Abstract
The mechanisms behind airway inflammation in chronic obstructive pulmonary disease (COPD) are still not well understood. Here we investigated lymphocyte subtypes in bronchoalveolar lavage fluid, likely to be involved in the pathogenesis of COPD, as well as exploring the effect of smoking cessation. Differential cell counts and T cell subsets were determined in BAL fluid from nineteen individuals with stable COPD (seven smokers, twelve ex-smokers) compared to twelve age-matched never-smokers and thirteen smoking-matched smokers with normal lung function. COPD-patients had higher percentages of airway CD8+ T cells compared to never-smokers. An increased population of CD4+ T cells expressed high levels of CD25 in smokers and COPD patients compared to never-smokers, suggesting the presence of regulatory T cells. As the T cell populations in smokers with normal lung function and COPD-patients were similar, the impact of current smoking in COPD was addressed in a subgroup analysis. Activation of CD8+ T cells was found regardless of smoking habits. In contrast, the enhanced expression of γ /δ T cells, was mainly associated with current smoking, whilst the increase in T regulatory cells appeared related to both smoking and COPD. Regardless of smoking habits, CD8+ T cell activation was found in COPD, supporting the contention that this T cell subset may play a role in the pathogenesis of COPD. As CD8+ T cells coexist with immunoregulatory CD4+ T cells in airways of COPD patients, it is likely that both cytotoxic T-cell responses and immunosuppressive mechanisms may be of importance in COPD pathogenesis.
INTRODUCTION
Chronic obstructive pulmonary disease, COPD, is characterized by a chronic airway inflammation and a progressive airway obstruction. Several cell types such as neutrophils, macrophages and T lymphocytes are likely to be involved in the airway inflammation in COPD (Citation[1]). The development and progression of COPD have also been suggested to be associated with increased oxidative stress and reduced antioxidant resources (Citation[2]). Still, the pathogenic mechanisms of COPD are not fully understood.
T-lymphocytes are believed to be key cells in regulating airway inflammation in COPD. T-cells are classified either as T cells receptor (TCR) α /β or γ /δ. TCR-α /β comprise 95% of the T-cell population and orchestrate the immune response by the secretion of activating and regulating cytokines, while TCR-γ /δ lymphocytes play a key role in mucosal homeostasis and in response to tissue damage at the epithelial surface (Citation[3]). Several studies have shown higher numbers of cytotoxic CD8+ T cells in patients with COPD than in smokers without COPD (Citation[4], Citation[5], Citation[6], Citation[7]). The underlying mechanism triggering the CD8+ T cells and their functional role in COPD are still elusive.
How smoking cessation affects airway inflammation in COPD is not fully understood, as only few studies address this issue. Rutgers et al. reported increased numbers of eosinophils in BAL fluid in ex-smoking subjects with COPD (Citation[8]). Another study indicated that ex-smokers with COPD had increased bronchial CD4+ and plasma cells compared to current smokers with COPD. However, the numbers of neutrophils, macrophages and CD8+ cells did not differ between the two groups (Citation[9]).
Activation of T cells is generally initiated by the recognition of an antigen presented by accessory cells together with cellular and soluble co-stimulatory molecules. One early marker of activated T cells is CD69, which is expressed on activated T, B and Natural killer (NK)-cells. HLA-DR is considered to be a more general marker of activated T-cells, whereas CD25, the α -chain of the IL-2 receptor, is presented on activated T, B-cells and monocytes (Citation[10]). High expression of CD25 on CD4+ lymphocytes is suggested to indicate the presence of regulatory T cells. This subset of immunoregulatory T cells is supposed to play a crucial role in suppressing immune responses to self-antigens and in preventing autoimmune diseases (Citation[11]) and recently published data suggest that cigarette smoke exposure up-regulates airway CD4+ regulatory cell numbers (Citation[12]).
