley R A. Chronic Obstructive Pulmonary Disease, inflammation and co-morbidity—a common inflammatory phenotype?. Respir Res 2006; 7: 70
- Di Francia M, Barbier D, Mege J L, Orehek J. Tumor necrosis factor-alpha levels and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1994; 150: 1453–1455, (5 Pt 1)
- Schols A MWJ, Buurman W A, Staal van den Brekel A J, Dentener M A, Wouters E F. Evidence for a relation between metabolic derangements and increased levels of inflammatory mediators in a subgroup of patients with chronic obstructive pulmonary disease. Thorax 1996; 51: 819–824
- Gan W Q, Man S F, Senthilselvan A, Sin D D. Association between chronic obstructive pulmonary disease and systemic inflammation: a systematic review and a meta-analysis. Thorax 2004; 59: 574–580
- Broekhuizen R, Wouters E F, Creutzberg E C, Schols A M. Raised CRP levels mark metabolic and functional impairment in advanced COPD. Thorax 2006; 61: 17–22
- Sauleda J, Noguera A, Busquets X, Miralles C, Villaverde J M, Agusti A GN. Systemic inflammation during exacerbations of chronic obstructive pulmonary disease. Lack of effect of steroid treatment [abstract]. Eur Respir J 1999; 14: 359s
- Asin J, Maesen B LP, van den Bosch J MM, Schramel F MNH, van Grinsven S MGE, Gerritsen W BM, Haas F JLM. Serum interleukin-8, circulating cell adhesion molecules and hydrogen peroxide in exhaled air in patients with unstable chronic obstructive pulmonary disease during treatment with corticosteroids [abstract]. Eur Respir J 1998; 12: 193s
- Wedzicha J A, Seemungal T A, MacCallum P K, Paul E A, Donaldson G C, Bhowmik A, Jeffries D J, Meade T W. Acute exacerbations of chronic obstructive pulmonary disease accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost 2000; 84: 210–215
- Spruit M A, Gosselink R, Troosters T, Kasran A, Gayan-Ramirez G, Bogaerts P, Bouillon R, Decramer M. Muscle force during an acute exacerbation in hospitalised patients with COPD and its relationship with CXCL8 and IGF-I. Thorax 2003; 58: 752–756
- Casaburi R, Mahler D A, Jones P W, Wanner A, San P G, Zu Wallack R L, Menjoge S S, Serby C W, Witek T, Jr. A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J 2002; 19: 217–224
- Vincken W, van Noord J A, Greefhorst A P, Bantje T A, Kesten S, Korducki L, Cornelissen P J. Dutch/Belgian Tiotropium Study Group. Improved health outcomes in patients with COPD during 1 yr's treatment with tiotropium. Eur Respir J 2002; 19: 209–216
- Niewoehner D E, Rice K, Cote C, Paulson D, Cooper J A, Jr, Korducki L, Cassino C, Kesten S. Prevention of exacerbations of chronic obstructive pulmonary disease with tiotropium, a once-daily inhaled anticholinergic bronchodilator: a randomized trial. Ann Intern Med 2005; 143: 317–326
- Calverley P, Pauwels R, Vestbo J, Jones P, Price N, Gulsvik A, Anderson J, Maden C, Trial of Inhaled Steroids And long-acting beta2 agonists study group. Combined salmeterol and fluticasone in the treatment of chronic obstructive pulmonary disease: a randomised controlled trial. Lancet 2003; 361: 449–456
- Calverley P M, Boonsawat W, Cseke Z, Zhong N, Peterson S, Olsson H. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J 2003; 22: 912–919
- Sin D D, McAlister F A, Man S F, Anthonisen N R. Contemporary management of chronic obstructive pulmonary disease: scientific review. JAMA 2003; 290: 2301–2312
- Anthonisen N R, Connett J E, Kiley J P, Altose M D, Bailey W C, Buist A S, Conway W A, Jr, Enright P L, Kanner R E, O'Hara P. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. JAMA 1994; 272: 1497–1505
- Anthonisen N R, Connett J E, Murray R P. Smoking and lung function of Lung Health Study participants after 11 years. Am J Respir Crit Care Med 2002; 166: 675–679
- Anthonisen N R, Skeans M A, Wise R A, Manfreda J, Kanner R E, Connett J E, Lung Health Study Research Group. The effects of a smoking cessation intervention on 14.5-year mortality: a randomized clinical trial. Ann Intern Med 2005; 142: 233–239
- Anzueto A, Tashkin D, Menjoge S, Kesten S. One-year analysis of longitudinal changes in spirometry in patients with COPD receiving tiotropium. Pulm Pharmacol Ther 2005; 18: 75–81
- Vestbo J. The TORCH (towards a revolution in COPD health) survival study protocol. Eur Respir J 2004; 24: 206–210
- Sutherland E R, Allmers H, Ayas N T, Venn A J, Martin R J. Inhaled corticosteroids reduce the progression of airflow limitation in chronic obstructive pulmonary disease: a meta-analysis. Thorax 2003; 58: 937–941
- Decramer M, Celli B, Tashkin D P, Pauwels R A, Burkhart D, Cassino C, Kesten S. Clinical trial design considerations in assessing long-term functional impacts of tiotropium in COPD: the UPLIFT® trial. COPD 2004; 1: 303–312
- Donaldson G C, Wilkinson T M, Hurst J R, Perera W R, Wedzicha J A. Exacerbations and time spent outdoors in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 171: 446–452
- Pitta F, Troosters T, Probst V S, Spruit M A, Decramer M, Gosselink R. Physical activity and hospitalization for exacerbation of COPD. Chest 2006; 129: 536–544
