Abstract
Approximately 15% of cases of COPD occur in non-smokers. Among the potential risk factors for COPD in non-smokers is second-hand smoke (SHS) exposure. However, the Surgeon General reported in 2006 that the evidence linking second hand smoke and COPD is insufficient to infer a causal relationship, largely because current evidence does not establish a biological link. The goal of this study was to determine whether SHS exposure can induce alveolar macrophage recruitment and expression of activation markers that we have previously demonstrated in human smokers and in mouse models of emphysema. To achieve these goals, we studied mice exposed to an ambient mixture of predominantly [89%] sidestream smoke at increasing doses over 3 months. We found that second hand smoke exposure induced a dose-dependent increase in alveolar macrophage recruitment (mean ± sd; 224,511 ± 52,330 vs 166,152 ± 47,989 macrophages/ml of bronchoalveolar lavage in smoke-exposed vs air-exposed controls at 3 months, p = 0.003). We also found increased expression of several markers of alveolar macrophage activation (PLA2g7, dkfzp434l142, Trem-2, and pirin, all p < 0.01 at 3 months) and increased lavage levels of two inflammatory mediators associated with COPD (CCL2 [MCP-1], 58 ± 12 vs. 43 ± 22 pg/ml, p = 0.03; and TNFα, 138 ± 43 vs 88 ± 78 pg/ml, p = 0.04 at 3 months). These findings indicate that second smoke exposure can cause macrophage recruitment and activation, providing a biological link between second-hand smoke exposure and the development of inflammatory processes linked to COPD.
INTRODUCTION
Chronic Obstructive Pulmonary Disease (COPD) affects 12.4 to 24 million people in the United States, is the sixth leading cause of death worldwide (Citation[1], Citation[2], Citation[3]) and is the only condition in the top 10 causes of death that has an increasing prevalence and mortality (Citation[1], Citation[4], Citation[5]). Although the majority of COPD occurs in smokers, approximately 15% of cases of COPD occur in non-smokers (Citation[6]). Thus, from a public health perspective, a relatively large number of cases of COPD occur in non-smokers. Among the potential risk factors for COPD in non-smokers is second-hand smoke (SHS) exposure. However, the 2006 U.S. Surgeon General Report on the health effects of SHS found that the evidence linking SHS and COPD is suggestive but not sufficient to infer a causal relationship. This is largely because current evidence is only epidemiologic (Citation[7], Citation[8], Citation[9], Citation[10], Citation[11], Citation[12], Citation[13], Citation[14], Citation[15], Citation[16]) and therefore does not establish a biological link (Citation[17]). Furthermore, all but one epidemiologic study (Citation[11]) combined asthma, chronic bronchitis, and emphysema to define COPD.
Although there has been a trend towards decreased exposure of the United States (US) population to SHS since 1988 (Citation[18]), the number of nonsmoking adults currently exposed to SHS in the US remains very large (Citation[17]). Overall, based on blood cotinine measurement, approximately 86 million nonsmoking adults aged 20 or more years in the US were exposed to SHS in 2000 (Citation[17]). Identification of SHS exposure as a risk factor for COPD in non-smokers would provide additional rationale for public health efforts aimed at control of SHS exposure and additional motivation for smoking cessation among smokers who live with non-smokers.
Given the paucity of biological evidence linking SHS exposure and COPD, the goal of this study was to determine whether ambient exposure to smoke that models the constituents of SHS can cause cellular and molecular abnormalities in the airway that are associated with COPD using a mouse model. Our primary endpoints included measurement of airway inflammation, macrophage recruitment to the lung, and markers of alveolar macrophage activation. The inflammatory mediators that we assayed included a chemokine and cytokine that have been linked to the pathogenesis of COPD, CCL2 (MCP-1) (Citation[19], Citation[20], Citation[21]), and tumor necrosis factor-α (TNF-α) (Citation[22], Citation[23], Citation[24], Citation[25], Citation[26]).
The gene expression markers of smoke-induced macrophage activation that we studied were those that we previously identified as induced in habitual human smokers, some of whom had emphysema, and in two mouse models of emphysema (Citation[27]). These markers included matrix metalloproteinase-12 (MMP-12), a proteinase linked to emphysema in mouse and human studies (Citation[27], Citation[28], Citation[29], Citation[30], Citation[31], Citation[32], Citation[33]). These markers also included four novel markers of macrophage activation, phospholipase A2, group VII (PLA2g7), triggering receptor expressed on myeloid cells 2 (Trem-2), an as yet unnamed transcript (dkfzp434l142, Entrez gene ID 51313) and pirin. Our secondary endpoints included quantitative assessment of emphysema. We studied these outcomes in C57Bl6/J mice exposed to an ambient mixture of predominantly (89%) sidestream smoke (SS) to model SHS exposure.
MATERIALS AND METHODS
Mice and second-hand smoke exposure
All exposures were conducted at the Institute of Toxicology and Environmental Health inhalation facilities (University of California, Davis) in accordance with institutional guidelines. C57Bl6/J mice were purchased from Jackson Laboratories (Bar Harbor ME, USA). We used a smoke exposure system that provided for whole body exposure (Citation[34]). Standard housing cages containing 4–5 mice each were placed into glass and stainless steel Hinners-type exposure chambers (volume: 0.44 m3) as described by Teague et al. (Citation[34]).
