
ABSTRACT
Alpha-1 antitrypsin deficiency (AATD) is associated with premature onset of emphysema resulting from low serum A1-PI levels. The only available pharmacological treatment affecting the underlying cause of AATD is A1-PI therapy. AATD-related emphysema is considered a good model to study disease-modifying effects of treatment as the causative process has been identified. Disease modification is a sustained improvement in disease state following therapeutic intervention that persists when therapy is discontinued. Appropriate trial design and the use of valid study endpoints are key to illustrating disease modification, particularly in clinical trials of rare diseases where it can be difficult to recruit sufficient numbers of patients. Delayed-start trials are advantageous ethically as all patients ultimately receive active treatment and imaging techniques have proven promising as valid study endpoints. Specifically, computed tomography (CT) measured lung density has been used to monitor emphysema and is considered a more sensitive outcome than pulmonary function tests to monitor disease progression. This review will discuss the importance of clinical endpoints and trial design to determine disease modification and will review the evidence for disease modification in AATD-related emphysema. Until recently, clinical studies have not shown a significant effect of A1-PI therapy, possibly due to insufficient numbers of patients, short duration of clinical trials and lack of appropriate trial design. A recently completed randomised trial and open-label extension study followed a larger study population for a longer duration and incorporated a delayed-start design. The results demonstrated clinical efficacy of A1-PI therapy and indicate that treatment is disease-modifying.
Introduction
Alpha-1 antitrypsin deficiency (AATD) is a co-dominant, hereditary condition caused by mutation of the SERPINA1 gene which encodes alpha-1 antitrypsin (also known as alpha1-proteinase inhibitor [A1-PI]). It is known to affect around 3.4 million individuals worldwide, although many of them are not yet diagnosed Citation(1). Severe AATD is characterised by low levels of A1-PI in blood and tissues and can lead to liver disease (cirrhosis or hepatocellular carcinoma), caused by aggregation of misfolded A1-PI as polymers in hepatocytes, and/or chronically progressing lung disease, including emphysema, as a result of uncontrolled degradation of lung tissue by proteases, mainly neutrophil elastase (NE) (Citation2, 3). Severe AATD presents with pulmonary symptoms, often including coughing, wheezing, shortness of breath and recurrent respiratory tract infections that are similar to those observed in typical chronic obstructive pulmonary disease (COPD) (Citation4–6). Whilst the pathomechanism of AATD-related COPD is distinct from COPD per se, recent evidence suggests that in both emphysema is a major contributor (Citation7, 8). As the causative process has been identified, AATD-associated emphysema is considered a good model to study a possible disease-modifying effect of therapy on the course of the disease. Disease modification can be broadly defined as the sustained change in a disease state that occurs following therapeutic intervention Citation(9), whereby the progression of emphysema is slowed. None of the currently approved pharmacological therapies for COPD has unequivocally demonstrated this effect.
The main target of A1-PI replacement therapy is to recover the normal physiological balance between proteases and antiproteases, and interrupt the cascade that perpetuates active pulmonary disease. In recent years, clinical trials have sought to provide evidence for clinical efficacy of A1-PI replacement therapy in patients with emphysema due to AATD. Following several inconclusive trials, the recent publication representing the differently designed and largest clinical trial completed to date provides clinical data on efficacy of A1-PI therapy and, importantly, on its disease-modifying effect Citation(10). This review will discuss the importance of clinical endpoints and trial design to determine disease modification and will review the evidence for disease modification in AATD. We will demonstrate that careful, unorthodox trial design is of key importance when studying relatively rare disorders, such as AATD, and slowly progressing diseases, such as emphysema, especially in the absence of sensitive and easily measurable biomarkers.
Disease modification in AATD-related emphysema
Disease modification can be described as an improvement or stabilisation of a disease state resulting from a reduction in the rate of disease progression that occurs following therapeutic intervention, which may persist after the intervention is discontinued Citation(9). As such, disease-modifying therapies should impact on the pathological and pathophysiological mechanisms underlying the disease rather than the clinical or symptomatic effects of the disease (Citation11, 12). Although disease state can be influenced by therapeutic interventions in several ways, current consensus is that disease-modifying treatments are those that alter the rate of disease progression (). Other treatments may provide symptomatic improvements that have either a sustained or temporary effect (). Whilst symptomatic treatments may ‘turn back the clock’ and provide a sustained improvement in disease symptoms which may persist after treatment has ceased, these are not considered to be truly disease-modifying Citation(9).
Figure 1. Impact of treatments on disease progression Citation(9). This figure shows the hypothetical effects of symptomatic and disease-modifying treatments on disease progression in early-start and delayed-start trial models. Disease-modifying treatments provide a sustained alteration in disease progression. Reprinted from COPD: Journal of Chronic Obstructive Pulmonary Disease. Halpin and Tashkin. Defining Disease Modification in Chronic Obstructive Pulmonary Disease. COPD 2009: 6: 211–213 with permission of the publisher Taylor & Francis Ltd (www.tandfonline.com).
