Abstract
Background. Augmentation with exogenous α1-antitrypsin (α1-AT) is the only specific therapy for α1-AT deficiency. Uncertainty persists concerning its effectiveness. Purpose. To test the hypothesis that augmentation therapy in patients with α1-AT deficiency slows the decline in FEV1. Study Selection. Randomized and nonrandomized clinical studies with either parallel-group design or single cohort pre-post design were eligible if they compared augmentation therapy with a control regimen and if long-term (> 1 y) longitudinal FEV1 follow-up data were collected. Data Synthesis. FEV1 data from five trials with 1509 patients were combined by random effects meta-analysis. The decline in FEV1 was slower by 23% (absolute difference, 13.4 ml/year; CI, 1.5 to 25.3 ml/year) among all patients receiving augmentation therapy. This overall protective effect reflected predominantly the results in the subset of patients with baseline FEV1 30–65% of predicted. In that subset, augmentation was associated with a 26% reduction in rate of FEV1 decline (absolute difference, 17.9 ml/year; CI, 9.6 to 26.1 ml/year). Similar trends amongst patients with baseline FEV1 percent of predicted < 30% or > 65% were not statistically significant. Conclusions. This meta-analysis supports the conclusion that augmentation can slow lung function decline in patients with AAT deficiency Patients with moderate obstruction are most likely to benefit.
INTRODUCTION
Intravenous administration of exogenous α1-AT harvested from pooled blood product is currently the only specific therapy available for α1-AT deficiency. Introduced in 1987, augmentation infusions given weekly, biweekly or monthly increase circulating levels of α1-AT to levels approximating those seen in heterozygotes. Antiprotease activity is increased in lung epithelial-lining fluid following infusion (Citation[1]) suggesting that such therapy will restore antiprotease protection to the lung will slow the accelerated decline in lung function typical of α1-AT deficiency emphysema. However, no single large prospective randomized placebo-controlled trial has been conducted to test this hypothesis. During the initial development of augmentation therapy for α1-AT deficiency, a placebo-controlled and randomized trial of sufficient duration and patient numbers to demonstrate preservation of spirometric indices was not thought to be feasible. Efficacy studies have been variable in design and generally small in scale. Various endpoints have been addressed including FEV1 (Citation[2], Citation[3], Citation[4], Citation[5], Citation[6], Citation[7], Citation[8]), survival (Citation[5]), lung density measured by computed tomography (7), diffusion capacity (Citation[2]), leukotriene B4 (Citation[9]), frequency of exacerbations (Citation[10]) and desmosine excretion (Citation[11]). Evidence furnished by these investigations is generally consistent with beneficial effects of augmentation therapy (Citation[12]). However, further confirmation is needed along with more precise estimates of the magnitude of the effects and more complete characterization of patient groups most likely to benefit. Thus far, there has been no systematic review encompassing all available evidence related to augmentation therapy. Current evidence has prompted a recommendation that augmentation therapy be reserved for patients with moderate airflow limitation (FEV1 35 to 50% of predicted) whose pulmonary function continues to deteriorate rapidly despite smoking cessation and optimal medical therapy (Citation[13], Citation[14]). Some data, however, suggest greater benefit in patients with better-preserved lung function (Citation[8]). We conducted a meta-analysis to test the hypothesis that augmentation therapy with exogenous α1-AT slows the accelerated decline in lung function, as measured by FEV1, among patients with α1-AT deficiency.
METHODS
A written protocol was developed prior to commencement of the meta-analysis. The protocol included an a priori statistical analysis plan.
Endpoints
The primary endpoint for the meta-analysis was the difference in FEV1 rate of decline during augmentation therapy as compared with a control regimen. (Survival was intended as a secondary endpoint but was abandoned due to insufficient available data).
Study inclusion criteria
Studies were eligible for inclusion if they compared augmentation therapy with a control regimen in patients with α1-AT deficiency and furnished long-term (greater than one year) longitudinal FEV1 follow-up data. Both parallel-group studies and single-cohort pre-post designs were acceptable. Unpublished as well as published data were sought in order to minimize the potential for publication bias, e.g., selective publication of results favoring a particular therapeutic intervention (Citation[15]).