Sidestream smoke was generated by burning conditioned (48 h in 60% humidity) Kentucky 2R4F reference cigarettes in a smoking machine with standardized 35 ml puffs of 2 sec duration, once every min, for a total of 8 puffs per cigarette. After dilution and aging in a conditioning chamber (2 min), the sidestream smoke produced between puffs was drawn into the exposure chambers. To simulate SHS, the sidestream smoke was reinforced every 58 sec with a 2 sec puff of mainstream smoke (representing 11% of the overall smoke mixture). The smoke generated was passed through two chambers oriented in series and large enough to house the experimental groups of mice. To create different exposure conditions in these two chambers, the smoke concentration was regulated through control of dilution with air and through loss of particulate matter on passing smoke into the second chamber.
Eight-week-old littermate mice balanced between groups for gender were exposed to filtered air or increasing levels of smoke for 5 days per week, 6 hours per day. Groups of mice were studied monthly for a total of 3 months (). For month 1, we took advantage of both exposure chambers by exposing mice to high dose SHS (chamber 1) and medium-dose SHS smoke (chamber 2). For the remainder of the study, we used only high-dose SHS exposure (chamber 1) and increased the dose of SHS to a target of 150 mg/m3 of total suspended particulate matter (TSP) gradually to ensure the health of the animals.
Table 1 Environmental conditions at each time point of exposure
This maximal dose was achieved at 6 weeks. The chamber atmosphere was monitored for temperature, humidity, carbon monoxide, TSP. TSP matter was measured by weighing material collected on filters and carbon monoxide was monitored with a model 880 non-dispersive-infrared (NDIR) analyzer (Beckmann Industries, La Habra CA, USA). Nicotine concentrations were measured by drawing air samples through sorbent tubes, extracting the nicotine and analysis with gas chromatography. Throughout the exposure period mice were weighed and monitored for general health.
Sample collection
Lungs were lavaged 5 times with 0.8-ml aliquots of sterile phosphate-buffered saline. An aliquot of 400 μ l was removed and processed for cell counts and cyto-centrifugation. The remainder of the lavage fluid was centrifuged, and the cell free supernatant was stored at −70°C until further processing. Macrophages were purified from lavage fluid by plating as previously described (Citation[27], Citation[35]). RNA was extracted from macrophages using the RNeasy kit (Qiagen, Valencia CA, USA) and DNAse-treated (Promega, Madison WI, USA). RNA concentration and quality were assessed using the Bioanalyzer 2100 (Agilent Technologies, Palo Alto CA, USA).
Real-time PCR
For analysis of alveolar macrophage gene expression levels, two-step qPCR was performed as described previously (Citation[27], Citation[36]). A panel of three housekeeping genes (GAPDH, S9, β -Actin) was measured during taqman profiling and the geometric mean value of the two housekeeping genes most stably expressed across the samples was used for normalization.
ELISA
ELISA for CCL2 and TNF-α was performed on cell-free BAL fluid per the manufacturer's protocol (R&D Systems, Minneapolis MN, USA).
Histology
For histologic staining, lungs from a subset of mice were inflated to 20 cm water pressure with buffered formaldehyde containing zinc (Z-FIX, Anatech Ltd, Battle Creek MI, USA). Fixed lungs were embedded in paraffin, sectioned at 5 μ m thickness, and stained with hematoxylin and eosin using standard protocols. Measurement of emphysema was made using stereological methods (Citation[37]). These analyses were performed with an Olympus light microscope (Olympus BX50, Olympus Optical Co. GMBH, Germany) equipped with a mechanical stage linked to a personal computer (IBM 300PL Personal Computer, IBM United Kingdom, England) running Computer Assisted Stereology Toolbox (C.A.S.T.-Grid) software (Olympus Danmark A/S, Denmark).
Using point counting, the volume of parenchymal tissue and air spaces within parenchymal tissue were estimated. Using intersection counting, the surface area of the alveolar epithelium was estimated in the same microscopic fields, and the alveolar surface area normalized to the volume of lung surveyed (parenchyma and air) was used as a measure of emphysema (Citation[38]). All measurements were made by a single investigator (AE) in a blinded fashion.
RESULTS
Exposure conditions
We exposed mice to increasing levels of SHS exposure and assessed them at monthly intervals for 3 months. For the first month, we exposed mice to either medium or high-dose SHS. Thereafter, SHS exposure levels were increased to a maximal level by 6 weeks and all remaining mice were exposed at this level. The average total suspended particle (TSP) concentration in the medium-dose exposure group at 1 month was 69 mg/m3. Mean daily TSP concentrations in the high-dose SHS exposure groups were higher at 3 months than at 2 months because of longer cumulative exposure to the maximal dose so that the average exposure level was 131 mg/m3 at 3 months ().
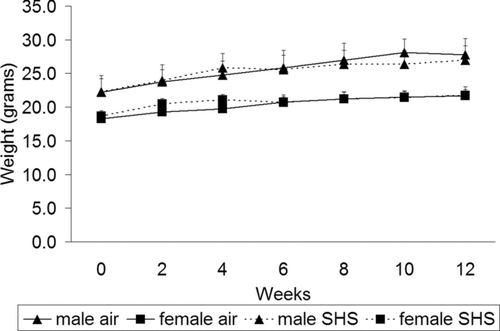
Average carbon monoxide levels increased progressively from 238 ppm in the medium dose group at 1 month to 394 ppm in the high dose group at 3 months. Average ambient nicotine levels ranged between 2.5 and 6.8 mg/m3. There was no smoke-related mortality during the study. Over the 3-month period of exposure, mice in the high SHS exposed group gained weight at a gender-appropriate rate throughout the exposure period () with no difference as compared to the healthy control group.