- Cooper C B. The connection between chronic obstructive pulmonary disease symptoms and hyperinflation and its impact on exercise and function. Am J Med 2006; 119: 21–31, (10 Suppl 1)
- Agusti A G, Noguera A, Sauleda J, Sala E, Pons J, Busquets X. Systemic effects of chronic obstructive pulmonary disease. Eur Respir J 2003; 21: 347–360
- Agusti A G. Systemic effects of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2: 367–370
- Wouters E F. Local and systemic inflammation in chronic obstructive pulmonary disease. Proc Am Thorac Soc 2005; 2: 26–33
- Vernooy J H, Küçükaycan M, Jacobs J A, Chavannes N H, Buurman W A, Dentener M A, Wouters E F. Local and systemic inflammation in patients with chronic obstructive pulmonary disease: soluble tumor necrosis factor receptors are increased in sputum. Am J Respir Crit Care Med 2002; 166: 1218–1224
- Rahman I, Morrison D, Donaldson K, MacNee W. Systemic oxidative stress in asthma, COPD, and smokers. Am J Respir Crit Care Med 1996; 154: 1055–1060, (4 Pt 1)
- Praticò D, Basili S, Vieri M, Cordova C, Violi F, Fitzgerald G A. Chronic obstructive pulmonary disease is associated with an increase in urinary levels of isoprostane F2alpha-III, an index of oxidant stress. Am J Respir Crit Care Med 1998; 158: 1709–1714
- Burnett D, Chamba A, Hill S L, Stockley R A. Neutrophils from subjects with chronic obstructive lung disease show enhanced chemotaxis and extracellular proteolysis. Lancet 1987; 2: 1043–1046
- Noguera A, Batle S, Miralles C, Iglesias J, Busquets X, MacNee W, Agusti A G. Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax 2001; 56: 432–437
- Noguera A, Busquets X, Sauleda J, Villaverde J M, MacNee W, Agusti A G. Expression of adhesion molecules and G proteins in circulating neutrophils in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 158: 1664–1668, (5 Pt 1)
- Li Y P, Schwartz R J, Waddell I D, Holloway B R, Reid M B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-kappaB activation in response to tumor necrosis factor alpha. FASEB J 1998; 12: 871–880
- Agusti A G, Sauleda J, Miralles C, Gomez C, Togores B, Sala E, Batle S, Busquets X. Skeletal muscle apoptosis and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166: 485–489
- Janssen S P, Gayan-Ramirez G, Van den Bergh A, Herijgers P, Maes K, Verbeken E, Decramer M. Interleukin-6 causes myocardial failure and skeletal muscle atrophy in rats. Circulation 2005; 111: 996–1005
- Booth F W, Gordon S E, Carlson C J, Hamilton M T. Waging war on modern chronic diseases: primary prevention through exercise biology. J Appl Physiol 2000; 88: 774–787
- Pitta F, Troosters T, Spruit M A, Probst V S, Decramer M, Gosselink R. Characteristics of physical activities in daily life in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 171: 972–977
- Belza B, Steele B G, Hunziker J, Lakshminaryan S, Holt L, Buchner D M. Correlates of physical activity in chronic obstructive pulmonary disease. Nurs Res 2001; 50: 195–202
- Decramer M, Gosselink R, Troosters T, Verschueren M, Evers G. Muscle weakness is related to utilization of health care resources in COPD patients. Eur Respir J 1997; 10: 417–423
- Marquis K, Debigaré R, Lacasse Y, LeBlanc P, Jobin J, Carrier G, Maltais F. Midthigh muscle cross-sectional area is a better predictor of mortality than body mass index in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002; 166: 809–813
- Nici L, Donner C, Wouters E, Zuwallack R, Ambrosino N, Bourbeau J, Carone M, Celli B, Engelen M, Fahy B, Garvey C, Goldstein R, Gosselink R, Lareau S, MacIntyre N, Maltais F, Morgan M, O'Donnell D, Prefault C, Reardon J, Rochester C, Schols A, Singh S, Troosters T, ATS/ERS Pulmonary Rehabilitation Writing Committee. American Thoracic Society/European Respiratory Society statement on pulmonary rehabilitation. Am J Respir Crit Care Med 2006; 173: 1390–1413
- Saey D, Debigare R, LeBlanc P, Mador M J, Cote C H, Jobin J, Maltais F. Contractile leg fatigue after cycle exercise: a factor limiting exercise in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 168: 425–430
- Sala E, Roca J, Marrades R M, Alonso J, Gonzalez de Suso J M, Moreno A, Barberá JÁ, Nadal J, do Jover L, Rodriguez-Roísin R, Wagner P D. Effects of endurance training on skeletal muscle bioenergetics in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159: 1726–1734
- Whittom F, Jobin J, Simard P M, LeBlanc P, Simard C, Bernard S, Belleau R, Maltais F. Histochemical and morphological characteristics of the vastus lateralis muscle in patients with chronic obstructive pulmonary disease. Med Sci Sports Exerc 1998; 30: 1467–1474
- Maltais F, LeBlanc P, Whittom F, Simard C, Marquis K, Bélanger M, Breton M J, Jobin J. Oxidative enzyme activities of the vastus lateralis muscle and the functional status in patients with COPD. Thorax 2000; 55: 848–853
- Rabinovich R A, Ardite E, Troosters T, Carbó N, Alonso J, Gonzalez de Suso J M, Vilaró J, Barberà J A, Polo M F, Argilés J M, Fernandez-Checa J C, Roca J. Reduced muscle redox capacity after endurance training in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164: 1114–1118