Search techniques
Studies conforming to the above inclusion criteria were sought by a variety of methods, including computer searches of bibliographic databases (MEDLINE and EMBASE), the Cochrane Library and Internet-resident conference reports, abstracts, compilations of references and full-text journal articles. Computer search terms included US National Library of Medicine Medical Subject Headings (MeSH) “alpha1-antitrypsin”, “alpha1-antitrypsin deficiency”, “trypsin inhibitors”, “fluid therapy”, and “forced expiratory volume”. Non-MeSH terms such as “augmentation” and “FEV1” were also used. Search techniques also included hand searching of selected journals and inquiries with investigators and α1-AT suppliers. Lists of reference citations in prior publications on α1-AT deficiency were examined. No language or time period restrictions were applied.
Data extraction
Studies for inclusion were selected and data extracted independently by two investigators. Differences in interpretation were resolved through discussion. Information in the trial reports as to authors, institutions, study time periods and treatment group characteristics were examined closely to avoid duplication and ensure completeness of data. Extracted patient data included FEV1 baseline, FEV1 percent predicted, patient characteristics, study design features, treatment regimen and duration of follow-up. Missing data and clarifications of unclear information in the study reports were sought as necessary through inquiry with the investigators.
Quality assessment
Study quality was assessed on the basis of prospective vs. retrospective data collection and study size in terms of total patient population. Larger studies are judged to be generally more rigorously designed and conducted (Citation[16]). In the case of randomized trials, blinding and adequacy of allocation concealment were additional quality criteria (Citation[17]).
Statistical analysis
Because of expected between-study heterogeneity, a random effects model was employed for meta-analysis (Citation[18], Citation[19], Citation[20]). Pooled differences in FEV1 slopes were estimated using the META program (Citation[21]). Publication bias was assessed by the method of Egger et al. (Citation[16]) with aid of the METABIAS program (Citation[22]). Individual patient data were analyzed by linear mixed modeling with cumulative duration of augmentation therapy as a time-dependent covariate, as elsewhere detailed (Citation[5]). HLM version 6.0 statistical software (Scientific Software International, Lincolnwood, Illinois, USA) was used for this purpose. This analytical approach is efficient for detecting therapy-related inflection points in FEV1 slopes and accounts appropriately for autocorrelation where data were available both prior to and during augmentation therapy.
RESULTS
Included studies
The study selection process is depicted in . Five studies with a total of 1509 patients fulfilled the selection criteria and were included (Citation[4], Citation[5], Citation[7], Citation[8], Citation[23]). Their attributes are summarized in . All were published. Longitudinal investigations not involving augmentation and uncontrolled studies or cases reports on augmentation, or otherwise eligible studies without long-term longitudinal FEV1 data were not included (Citation[1], Citation[2], Citation[3], Citation[10], Citation[11], Citation[24], Citation[25], Citation[26], Citation[27], Citation[28], Citation[29], Citation[30], Citation[31], Citation[32], Citation[33], Citation[34], Citation[35], Citation[36]). Findings from the included studies have also appeared in other publications (Citation[6], Citation[37], Citation[38], Citation[39], Citation[40], Citation[41], Citation[42], Citation[43], Citation[44]). One study was a randomized controlled trial, three studies were non-randomized parallel-group comparisons (Citation[4], Citation[5], Citation[23]), and one compared FEV1 slopes longitudinally before and during augmentation in a patient cohort (Citation[8]). The median number of patients per study was 164 (interquartile range, 96–295). Prolastin® (Talecris Biotherapeutics, formerly a part of Bayer Healthcare, Clayton, North Carolina, USA) was the α1-AT preparation used for augmentation in 4 of the studies (Citation[4], Citation[5], Citation[8], Citation[23]), while α1-AT was supplied by the Laboratoire Français du Fractionnement et des Biotechnologies (Lille, France) in one study (Citation[7]). Follow-up exceeded 3 years in all studies.
Table 1 Attributes of included studies
Figure 1 Consort diagram of literature retrieval and reviewed used in the meta-analysis.
Summary data were extracted from the reports of four studies (Citation[4], Citation[5], Citation[7], Citation[8]). Individual patient data were obtained and analyzed from the Alpha One International Registry (AIR) in Canada and the UK. AIR is a multinational research program with the participation of nearly 20 countries (Citation[45]). Chapman et al. (Citation[23]) described findings in 21 augmentation and 42 control patients from the Canadian AIR. In this meta-analysis those data were pooled with those of 101 additional control patients with 3 year follow-up from the UK AIR. A report has appeared on longitudinal FEV1 changes in 43 of the UK AIR patients followed for 2 years (Citation[43]). In addition to FEV1 measurements during augmentation, 16 of the 21 Canadian augmentation patients were followed with serial FEV1 determinations for an average duration of 2.5 years (standard deviation, 1.8 years) prior to commencement of augmentation therapy.