The hypothesis of the present study was that patients with moderate to severe, but clinically stable, COPD would have differences in lymphocyte subsets in BAL compared to never-smokers and smokers with normal lung function, and that lymphocytes may be of importance in the pathogenesis of COPD. The primary aim was to compare airway T cells population and their activation between patients with COPD, never-smokers and smokers with normal lung function. Furthermore, a second aim was to investigate whether smoking cessation in COPD patients would affect airway T-cell subsets.
MATERIALS AND METHODS
Subjects
Forty-four subjects participated in this study; 19 patients with moderate to severe COPD according to GOLD-criteria (FEV1 37-60% of predicted), 12 healthy volunteers with no smoking history (NS) and 13 smokers with normal lung function (S). Subjects' demographics are given in . Of the COPD patients, 7 were current smokers and 12 were ex-smokers with smoking cessation more than 5 years prior to inclusion. Both COPD-groups had significantly reduced lung function described as FEV1/FVC% ratio and FEV1% of predicted post-bronchodilation compared to both smokers with normal lung function (p = 0.001 + p = 0.001) and never-smokers (p = 0.001 + p = 0.001).
Table 1 Demographics and spirometry values of the four groups
No differences in smoking habits were found between smokers, COPD ex-smokers and COPD smokers. The COPD patients received no treatment with inhaled corticosteroids during at least four weeks prior to study start and neither long-acting β2-agonists nor long-acting anti-cholinergic drugs were allowed within two weeks prior to bronchoscopy. Short-acting β2-agonists and/or anti-cholinergic drugs were used on demand. All the subjects were non-atopic and free from symptomatic respiratory infection within a 6-week period prior to and during the study. No history of frequent exacerbations. No anti-inflammatory drugs, i.e., non-steroidal anti-inflammatory drugs, oral steroids or any additional intake of vitamin C or E were permitted. Informed consent was obtained from all volunteers after verbal and written information and the study was approved by the local Ethics Committee at Umeå University, Sweden, and performed according to the declaration of Helsinki.
METHODS
Spirometry
Dynamic spirometry variables (FVC and FEV1) were measured post bronchodilation with 1 mg Bricanyl, using a Vitalograph spirometer (Vitalograph Ltd. Buckingham, UK). At least three satisfactorily performed and well-co-operated measurements of each variable were carried out, according to the recommendations of the American Thoracic Society (Citation[13]).
Bronchoscopy
Atropine was given subcutaneously before bronchoscopy and topical anaesthesia of the airways was obtained using lidocaine. The subjects were examined in the supine position using an Olympus BF T240 video bronchoscope (Olympus, Tokyo, Japan). Bronchoalveolar lavage (BAL) was performed by infusing three aliquots of 60 ml of sterile sodium chloride (NaCl), pH 7.3 at 37°C that were gently sucked back after each infusion and pooled into a tube placed in iced water. The recovered fluid was immediately transported to the laboratory for analysis.
Cell preparation
The chilled bronchoalveolar lavage (BAL) was processed by filtration through a nylon filter (pore diameter 100 μ m, Syntab Product AB, Malmö, Sweden) and by centrifugation (400 g, 15 minutes, at 4°C). The cell pellet was resuspended in PBS. The total number of cells was counted and adjusted to a final concentration of 106 cells/ml.
Cytocentrifuged specimens with 50 μ l suspended cells from 106/ml non-epihelial cells/slide were prepared using a Cytospin 2® (Shandon Southern Instruments Ins., Sewikly, PA, USA) 1,000 rpm for 5 minutes. Slides were stained according to May-Grünwald Giemsa for standard total and cell differential counts and 500 cells per slide were counted.
Flow cytometry analysis
Subpopulations of lymphocytes in BAL were determined by flow cytometry. BAL cells were centrifuged and diluted to a final concentration of 106 cells/ml. For each test, 10 μ l of antibody solution was added to 200 μ l of cell suspension and allowed to bind for 30 minutes at 4°C in darkness. Red blood cells were lysed with 2 ml FACS™ Lysing solution (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) for 10 minutes at room temperature. After lysing red cells, the remaining cells were washed by adding PBS to the tubes and centrifuged at 4°C for 10 minutes, 300 g. This washing procedure was performed twice. Cells were then fixed with 500 μ l CellFIX™ (Becton Dickinson Immunocytometry Systems, San Jose, CA, USA) before analysis using a FACScan™ (Becton Dickinson) flow cytometer. Up to 10,000 total events were collected per sample. The lymphocyte population was gated on their physical characteristics in a region according to their characteristic forward scatter (FCS) and side scatter (SSC) profiles.