- Rabinovich R A, Ardite E, Mayer A M, Polo M F, Vilaró J, Argilés J M, Roca J. Training depletes muscle glutathione in patients with chronic obstructive pulmonary disease and low body mass index. Respiration 2006; 73(6)757–761
- Rabinovich R A, Bastos R, Ardite E, Llinàs L, Orozco-Levi M, Gea J, Vilaró J, Barberà J A, Rodriquez-Roísin R, Fernandez-Checa J C, Roca J. Mitochondrial dysfunction in COPD patients with low body mass index. Eur Respir J 2007; 29: 643–650
- Smorawiński J, Nazar K, Kaciuba-Uscilko H, Kamińska E, Cybulski G, Kodrzycka A, Bicz J E. Greenleaf Effects of 3-day bed rest on physiological responses to graded exercise in athletes and sedentary men. J Appl Physiol 2001; 91: 249–257
- Decramer M, Lacquet L M, Fagard R, Rogiers P. Corticosteroids contribute to muscle weakness in chronic airflow obstruction. Am J Respir Crit Care Med 1994; 150: 11–16
- Koechlin C, Couillard A, Simar D, Cristol J P, Bellet H, Hayot M, Prefaut C. Does oxidative stress alter quadriceps endurance in chronic obstructive pulmonary disease?. Am J Respir Crit Care Med 2004; 169: 1022–1027
- Powers S K, Kavazis A N, Deruisseau K C. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol 2005; 288: R337–R344
- Bruunsgaard H. Physical activity and modulation of systemic low-level inflammation. J Leukoc Biol 2005; 78: 819–835
- Van Vliet M, Spruit M A, Verleden G, Kasran A, Van Herck E, Pitta F, Bouillon R, Decramer M. Hypogonadism, quadriceps weakness, and exercise intolerance in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2005; 172: 1105–1111
- Jones R CM, Hyland M E, Hanney K, Erwin J. A qualitative study of compliance with medication and lifestyle modification in chronic obstructive pulmonary disease (COPD). Prim Care Respir J 2004; 13: 149–154
- De Peuter S, Van Diest I, Lemaigre V, Verleden G, Demedts M, Van den Bergh O. Dyspnea: the role of psychological processes. Clin Psychol Rev 2004; 24: 557–581
- NIH Consensus development program. Osteoporosis prevention, diagnosis and therapy. NIH consensus statement, Washington, DC 2000; 17: 1–45
- Lorentzon M, Mellström D, Haug E, Ohlsson C. Smoking is associated with lower bone mineral density and reduced cortical thickness in young men. J Clin Endocrinol Metab 2007; 92: 497–503
- Rochira V, Balestrieri A, Madeo B, Birilli L, Granata A R, Carani C. Osteoporosis and male age-related hypogonadism: role of sex steroids on bone (patho)physiology. Eur J Endocrinol 2006; 154: 175–185
- Oga T, Nishimura K, Tsukino M, Sato S, Hajiro T, Mishima M. Longitudinal deteriorations in patient reported outcomes in patients with COPD. Respir Med 2007; 101: 146–153
- O'Donnell D E, Voduc N, Fitzpatrick M, Webb K A. Effect of salmeterol on the ventilatory response to exercise in chronic obstructive pulmonary disease. Eur Respir J 2004; 24: 86–94
- O'Donnell D E, Sciurba F, Celli B, Mahler D A, Webb K A, Kalberg C J, Knobil K. Effect of fluticasone propionate/salmeterol on lung hyperinflation and exercise endurance in COPD. Chest 2006; 130: 647–656
- Couillard A, Maltais F, Saey D, Debigaré R, Michaud A, Koechlin C, LeBlanc P, Préfaut C. Exercise-induced quadriceps oxidative stress and peripheral muscle dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 167: 1664–1669
- Mador M J, Kufel T J, Pineda L A, Steinwald A, Aggarwal A, Upadhyay A M, Khan M A. Effect of pulmonary rehabilitation on quadriceps fatiguability during exercise. Am J Respir Crit Care Med 2001; 163: 930–935
- Casaburi R, Porszasz J, Burns M R, Carithers E R, Chang R S, Cooper C B. Physiologic benefits of exercise training in rehabilitation of patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997; 155: 1541–1551
- Porszasz J, Emtner M, Goto S, Somfay A, Whipp B J, Casaburi R. Exercise training decreases ventilatory requirements and exercise-induced hyperinflation at submaximal intensities in patients with COPD. Chest 2005; 128: 2025–2034
- Somfay A, Porszasz J, Lee S M, Casaburi R. Dose-response effect of oxygen on hyperinflation and exercise endurance in nonhypoxaemic COPD patients. Eur Respir J 2001; 18: 77–84
- Probst V S, Heyvaert H, Coosemans I, Pitta F, Spruit M A, Troosters T, Gosselink R, Decramer M. Effects of a rollator on exercise capacity, gas exchange and ventilation in COPD patients [abstract]. Am J Respir Crit Care Med 2003; 167: A669
- Fishman A, Martinez F, Naunheim K, Piantadosi S, Wise R, Ries A, Weinmann G, Wood D E, National Emphysema Treatment Trial Research Group. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N Engl J Med 2003; 348: 2059–2073
- Casaburi R, Kukafka D, Cooper C B, Witek T J, Jr, Kesten S. Improvement in exercise tolerance with the combination of tiotropium and pulmonary rehabilitation in patients with COPD. Chest 2005; 127: 809–817
- Peters M M, Webb K A, O'Donnell D E. Combined physiological effects of bronchodilators and hyperoxia on exertional dyspnoea in normoxic COPD. Thorax 2006; 61: 559–567
- Ram F S, Wedzicha J A. Ambulatory oxygen for chronic obstructive pulmonary disease. Cochrane Database Syst Rev 2002, (2):CD000238