Study quality
Data collection was partially or entirely prospective in all five included studies. The single randomized trial was double-blind, although method of allocation concealment was unclear, and the demographic comparability of the randomized groups was not reported. Clarification on these issues could not be obtained from the investigators. The four nonrandomized studies each involved approximately 100 patients or more. With 56 patients the randomized trial was the smallest included study.
FEV1 slope differences
presents the individual study and pooled FEV1 slopes and slope differences. Among all patients, augmentation was associated with a 23% slower decline in FEV1 (absolute difference, 13.4 ml/year; CI, 1.5 to 25.3 ml/year). As expected, significant heterogeneity was apparent (p = 0.012), confirming the appropriateness of a random effects model. There was no evidence of publication bias (p = 0.90).
Figure 2 Forest plot of studies included in the meta-analysis.
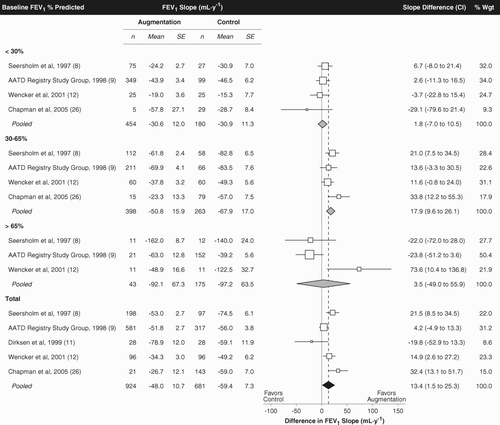
The protective effect of augmentation was primarily attributable to the subset of patients with baseline FEV1 of 30–65% of predicted. In that subset, FEV1 decline was slower during augmentation by 26% (absolute difference, 17.9 ml/year; CI, 9.6 to 26.1 ml/year). Statististically significant effects could not be demonstrated in the subsets with baseline FEV1 < 30% or > 65% of predicted.
Sensitivity analyses
Several sensitivity analyses indicated the robustness of the meta-analysis in the face of alterations in assumptions. The primary endpoint of the single randomized trial was FEV1 determined by daily self-administered spirometry. In addition, laboratory FEV1 measurements were performed every 3 months, and the laboratory-derived data were used in the meta-analysis due to their greater comparability to the measurements reported for the other included studies. If the daily values are substituted for the 3 month data, the pooled FEV1 slope difference would increase from 13.4 ml/year (CI, 1.5 to 25.3ml/year) to 15.6 ml/year (CI, 5.2 to 26.1 ml/year). In the study of Wencker et al. (Citation[8]), the data from the initial follow-up period during augmentation at least partially overlapped those reported by Seersholm et al. 1997 (Citation[4]), although no overlap existed for the control period.
If the Wencker et al. study is excluded from the meta-analysis the pooled FEV1 slope difference would become 12.5 ml/year (−3.9 to 28.9 ml/year). Thus, in this scenario there would be negligible impact on the point estimate of slope difference, although due to the reduced data set overall statistical significance would no longer be demonstrable. Significance would persist for the subgroup with baseline FEV1 30–65% predicted, however (pooled FEV1 slope difference, 21.2 ml/year; CI, 11.5 to 30.9 ml/year). shows that exclusion of the German/Danish study (effect size, 21.5 mL/y) would have the effect of reducing the observed overall treatment benefit (13.4 mL/y pooled effect size for all 5 studies), while excluding the NHLBI Registry study (effect size, 4.2 mL/y) would have the opposite impact.
A baseline FEV1 percent predicted stratum of 35–49% was also evaluated by the AATD Registry Study Group (Citation[5]). Substitution of those results in the 30–65% category of the meta-analysis would change the pooled FEV1 slope difference for the category to 20.1 ml/year (CI, 11.1 to 29.2 ml/year).
Due to the relatively small number of studies and limitations of the reported data it was not feasible to model formally various patient-, therapy- and study-related factors, for instance by metaregression. Thus, we could not assess adequately the impact on rate of FEV1 decline of age, sex, pre-existing atopy (allergy), baseline responsiveness to bronchodilators, α1-AT dose, duration and schedule of therapy, and study design and size.