Figure 1 No differences in weight gain were measured in mice exposed to high dose SHS. Filtered air- and high-dose SHS-exposed mice were weighed at 2-week intervals throughout the exposure. Male mice weighed more than female mice at every time point. There were no statistically significant differences in the amount of weight gain between filtered air- and SHS-exposed mice. Data are expressed as mean weight in grams ± SD; n = 13–15 for each group at weeks 0–4, n = 9–12 for each group at weeks 6–8 and n = 7–9 for each group at week 10.
Bronchoalveolar lavage cell differentials
Since chronic cigarette smoking leads to increased numbers of alveolar macrophages in the lung as assessed by BAL (Citation[39], Citation[40]), we measured the degree of pulmonary inflammation induced by chronic SHS exposure in our exposed mice. We found consistent increases in macrophage numbers in BAL fluid over time (). Total BAL inflammatory cell and alveolar macrophage numbers increased significantly with high dose SHS exposure at 1 and 3 months, with a trend for an increase at 2 months. At 1 month of exposure, total inflammatory cell and macrophage numbers in BAL fluid increased 33% in high dose SHS-exposed mice as compared to the air-exposed controls (p = 0.049, based on 6 air and 6 smoke exposed mice). Medium dose SHS had a smaller effect (25% increase) which was non-statistically significant (p = 0.18, based on 6 air and 5 smoke exposed mice).
Figure 2 BAL total inflammatory cell and macrophage numbers were increased in high dose SHS-exposed mice compared to filtered air-exposed. Cell counts and differentials were determined in BAL fluid from filtered air- and medium- or high- dose SHS-exposed mice after 1, 2 or 3 months of exposure. p-values are for comparison with filtered air-exposed control at that time point. Data are expressed as median (line), interquartile range (box) and range (whiskers).
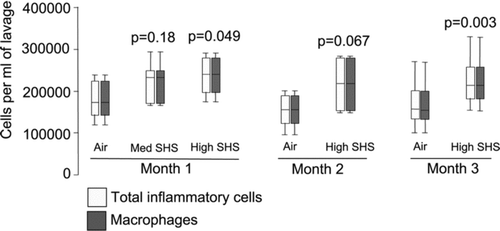
At 3 months of exposure, inflammatory cell and macrophage numbers in BAL increased 35% and the effect was statistically significant (p = 0.003, based on 15 air and 16 smoke exposed mice). BAL neutrophil, eosinophil and lymphocyte percentages remained < 2% in all groups, and there was no effect of SHS exposure at any time point in numbers of these cells in BAL. These data indicate that exposure to components of SHS in mice result in recruitment of alveolar macrophages with as little as 1 month of exposure.
Macrophage activation
In prior studies of alveolar macrophages from two mouse models of emphysema and from habitual human smokers, some of whom had emphysema, we identified several markers of macrophage activation that were induced at the gene expression level (Citation[27]). We used expression levels of a panel of 5 of these markers to assess alveolar macrophage activation in this study.
In descending order of level of induction in macrophages from human smokers by microarray (Citation[27]), these 5 markers included PLA2g7 (the 2nd most highly induced gene in human smokers), MMP-12 (3rd), dkfzp434l142 (7th), Trem-2 (40th) and pirin (48th). We picked these markers either based on their known association with emphysema (MMP-12), or based on data showing that they are directly up-regulated in macrophages when stimulated in vitro with components of SHS (data not shown). In contrast with our prior findings in macrophages from human smokers, MMP-12 expression was not induced in alveolar macrophages with any level of SHS exposure as compared to control nor at any time point. However, PLA2g7 was induced with both medium and high-dose SHS exposure and at all time points (all p < 0.0001, ).
Table 2 Induction of selected markers of alveolar macrophage activation with SHS exposure.
High-level SHS exposure also induced expression of pirin at 1 month (p = 0.028), Trem-2 and dkfzp434l142 at 2 months (both p < 0.002) and pirin, Trem-2 and dkfzp434l142 at 3 months (all p < 0.01, ). Taken together, these data suggest that SHS exposure not only recruits alveolar macrophages to the lung, but also induces a state of macrophage activation that shares some gene expression profiles in common with macrophages from habitual smokers and genetic mouse models of emphysema.
Development of emphysema
We evaluated the development of emphysema in SHS exposed mice at 3 months (our final time point) by measuring the surface area of alveoli normalized to the volume of lung surveyed as previously described (Citation[38]). We found no difference in surface to volume ratios in 8 high-dose SHS exposed mice as compared to 8 controls (mean ± se, 1434 ± 82 vs 1338 ± 59 cm2/cm3, p = 0.36).
Airway inflammatory mediators
We next evaluated whether exposure to SHS induced chemokines and/or cytokines that have previously been associated with the development of COPD. Using BAL fluid from SHS exposed and control mice, we performed ELISA for the following mediators: CCL2 (MCP-1) and TNFα. We found significant increases in CCL2 and TNFα in mice with high dose SHS exposure at a range of time points (). For CCL2, we found a significant increase in high dose SHS-exposed mice as compared to control mice at month 1 (mean ± sd, 42 ± 5 vs 28 ± 11 pg/ml, p = 0.02) but a smaller and non-significant increase with medium-level SHS exposure (38 ± 15 vs 28 ± 1 pg/ml, p = 0.25).
Figure 3 Protein levels of CCL2 and TNFα were increased in BAL fluid from mice exposed to high dose SHS. CCL2 and TNFα were measured using ELISA in BAL fluid from filtered air- and medium- or high- dose SHS-exposed mice at after 1, 2 or 3 months of exposure. N = 5–6 for each group except for month 3, at which n = 16 for each group. Data are expressed as median (line), interquartile range (box) and range (whiskers, with extreme values as open circles); *denotes p < 0.05 as compared to air-exposed control. Med SHS = medium dose SHS.