To obtain CD4+ and CD8+ cells, the cells were stained with Peridinin Chlorophyll Protein (PerCP) conjugated anti-human CD3, fluorescein isothiocyanate (FITC) conjugated anti-human CD4 and phycoerytrin (PE) conjugated anti-human CD8 in the same test tube. To obtain NK cells and NK/T-cells, the cells were stained with FITC conjugated anti-human CD3, PE conjugated anti-human CD56 and PE conjugated anti-human CD16 in the same tube. To obtain TCR-α /β and TCR-γ /δ, cells were stained with PerCP conjugated anti-human CD3, FITC conjugated anti-human TCR-α /β and PE conjugated anti-human TCR-γ /δ in the same test tube.
To detect activation markers, cells were stained with FITC conjugated anti-human CD4, PerCP conjugated anti-human CD8 in combination with either PE conjugated anti-human CD69, anti-human HLA-DR or anti-human CD25. PE, FITC and PerCP conjugated IgG matched antibody subtypes served as controls. The percentage of different cell types was counted out of gated lymphocytes. The quantification of CD25bright cells was performed as previously described (Citation[12], Citation[14]). Source of antibodies was Becton Dickinson Immunocytometry Systems, San Jose, CA, USA.
Statistical analysis
Flow cytometry data were acquired and analysed using CellQuest Software (Becton Dickinson, San Jose, CA, USA). Differences between COPD, never-smokers and smokers with normal lung function were tested using Kruskal-Wallis test and a p-value of less than 0.05 was considered significant.
If the Kruskal-Wallis test indicated significance, the Mann-Whitney U-test was used for post-hoc analysis for comparison between two groups, with corrections of p-values according to Bonferroni (a p-value less than 0.017 was considered significant). In order to distinguish between COPD and smoking-related effects, a subgroup analysis was carried out within the COPD group. The ex-smoking COPD group was compared to both the smoking COPD group and the never smoking group, using Mann-Whitney U-test. Here, a p-value of less than 0.05 was considered significant. Correlation analyses were performed with Pearson correlation test.
RESULTS
Differential cell counts in BALF
Smokers with normal lung function had increased number of total leukocytes in BALF compared to never-smokers (p = 0.002). Among leukocytes, the number of macrophages (p < 0.001), mast cells (p = 0.001) and neutrophils (p = 0.012) were increased, .
Table 2 Differential cell count of white blood cells in BAL fluid, given in number cells/ml*104
COPD patients had decreased number of leukocytes in BALF compared to the population of smokers with normal lung function (p = 0.001). The differential cell count demonstrated that this difference was due to a lower number of macrophages in COPD (p = 0.001), .
To examine whether the difference in airway inflammation between COPD patients and smokers with normal lung function was due to smoking habits, the group of COPD patients was divided into current smokers and ex-smokers. The results of this subgroup analysis showed that smoking COPD patients had increased numbers of BAL macrophages (p = 0.003) and total leukocytes (p = 0.014) compared to ex-smoking COPD patients, whereas there was no significant difference between COPD ex-smokers and never-smokers, .