DISCUSSION
This meta-analysis supports the conclusion that augmentation can slow lung function decline in patients with α1-AT deficiency. Systematic review and quantitative combination of data by meta-analysis are well-established methods for obtaining the most reliable and precise answers to unresolved health care questions (Citation[46], Citation[47]). Meta-analysis can borrow strength from multiple small studies and thereby increase the precision of effect size estimates. As shown in , most of the available individual studies revealed nonsignficant trends toward preservation of lung function due to augmentation while one study showed no FEV1 benefit. These inconclusive findings have left the question of augmentation therapy's effect unresolved. The present meta-analysis provides the first demonstration derived from all the available studies that the net quantitative effect of augmentation is to slow the progressive decline in FEV1.
Significant benefit was demonstrable only in the subset of patients with moderate pulmonary impairment. Patients with severe impairment may be refractory to augmentation but other possibilities must be considered (Citation[47]). Suissa has outlined the bias resulting from regression to the mean in longitudinal studies of FEV1 decline (Citation[48]). On the other hand, it may simply be more difficult to detect an effect of therapy when airflow limitation is severe and comorbidity has developed, since patients of this severity are more likely to be censored due to lung transplantation, morbidity or death. In accord with this interpretation, the average duration of observation in the severe AIR patients was 3.2 y compared with 4.7 y in mild or moderate cases. Our data leave unanswered the question whether patients with mild impairment might benefit from augmentation. Although a significant treatment effect was not apparent, the numbers of patients in this subset accounted for only 14% of the total, and the confidence interval around the pooled FEV1 slope difference was wide.
The difference in rate of FEV1 decline between augmentation and control therapy appears small in absolute terms; in an α1-AT deficient patient with moderate obstruction, the difference of 18 ml over the course of one year would not be discernable. However, sustained over several years of therapy, this 26% slower rate of lung function decline would result in substantial preservation of FEV1. The minimum clinically important difference for rate of FEV1 decline has not been established for studies in COPD (Citation[49]). However, the investigators of the UPLIFT study have suggested that a difference between treatment groups of 15 ml per year is clinically important, having powered their study to detect this magnitude of difference (Citation[50]). Using another major contemporary COPD trial as a yardstick, the TORCH trial's likely mortality reduction (p = 0.052) with combined salmeterol/fluticasone versus placebo was accompanied by a 16 ml per year slower rate of FEV1 decline in the combination treatment versus placebo arms (Citation[51]).
Certain limitations should prompt caution in interpreting our results. Although the meta-analysis encompassed more than 1500 patients, the number studies included was relatively small, limiting our ability to assess the impact of study design features and other covariates on outcome. Four of the five included studies were nonrandomized. Such studies are vulnerable to well-known imbalances that multivariate statistical analysis may be unable to control for completely due to the influence of latent (unobserved or unknown) variables. Such imbalances are less likely in randomized trials, though they can arise due to chance particularly in small randomized trials. While randomized trials provide an excellent source of data for meta-analysis, nonrandomized studies are often the sole or predominant available source of evidence to support timely and informed clinical decisions. Consequently, meta-analyses of nonrandomized studies have markedly increased over the past 40 years, with over 400 nonrandomized study meta-analyses being reported in 1996 alone (Citation[52]).
Bias due to nonrandom allocation might actually lead to underestimation of the true augmentation effect, since augmentation is more likely to be reserved for patients experiencing very rapid decline in FEV1. If indeed the rate of FEV1 decline prior to the institution of augmentation therapy exceeds that in control patients, the average apparent effect of therapy would be diminished. A rate of FEV1 decline > 120 ml/year was an explicit criterion to commence augmentation for two studies in the meta-analysis (Citation[4], Citation[8]), as summarized in . The Canadian Thoracic Society has advocated restricting augmentation to patients whose FEV1 is decreasing by > 80 ml/year (Citation[13], Citation[14]), and this recommendation is likely to have affected decisions to initiate therapy in the Canadian AIR patients. Consistent with this expectation, the initial rate of FEV1 decline in the AIR augmentation group prior to commencement of therapy was greater by 39.1 ml/year (CI, −14.3 to 92.6 ml/year) than the control group rate. With multivariate adjustment for this difference, the reduction in FEV1 slope attributable to augmentation therapy increases from 32.4 ml/year (CI, 13.1 to 51.7 ml/year) to 64.5 ml/year (CI, 14.6 to 114.4 ml/year) in the AIR study.