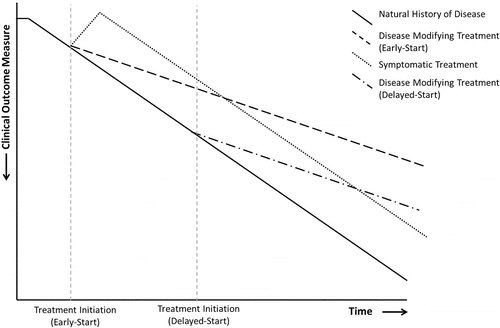
In AATD-related emphysema, as in typical COPD, symptomatic treatments such as bronchodilators or corticosteroids do not affect the underlying cause of the disease and the progressive destruction of lung tissue (Citation3, 13). Only lifestyle modifications, such as smoking cessation, have demonstrated some effect on disease progression; however, it is not entirely clear to what extent and after what period the deteriorating effect of smoking ceases Citation(14). The only available treatment for the underlying cause of AATD is treatment with A1-PI, which was demonstrated by early biochemical studies to be able to partially restore A1-PI levels in the serum (to above the 11-µM protective threshold) and importantly in the epithelial lining fluid of the lungs (Citation15, 16). Consequently, it has been expected that therapy with A1-PI might slow the rate of lung tissue destruction and reduce the frequency and severity of lung infections (Citation15, 17, 18). Accordingly, retrospective analysis of data from the US AATD registry demonstrated significantly lower relative risk of death in A1-PI treated subjects (RR = 0.64, p = 0.02) and slower rate of lung function loss (assessed by forced expiratory volume in 1 second [FEV1]) in a subgroup with baseline FEV1of 31–49% predicted Citation(19). Within this analysis, study design was the major confounding factor. The observational registry study was not a prospective, placebo-controlled, randomised study and therefore it was susceptible to numerous limitations resulting from differences in socioeconomic factors, health delivery systems and varying length of treatment. Despite these limitations, a similar conclusion regarding the effect of A1-PI replacement therapy on FEV1 decline rate was provided by a smaller observational European study Citation(20).
Subsequent randomised clinical studies, Danish-Dutch Citation(21) and EXACTLE Citation(22), also failed to demonstrate clinical efficacy of A1-PI in AATD-related emphysema or provide evidence of disease modification. Both demonstrated a clear favorable trend on the lung density loss assessed by computed tomography (CT) densitometry, but no benefit on less sensitive endpoints, such as FEV1, was observed. Additionally, both trials were not designed to address mortality. It was generally considered that both studies were underpowered with cohorts of 58 and 77 patients, respectively, and a much larger group of patients (150–500) would be necessary to demonstrate efficacy of A1-PI therapy within the classic double-arm, randomised design. This requirement would be difficult to fulfill considering the relatively low prevalence of AATD and the low number of potentially available patients with AATD-related emphysema. As in other orphan disorders, obtaining the optimal sample size in AATD clinical trials proves difficult. Hence, careful trial design and selection of clinical endpoints seems to be of crucial importance as demonstrated by recently published data Citation(10).
Trial design in disease modification studies
In order to demonstrate a disease-modifying effect in AATD and other chronic and slowly progressing diseases, the design of the clinical trial is important. Standard trial models, such as parallel group randomised controlled trials where patients receive either placebo or active treatment, are characterised by minimal bias and therefore considered the gold standard. However, in addition to ethical issues around running a long-duration placebo-controlled trial, particularly in rare disorders such as AATD, they do not provide clear distinction between temporary symptomatic and disease-modifying effects.
Studies of disease modification performed in other fields have led to a greater understanding of the concept of disease modification and consequently improvements in trial design. Disease modification has been most extensively studied in neurological diseases such as Alzheimer's disease, Parkinson's disease and multiple sclerosis which, like AATD, are chronic, slowly progressing diseases. Some of these studies utilised a withdrawal trial design where patients are randomised to either placebo or active treatments and subsequently all patients are switched to placebo (Citation23, 24). For example, in one study patients with Parkinson's disease were followed for 3 months during washin and for 2 months during washout (withdrawal period) Citation(25). However, evaluation of disease-modifying effects was confounded by short-term symptomatic effects and the short duration of washout was insufficient to demonstrate if any effects of treatment were sustained and disease-modifying. This is an important issue to be considered as the majority of disease modification studies follow an add-on design, i.e., concurrent symptomatic optimal therapy is not modified.
More recently, delayed-start trial designs have been utilised to demonstrate disease modification, which have two phases. In the first phase patients are randomised to receive either placebo or active treatment, as in classical placebo-controlled, parallel group study design, but differences in study endpoints between the two groups at the end of this phase could be due to symptomatic effects or short-term benefits and not due to disease modification. In phase 2, both groups receive active treatment; if a difference in a study endpoint remains at the end of this stage a treatment may be considered to be disease-modifying (Citation23, 24). This trial design has been used effectively to study the effect of rasagiline, a monoamine oxidase type-B inhibitor, in the treatment of Parkinson's disease Citation(26).