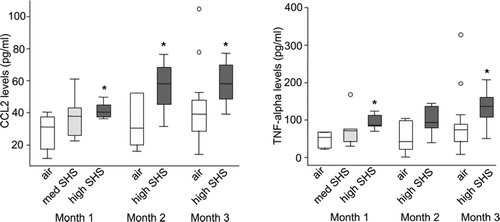
CCL2 levels were also significantly elevated with high dose SHS at month 2 (56 ± 16 vs 34 ± 16 pg/ml, p = 0.03) and month 3 (58 ± 12 vs. 43 ± 22 pg/ml, p = 0.03). For TNFα, we found a significant increase in high-dose SHS exposed mice at month 1 (94 ± 20 vs 48 ± 20 pg/ml, p = 0.003) but no significant increase with medium dose SHS (78 ± 54 vs 48 ± 20 pg/ml, p = 0.24) There was also a trend towards an increase with high-dose SHS at month 2 (98 ± 39 vs 52 ± 42 pg/ml, p = 0.08) and a further statistically significant increase with high-dose SHS at month 3 (138 ± 43 vs 88 ± 78 pg/ml, p = 0.04). These data indicate that exposure to SHS is sufficient to induce a pro-inflammatory mileu in the lung that did not wane over time.
Serum markers of inflammation
Based on our BAL fluid results indicating an increased in BAL fluid levels of CCL2 and TNFα, we performed ELISA for both of these mediators in serum from SHS exposed and control mice. However, we found that levels of both mediators were below the limit of detection in serum from all groups.
DISCUSSION
SHS exposure differs from active smoking in both the dose and composition of the smoke. Given the paucity of biological evidence linking SHS exposure and COPD, the chief aim of this study was to model the composition of SHS in an experimental setting and determine whether SHS exposure can cause changes in alveolar macrophage recruitment, activation and airway inflammation that are associated with COPD.
Using a mouse model, we found that exposure to the sidestream smoke which constitutes SHS induced a dose-dependent increase in alveolar macrophage recruitment and expression of several markers of alveolar macrophage activation that we have observed in habitual human smokers and mouse models of emphysema (Citation[27]). We also found that SHS exposure increased protein levels of two inflammatory mediators in BAL fluid that are associated with COPD (CCL2 and TNFα). These effects occurred without any measurable effects on weight gain or general health of the mice. In addition, these effects persisted over the 3 month time period without any evidence of waning due to adaptation to the noxious effects of SHS. We found no measurable effect of SHS on the development of emphysema in this 3-month study, which would be expected since mouse models of emphysema with mainstream tobacco smoke require at least 4 months (Citation[41]).
Our study was primarily focused on the effects of SHS on alveolar macrophages because growing evidence suggests that alveolar macrophages contribute to the development of emphysema in human smokers and in mouse models. Several studies have shown increased numbers of alveolar macrophages in the lung and BAL fluid in human smokers (Citation[39], Citation[40]). In addition, the expression of inflammatory mediators that recruit or activate macrophages, such as CCL2 and TNFα, are increased in COPD (Citation[19], Citation[42]). Furthermore, the expression and/or function of several products of activated macrophages, including the matrix metalloproteinases (MMP)-1, 2, 9, 12, and 14 (Citation[31], Citation[32], Citation[33], Citation[43], Citation[44], Citation[45]), and cathepsins L and C (Citation[46], Citation[47], Citation[48]) have been found to be increased in smokers and subjects with COPD.
One current hypothesis concerning the role of these macrophage-associated proteinases in the development of COPD is that they are consistently induced by smoking, leading to tissue injury. Subsequently, COPD occurs in the subset of smokers who are unable to sufficiently repair that tissue injury (Citation[49]). Thus, macrophage activation is thought to be a necessary initial step in the development of COPD. Mouse models have further delineated a specific macrophage-associated pathway in the development of emphysema by demonstrating that the macrophage metalloelastase, MMP-12, is required for the development of emphysema in response to high-dose mainstream smoke exposure (Citation[28]).
Other mouse models, such as airway over-expression of the cytokine interleukin-13 (IL-13) (Citation[30]) and deletion of the epithelial integrin subunit β 6 (Citation[29]), develop age-related emphysema; in each case, alveolar macrophage activation plays a critical role in the destruction of alveolar walls through the production of proteinases.
Our data show that second hand smoke exposure is sufficient to cause macrophage recruitment to the lung. However, there appear to be important differences in the mechanisms underlying macrophage recruitment in our study as compared to studies of mice exposed to high-dose mainstream smoke. A recent study by Houghton et al. exposing mice to mainstream cigarette smoke from 2 unfiltered cigarettes, 6 days a week for 2 months, found that elastin fragments were responsible for monocyte/macrophage chemotaxis and recruitment to the lung (Citation[50]).
Further, they showed that the generation of elastin fragments was due to induction and cleavage by MMP-12 (Citation[50]). Although we found macrophage recruitment to the lung, we did not find an increase in elastin fragments in BAL fluid (data not shown), nor did we find induction of MMP-12. In contrast, our results suggest that the mechanism of alveolar macrophage recruitment to SHS exposure may be due to CCL2 up-regulation, which we found at the protein level in BAL fluid from SHS-exposed mice. CCL2 is a potent monocyte/macrophage chemoattractant that has been measured in BAL fluid from chronic cigarette smokers (Citation[51]). In addition to its role as a chemoattractant, CCL2 can also induce activation of inflammatory cells (Citation[52], Citation[53], Citation[54]) and enhance certain macrophage functions such as anti-bacterial activity (Citation[55]).