The view has been expressed that only randomized trials of augmentation therapy should be considered valid (Citation[53]). The concern is that nonrandomized studies may exaggerate therapeutic benefits (Citation[54]). Nevertheless, recent empirical investigations have failed to reveal systematic differences in the results of randomized vs nonrandomized investigations (Citation[54], Citation[55], Citation[56]). In any case, randomization is only one of multiple attributes indicative of study quality. For instance, the included National Heart, Lung, and Blood Institute Registry study by the AATD Registry Study Group (Citation[5]) has been recognized for its high quality based upon prospective design, large scale and attention to quality assurance of pulmonary function testing (Citation[57]). Conversely, the included randomized trial was small, affording only limited statistical power. Further complicating interpretation was the use of albumin in the placebo arm of that trial (Citation[7]). A number of randomized trials have indicated that exogenous albumin itself can improve pulmonary function, at least in critically ill patients (Citation[58], Citation[59], Citation[60], Citation[61], Citation[62]). Other significant differences between trials may confound the interpretation of results. The dose and schedule of infusions was not uniform amongst studies or within studies. Although the most common infusion schedule was weekly, in the largest dataset from the NHLBI registry only half of the patients received infusions at this frequency (Citation[5]) and in the Danish-Dutch trial which showed a CT scan measured lung density benefit of augmentation but no detectable spirometric benefit, infusions were give at four week intervals (Citation[7]). Soy and colleagues have recently explored dosing schedules for augmentation therapy finding feasible dosing intervals as long as 14 to 21 days but not as long as 28 days (Citation[63]).
Another limitation of our meta-analysis concerned the stratification scheme for FEV1 percent predicted. The 30–65% category in which the primary therapeutic benefit could be discerned is broad in clinical terms. Patients with 65% predicted FEV1 are likely to be asymptomatic, whereas their 30% counterparts would be severely affected by their disease. Unfortunately, while the AIR individual patient data would have allowed narrower strata to be examined, the stratification schemes adopted in the other studies constrained our metaanalysis to broad FEV1 percent predicted categories. The importance of conducting a definitive randomized trial of augmentation therapy has been a topic of lively and enduring debate and discussion (Citation[1], Citation[12], Citation[47], Citation[64]). Our results suggest such a trial would be likely to confirm the effectiveness of augmentation, since a high frequency of concordance has been demonstrated between meta-analyses and definitive randomized trials. Thus, in 88% of comparisons there was no significant difference between the results of meta-analyses and those of large-scale randomized trials (Citation[65]).
Our findings imply that exogenous α1-AT slows the loss of lung function seen in patients with α1-AT deficiency. Therapeutic benefit has been shown in patients with 30–65% FEV1 percent predicted. The effect of augmentation in patients with mild (> 65%) or severe (< 30%) obstructive requires further study.
ACKNOWLEDGEMENT
Professor Chapman is a recipient of the GSK-CIHR Research Chair in Respiratory Health Care Delivery.