Expert consensus and guidance provided by the US Food and Drug Administration (FDA) suggests that trial designs such as withdrawal or delayed-start represent appropriate means to illustrate sustained effects on disease progression (Citation27–29). Ethically, delayed-start trial designs may be more appropriate and within such trials disease modification is shown if patients initially on placebo and switched to active treatment fail to ‘catch up’ with patients who received active treatment for the entire duration ().
Endpoints in disease modification studies
In addition to careful clinical trial design, key considerations for disease modification studies include the use of reliable biomarkers that correlate with clinical and/or surrogate endpoints, and the use of highly sensitive and reliable endpoints.
Guidance on the use of biomarkers and study endpoints has been provided by the FDA Citation(30). To date numerous biomarkers are under development for use in cardiovascular disease, infectious diseases, cancer, neuropsychiatric disease, and autoimmune and inflammatory disease Citation(30). In asthma, β-adrenergic receptor polymorphisms have been shown to predict short-term patient outcome Citation(30) and in COPD, elastin degradation products are one of the many potential effective biomarkers currently under investigation Citation(31). As demonstrated in the ECLIPSE trial numerous study outcome measures, including pulmonary function tests (Citation32, 33), biomarkers (Citation34, 35), health outcomes (Citation32, 36) and exacerbations (Citation32, 37) have been evaluated as markers in COPD. However, a key challenge in COPD is the identification of markers that can accurately measure disease progression, are responsive to treatment and are superior to the gold standard, FEV1 (Citation32, 38). The utilisation of imaging techniques as valid study endpoints has shown promising results. In particular, serial CT scanning to monitor quantifiable changes in lung density has been used to monitor disease progression in emphysema (Citation30, 35).
Likewise, the study of disease modification in AATD-related emphysema is confounded by the lack of a standard definition and agreement of appropriate markers by which modification should be assessed. As in COPD, neither lung function (FEV1) nor biochemical biomarkers (elastin degradation) provide adequate sensitivity to conclusively demonstrate the effect of A1-PI therapy, or lack of it, within the standard double arm randomised trial. Furthermore, these outcomes along with CT lung density have been underpowered in previous clinical studies to show a treatment effect of A1-PI therapy (Citation17, 21, 22).
Problems with traditional outcome measures
FEV1 has traditionally been used in patients with COPD to monitor expiratory flow limitation as a surrogate marker for disease progression. However, our understanding of the pathogenesis of COPD is evolving and evidence indicates that several pathological processes, including narrowing of small airways, bronchiolar fibrosis, and destruction of alveolar tissue, contribute to the clinical picture of COPD Citation(39). As FEV1 is a surrogate marker, it may not be the most appropriate measure to monitor disease progression. This may also explain why changes in FEV1 have been shown to be poorly correlated with other outcome measures such as dyspnoea, exercise capacity, exacerbation and mortality Citation(32). Furthermore, as changes in FEV1 reflecting lung tissue destruction or remodelling typically occur slowly over time, and given the intra-subject variability of FEV1 measures Citation(40), a large and lengthy clinical trial would be required to demonstrate modest effects of treatment on FEV1 (Citation39,40). Demonstrating a therapeutic reduction in FEV1 decline of around 20 mL/year would take several years and require the enrollment of at least 1,000 subjects Citation(39). Consequently, in clinical studies to date only smoking cessation has been shown to have a significant impact on FEV1 decline and alter disease progression (Citation39, 40); whilst treatment with corticosteroids or bronchodilators have been shown to provide improvements in FEV1 which may last for several years if continuously applied, they do not alter the progression of disease and decline in lung function in the long term (Citation39, 41). Following initial trials conducted in the 1990s, it was generally accepted that FEV1, then the gold standard endpoint in monitoring COPD progression, is not suitable in AATD research and no study on augmentation therapy could be adequately powered to demonstrate a significant treatment effect. A meta-analysis of five clinical studies and 1,509 AATD patients demonstrated a significant reduction in the decline in lung function with A1-PI therapy. However, an important bias of this analysis was that only a single study containing 58 subjects was designed as a placebo-controlled trial Citation(42).