- Kannus P, Palvanen M, Niemi S, Parkkari J, Järvinen M, Vuori I. Osteoporotic fractures of the proximal humerus in elderly Finnish persons: sharp increase in 1970-1998 and alarming projections for the new millennium. Acta Orthop Scand 2000; 71: 465–470
- Wehren L E. The epidemiology of osteoporosis and fractures in geriatric medicine. Clin Geriatr Med 2003; 19: 245–258
- O'Neill T W, Felsenberg D, Varlow J, Cooper C, Kanis J A, Silman A J. The prevalence of vertebral deformity in european men and women: the European Vertebral Osteoporosis Study. J Bone Miner Res 1996; 11: 1010–1018
- NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. National Institutes of Health, Accessed 1 December 2006, NIH Consensus Statement Online
- Consensus development conference. Diagnosis, prophylaxis, and treatment of osteoporosis. Am J Med 1993; 94: 646–650
- World Health Organization. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. 843. World Health Organization, GenevaSwitzerland 1994, WHO Technical Report Series
- Rosen C J, Brown S A. A rational approach to evidence gaps in the management of osteoporosis. Am J Med 2005; 118: 1183–1189
- Leech J A, Dulberg C, Kellie S, Pattee L, Gay J. Relationship of lung function to severity of osteoporosis in women. Am Rev Respir Dis 1990; 141: 68–71
- Schlaich C, Minne H W, Bruckner T, Wagner G, Gebest H J, Grunze M, Ziegler R, Leidig-Bruckner G. Reduced pulmonary function in patients with spinal osteoporotic fractures. Osteoporos Int 1998; 8: 261–267
- Culham E G, Jimenez H A, King C E. Thoracic kyphosis, rib mobility, and lung volumes in normal women and women with osteoporosis. Spine 1994; 19: 1250–1255
- Moffett S P, Zmuda J M, Oakley J I, Beck T J, Cauley J A, Stone K L, Lui L Y, Ensrud K E, Hillier T A, Hochberg M C, Morin P, Peltz G, Greene D, Cummings S R. Tumor necrosis factor-alpha polymorphism, bone strength phenotypes, and the risk of fracture in older women. J Clin Endocrinol Metab 2005; 90: 3491–3497
- Gourlay M, Richy F, Reginster J Y. Strategies for the prevention of hip fracture. Am J Med 2003; 115: 309–317
- Filley G F, Beckwitt H J, Reeves J T, Mitchell R S. Chronic obstructive bronchopulmonary disease. II. Oxygen transport in two clinical types. Am J Med 1968; 44: 26–38
- Vandenbergh E, Van de Woestijne K P, Gyselen A. Weight changes in the terminal stages of chronic obstructive pulmonary disease. Relation to respiratory function and prognosis. Am Rev Respir Dis 1967; 95: 556–566
- Braun S R, Keim N L, Dixon R M, Glagnaz P, Anderegg A, Shrago E S. The prevalence and determinants of nutritional changes in chronic obstructive pulmonary disease. Chest 1984; 86: 558–563
- Mitchell R S, Filley G F. Chronic obstructive bronchopulmonary disease. I. Clinical features. Am Rev Respir Dis 1964; 89: 360–371
- Hunter A M, Carey M A, Larsh H W. The nutritional status of patients with chronic obstructive pulmonary disease. Am Rev Respir Dis 1981; 124: 376–381
- Wilson D O, Rogers R M, Wright E C, Anthonisen N R. Body weight in chronic obstructive pulmonary disease. The National Institutes of Health Intermittent Positive-Pressure Breathing Trial. Am Rev Respir Dis 1989; 139: 1435–1438
- Sahebjami H, Doers J T, Render M L, Bond T L. Anthropometric and pulmonary function test profiles of outpatients with stable chronic obstructive pulmonary disease. Am J Med 1993; 94: 469–474
- Schols A M, Soeters P B, Dingemans A M, Mostert R, Frantzen P J, Wouters E F. Prevalence and characteristics of nutritional depletion in patients with stable COPD eligible for pulmonary rehabilitation. Am Rev Respir Dis 1993; 147: 1151–1156
- Openbrier D R, Irwin M M, Rogers R M, Gottlieb G P, Dauber J H, Van Thiel D H, Pennock B E. Nutritional status and lung function in patients with emphysema and chronic bronchitis. Chest 1983; 83: 17–22
- Vestbo J, Prescott E, Almdal T, Dahl M, Nordestgaard B G, Andersen T, Sørensen T I, Lange P. Body mass, fat-free body mass, and prognosis in patients with chronic obstructive pulmonary disease from a random population sample: findings from the Copenhagen City Heart Study. Am J Respir Crit Care Med 2006; 173(1)79–83
- Vermeeren M A, Creutzberg E C, Schols A M, Postma D S, Pieters W R, Roldaan A C, Wouters E F, COSMIC Study Group. Prevalence of nutritional depletion in a large out-patient population of patients with COPD. Respir Med 2006; 100: 1349–1355
- Schols A M, Broekhuizen R, Weling-Scheepers C A, Wouters E F. Body composition and mortality in chronic obstructive pulmonary disease. Am J Clin Nutr 2005; 82: 53–59
- Engelen M P, Schols A M, Does J D, Wouters E F. Skeletal muscle weakness is associated with wasting of extremity fat-free mass but not with airflow obstruction in patients with chronic obstructive pulmonary disease. Am J Clin Nutr 2000; 71: 733–738
- Creutzberg E C, Schols A M, Bothmer-Quaedvlieg F C, Wouters E F. Prevalence of an elevated resting energy expenditure in patients with chronic obstructive pulmonary disease in relation to body composition and lung function. Eur J Clin Nutr 1998; 52: 396–401