We also found that exposure to SHS is sufficient to induce expression of several markers of macrophage activation that we previously identified as highly induced in two mouse models of emphysema and in human smokers, including PLA2g7, dkfzp434l142, Trem-2 and pirin (Citation[27]). In addition to serving as relevant markers of smoke exposure, the protein products of these genes may also contribute to pathophysiology of COPD. PLA2G7 (aka Lp-PLA2) is thought to play an important role in atherosclerosis by oxidizing lipids into reactive intermediates and PLA2g7 levels are an independent risk factor for cardiovascular events (i.e. myocardial infarction and stroke) (Citation[56], Citation[57], Citation[58]). Pirin has been found to be induced in both alveolar macrophages (Citation[27]) and in airway epithelial cells (Citation[59]) in smokers.
Over-expression of pirin in human bronchial epithelial cells leads to apoptosis (Citation[59]), a process which is thought to contribute to the development of emphysema (Citation[60]). TREM-2 is a cell surface receptor which is thought to modulate macrophage activation by signaling through the immunoreceptor protein DAP12 (Citation[61]). The function of dkfzp434l142 remains unknown at this time. Induction of these four genes by SHS exposure without induction of MMP-12 suggests either that induction of MMP-12 requires longer-term SHS exposure or higher levels, or that there are important differences in the type of macrophage activation induced by sidestream as compared to mainstream smoke. Nonetheless, these data indicate that SHS exposure is sufficient to induce the expression of genes in macrophages that plausibly contribute to inflammatory processes important in COPD, as well as cardiovascular diseases (e.g., PLA2g7).
Although there is little prior data on the effects of SHS on alveolar macrophage recruitment and activation, there are prior studies of the effects of SHS on systemic inflammatory cytokine production, oxidative stress, and vascular endothelial injury (Citation[62], Citation[63], Citation[64], Citation[65], Citation[66], Citation[67], Citation[68], Citation[69]). On comparison with these studies, our study differs in that we did not find increased serum inflammatory cytokine levels. This is most likely explained by differences in our assays and time courses of analyses. For example, in one prior study, systemic inflammatory cytokine production was identified by ex vivo stimulation of spleen cells (Citation[62]), whereas we assayed for cytokines directly in serum by ELISA. In a second prior study, plasma CCL2 and TNFα levels were found to be increased with long-term SHS exposure (1 year) but not with short-term exposure (14 weeks) (Citation[63]). These results are consistent with our findings in serum over 12 weeks. Of note, that study also found increased macrophage numbers in the arterial wall in SHS exposed mice suggesting that SHS exposure can affect macrophage recruitment to multiple relevant end-organs (Citation[63]).
SHS contains both sidestream smoke released from the smoldering cigarette and a small percentage of mainstream smoke exhaled by the smoker. Because sidestream smoke is generated at lower temperatures and under different combustion conditions, its composition differs from that of mainstream smoke (Citation[17]). Thus, our use of 89% sidestream smoke supplemented by 11% mainstream smoke mimicked the composition of SHS. In so doing, we chose ambient exposure levels higher than those typically observed in human SHS exposure environments, but significantly lower than those observed in active smokers (Citation[17]).
Additional factors, such as cumulative exposure and anatomical differences in airways affect interspecies scaling of equitoxic doses of SHS. Detailed discussion of interspecies scaling using an identical smoke exposure system are presented by Bogen et al. (Citation[75]), who concluded that the carcinogenic effects of these mouse exposure levels were similar to the increase in lifetime lung cancer risk observed with typical US residential SHS exposure. We modeled our exposure levels after this prior publication to facilitate comparison with the published literature on SHS exposure using our exposure system, recognizing that the ambient levels are high compared to SHS levels in human environments.
Our SHS exposure model also differs from mouse models of mainstream smoke exposure which typically expose mice to mainstream smoke only and which can achieve significantly higher doses (Citation[76]). Optimal contextual interpretation of our smoke exposure levels will depend on improved standardization and reporting of exposure levels in the various published models, which remains a challenge for the field.
In conclusion, we have found that short-term exposure to SHS in mice induces alveolar macrophage recruitment and expression of several markers of alveolar macrophage activation that we have observed in habitual human smokers and in mouse models of emphysema (Citation[27]).
These findings contribute to our understanding of the health effects of SHS exposure by providing a plausible biological link between SHS exposure and the development of emphysema. Our data also highlight that there may be several mechanisms underlying macrophage recruitment and activation with smoke exposure, including pathways independent of MMP-12 induction. These alternative pathways may relate to both the type of smoke exposure (sidestream versus mainstream) and dose or duration of smoke exposure. Given these findings in mice, future studies should assess the effects of long term SHS exposure in humans on alveolar macrophage recruitment, macrophage activation and other biological processes linked to COPD.
Declaration of interest
The authors report no conflict of interest. The authors alone are responsible for the content and writing of the paper.
ACKNOWLEDGMENTS
We thank Dale Uyeminami for assistance with the tobacco smoke exposure.
Funded by NIH grants RR17002, HL072915. Previously presented in abstract form at the American Thoracic Society International Conference, 2008.