Given the limitations of FEV1 for monitoring COPD, several alternatives, including physiological parameters and patient-centric outcome measures, have been suggested as means to monitor COPD progression. Several surrogate outcome measures have been suggested for COPD as shown in (Citation9, 41, 43, 44); these include dyspnoea, health-related quality of life (HRQL), exercise capacity, exacerbations and mortality. Improvements in dyspnoea have been observed for several long-lasting bronchodilators, long-acting antimuscarinic agents and inhaled corticosteroids. Significant HRQL improvements have been observed with numerous treatment options, although these changes have not been clinically meaningful Citation(41). Similarly, several treatments have been shown to improve exercise capacity, as measured by the shuttle walk or cycling test, and reduce the rate of common COPD exacerbations Citation(41). Mortality rate was measured during both the TORCH Citation(45) and UPLIFT Citation(46) studies whereby treatment with salmeterol and fluticasone combined or tiotropium, respectively, reduced mortality rates in COPD patients although these changes did not reach significance (Citation41, 45, 46). The body mass index, airflow obstruction, dyspnoea and exercise (BODE) index combines several outcome measures to provide a multi-dimensional tool to monitor disease progression in COPD Citation(47). A recent study by de Torres et al. Citation(48) illustrates that the BODE index is a useful tool in COPD that is more sensitive at predicting survival than ABCD GOLD categories or FEV1 alone (Citation48,49). In patients with severe emphysema, lung volume reduction surgery resulted in a decrease in the BODE index that correlated with increased survival and reduced mortality Citation(50). In studies of AATD patients, utilisation of alternative pulmonary function measures, mortality outcomes, HRQL assessments and exacerbations has proven to be insufficient to demonstrate significant effects of A1-PI treatment (Citation10, 19, 22, 51). As with FEV1, this is due to poor sensitivity of these measures to detect changes in AATD-related emphysema and the need for studies to be sufficiently powered, i.e., with larger patient populations and/or longer duration.
Table 1. Summary of outcome measures used to monitor COPD and emphysema progression (Citation9, 39, 41, 43, 44, 69).
Computed tomography
Whilst surrogate markers such as dyspnoea, HRQL and exercise capacity may be beneficial in monitoring disease progression in COPD, they can be affected by external influences such as co-morbidities and may not accurately represent pathological changes which may be ongoing in the lung. Instead alternative measures that can recognise the two major disease processes which affect disease progression, namely airway disease and alveolar disease, are more suitable to gain an insight into the disease-modifying behavior of a therapy.
Only highly sensitive, objectively assessed measures that allow a clear distinction between airway and alveolar disease might provide a satisfactory tool to assess therapeutic effect and provide evidence of disease modification. Traditional lung function measures are indirect by nature, e.g., FEV1, forced vital capacity (FVC) and diffusing capacity of carbon monoxide (DLCO) assess the changes that result from emphysema progression but do not represent the changes in lung structure per se. Therefore these measures are not sensitive enough to detect emphysema progression over short periods of time, i.e., a couple of years. Imaging techniques, such as CT lung densitometry, can not only help to distinguish emphysema from other airway disease but can be used to quantify the severity of disease and may also be used to characterise the distribution of emphysema in the lungs. This methodology best reflects the pathological changes that occur in the lung with AATD-related emphysema. However, CT densitometry is not able to distinguish between lobular forms of emphysema, e.g., centrilobular and panlobular emphysema, and so for diseases such as COPD in general and cystic fibrosis, other CT-based methodologies are used, such as pattern recognition-based analysis and measuring bronchial wall thickening.
Compared to traditional lung function measures CT lung densitometry is a more reliable, reproducible and 2.5-fold more sensitive measure to monitor emphysema progression (Citation51, 52). CT lung densitometry has been shown to correlate with other outcome measures including FEV1, health status and exercise capacity (Citation53, 54). Consequently CT lung densitometry has been accepted by the FDA as a clinically meaningful tool for assessing the impact of treatment on emphysema disease progression in AATD Citation(55) and a 2003 statement from the ATS and ERS highlighted the need for clinical trials utilising CT Citation(2).
In a clinical trial setting, CT methodology is standardised to ensure that data can be collected and analysed consistently between different centers. Despite this, in routine clinical practice there remains a requirement for standardisation, including alignment of measured parameters, CT scanner setup and software algorithms, and there is a lack of evidence to guide the use of different methodological approaches Citation(52). Widespread use of CT lung densitometry is also limited by expense, availability, and the need for specialised software, e.g., for densitometry analyses, and experienced personnel to analyse the data (Citation56, 57). Moreover, although CT has been shown to correlate with other clinical endpoints (Citation53, 54), a clear correlation with mortality is lacking; presently, only a single study has suggested that CT may better predict mortality in AATD patients than lung function parameters Citation(58). The lack of data demonstrating function or survival benefit arising from therapy has hindered widespread acceptance of CT as an outcome measure, and patients and health authorities frequently prefer cheaper, easily accessible outcomes with clearer physical effects, such as improvements in FEV1.