BALF lymphocyte subpopulations
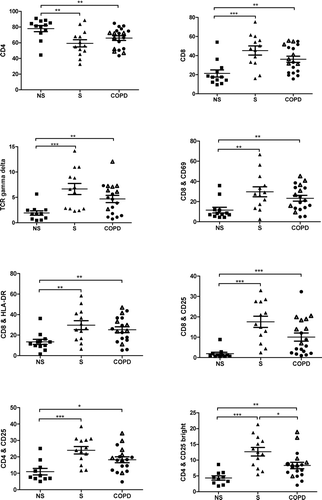
COPD patients and smokers with normal lung function had significantly increased percentages of CD8+ and TCR-γ /δ+ among gated lymphocytes as well as increased expression of CD69, HLA-DR and CD25 on CD8+ cells, compared to never-smokers, . The percentage of CD4+ was significantly decreased. CD25 expression on CD4+ T cells was significantly enhanced in COPD patients and smokers with normal lung function compared to never-smokers, whilst the expression of CD69 and HLA-DR was not significantly altered on CD4+ T cells. The total CD25+ cell population was further divided into a high expression (bright) and a low expression (dim) population, . The percentage of CD4+CD25bright cells was enhanced among COPD patients and smokers with normal lung function, compared to never-smokers. In contrast to all other markers, the percentage of CD4+CD25bright cells was significantly decreased in COPD patients compared to smokers with normal lung function, .
Figure 1 Lymphocytes derived from BAL fluid were gated according to forward and side scatter (FSC/SSC) and screened by flow cytometry for the presence of CD4+CD25+ and CD4+CD25bright cells. Representative FACS stainings from one individual with COPD and one control subject are shown. To the right of each figure, percentages of bright, dim and negative population of CD4+ and CD8+ cells are shown.
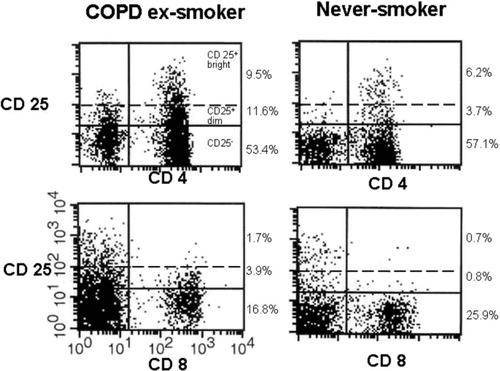
Figure 2 Flow cytometry of never-smokers (NS), smokers (S) and COPD. CD4, CD8 and TCR gamma delta are given as percent of CD3. Among the COPD group • indicates ex-smokers, whilst Δ indicates smokers. A p-value below 0.017 is considered significant. Significance levels are noted as * p < 0.017, ** p < 0.01 and *** p < 0.001.
To examine whether the comparison in T-cells responses between smokers and ex-smokers with COPD and smokers with normal lung function was affected by smoking cessation, the group of COPD patients was divided into current smokers and ex-smokers. Ex-smoking COPD patients expressed decreased percentage of TCR-γ /δ+, CD8+ CD25+ and CD4+ CD25bright cells in BALF compared to smoking COPD patients, .
Figure 3 Sub-groups analysis of never-smokers (NS), COPD ex-smokers (COPD ex-s) and COPD smokers (COPD s). CD4, CD8 and TCR gamma delta are given as percent of CD3. A p-value below 0.05 is considered significant. Significance levels are noted as * p < 0.05 and ** p < 0.01.
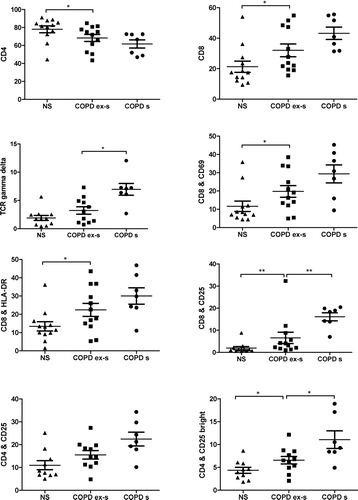
In contrast, the percentage of CD8+ cells was increased in COPD ex-smokers compared to never-smokers. Activation markers, such as CD69, HLA-DR and CD25 on CD8+ cells were also significantly enhanced in ex-smokers with COPD compared to never-smokers. There was no significant difference in the ratio of CD8/CD4CD25 between the four groups. No significant correlation was found between pack year history and lung function, in terms of FEV1, in the smoking and ex-smoking subjects (data not shown).