- Hugli O, Schutz Y, Fitting J W. The cost of breathing in stable chronic obstructive pulmonary disease. Clin Sci (Lond) 1995; 89: 625–632
- Baarends E M, Schols A M, Westerterp K R, Wouters E F. Total daily energy expenditure relative to resting energy expenditure in clinically stable patients with COPD. Thorax 1997; 52: 780–785
- Baarends E M, Schols A M, Pannemans D L, Westerterp K R, Wouters E F. Total free living energy expenditure in patients with severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1997; 155: 549–554
- Vermeeren M A, Wouters E F, Geraerts-Keeris A J, Schols A M. Nutritional support in patients with chronic obstructive pulmonary disease during hospitalization for an acute exacerbation; a randomized controlled feasibility trial. Clin Nutr 2004; 23: 1184–1192
- Creutzberg E C, Wouters E F, Vanderhoven-Augustin I M, Dentener M A, Schols A M. Disturbances in leptin metabolism are related to energy imbalance during acute exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2000; 162: 1239–1245, (4 Pt 1)
- Karajgi B, Rifkin A, Doddi S, Kolli R. The prevalence of anxiety disorders in patients with chronic obstructive pulmonary disease. Am J Psychiatry 1990; 147: 200–201
- Moore M C, Zebb B J. The catastrophic misinterpretation of physiological distress. Behav Res Ther 1999; 37: 1105–1118
- Yellowlees P M, Haynes S, Potts N, Ruffin R E. Psychiatric morbidity in patients with life-threatening asthma: initial report of a controlled study. Med J Aust 1988; 149: 246–249
- Borak J, Chodosowska E, Matuszewski A, Zielinski J. Emotional status does not alter exercise tolerance in patients with chronic obstructive pulmonary disease. Eur Respir J 1998; 12: 370–373
- Engström C P, Persson L O, Larsson S, Rydén A, Sullivan M. Functional status and well being in chronic obstructive pulmonary disease with regard to clinical parameters and smoking: a descriptive and comparative study. Thorax 1996; 51: 825–830
- Kim H F, Kunik M E, Molinari V A, Hillman S L, Lalani S, Orengo C A, Petersen N J, Nahas Z, Goodnight-White S. Functional impairment in COPD patients: the impact of anxiety and depression. Psychosomatics 2000; 41: 465–471
- Prigatano G P, Wright E C, Levin D. Quality of life and its predictors in patients with mild hypoxemia and chronic obstructive pulmonary disease. Arch Intern Med 1984; 144: 1613–1619
- Withers N J, Rudkin S T, White R J. Anxiety and depression in severe chronic obstructive pulmonary disease: the effects of pulmonary rehabilitation. J Cardiopulm Rehabil 1999; 19: 362–365
- Yohannes A M, Baldwin R C, Connolly M J. Depression and anxiety in elderly outpatients with chronic obstructive pulmonary disease: prevalence, and validation of the BASDEC screening questionnaire. Int J Geriatr Psychiatry 2000; 15: 1090–1096
- Kvaal K, Macijauskiene J, Engedal K, Laake K. High prevalence of anxiety symptoms in hospitalized geriatric patients. Int J Geriatr Psychiatry 2001; 16: 690–693
- van Manen J G, Bindels P J, Dekker F WI, Jzermans C J, van der Zee J S, Schadé E. Risk of depression in patients with chronic obstructive pulmonary disease and its determinants. Thorax 2002; 57: 412–416
- van Ede L, Yzermans C J, Brouwer H J. Prevalence of depression in patients with chronic obstructive pulmonary disease: a systematic review. Thorax 1999; 54: 688–692
- Felker B, Katon W, Hedrick S C, Rasmussen J, McKnight K, McDonnell M B, Fihn S D. The association between depressive symptoms and health status in patients with chronic pulmonary disease. Gen Hosp Psychiatry 2001; 23: 56–61
- Cully J A, Graham D P, Stanley M A, Ferguson C J, Sharafkhaneh A, Souchek J, Kunik M E. Quality of life in patients with chronic obstructive pulmonary disease and comorbid anxiety or depression. Psychosomatics 2006; 47: 312–319
- Beck J G, Scott S K, Teague R B, Perez F I, Brown G A. Correlates of daily impairment in COPD. Rehabil Psychol 1988; 33: 77–84
- Dahlen I, Janson C. Anxiety and depression are related to the outcome of emergency treatment in patients with obstructive pulmonary disease. Chest 2002; 122: 1633–1637
- Almagro P, Calbo E, Ochoa de Echagüen A, Barreiro B, Quintana S, Heredia J L, Garau J. Mortality after hospitalization for COPD. Chest 2002; 121: 1441–1448
- Di Marco F, Verga M, Reggente M, Maria Casanova F, Santus P, Blasi F, Allegra L, Centanni S. Anxiety and depression in COPD patients: The roles of gender and disease severity. Respir Med 2006; 100: 1767–1774
- de Torres J P, Casanova C, Hernández C, Abreu J, Aguirre-Jaime A, Celli B R. Gender and COPD in patients attending a pulmonary clinic. Chest 2005; 128: 2012–2016
- de Torres J P, Casanova C, Hernández C, Abreu J, Montejo de Garcini A, Aguirre-Jaime A, Celli B R. Gender associated differences in determinants of quality of life in patients with COPD: a case series study. Health Qual Life Outcomes 2006; 4: 72
- Thomsen D K, Mehlsen M Y, Viidik A, Sommerlund B, Zachariae R. Age and gender differences in negative affect. Is there a role for emotion regulation? Personality Individ Diff 2005; 38: 1935–1946