Clinical studies of disease modification in AATD-related emphysema
Limitations of randomised placebo-controlled trial design
In order to demonstrate a disease-modifying effect in AATD-related emphysema, appropriate trial design and choice of study endpoints is critical. Lung function measures, i.e., FEV1, have inherent methodological issues that limit their ability to demonstrate clinical efficacy of A1-PI therapy: lung function generally declines slowly in emphysema and consequently demonstrating disease-modifying effects in a standard two-arm trial setup would necessitate unfeasibly long follow-up and large patient cohorts Citation(21). To overcome this issue, the two relatively small randomised placebo-controlled trials were the first to utilise CT lung density as a more sensitive endpoint to assess the effect of A1-PI therapy on emphysema progression (Citation21, 22). First, in a study of Danish and Dutch ex-smokers with AATD the efficacy of A1-PI therapy was assessed using CT scan measurements taken at forced residual capacity. Whilst the study demonstrated a protective effect of A1-PI against loss of lung density, this did not reach statistical significance (p = 0.07) Citation(21). Subsequently, the EXAcerbations and Computed Tomography scan as Lung End-points (EXACTLE) study followed 77 patients randomised 1:1 to A1-PI or placebo over 2–2.5 years Citation(22). The efficacy of A1-PI therapy in AATD patients was assessed using CT measurements taken at total lung capacity (TLC) and the number of exacerbations. Similar to earlier studies a trend towards a beneficial effect of A1-PI treatment compared to placebo was observed with CT densitometry measurements (p-values ranged between 0.049 and 0.084); however, no significant differences in the frequency of lung exacerbations were observed between the groups (A1-PI = 2.55 ± 2.14; Placebo = 2.19 ± 1.33, p = 0.265) although the severity of exacerbations was reduced (Citation22, 52). Although results from the two studies did not reach significance when analysed alone, the trials were of comparable design and a combined analysis of results from these two studies suggested that A1-PI therapy may significantly reduce the decline in lung density in patients with AATD (p = 0.006) Citation(54).
It is generally acknowledged that both trials were not adequately powered to demonstrate a treatment effect as they were not conducted over a sufficient period of time, and did not include enough patients Citation(21). Consequently, disease-modifying effects of A1-PI therapy could not be ascertained. However, it is important to note that the designs of the Danish-Dutch and EXACTLE studies of disease modification in AATD did not include delayed or withdrawal periods and therefore may not have been optimal (Citation21, 22). Furthermore, the findings from the Danish-Dutch and EXACTLE studies highlight that standard trial designs are often inadequate to demonstrate disease modification. From these studies it was concluded that in order to determine whether A1-PI therapy demonstrates a significant protective effect, patients would need to be followed for a longer duration ≥4 years and a greater number of patients (150–500) would need to be included.
When planning clinical trials in orphan diseases a major difficulty is recruiting sufficient numbers of patients to provide adequate statistical power for the study. In rare diseases such as AATD, utilising a delayed-start trial design, where patients receive the same active treatment but at different starting times, may overcome the need for a large patient population. Also, all patients ultimately receive active treatment, and so ethically it may be more appropriate than withdrawal or washout trial designs and trials where patients spend extended time on placebo. The accuracy of this approach was clearly demonstrated by a recent study by Chapman and colleagues. They not only enrolled a larger study population that was assessed over 4 years but also included both an Early-Start and Delayed-Start group () that permitted disease-modifying effects to be assessed Citation(10).
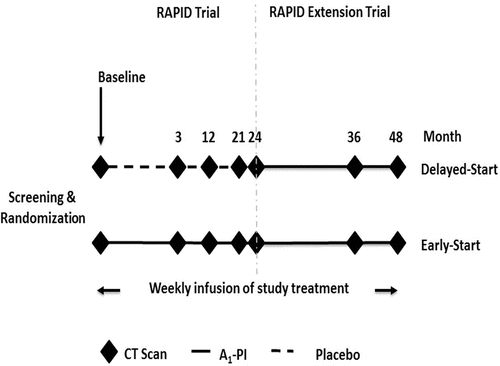
Figure 2. RAPID and RAPID Extension trial design. This figure shows the design of the RAPID trial; subjects in RAPID received A1-PI or placebo for 2 years. During the RAPID Extension trial all non-US subjects received A1-PI for a further 2 years.
Evidence from the delayed-start trial
The Randomised, placebo-controlled trial of augmentation therapy in Alpha-1 Proteinase Inhibitor Deficiency (RAPID) and the RAPID Extension trial represent the largest completed clinical study to date of A1-PI therapy in AATD patients and the only trial specifically designed to look at the disease-modifying effect of treatment Citation(10). RAPID investigated A1-PI therapy in 180 patients with AATD; 93 received A1-PI and 87 received placebo Citation(10). The primary endpoint was the change in CT lung density followed over 24 months (). Following completion of the main two-arm, placebo-controlled RAPID trial, non-US subjects were invited to join RAPID Extension – a 2-year, open, non-controlled study in which all patients received A1-PI therapy () from which interim data has recently been published Citation(10). Due to the delayed-start trial design the potential disease-modifying effects of A1-PI therapy could be investigated. Patients on active therapy who received A1-PI substitution for 4 years throughout the RAPID and RAPID Extension trial were designated as the Early-Start cohort whilst those who initially received placebo and switched to active treatment during the RAPID Extension trial were termed the Delayed-Start cohort.