REFERENCES
- Wewers M D, Casolaro M A, Sellers S E, Swayze S C, Mcphaul K M, Wittes J T, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med 1987; 316: 1055–1062
- Schwaiblmair M, Vogelmeier C, Fruhmann G. Long-term augmentation therapy in twenty patients with severe alpha-1-antitrypsin deficiency–three-year follow-up. Respiration 1997; 64(1)10–15
- Wencker M, Denker J, Konietzko N. Serial measurements of FEV1 over 12 years in a patient with alpha-1-protease inhibitor deficiency: Influence of augmentation therapy and infections. Respiration 1994; 61(4)195–198
- Seersholm N, Wencker M, Banik N, Viskum K, Dirksen A, Kok-Jensen A, et al. Does alpha12-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency?. Eur Respir J 1997; 10: 2260–2263
- Vreim C E, Wu M, Crystal R G, Buist A S, Burrows B, Cohen A B, et al. Survival and FEV1 decline in individuals with severe deficiency of A1-antitrypsin. Am J Respir Crit Care Med 1998; 158(1)49–59
- Wencker M, Banik N, Buhl R, Seidel R, Konietzko N. Long-term treatment of alpha1-antitrypsin deficiency-related pulmonary emphysema with human alpha1-antitrypsin. Wissenschaftliche arbeitsgemeinschaft zur therapie von lungenerkrankungen (WATL)-alpha1-AT-study group. Eur Respir J 1998; 11(2)428–433
- Dirksen A, Dijkman J H, Madsen F, Stoel B, Hutchison D CS, Ulrik C S, et al. A randomized clinical trial of A1-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160(5)1468–1472
- Wencker M, Fuhrmann B, Banik N, Konietzko N, Wissenschaftliche A. Longitudinal follow-up of patients with A1-protease inhibitor deficiency before and during therapy with IV A1-protease inhibitor. Chest 2001; 119(3)737–744
- Stockley R A, Bayley D L, Unsal I, Dowson L J. The effect of augmentation therapy on bronchial inflammation in A1-antitrypsin deficiency. Am J Respir Crit Care Med 2002; 165(11)1494–1498
- Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: A new hypothesis with supporting data. Chest 2000; 118(5)1480–1485
- Gottlieb D J, Luisetti M, Stone P J, Allegra L, Cantey-Kiser J M, Grassi C, et al. Short-term supplementation therapy does not affect elastin degradation in severe alpha(1)-antitrypsin deficiency. The american-italian AATD study group. Am J Respir Crit Care Med 2000; 162(6)2069–2072
- Stoller J K. Augmentation therapy for severe a1-antitrypsin deficiency: Is the jury still out on a trial?. Thorax 1998; 53(12)1007–1009
- O'Donnell D E, Aaron S, Bourbeau J, Hernandez P, Marciniuk D, Balter M, et al. Canadian thoracic society recommendations for management of chronic obstructive pulmonary disease–2003. Can Respir J 2003; 10(Suppl A)11A–65A
- Abboud R T, Ford G T, Chapman K R. Alpha1-antitrypsin deficiency: A position statement of the canadian thoracic society. Can Respir J 2001; 8(2)81–88
- Egger M, Smith G D. Bias in location and selection of studies. BMJ 1998; 316(7124)61–66
- Egger M, Davey S G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997; 315(7109)629–634
- Schulz K F, Chalmers I, Hayes R J, Altman D G. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995; 273(5)408–412
- Stram D O. Meta-analysis of published data using a linear mixed-effects model. Biometrics 1996; 52(2)536–544
- Shea S, Dumouchel W, Bahamonde L. A Meta-analysis of 16 randomized controlled trials to evaluate computer-based clinical reminder systems for preventive care in the ambulatory setting. J Am Med Inform Assoc 1996; 3(6)399–409
- Lau J, Ioannidis J P, Schmid C H. Summing up evidence: One answer is not always enough. Lancet 1998; 351(9096)123–127
- Sharp S, Sterne J. Meta-analysis. 1997; 100–106, Stata Tech Bull STB-38
- Steichen T J, Egger M, Sterne J. Tests for publication bias in meta-analysis. 1998; 84–85, Stata Tech Bull STB-44
- Chapman K R, Bradi A C, Paterson D, Navickis R J, Wilkes M M. Slower lung function decline during augmentation therapy in patients with alpha1-antitrypsin deficiency (A1ATD): Results from the canadian AIR registry. Proc.Am.Thorac.Soc. 2005; 2: A808
- Buist A S, Burrows B, Eriksson S, Mittman C, Wu M. The natural history of air-flow obstruction in piz emphysema. Report of an NHLBI workshop. Am Rev Respir Dis 1983; 127(2)S43–S45