DISCUSSION
This is the first study demonstrating that CD8+ lymphocytes in BAL fluid from COPD patients are highly activated, regardless of current smoking habits. The increase in T regulatory cells seems to be both smoking-related and disease-dependent, whereas TCR-γ /δ expression is largely related to smoking habits. As CD8+ cells coexist with immunoregulatory CD4+ T cells in the airways of COPD patients, it is likely that both cytotoxic T-cell responses and immunosuppressive mechanisms may play an important role in COPD pathogenesis.
In this study, bronchoscopy was performed in clinically stable COPD patients with no history of present or recent exacerbation or concomitant disease. This gave an opportunity to minimize the risk of influence by i.e., co-morbidity, medication and exacerbations. It was also possible to compare COPD patients with smoking-matched and never smoking controls.
We demonstrate an increase in total airway leukocytes of smokers compared to never smokers. The changes in BAL cells were mainly due to an expansion of alveolar macrophages and, to a lesser extent, to an increase in neutrophils and mast cells, indicating a prominent role of macrophages in the smoke-induced defence mechanism. The present data are thus in concordance with other published studies showing an increase in total leukocytes in airways of smokers with normal lung function (Citation[15], Citation[16]).
We did not observe any significant difference in the absolute numbers of BAL neutrophils between COPD patients and controls. This differs from an increase in total BAL neutrophils reported in both smoking and ex-smoking COPD patients, compared to smokers and never-smokers (Citation[17]). Earlier studies have shown that COPD-patients display increased numbers of airway neutrophils (PMN) (Citation[18]). In BAL, Hodge et al. showed no significant difference in PMN percentages between COPD patients and healthy individuals. However, when COPD was divided into mild and moderately severe disease, a significant increase in PMN percentages was observed with more severe disease (Citation[19]). This may explain the discrepancy between our and previous data (Citation[17]), as only clinically stable COPD patients were included in the present study. Other factors such as medication or exacerbation frequency may also influence the neutrophilic response.
Smokers with normal lung function and COPD patients had significantly higher percentages of cytotoxic T cells expressing CD8 or TCR-γ /δ compared to never smokers. Several studies have reported that CD8+ T cells are increased in lung tissue from large, peripheral airways, lung parenchyma, smooth muscle and bronchial arteries of COPD patients compared to smokers or non-smokers (Citation[5], Citation[20], Citation[21], Citation[22], Citation[23], Citation[24]). Earlier studies have shown an inverse relationship between the number of CD8+ cells in bronchial biopsies and FEV1 in both COPD patients and smokers (Citation[5], Citation[16], Citation[20]), indicating that CD8+ cells may play a role in tissue destruction and emphysema progression. However, Pons et al. have compared BAL fluid from COPD patients with smokers and non-smokers but did not find any significant differences between the groups when it came to CD4+ and CD8+ cells (Citation[25]).
Of importance, we demonstrated that even 5 years after smoking cessation, the COPD patients had significantly higher percentages of CD8+ cells compared to never-smokers. The reason for the persistent increase of CD8+ cells in COPD is still unknown. Hypothetically, cytotoxic T cells may be recruited or expanded as a result of cigarette smoke-induced cell stress and pro-inflammatory responses triggered in the lung epithelium. One possible mechanism for such activation is the stress-induced expression of the MHC class I-like MIC-receptors on epithelial cells. This stress response is previously shown to activate CD8+ T cells and γ δ T cells through the counter receptor NKG2D (Citation[26]). Alternatively, it is suggested that the frequent infections in COPD patients may trigger a sustained CD8+ T cell response (Citation[27], Citation[28]). The increased numbers of CD8+ cells in COPD may thus be explained as a defence mechanism to infectious agents such as bacterial (Citation[29]) or persistent viral infections (Citation[28]). There are also alternative theories suggesting that CD8+cells recognize self antigens, indicating an autoimmune component in COPD (Citation[30]). Lee et al. have implied that emphysema is an autoimmune disease characterized by the presence of anti-elastin antibodies and a T-helper type 1 response, which correlate with emphysema severity (Citation[31]).