REFERENCES
- Petty T L. Scope of the COPD problem in North America: early studies of prevalence and NHANES III data: basis for early identification and intervention. Chest 2000; 117(5 Suppl 2)326S–31S
- Ward M M, Javitz H S, Smith W M, Bakst A. Direct medical cost of chronic obstructive pulmonary disease in the U.S.A. Respir Med 2000; 94(11)1123–1129
- Gulsvik A. The global burden and impact of chronic obstructive pulmonary disease worldwide. Mondaldi Arch Chest Dis 2001; 56: 261–264
- Centers for Disase Control and Prevention. Chronic Obstructive Pulmonary Disease Surveillance-United States, 1971–2000. MMWR 2002; 51(SS-6)1–7
- Pauwels R A, Buist A S, Calverley P M, Jenkins C R, Hurd S S. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 2001; 163(5)1256–1276
- Thun M J, Myers D G, Day-Lally C, Namboodiri M M, Calle E E, Flanders W D, Adams S L, Heath C W. Age and the exposure-response relationships between cigarette smoking and premature death in Cancer Prevention Study II. Changes in Cigarette-Related Disease Risks and Their Implication for Prevention and Control, D R Shopland, D M Burns, L Garfinkel, J M Samet. U. S. Department of Health and Human Services, Public Health Service, National Institutes of Health, National Cancer Institute, Bethesda, MD 1997; 383–413, Smoking and Tobacco Control Monograph No. 8
- Robbins A S, Abbey D E, Lebowitz M D. Passive smoking and chronic respiratory disease symptoms in non-smoking adults. Int J Epidemiol 1993; 22(5)809–817
- Dayal H H, Khuder S, Sharrar R, Trieff N. Passive smoking in obstructive respiratory disease in an industrialized urban population. Environ Res 1994; 65(2)161–171
- Leuenberger P, Schwartz J, Ackermann-Liebrich U, Blaser K, Bolognini G, Bongard J P, Brandli O, Braun P, Bron C, Brutsche M, et al. Passive smoking exposure in adults and chronic respiratory symptoms (SAPALDIA Study). Swiss Study on Air Pollution and Lung Diseases in Adults, SAPALDIA Team. Am J Respir Crit Care Med 1994; 150: 1222–1228, (5 Pt 1)
- Piitulainen E, Tornling G, Eriksson S. Environmental correlates of impaired lung function in non-smokers with severe alpha 1-antitrypsin deficiency (PiZZ). Thorax 1998; 53(11)939–943
- Forastiere F, Mallone S, Lo Presti E, Baldacci S, Pistelli F, Simoni M, Scalera A, Pedreschi M, Pistelli R, Corbo G, Rapiti E, Agabiti N, Farchi S, Basso S, Chiaffi L, Matteelli G, Di Pede F, Carrozzi L, Viegi G. Characteristics of nonsmoking women exposed to spouses who smoke: epidemiologic study on environment and health in women from four Italian areas. Environ Health Perspect 2000; 108(12)1171–1177
- Lee P N, Chamberlain J, Alderson M R. Relationship of passive smoking to risk of lung cancer and other smoking-associated diseases. Br J Cancer 1986; 54(1)97–105
- Kalandidi A, Trichopoulos D, Hatzakis A, Tzannes S, Saracci R. Passive smoking and chronic obstructive lung disease. Lancet 1987; 2(8571)1325–1326
- Hirayama T. Non-smoking wives of heavy smokers have a higher risk of lung cancer: a study from Japan. Br Med J (Clin Res Ed) 1981; 282(6259)183–185
- Sandler D P, Comstock G W, Helsing K J, Shore D L. Deaths from all causes in non-smokers who lived with smokers. Am J Public Health 1989; 79(2)163–167
- Berglund D J, Abbey D E, Lebowitz M D, Knutsen S F, McDonnell W F. Respiratory symptoms and pulmonary function in an elderly nonsmoking population. Chest 1999; 115(1)49–59
- U. S. Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. U. S. Department of Health and Human Services, Centers for Disease Control and Prevention, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health, Atlanta, GA 2006
- Pirkle J L, Bernert J T, Caudill S P, Sosnoff C S, Pechacek T F. Trends in the exposure of nonsmokers in the U. S. population to secondhand smoke: 1988–2002. Environ Health Perspect 2006; 114(6)853–858
- Traves S L, Culpitt S V, Russell R E, Barnes P J, Donnelly L E. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax 2002; 57(7)590–595
- Aldonyte R, Eriksson S, Piitulainen E, Wallmark A, Janciauskiene S. Analysis of systemic biomarkers in COPD patients. COPD 2004; 1(2)155–164
- Aldonyte R, Jansson L, Piitulainen E, Janciauskiene S. Circulating monocytes from healthy individuals and COPD patients. Respir Res 2003; 4: 11
- Di Francia M, Barbier D, Mege J L, Orehek J. Tumor necrosis factor-alpha levels and weight loss in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1994; 150: 1453–1455, (5 Pt 1)
- Yasuda N, Gotoh K, Minatoguchi S, Asano K, Nishigaki K, Nomura M, Ohno A, Watanabe M, Sano H, Kumada H, Sawa T, Fujiwara H. An increase of soluble Fas, an inhibitor of apoptosis, associated with progression of COPD. Respir Med 1998; 92(8)993–999
- Takabatake N, Nakamura H, Abe S, Hino T, Saito H, Yuki H, Kato S, Tomoike H. Circulating leptin in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999; 159: 1215–1219, (4 Pt 1)