- Nolen-Hoeksema S, Larson J, Grayson C. Explaining the gender difference in depressive symptoms. J Pers Soc Psychol 1999; 77: 1061–1072
- Musselman D L, Miller A H, Porter M R, Manatunga A, Gao F, Penna S, Pearce B D, Landry J, Glover S, McDaniel J S, Nemeroff C B. Higher than normal plasma interleukin-6 concentrations in cancer patients with depression: preliminary findings. Am J Psychiatry 2001; 158: 1252–1257
- Maes M. Major depression and activation of the inflammatory response system. Adv Exp Med Biol 1999; 461: 25–46
- Raison C L, Demetrashvili M, Capuron L, Miller A H. Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs 2005; 19: 105–123
- Berkenbosch F, van Oers J, del Rey A, Tilders F, Besedovsky H. Corticotropin-releasing factor-producing neurons in the rat activated by interleukin-1. Science 1987; 238: 524–526
- Borson S, Claypoole K, McDonald G J. Depression and chronic obstructive pulmonary disease: treatment trials. Semin Clin Neuropsychiatry 1998; 3: 115–130
- Sanders S K, Morzorati S L, Shekhar A. Priming of experimental anxiety by repeated subthreshold GABA blockade in the rat amygdala. Brain Res 1995; 699: 250–259
- Shekhar A, Sajdyk T J, Gehlert D R, Rainnie D G. The amygdala, panic disorder, and cardiovascular responses. Ann N Y Acad Sci 2003; 985: 308–325
- Porzelius J, Vest M, Nochomovitz M. Respiratory function, cognitions, and panic in chronic obstructive pulmonary patients. Behav Res Ther 1992; 30: 75–77
- Breslau N, Klein D F. Smoking and panic attacks: an epidemiologic investigation. Arch Gen Psychiatry 1999; 56: 1141–1147
- Kendler K S, Neale M C, MacLean C J, Heath A C, Eaves L J, Kessler R C. Smoking and major depression. A causal analysis. Arch Gen Psychiatry 1993; 50: 36–43
- Hanania N A, Darken P, Horstman D, Reisner C, Lee B, Davis S, Shah T. The efficacy and safety of fluticasone propionate (250 microg)/salmeterol (50 microg) combined in the Diskus inhaler for the treatment of COPD. Chest 2003; 124: 834–843
- Kardos P, Wencker M, Glaab T, Vogelmeier C. Impact of salmeterol/fluticasone propionate versus salmeterol on exacerbations in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007; 175: 144–149
- Griffiths T L, Burr M L, Campbell I A, Lewis-Jenkins V, Mullins J, Shiels K, Tumer-Lawlor P J, Payne N, Newcombe R G, Ionescu A A, Thomas J, Tunbridge J. Results at 1 year of outpatient multidisciplinary pulmonary rehabilitation: a randomised controlled trial. Lancet 2000; 355: 362–368
- Kayahan B, Karapolat H, Atýntoprak E, Atasever A, Oztürk O. Psychological outcomes of an outpatient pulmonary rehabilitation program in patients with chronic obstructive pulmonary disease. Respir Med 2006; 100: 1050–1057
- Eaton T, Garrett J E, Young P, Fergusson W, Kolbe J, Rudkin S, Whyte K. Ambulatory oxygen improves quality of life of COPD patients: a randomised controlled study. Eur Respir J 2002; 20: 306–312
- Lorig K R, Sobel D S, Ritter P L, Laurent D, Hobbs M. Effect of a self-management program on patients with chronic disease. Eff Clin Pract 2001; 4: 256–262
- Harte J L, Eifert G H, Smith R. The effects of running and meditation on beta-endorphin, corticotropin-releasing hormone and cortisol in plasma, and on mood. Biol Psychol 1995; 40: 251–265
- Nabkasorn C, Miyai N, Sootmongkol A, Junprasert S, Yamamoto H, Arita M, Miyashita K. Effects of physical exercise on depression, neuroendocrine stress hormones and physiological fitness in adolescent females with depressive symptoms. Eur J Public Health 2006; 16: 179–184
- Troosters T, Gosselink R, Decramer M. Short- and long-term effects of outpatient rehabilitation in patients with chronic obstructive pulmonary disease: a randomized trial. Am J Med 2000; 109: 207–212
- Bourbeau J, Julien M, Maltais F, Rouleau M, Beaupré A, Bégin R, Renzi P, Nault D, Borycki E, Schwartzman K, Singh R, Collet J P. Chronic Obstructive Pulmonary Disease axis of the Respiratory Network Fonds de la Recherche en Santé du Québec. Reduction of hospital utilization in patients with chronic obstructive pulmonary disease: a disease-specific self-management intervention. Arch Intern Med 2003; 163: 585–591
- Janowsky D S, Overstreet D H, Nurnberger J I, Jr. Is cholinergic sensitivity a genetic marker for the affective disorders?. Am J Med Genet 1994; 54: 335–344
- Mannino D M, Ford E S, Redd S C. Obstructive and restrictive lung disease and functional limitation: data from the Third National Health and Nutrition Examination. J Intern Med 2003; 254: 540–547
- Andreas S, Anker S D, Scanlon P D, Somers V K. Neurohumoral activation as a link to systemic manifestations of chronic lung disease. Chest 2005; 128: 3618–3624
- Mannino D M, Homa D M, Akinbami L J, Ford E S, Redd S C. Chronic obstructive pulmonary disease surveillance—United States, 1971-2000. Respir Care 2002; 47: 1184–1199
- Mannino D M, Brown C, Giovino G A. Obstructive lung disease deaths in the United States from 1979 through 1993. An analysis using multiple-cause mortality data. Am J Respir Crit Care Med 1997; 156: 814–818, (3 Pt 1)