The proportion of γ /δ T cells was increased in healthy smokers and individuals with COPD compared to never-smokers. The role of γ /δ T cells in COPD pathogenesis is unclear, but it has been suggested that these cells are important regulators of airway immune responses (Citation[32]). Cigarette smokers have increased γ /δ T cell numbers in bronchial glands compared to non-smokers (Citation[33]). Pons et al. have reported that γ /δ T lymphocytes were significantly increased in BAL from smokers with preserved lung function compared with never-smokers and COPD patients (Citation[25]). Whether the expansion of γ δ T cells in COPD reflects a common triggering mechanism of cytotoxic T cells, such as the MIC/NKG2D pathway, or is due to any specific responsiveness of this T cell subset is still unknown.
This is the first study demonstrating that CD8+ lymphocytes in BAL fluid from COPD patients are highly activated, in terms of increased CD69 and HLA-DR expression. In lung tissue, an increased expression of HLA-DR was shown in COPD patients compared to normal subjects (Citation[20]). The increase in CD69 and HLA-DR on CD8+ lymphocytes is consistent with an active participation of cytotoxic T cells in the pathogenic response to cigarette smoke, further supporting that these T cells are triggered by a specific recognition of antigenic determinants from self antigens or infectious agents, and/or by co-stimulatory mechanisms such as the MIC/NKG2D interactions.
The expression of CD25 was enhanced on both CD4+ and CD8+ cells in patients with COPD and smokers with normal lung function compared to never-smokers. The increase in CD25 on CD4+ cells may indicate the presence of regulatory T cells, which was further supported by the observed increase in the proportion of CD25bright T cells in these groups. Recent data support this finding, even though the studies cannot be fully compared, due to significant differences in study design and methodology (Citation[12]). Lee et al. showed a decrease in regulatory T cells in lungs of subjects with emphysema, suggesting that emphysema is an autoimmune disease (Citation[31]). Adaptive TReg cells display a variable CD25 expression and mediate their inhibitory activities by producing immunosuppressive cytokines, such as transforming growth factor-β (TGF- β) and IL-10 (Citation[34]). TGF-β inhibits proliferation and cytokine secretion by T-cells and IL-10 may act as an immunosuppressive cytokine in type 1 inflammatory response (Citation[35]). It is thus likely that a high CD25 expression on CD4+ cells indicates the presence of immunosuppressive mechanisms along with pro-inflammatory activity at the site of inflammation in COPD.
The main difference in lymphocyte pattern in BAL fluid from ex-smoking COPD patients compared to currently smoking COPD patients was the lower percentages of macrophages and γ /δ cells, indicating that the increase in these leukocyte subsets was related to cigarette smoking rather than to the chronic immune response in stable COPD. There was no significant difference in the proportion of CD8+ T cells and the expression of CD69 and HLA-DR on CD8+ cells in ex-smoking COPD patients compared to smoking COPD patients. The former smokers among COPD patients expressed more activated CD8+ lymphocytes, when it came to CD69, HLA-DR and CD25, compared to never-smokers. Thus, still 5 years after smoking cessation the cytotoxic T lymphocytes were activated, demonstrating that their expansion and activation were not only smoking-related, but also associated with a chronic immune response in COPD.
However, no correlation was found between the lymphocyte markers and time since smoking cessation. In individuals with COPD, the present data indicate that cytotoxic T-lymphocytes are activated in the distal part of the lung, as reflected in BAL fluid, in terms of increased expression of the activation markers CD69 and HLA-DR on CD8+ T cells. CD8+CD25+ T cells and CD25bright expression on CD4+ T cells decreased in COPD-patients after smoking cessation, but were still enhanced compared to non-smoking healthy individuals. Taken together, these findings indicate that, despite smoking cessation, COPD-patients exhibit signs of increased percentages of regulatory T cells along with enhanced activation of cytotoxic T-cells.