During the RAPID trial a significant difference in the annual rate of decline in lung density (0.74 g/L; p = 0.033, two-sided test, ) was observed between A1-PI and placebo (−1.45 and −2.19 g/L/year, respectively). This corresponds to a reduction of 34% in the annual rate of lung density decline when compared with placebo. Interim analysis of the RAPID Extension trial showed that switching patients from placebo to A1-PI resulted in a reduction in the slope of decline in lung density (from −2.19 g/L/year with placebo to −1.31 g/L/year with A1-PI). The rate of decline observed with A1-PI was distinctly slower than that observed during the placebo treatment phase in the RAPID trial and between 24 and 48 months the rate of annual decline was similar between the Early-Start and Delayed-Start cohorts. However, the lung density loss that occurred in the Delayed-Start cohort prior to A1-PI therapy (dashed line) was not regained upon initiation of A1-PI therapy (, lower solid line).
Figure 3. Disease modification in the RAPID and RAPID Extension trials Citation(10). Graph showing the annualised rate of decline in physiologically adjusted P15 (g/L) at TLC over 48 months (ITT population). Reprinted from The Lancet, Vol 386. Chapman, KR et al., Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial, pp. 360–368. Copyright (2015), with permission from Elsevier.
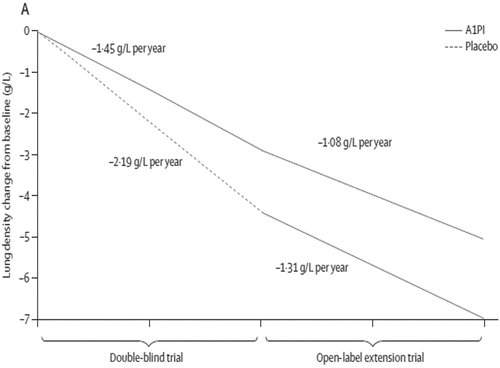
Analysis of both trials demonstrates that A1-PI therapy is able to slow the progression of AATD-related emphysema and the rate of lung density loss by 50% compared with placebo. The findings from the RAPID trials strengthen those from the EXACTLE trial and the Danish-Dutch trial, which suggested a trend towards a protective effect of A1PI but did not reach statistical significance. Furthermore treatment with A1-PI therapy may delay the time to terminal lung function by several years Citation(10), and this association with a survival benefit may also improve the acceptance of CT as a valid outcome. Due to the careful implementation of the appropriate delayed-start study design the combined RAPID and RAPID Extension trial data are the first to show a disease-modifying effect of specific treatment in pulmonary emphysema, i.e., A1-PI therapy in AATD. Whilst the RAPID trial and RAPID Extension trial indicate that A1-PI therapy is effective irrespective of the time of intervention, lung tissue lost during periods of no treatment cannot be regained. This is consistent with the impact of delayed-start treatment in that the clinical outcome measure in the Delayed-Start group never fully catches up with the Early-Start group ().
Results from the RAPID and RAPID Extension trial highlight the need for more timely diagnosis of AATD to enable patients to receive appropriate treatment. As AATD-related emphysema is a progressive lung disease, earlier diagnosis may facilitate implementation of lifestyle changes and specific treatment options to slow lung density decline in affected patients. However, AATD-related emphysema may be difficult to differentiate from other COPD phenotypes solely on the basis of clinical symptoms, and on average the delay between symptom onset and a correct diagnosis of AATD is 5–7 years (Citation6, 59). Given that lung tissue lost cannot be regained there is a need for both accurate and early diagnosis to allow the potential for early commencement of A1-PI treatment to prevent the loss of lung tissue. Likewise, it opens new perspectives for patients with non-AATD-related emphysema by demonstrating that with better understanding of disease mechanisms more effective, targeted therapies might be able to prevent/slow disease progression.
Future perspectives in AATD
Beyond the use of A1-PI in AATD-related emphysema, there is growing interest in disease modification of AATD that has future potential and requires acknowledgement. Experimental in vitro and in vivo AAT gene transfer with viral vectors has resulted in enhanced A1-PI serum levels and a promising safety profile. However, human clinical trials with adeno-associated viral vectors did not achieve a protective level of A1-PI in serum Citation(60). Retinoic acid and other retinoids activate nuclear hormone receptors, Retinoic Acid Receptor (RAR) and Retinoid X Receptor (RXR), to activate and/or repress gene expression to modulate structure and function of tissues Citation(61). Additionally, retinoids regulate growth factor receptors associated with alveolar septation and epithelial repair Citation(61). As such, there is interest in the potential for retinoids to restore lung function and slow the progression of emphysema; however, studies of retinoids in humans Citation(62) have thus far failed to replicate findings in animals (Citation63, 64). Also, severe AATD may present as liver disease (cirrhosis), and there is growing interest in disease modification that protects proteostasis mechanisms. Potential molecules include carbamazepine, which has shown promise in mouse models Citation(65), and is currently being investigated in a phase II clinical trial for its use in AATD-related liver disease Citation(66). An alternative approach is to use antisense oligonucleotides to silence the mutated AAT gene and reduce the hepatic production of the mutant Z-A1-PI protein and polymer formation. This may potentially prevent liver damage or allow the liver to repair. This approach has proven effective in animals Citation(67) and is currently under investigation in a phase I clinical trial Citation(68).