- Eid A A, Ionescu A A, Nixon L S, Lewis-Jenkins V, Matthews S B, Griffiths T L, Shale D J. Inflammatory response and body composition in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2001; 164: 1414–1418, (8 Pt 1)
- Keatings V M, Collins P D, Scott D M, Barnes P J. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996; 153(2)530–534
- Woodruff P G, Koth L L, Yang Y H, Rodriguez M W, Favoreto S, Dolgano G M, Paquet A C, Erle D J. A distinctive alveolar macrophage activation state induced by cigarette smoking. Am J Respir Crit Care Med 2005; 172(11)1383–1392
- Hautamaki R D, Kobayashi D K, Senior R M, Shapiro S D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997; 277(5334)2002–2004
- Morris D G, Huang X, Kaminski N, Wang Y, Shapiro S D, Dolganov G, Glick A, Sheppard D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 2003; 422(6928)169–173
- Zheng T, Zhu Z, Wang Z, Homer R J, Ma B, Riese R J, Jr, Chapman H A, Jr, Shapiro S D, Elias J A. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 2000; 106(9)1081–1093
- Molet S, Belleguic C, Lena H, Germain N, Bertrand C P, Shapiro S D, Planquois J M, Delaval P, Lagente V. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res 2005; 54(1)31–36
- Demedts I K, Morel-Montero A, Lebecque S, Pacheco Y, Cataldo D, Joos G F, Pauwels R A, Brusselle G G. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax 2006; 61(3)196–201
- Grumelli S, Corry D B, Song L Z, Song L, Green L, Huh J, Hacken J, Espada R, Bag R, Lewis D E, Kheradmand F. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med 2004; 1(1)e8
- Teague S V, Pinkerton K E, Goldsmith M, Gebremichael A, Chang S, Jenkins R A, Moneyhun J H. Sidestream cigarette smoke generation and exposure system for environmental tobacco smoke studies. Inhal Toxicol 1994; 6: 79–93
- Koth L L, Alex B, Hawgood S, Nead M A, Sheppard D, Erle D J, Morris D G. Integrin beta6 mediates phospholipid and collectin homeostasis by activation of latent TGF-beta1. Am J Respir Cell Mol Biol 2007; 37(6)651–659
- Dolganov G M, Woodruff P G, Novikov A A, Zhang Y, Ferrando R E, Szubin R, Fahy J V. A novel method of gene transcript profiling in airway biopsy homogenates reveals increased expression of a Na+ -K+-Cl− cotransporter (NKCC1) in asthmatic subjects. Genome Res 2001; 11(9)1473–1483
- Ochs M. A brief update on lung stereology. J Microsc 2006; 222: 188–200, (Pt 3)
- Ochs M, Knudsen L, Allen L, Stumbaugh A, Levitt S, Nyengaard J R, Hawgood S. GM-CSF mediates alveolar epithelial type II cell changes, but not emphysema-like pathology, in SP-D-deficient mice. Am J Physiol Lung Cell Mol Physiol 2004; 287(6)L1333–41
- Barbers R G, Gong H, Jr, Tashkin D P, Oishi J, Wallace J M. Differential examination of bronchoalveolar lavage cells in tobacco cigarette and marijuana smokers. Am Rev Respir Dis 1987; 135(6)1271–1275
- Kuschner W G, D'Alessandro A, Wong H, Blanc P D. Dose-dependent cigarette smoking-related inflammatory responses in healthy adults. Eur Respir J 1996; 9(10)1989–1994
- Wright J L, Churg A. Animal models of cigarette smoke-induced COPD. Chest 2002; 122(6 Suppl)301S–306S
- Chung K F. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl 2001; 34: 50s–59s
- Finlay G A, Russell K J, McMahon K J, D'Arcy E M, Masterson J B, Fitz Gerald M X, O'Connor C M. Elevated levels of matrix metalloproteinases in bronchoalveolar lavage fluid of emphysematous patients. Thorax 1997; 52(6)502–506
- Finlay G A, O'Driscoll L R, Russell K J, D'Arcy E M, Masterson J B, Fitz Gerald M X, O'Connor C M. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med 1997; 156(1)240–247
- Segura-Valdez L, Pardo A, Gaxiola M, Uhal B D, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000; 117(3)684–694
- Takeyabu K, Betsuyaku T, Nishimura M, Yoshioka A, Tanino M, Miyamoto K, Kawakami Y. Cysteine proteinases and cystatin C in bronchoalveolar lavage fluid from subjects with subclinical emphysema. Eur Respir J 1998; 12(5)1033–1039
- Takahashi H, Ishidoh K, Muno D, Ohwada A, Nukiwa T, Kominami E, Kira S. Cathepsin L activity is increased in alveolar macrophages and bronchoalveolar lavage fluid of smokers. Am Rev Respir Dis 1993; 147: 1562–1568, (6 Pt 1)
- Reilly J J, Jr, Chen P, Sailor L Z, Wilcox D, Mason R W, Chapman H A, Jr. Cigarette smoking induces an elastolytic cysteine proteinase in macrophages distinct from cathepsin L. Am J Physiol 1991; 261: L41–48, (2 Pt 1)
- Shapiro S D, Ingenito E P. The pathogenesis of chronic obstructive pulmonary disease: advances in the past 100 years. Am J Respir Cell Mol Biol 2005; 32(5)367–372
- Houghton A M, Quintero P A, Perkins D L, Kobayashi D K, Kelley D G, Marconcini L A, Mecham R P, Senior R M, Shapiro S D. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest 2006; 116(3)753–759
- Capelli A, Di Stefano A, Gnemmi I, Balbo P, Cerutti C G, Balbi B, Lusuardi M, Donner C F. Increased MCP-1 and MIP-1beta in bronchoalveolar lavage fluid of chronic bronchitics. Eur Respir J 1999; 14(1)160–165
- Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. Activation of NK cells by CC chemokines. Chemotaxis, Ca2 + mobilization, and enzyme release. J Immunol 1996; 156(1)322–327
- Bischoff S C, Krieger M, Brunner T, Dahinden C A. Monocyte chemotactic protein 1 is a potent activator of human basophils. J Exp Med 1992; 175(5)1271–1275
- Karpus W J, Lukacs N W, Kennedy K J, Smith W S, Hurst S D, Barrett T A. Differential CC chemokine-induced enhancement of T helper cell cytokine production. J Immunol 1997; 158(9)4129–4136
- Ritter U, Moll H. Monocyte chemotactic protein-1 stimulates the killing of leishmania major by human monocytes, acts synergistically with IFN-gamma and is antagonized by IL-4. Eur J Immunol 2000; 30(11)3111–3120
- Hakkinen T, Luoma J S, Hiltunen M O, Macphee C H, Milliner K J, Patel L, Rice S Q, Tew D G, Karkola K, Yla-Herttuala S. Lipoprotein-associated phospholipase A(2), platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler Thromb Vasc Biol 1999; 19(12)2909–2917
- Tselepis A D, John Chapman M. Inflammation, bioactive lipids and atherosclerosis: potential roles of a lipoprotein-associated phospholipase A2, platelet activating factor-acetylhydrolase. Atheroscler Suppl 2002; 3(4)57–68
- Macphee C H, Nelson J J, Zalewski A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr Opin Lipidol 2005; 16(4)442–446
- Gelbman B D, Heguy A, O'Connor T P, Zabner J, Crystal R G. Upregulation of pirin expression by chronic cigarette smoking is associated with bronchial epithelial cell apoptosis. Respir Res 2007; 8: 10
- Tuder R M, Petrache I, Elias J A, Voelkel N F, Henson P M. Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol 2003; 28(5)551–554
- Hamerman J A, Jarjoura J R, Humphrey M B, Nakamura M C, Seaman W E, Lanier L L. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 2006; 177(4)2051–2055
- Zhang J, Liu Y, Shi J, Larson D F, Watson R R. Side-stream cigarette smoke induces dose-response in systemic inflammatory cytokine production and oxidative stress. Exp Biol Med (Maywood) 2002; 227(9)823–829
- Yuan H, Wong L S, Bhattacharya M, Ma C, Zafarani M, Yao M, Schneider M, Pitas R E, Martins-Green M. The effects of second-hand smoke on biological processes important in atherogenesis. BMC Cardiovasc Disord 2007; 7: 1
- Yu M, Pinkerton K E, Witschi H. Short-term exposure to aged and diluted sidestream cigarette smoke enhances ozone-induced lung injury in B6C3F1 mice. Toxicol Sci 2002; 65(1)99–106
- Zhang Z, Araghiniknam M, Inserra P, Jiang S, Lee J, Chow S, Breceda V, Balagtas M, Witten M, Watson R R. Vitamin E supplementaion prevents lung dysfunction and lipid peroxidation in nude mice exposed to side-stream cigarette smoke. Nutr Res 1999; 19: 75–84
- Zhang J, Jiang S, Watson R R. Antioxidant supplementation prevents oxidation and inflammatory responses induced by sidestream cigarette smoke in old mice. Environ Health Perspect 2001; 109(10)1007–1009
- Mullick A E, McDonald J M, Melkonian G, Talbot P, Pinkerton K E, Rutledge J C. Reactive carbonyls from tobacco smoke increase arterial endothelial layer injury. Am J Physiol Heart Circ Physiol 2002; 283(2)H591–597
- Gairola C G, Drawdy M L, Block A E, Daugherty A. Sidestream cigarette smoke accelerates atherogenesis in apolipoprotein E-/- mice. Atherosclerosis 2001; 156(1)49–55
- Torok J, Gvozdjakova A, Kucharska J, Balazovjech I, Kysela S, Simko F, Gvozdjak J. Passive smoking impairs endothelium-dependent relaxation of isolated rabbit arteries. Physiol Res 2000; 49(1)135–41
- Witschi H, Espiritu I, Maronpot R R, Pinkerton K E, Jones A D. The carcinogenic potential of the gas phase of environmental tobacco smoke. Carcinogenesis 1997; 18(11)2035–2042
- Witschi H, Espiritu I, Peake J L, Wu K, Maronpot R R, Pinkerton K E. The carcinogenicity of environmental tobacco smoke. Carcinogenesis 1997; 18(3)575–86
- Witschi H, Espiritu I, Uyeminami D. Chemoprevention of tobacco smoke-induced lung tumors in A/J strain mice with dietary myo-inositol and dexamethasone. Carcinogenesis 1999; 20(7)1375–1378
- Witschi H, Espiritu I, Yu M, Willits N H. The effects of phenethyl isothiocyanate, N-acetylcysteine and green tea on tobacco smoke-induced lung tumors in strain A/J mice. Carcinogenesis 1998; 19(10)1789–1794
- Witschi H, Uyeminami D, Moran D, Espiritu I. Chemoprevention of tobacco-smoke lung carcinogenesis in mice after cessation of smoke exposure. Carcinogenesis 2000; 21(5)977–982
- Bogen K T, Witschi H. Lung tumors in A/J mice exposed to environmental tobacco smoke: estimated potency and implied human risk. Carcinogenesis 2002; 23(3)511–519
- Hahn F F, Gigliotti A P, Hutt J A, March T H, Mauderly J L. A review of the histopathology of cigarette smoke-induced lung cancer in rats and mice. Int J Toxicol 2007; 26(4)307–13