- Camilli A E, Robbins D R, Lebowitz M D. Death certificate reporting of confirmed airways obstructive disease. Am J Epidemiol 1991; 133: 795–800
- Vilkman S, Keistinen T, Tuuponen T, Kivelä S L. Survival and cause of death among elderly chronic obstructive pulmonary disease patients after first admission to hospital. Respiration 1997; 64: 281–284
- Zielinski J, MacNee W, Wedzicha J, Ambrosino N, Braghiroli A, Dolensky J, Howard P, Gorzelak K, Lahdensuo A, Strom K, Tobiasz M, Weitzenblum E. Causes of death in patients with COPD and chronic respiratory failure. Monaldi Arch Chest Dis 1997; 52: 43–47
- Holguin F, Folch E, Redd S C, Mannino D M. Comorbidity and mortality in COPD-related hospitalizations in the United States, 1979 to 2001. Chest 2005; 128: 2005–2011
- Mannino D M, Doherty D E, Sonia B A. Global Initiative on Obstructive Lung Disease (GOLD) classification of lung disease and mortality: findings from the Atherosclerosis Risk in Communities (ARIC) study. Respir Med 2006; 100: 115–122
- Mannino D M, Buist A S, Petty T L, Enright P L, Redd S C. Lung function and mortality in the United States: data from the First National Health and Nutrition Examination Survey follow up study. Thorax 2003; 58: 388–393
- Calverley P M, Anderson J A, Celli B, Ferguson G T, Jenkins C, Jones P W, Yates J C, Vestbo J, TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356: 775–789
- Doll R, Peto R, Boreham J, Sutherland I. Mortality in relation to smoking: 50 years' observations on male British doctors. BMJ 2004; 328: 1519
- The nocturnal oxygen therapy trial–NOTT. Is 12-hour oxygen as effective as 24-hour oxygen in advanced chronic obstructive pulmonary disease with hypoxemia? (The nocturnal oxygen therapy trial–NOTT). Chest 1980; 78: 419–420
- Medical Research Council Working Party. Long term domiciliary oxygen therapy in chronic hypoxic cor pulmonale complicating chronic bronchitis and emphysema. Report of the Medical Research Council Working Party. Lancet 1981; 1: 681–686
- Piccioni P, Caria E, Bignamini E, Forconi G, Nebiolo F, Arossa W, Bugiani M. Predictors of survival in a group of patients with chronic airflow obstruction. J Clin Epidemiol 1998; 51: 547–555
- Kinnunen T, Säynäjäkangas O, Keistinen T. The COPD-induced hospitalization burden from first admission to death. Respir Med 2007; 101: 294–299
- Cote C G, Celli B R. Pulmonary rehabilitation and the BODE index in COPD. Eur Respir J 2005; 26: 630–636
- Wilt T J, Niewoehner D, Kim C, Kane R L, Linabery A, Tacklind J, Macdonald R, Rutks I. Use of spirometry for case finding, diagnosis, and management of chronic obstructive pulmonary disease (COPD). Evid Rep Technol Assess (Summ) 2005; 121: 1–7
- Bednarek M, Gorecka D, Wielgomas J, Czajkowska-Malinowska M, Regula J, Mieszko-Filipczyk G, Jasionowicz M, Bijata-Bronisz R, Lempicka-Jastrzebska M, Czajkowski M, Przybylski G, Zielinski J. Smokers with airway obstruction are more likely to quit smoking. Thorax 2006; 61: 869–873
- Tashkin D P. The role of patient-centered outcomes in the course of chronic obstructive pulmonary disease: how long-term studies contribute to our understanding. Am J Med 2006; 119: 63–72, (10 Suppl 1)
- Huiart L, Ernst P, Ranouil X, Suissa S. Low-dose inhaled corticosteroids and the risk of acute myocardial infarction in COPD. Eur Respir J 2005; 250: 634–639
- Papi A, Bellettato C M, Braccioni F, Romagnoli M, Casolari P, Caramori G, Fabbri L M, Johnston S L. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am J Respir Crit Care Med 2006; 173: 1114–1121
- Oudijk E J, Gerritsen W B, Nijhuis E H, Kanters D, Maesen B L, Lammers J W, Koenderman L. Expression of priming-associated cellular markers on neutrophils during an exacerbation of COPD. Respir Med 2006; 100: 1791–1799
- Tug T, Karatas F, Terzi S M. Antioxidant vitamins (A, C and E) and malondialdehyde levels in acute exacerbation and stable periods of patients with chronic obstructive pulmonary disease. Clin Invest Med 2004; 27: 123–128
- Sethi S, Wrona C, Veeramachaneni S, Eschbeger K, Muphy T F. Changes in serum C-reactive protein (CRP) with onset and resolution of exacerbations of COPD and relationship with etiology. [Abstract] Proc Am Thorac Soc 2006; 3: A271
- Scanlon P D, Connett J E, Wise R A, Tashkin D P, Madhok T, Skeans M, Carpenter P C, Bailey W C, Buist A S, Eichenhorn M, Kanner R E, Weinmann G, Lung Health Study Research Group. Loss of bone density with inhaled triamcinolone in Lung Health Study II. Am J Respir Crit Care Med 2004; 170: 1302–1309
- Burge P S, Calverley P M, Jones P W, Spencer S, Anderson J A. Prednisolone response in patients with chronic obstructive pulmonary disease: results from the ISOLDE study. Thorax 2003; 58: 654–658
- Celli B R, MacNee W, committee members. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004; 23: 932–946
- Burge S, Wedzicha J A. COPD exacerbations: definitions and classifications. Eur Respir J Suppl 2003; 41: S46–S53
- Niewoehner D E. The impact of severe exacerbations on quality of life and the clinical course of chronic obstructive pulmonary disease. Am J Med 2006; 119: 38–45, (10 Suppl 1)