- Brantly M L, Paul L D, Miller B H, Falk R T, Wu M, Crystal R G. Clinical features and history of the destructive lung disease associated with alpha-1-antitrypsin deficiency of adults with pulmonary symptoms. Am Rev Respir Dis 1988; 138(2)327–336
- Hubbard R C, Sellers S, Czerski D, Stephens L, Crystal R G. Biochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiency. JAMA 1988; 260: 1259–1264
- Konietzko N, Becker M, Schmidt E W, Rasche B, Ulmer W T, Ferlinz R, et al. [Substitution therapy with alpha-1-Pi in patients with alpha-1-Pi deficiency and progressive pulmonary edema]. Dtsch Med Wochenschr 1988; 113(10)369–373
- Schmidt E W, Rasche B, Ulmer W T, Konietzko N, Becker M, Fallise J P, et al. Replacement therapy for alpha-1-protease inhibitor deficiency in piz subjects with chronic obstructive lung disease. Am J Med 1988; 84(6A)63–69
- Ulmer W T, Schmidt E W, Rasche B. Long term effect on lung function of alpha 1-protease inhibitor substitution therapy in COPD patients with Pi ZZ phenotype. Eur Respir J Suppl 1990; 9: 21s–22s
- Miravitlles M, Vidal R, Torrella M, Bofill J M, Cotrina M, De G J. [Evaluation of replacement therapy in emphysema caused by alpha 1-antitrypsin deficiency]. Arch Bronconeumol 1994; 30(10)479–484
- Stone P J, Morris T A, III, Franzblau C, Snider G L. Preliminary evidence that augmentation therapy diminishes degradation of cross-linked elastin in alpha-1-antitrypsin-deficient humans. Respiration 1995; 62(2)76–79
- Barker A F, Iwata-Morgan I, Oveson L, Roussel R. Pharmacokinetic study of A1-antitrypsin infusion in A1-antitrypsin deficiency. Chest 1997; 112(3)607–613
- Stoller J K, Rouhani F, Brantly M, Shahin S, Dweik R A, Stocks J A, et al. Biochemical efficacy and safety of a new pooled human plasma a1-antitrypsin, respitin. Chest 2002; 122(1)66–74
- Kjus T, Lutzow-Holm C, Christensen O B. Treatment of panniculitis associated with alpha-1-antitrypsin deficiency with alpha-1-protease inhibitor. Acta Derm Venereol 2003; 83(6)462–463
- Piitulainen E, Bernspang E, Bjorkman S, Berntorp E. Tailored pharmacokinetic dosing allows self-administration and reduces the cost of IV augmentation therapy with human alpha(1)-antitrypsin. Eur J Clin Pharmacol 2003; 59(2)151–156
- Silva G E, Sherrill D L, Guerra S, Barbee R A. A longitudinal study of alpha1-antitrypsin phenotypes and decline in FEV1 in a community population. Chest 2003; 123(5)1435–1440
- Graf H J, Drenker J, Konietzko N. Lung function study of the german multicenter group (WATL) in Á1-PI deficient patients with severe lung emphysema before and under substitution therapy. Am Rev Respir Dis 1991; 143(Suppl.)A672
- Barker A F, Siemsen F, Pasley D, D'Silva R, Buist A S. Replacement therapy for hereditary alpha1-antitrypsin deficiency. A program for long-term administration. Chest 1994; 105(5)1406–1410
- Seersholm N, Kok-Jensen A, Dirksen A. Decline in FEV1 among patients with severe hereditary a1-antitrypsin deficiency type Piz. Am J Respir Crit Care Med 1995; 152: 1922–1925, (6 PT 1)
- Dirksen A, Friis M, Olesen K P, Skovgaard L T, Sorensen K. Progress of emphysema in severe a1-antitrypsin deficiency as assessed by annual CT. Acta Radiol 1997; 38(5)826–832
- Mcelvaney N G, Stoller J K, Buist A S, Prakash U BS, Brantly M L, Schluchter M D, et al. Baseline characteristics of enrollees in the national heart, lung and blood institute registry of a1-antitrypsin deficiency. Chest 1997; 111(2)394–403
- Dirksen A, Holstein-Rathlou N H, Madsen F, Skovgaard L T, Ulrik C S, Heckscher T, et al. Long-range correlations of serial FEV1 measurements in emphysematous patients and normal subjects. J Appl Physiol 1998; 85(1)259–265
- Dowson L J, Guest P J, Stockley R A. Longitudinal changes in physiological, radiological, and health status measurements in alpha(1)-antitrypsin deficiency and factors associated with decline. Am J Respir Crit Care Med 2001; 164: 1805–1809, (10 Pt 1)