Conclusions
When considering chronic progressive diseases simply treating the associated symptoms is not sufficient to provide long-term improvements on disease progression. Disease-modifying treatments that affect the underlying cause of the disease can slow the progression of disease. Crucial to the success of clinical trials of potential disease-modifying therapies is careful trial design incorporating the use of reliable and sensitive study endpoints and/or markers of disease progression.
An important factor for consideration is that clinical outcome measures never truly catch up if disease-modifying treatment is delayed compared with initiated early, which emphasises the importance of early treatment. In the context of AATD-related emphysema appropriate and timely treatment is critical to prevent loss of lung tissue, particularly as the lung tissue lost prior to treatment cannot be regained. A1-PI therapy is the only pharmacological treatment that currently addresses the underlying cause of disease in AATD and has been demonstrated to be disease-modifying, i.e., able to reduce the decline in lung tissue loss and delay the time to terminal respiratory failure and lung transplantation. The mechanism of action of A1-PI is consistent with a disease-modifying effect; A1-PI therapy preserves lung function by protecting the lung parenchymal tissue and air exchange membranes (alveoli) from degradation by NE.
Well-designed clinical trials are needed to conclusively prove the efficacy of disease-modifying therapies and careful study design in the RAPID trial has led to disease-modifying therapy recently being proven for AATD-related emphysema. Future clinical trials should incorporate careful trial design, such as delayed-start trial design, along with the use of highly sensitive outcome measures and/or reliable biomarkers that correlate with clinical outcomes and other surrogate endpoints.
Declaration of interest
Dr. Chorostowska-Wynimko has received grants, personal fees and non-financial support from Grifols, grants from Baxalta US Inc, and personal fees from CSL Behring. Editorial assistance was provided by Meridian HealthComms (Plumley, UK), funded by CSL Behring.
References
- Blanco I, de Serres FJ, Carcaba V, et al. Alpha-1 antitrypsin deficiency PI*Z and PI*S gene frequency distribution using on maps of the world by an inverse distance weighting (IDW) multivariate interpolation method. Hepat Mon 2012; 12(10 HCC):e7434.
- American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med 2003; 168(7):818–900.
- Janciauskiene SM, Bals R, Koczulla R, et al. The discovery of alpha1-antitrypsin and its role in health and disease. Respir Med 2011; 105(8):1129–1139.
- Bernspång E, Sveger T, Piitulainen E. Respiratory symptoms and lung function in 30-year-old individuals with alpha-1-antitrypsin deficiency. Respir Med 2007; 101(9):1971–1976.
- Needham M, Stockley RA. Alpha 1-antitrypsin deficiency. 3: Clinical manifestations and natural history. Thorax 2004; 59(5):441–445.
- Stoller JK, Smith P, Yang P, Spray J. Physical and social impact of alpha 1-antitrypsin deficiency: results of a survey. Cleve Clin J Med 1994; 61(6):461–467.
- Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350(26):2645–2653.
- McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 2011; 365(17):1567–1575.
- Halpin DM, Tashkin DP. Defining disease modification in chronic obstructive pulmonary disease. COPD 2009; 6(3):211–225.
- Chapman KR, Burdon JGW, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet 2015; 386:360–368.
- Bespalov A, Klein C, Behl B, et al. Development of disease-modifying treatment of schizophrenia. Handb Exp Pharmacol 2012; 213:419–442.
- Cummings JL. Challenges to demonstrating disease-modifying effects in Alzheimer's disease clinical trials. Alzheimers Dement 2006; 2(4):263–271.
- Köhnlein T, Welte T. Alpha-1 antitrypsin deficiency: pathogenesis, clinical presentation, diagnosis, and treatment. Am J Med 2008; 121(1):3–9.
- O'Brien ME, Pennycooke K, Carroll TP, et al. The impact of smoke exposure on the clinical phenotype of alpha-1 antitrypsin deficiency in Ireland: exploiting a national registry to understand a rare disease. COPD 2015; 12 Suppl 1:2–9.
- Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest 1981; 68(5):1158–1165.
- Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med 1987; 316(17):1055–1062.
- Wewers MD, Crystal RG. Alpha-1 antitrypsin augmentation therapy. COPD 2013; 10 Suppl 1:64–67.
- Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest 2000; 118(5):1480–1485.
- The Alpha-1-Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha1-antitrypsin. Am J Respir Crit Care Med 1998; 158(1):49–59.
- Seersholm N, Wencker M, Banik N, et al. Does alpha1-antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha1-antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) alpha1-AT study group. Eur Respir J 1997; 10(10):2260–2263.
- Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med 1999; 160(5 Pt 1):1468–1472.
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J 2009; 33(6):1345–1353.