The lack of significant association between lung function and pack-year history may be due to the small number of smoking/ex-smoking subjects and that smoking cessation in the ex-smoking COPD-group was in average more than 5 years ago.
In summary, we have demonstrated that activated CD8+ T lymphocytes were increased in BAL fluid from clinically stable COPD-patients. The increase in CD8 activation markers in COPD was present regardless of current smoking habits and suggests that changes in CD8+ T cells are associated with a persistent immune response in COPD and may therefore be of importance in COPD pathogenesis. In contrast, γ /δ T cells were mainly related to current smoking. In airways of COPD patients, CD8+ cells co-exist with immunoregulatory CD4+ T cells. It is thus likely that pro-inflammatory responses are balanced by immunosuppressive mechanisms in stable COPD. Whether these findings also are applicable to patients with more severe COPD with frequent exacerbations, needs to be further evaluated.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
ACKNOWLEDGMENT
This study was supported by Swedish Heart-Lung Foundation, King Gustaf V:s and Queen Victorias foundation and the Umeå University. The authors would like to thank A M Wallström, Ann-Britt Lundström, Elisabeth Åslund, Annica Johansson, Helena Tjällgren-Bogseth and Frida Holmström for their contribution to the project.
REFERENCES
- Hogg J C, Chu F, Utokaparch S, Woods R, Elliott W M, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med Jun 24, 2004; 350: 2645–2653
- Betsuyaku T. [Oxidative stress in pathogenesis of COPD]. Nippon Rinsho Apr, 2007; 65: 633–636
- Jameson J, Witherden D, Havran W L. T-cell effector mechanisms: gammadelta and CD1d-restricted subsets. Curr Opin Immunol Jun, 2003; 15: 349–353
- Saetta M. Airway inflammation in chronic obstructive pulmonary disease. Amer J Respir Crit Care Med Nov, 1999; 160: S17–20
- Saetta M, Di Stefano A, Turato G, Facchini F M, Corbino L, Mapp C E, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Amer J Respir Criti Care Med Mar, 1998; 157: 822–826
- Saetta M, Baraldo S, Turato G, Beghé B, Casoni G L, Bellettato C M, Rea F, Zuin R, Fabbri L M, Papi A. Increased proportion of CD8+ T-lymphocytes in the paratracheal lymphnodes of smokers with mild COPD. Sarcoidosis Vasculitis Diffuse Lung Dis 2003; 20: 28–32
- Chrysofakis G, Tzanakis N, Kyriakoy D, Tsoumakidou M, Tsiligianni I, Klimathianaki M, et al. Perforin expression and cytotoxic activity of sputum CD8+ lymphocytes in patients with COPD. Chest Jan, 2004; 125: 71–76
- Rutgers S R, Postma D S, ten Hacken N H, Kauffman H F, van Der Mark T W, Koeter G H, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax Jan, 2000; 55: 12–18
- Lapperre T S, Postma D S, Gosman M M, Snoeck-Stroband J B, ten Hacken N H, Hiemstra P S, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax Feb, 2006; 61: 115–121
- Janeway C, Travers P, Walport M, Shlomchik M. Immunobiology. 6 ed. Garland Science Publishing. 2005
- Mills K H. Regulatory T cells: friend or foe in immunity to infection?. Nat Rev Immunol Nov, 2004; 4: 841–855
- Smyth L J, Starkey C, Vestbo J, Singh D. CD4-regulatory cells in COPD patients. Chest Jul, 2007; 132: 156–163
- American Thoracic Society. Lung function testing:selection of reference values and interpretative strategies. Am Rev Respir Dis 1991; 144: 1202–1218
- Crispin J C, Martinez A, Alcocer-Varela J. Quantification of regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun Nov, 2003; 21: 273–276
- Barcelo B, Pons J, Ferrer J M, Sauleda J, Fuster A, Agusti A G. Phenotypic characterisation of T-lymphocytes in COPD: abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J Mar, 2008; 31: 555–562