- Stoller J K, Fallat R, Schluchter M D, O'Brien R G, Connor J T, Gross N, et al. Augmentation therapy with alpha1-antitrypsin: Patterns of use and adverse events. Chest 2003; 123(5)1425–1434
- Luisetti M, Miravitlles M, Stockley R A. Alpha1-antitrypsin deficiency: A report from the 2nd meeting of the alpha one international registry, Rapallo (Genoa, Italy), 2001. Eur Respir J 2002; 20(4)1050–1056
- Bailar J C, III. The promise and problems of meta-analysis. N Engl J Med 1997; 337(8)559–561
- Burrows B. A clinical trial of efficacy of antiproteolytic therapy: Can it be done?. Am Rev Respir Dis 1983; 127(2)S42–S43
- Suissa S. Lung function decline in COPD trials: Bias from regression to the mean. Eur Respir J 2008; 32(4)829–831
- Rennard S I. Minimal clinically important difference, clinical perspective: An opinion. COPD 2005; 2(1)51–55
- Decramer M, Celli B, Tashkin D P, Pauwels R A, Burkhart D, Cassino C, et al. Clinical trial design considerations in assessing long-term functional impacts of tiotropium in COPD: The UPLIFT trial. COPD 2004; 1(2)303–312
- Celli B, Ferguson G T, Anderson J A, Jenkins C R, Jones P W, Vestbo J, et al. Salmeterol/fluticasone propionate (SFC) improves lung function and reduces the rate of decline over three years in the TORCH survival study. Proc Am Thorac Soc 2006; 175: A763
- Stroup D F, Berlin J A, Morton S C, Olkin I, Williamson G D, Rennie D, et al. Meta-analysis of observational studies in epidemiology: A proposal for reporting. meta-analysis of observational studies in epidemiology (MOOSE) group. JAMA 2000; 283(15)2008–2012
- Hutchison D C, Cooper D. Alpha-1-antitrypsin deficiency: Smoking, decline in lung function and implications for therapeutic trials. Respir Med 2002; 96(11)872–880
- Benson K, Hartz A J. A comparison of observational studies and randomized, controlled trials. N Engl J Med 2000; 342(25)1878–1886
- Concato J, Shah N, Horwitz R I. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med 2000; 342(25)1887–1892
- Ioannidis J P, Haidich A B, Pappa M, Pantazis N, Kokori S I, Tektonidou M G, et al. Comparison of evidence of treatment effects in randomized and nonrandomized studies. JAMA 2001; 286(7)821–830
- Schluchter M D, Stoller J K, Barker A F, Buist A S, Crystal R G, Donohue J F, et al. Feasibility of a clinical trial of augmentation therapy for alpha(1)-antitrypsin deficiency. The alpha 1-antitrypsin deficiency registry study group. Am J Respir Crit Care Med 2000; 161: 796–801, (3 Pt 1)
- Jelenko C, III, Williams J B, Wheeler M L, Callaway B D, Fackler V K, Albers C A, et al. Studies in shock and resuscitation, I: Use of a hypertonic, albumin-containing, fluid demand regimen (HALFD) in resuscitation. Crit Care Med 1979; 7(4)157–167
- Rackow E C, Falk J L, Fein I A, Siegel J S, Packman M I, Haupt M T, et al. Fluid resuscitation in circulatory shock: A comparison of the cardiorespiratory effects of albumin, hetastarch, and saline solutions in patients with hypovolemic and septic shock. Crit Care Med 1983; 11(11)839–850
- Metildi L A, Shackford S R, Virgilio R W, Peters R M. Crystalloid versus colloid in fluid resuscitation of patients with severe pulmonary insufficiency. Surg Gynecol Obstet 1984; 158(3)207–212
- Hoeft A, Korb H, Mehlhorn U, Stephan H, Sonntag H. Priming of cardiopulmonary bypass with human albumin or ringer lactate: Effect on colloid osmotic pressure and extravascular lung water. Br J Anaesth 1991; 66(1)73–80
- Martin G S, Mangialardi R J, Wheeler A P, Dupont W D, Morris J A, Bernard G R. Albumin and furosemide therapy in hypoproteinemic patients with acute lung injury. Crit Care Med 2002; 30(10)2175–2182
- Soy D, De La R C, Lara B, Esquinas C, Torres A, Miravitlles M. Alpha-1-antitrypsin deficiency: Optimal therapeutic regimen based on population pharmacokinetics. Thorax 2006; 61(12)1059–1064
- Hutchison D CS, Hughes M D. Alpha-1-antitrypsin replacement therapy: Will its efficacy ever be proved?. Eur Respir J 1997; 10: 2191–2193
- Lelorier J, Gregoire G, Benhaddad A, Lapierre J, Derderian F. Discrepancies between meta-analyses and subsequent large randomized, controlled trials. N Engl J Med 1997; 337(8)536–542