- Cooper C B, Tashkin D P. Recent developments in inhaled therapy in stable chronic obstructive pulmonary disease. BMJ 2005; 330: 640–644
- Rennard S I, Calverley P. Rescue! Therapy and the paradox of the Barcalounger. Eur Respir J 2003; 21: 916–917
- Chrystyn H, Mulley B A, Peake M D. Dose response relation to oral theophylline in severe chronic obstructive airways disease. BMJ 1988; 297: 1506–1510
- Belman M J, Botnick W C, Shin J W. Inhaled bronchodilators reduce dynamic hyperinflation during exercise in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1996; 153: 967–975
- Newton M F, O'Donnell D E, Forkert L. Response of lung volumes to inhaled salbutamol in a large population of patients with severe hyperinflation. Chest 2002; 121: 1042–1050
- O'Donnell D E, Lam M IU, Webb K A. Measurement of symptoms, lung hyperinflation, and endurance during exercise in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998; 158: 1557–1565
- Celli B, ZuWallack R, Wang S, Kesten S. Improvement in resting inspiratory capacity and hyperinflation with tiotropium in COPD patients with increased static lung volumes. Chest 2003; 124: 1743–1748
- O'Donnell D E, Flüge T, Gerken F, Hamilton A, Webb K, Aguilaniu B, Make B, Magnussen H. Effects of tiotropium on lung hyperinflation, dyspnea and exercise tolerance in patients with COPD. Eur Respir J 2004; 23: 832–840
- Maltais F, Hamilton A, Marciniuk D, Hernandez P, Sciurba F C, Richter K, Kesten S, O'Donnell D. Improvements in symptom-limited exercise performance over 8 h with once-daily tiotropium in patients with COPD. Chest 2005; 128: 1168–1178
- Palange P, Valli G, Onorati P, Antonucci R, Paoletti P, Rosato A, Manfredi F, Serra P. Effect of heliox on lung dynamic hyperinflation, dyspnea, and exercise endurance capacity in COPD patients. J Appl Physiol 2004; 97: 1637–1642
- Pecchiari M, Pelucchi A, D'Angelo E, Foresi A, Milic-Emili J, D'Angelo E. Effect of heliox breathing on dynamic hyperinflation in COPD patients. Chest 2004; 125: 2075–2082
- Hopkinson N S, Toma T P, Hansell D M, Goldstraw P, Moxham J, Geddes D M, Polkey M I. Effect of bronchoscopic lung volume reduction on dynamic hyperinflation and exercise in emphysema. Am J Respir Crit Care Med 2005; 171: 453–460
- Koyama S, Rennard S I, Robbins R A. Acetylcholine stimulates bronchial epithelial cells to release neutrophil and monocyte chemotactic activity. Am J Physiol 1992; 262: L466–L471, (4 Pt 1)
- Sato E, Koyama S, Okubo Y, Kubo K, Sekiguchi M. Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am J Physiol 1998; 274: 970–979, (6 Pt 1)
- Buhling F, Lieder N, Reisenauer A, Welte T. Anti-inflammatory function of tiotropium mediated by suppression of acetylcholine-induced release of chemotactic activity. [Abstract] Eur Respir J 2004; 24: 318s
- Fukuchi K, Zushi H, Kikuchi Y, Hase M, Yasue T, Kishi T. Involvement of muscarinic receptor activation in cigarette smoke induced pulmonary inflammation in mice. [Abstract] Proc Am Thorac Soc 2005; 2: A142
- Lipworth B J. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet 2005; 365: 167–175
- Gamble E, Grootendorst D C, Brightling C E, Troy S, Qiu Y, Zhu J, Parker D, Matin D, Majumdar S, Vignola A M, Kroegel C, Morell F, Hansel T T, Rennard S I, Compton C, Amit O, Tat T, Edelson J, Pavord I D, Rabe K F, Barnes N C, Jeffery P K. Antiinflammatory effects of the phosphodiesterase-4 inhibitor cilomilast (Ariflo) in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003; 168: 976–982
- Martorana P A, Beume R, Lucattelli M, Wollin L, Lungarella G. Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. Am J Respir Crit Care Med 2005; 172: 848–853
- Hanania N A, Ambrosino N, Calverley P, Cazzola M, Donner C F, Make B. Treatments for COPD. Respir Med 2005; 99(Suppl B)S28–S40
- Foglio K, Carone M, Pagani M, Bianchi L, Jones P W, Ambrosino N. Physiological and symptom determinants of exercise performance in patients with chronic airway obstruction. Respir Med 2000; 94: 256–263
- Oga T, Nishimura K, Tsukino M, Sato S, Hajiro T, Mishima M. Exercise capacity deterioration in patients with COPD: longitudinal evaluation over 5 years. Chest 2005; 128: 62–69
- Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood J E. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med 2002; 346: 793–801
- Manini T M, Everhart J E, Patel K V, Schoeller D A, Colbert L H, Visser M, Tylavsky F, Bauer D C, Goodpaster B H, Harris T B. Daily activity energy expenditure and mortality among older adults. JAMA 2006; 296: 171–179
- Barr R G, Bourbeau J, Camargo C A, Ram F S. Tiotropium for stable chronic obstructive pulmonary disease: a meta-analysis. Thorax 2006; 61: 854–862
- Alsaeedi A, Sin D D, McAlister F A. The effects of inhaled corticosteroids in chronic obstructive pulmonary disease: a systematic review of randomized placebo-controlled trials. Am J Med 2002; 113: 59–65
- Donaldson G C, Seemungal T AR, Bhowmik A, Wedzicha J A. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax 2002; 57: 847–852