- Ekberg-Jansson A, Arva E, Nilsson O, Lofdahl C G, Andersson B. A comparison of the expression of lymphocyte activation markers in blood, bronchial biopsies and bronchoalveolar lavage: evidence for an enrichment of activated T lymphocytes in the bronchoalveolar space. Respir Med Aug, 1999; 93: 563–570
- Babusyte A, Stravinskaite K, Jeroch J, Lotvall J, Sakalauskas R, Sitkauskiene B. Patterns of airway inflammation and MMP-12 expression in smokers and ex-smokers with COPD. Respir Res 2007; 8: 81
- Barnes P J, Shapiro S D, Pauwels R A. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J Oct, 2003; 22: 672–688
- Hodge S J, Hodge G L, Holmes M, Reynolds P N. Flow cytometric characterization of cell populations in bronchoalveolar lavage and bronchial brushings from patients with chronic obstructive pulmonary disease. Cytometry B Clin Cytom Sep, 2004; 61: 7–34
- O'Shaughnessy T C, Ansari T W, Barnes N C, Jeffery P K. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Amer J Respir Crit Care Med Mar, 1997; 155: 852–857
- Turato G, Zuin R, Miniati M, Baraldo S, Rea F, Beghe B, et al. Airway inflammation in severe chronic obstructive pulmonary disease: relationship with lung function and radiologic emphysema. Amer J Respir Crit Care Med Jul 1, 2002; 166: 105–110
- Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, et al. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Amer J Respir Crit Care Med Aug, 1999; 160: 711–717
- Baraldo S, Turato G, Badin C, Bazzan E, Beghe B, Zuin R, et al. Neutrophilic infiltration within the airway smooth muscle in patients with COPD. Thorax Apr, 2004; 59: 308–312
- Peinado V I, Barbera J A, Abate P, Ramirez J, Roca J, Santos S, et al. Inflammatory reaction in pulmonary muscular arteries of patients with mild chronic obstructive pulmonary disease. Amer J Respir Crit Care Med May, 1999; 159: 1605–1611
- Pons J, Sauleda J, Ferrer J M, Barcelo B, Fuster A, Regueiro V, et al. Blunted gamma delta T-lymphocyte response in chronic obstructive pulmonary disease. Eur Respir J Mar, 2005; 25: 441–446
- Bauer S, Groh V, Wu J, Steinle A, Phillips J H, Lanier L L, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (New York, NY) Jul 30, 1999; 285: 727–729
- de Bree G J, Heidema J, van Leeuwen E M, van Bleek G M, Jonkers R E, Jansen H M, et al. Respiratory syncytial virus-specific CD8+ memory T cell responses in elderly persons. J Infect Dis May 15, 2005; 191: 1710–1718
- Retamales I, Elliott W M, Meshi B, Coxson H O, Pare P D, Sciurba F C, et al. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Amer J Respir Crit Care Med Aug 1, 2001; 164: 469–473
- Sethi S, Murphy T F. Bacterial infection in chronic obstructive pulmonary disease in 2000: a state-of-the-art review. Clin Microbiol Rev Apr, 2001; 14: 336–363
- Cosio M G, Majo J, Cosio M G. Inflammation of the airways and lung parenchyma in COPD: role of T cells. Chest May, 2002; 121: 160S–165S
- Lee S H, Goswami S, Grudo A, Song L Z, Bandi V, Goodnight-White S, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nature Med May, 2007; 13: 567–569
- Lahn M. The role of gammadelta T cells in the airways. J Mol Med 2000; 78: 409–425
- Richmond I, Pritchard G E, Ashcroft T, Corris P A, Walters E H. Distribution of gamma delta T-cells in the bronchial tree of smokers and non-smokers. J Clin Pathol Oct, 1993; 46: 926–930
- Bluestone J A, Abbas A K. Natural versus adaptive regulatory T cells. Nat Rev Immunol Mar, 2003; 3: 253–257
- van Oosterhout A J, Bloksma N. Regulatory T-lymphocytes in asthma. Eur Respir J Nov, 2005; 26: 918–932