- Bhattaram VA, Siddiqui O, Kapcala LP, Gobburu JV. Endpoints and analyses to discern disease-modifying drug effects in early Parkinson's disease. AAPS J 2009; 11(3):456–464.
- D'Agostino RB, Sr. The delayed-start study design. N Engl J Med 2009; 361(13):1304–1306.
- Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 1993; 328(3):176–183.
- Olanow CW, Rascol O, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson's disease. N Engl J Med 2009; 361(13):1268–1278.
- Vellas B, Andrieu S, Sampaio C, et al. Endpoints for trials in Alzheimer's disease: a European task force consensus. Lancet Neurol 2008; 7(5):436–450.
- Vellas B, Andrieu S, Sampaio C, et al. Disease-modifying trials in Alzheimer's disease: a European task force consensus. Lancet Neurol 2007; 6(1):56–62.
- U.S. Food and Drug Administration (FDA). Guidance for industry—Alzheimer's disease: developing drugs for the treatment of early stage disease. Available from: http://wwwfdagov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338287pdf (published February 2013, accessed April 2016).
- U.S. Food and Drug Administration (FDA). Critical path opportunities list. Available from: http://wwwfdagov/downloads/Science-Research/SpecialTopics/CriticalPathInitiative/CriticalPathOpport-unitiesReports/UCM077258pdf (published March 2006, accessed April 2016).
- Turino GM, Lin YY, He J, et al. Elastin degradation: an effective biomarker in COPD. COPD 2012; 9(4):435–438.
- Agusti A, Edwards LD, Celli B, et al. Characteristics, stability and outcomes of the 2011 GOLD COPD groups in the ECLIPSE cohort. Eur Respir J 2013; 42(3):636–646.
- Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011; 365(13):1184–1192.
- Faner R, Tal-Singer R, Riley JH, et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax 2014; 69(7):666–672.
- Coxson HO, Dirksen A, Edwards LD, et al. The presence and progression of emphysema in COPD as determined by CT scanning and biomarker expression: a prospective analysis from the ECLIPSE study. Lancet Respir Med 2013; 1(2):129–136.
- Wilke S, Jones PW, Mullerova H, et al. One-year change in health status and subsequent outcomes in COPD. Thorax 2015; 70(5):420–425.
- Mullerova H, Maselli DJ, Locantore N, et al. Hospitalized exacerbations of COPD: risk factors and outcomes in the ECLIPSE cohort. Chest 2015; 147(4):999–1007.
- Vestbo J, Anderson W, Coxson HO, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur Respir J 2008; 31(4):869–873.
- Rennard SI. Chronic obstructive pulmonary disease: linking outcomes and pathobiology of disease modification. Proc Am Thorac Soc 2006; 3(3):276–280.
- Anthonisen NR, Connett JE, Kiley JP, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The lung health study. JAMA 1994; 272(19):1497–1505.
- Zuwallack RL, Nici L. Modifying the course of chronic obstructive pulmonary disease: looking beyond the FEV1. COPD 2012; 9(6):637–648.
- Chapman KR, Stockley RA, Dawkins C, et al. Augmentation therapy for alpha1 antitrypsin deficiency: a meta-analysis. COPD 2009; 6(3):177–184.
- Shaw JG, Vaughan A, Dent AG, et al. Biomarkers of progression of chronic obstructive pulmonary disease (COPD). J Thorac Dis 2014; 6(11):1532–1547.
- Glaab T, Vogelmeier C, Buhl R. Outcome measures in chronic obstructive pulmonary disease (COPD): strengths and limitations. Respir Res 2010; 11:79.
- Calverley PM, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007; 356(8):775–789.
- Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359(15):1543–1554.
- Celli BR. Predictors of mortality in COPD. Respir Med 2010; 104(6):773–779.
- de Torres JP, Casanova C, Marin JM, et al. Prognostic evaluation of COPD patients: GOLD 2011 versus BODE and the COPD comorbidity index COTE. Thorax 2014; 69(9):799–804.
- Celli BR. Change in the BODE index reflects disease modification in COPD: lessons from lung volume reduction surgery. Chest 2006; 129(4):835–836.
- Imfeld S, Bloch KE, Weder W, Russi EW. The BODE index after lung volume reduction surgery correlates with survival. Chest 2006; 129(4):873–878.
- Stolk J, Putter H, Bakker EM, et al. Progression parameters for emphysema: a clinical investigation. Respir Med. 2007; 101(9):1924–1930.
- Parr DG, Dirksen A, Piitulainen E, et al. Exploring the optimum approach to the use of CT densitometry in a randomised placebo-controlled study of augmentation therapy in alpha 1-antitrypsin deficiency. Respir Res 2009; 10:75.
- Parr DG, Stoel BC, Stolk J, Stockley RA. Validation of computed tomographic lung densitometry for monitoring emphysema in alpha1-antitrypsin deficiency. Thorax 2006; 61(6):485–490.
- Stockley RA, Parr DG, Piitulainen E, et al. Therapeutic efficacy of alpha-1 antitrypsin augmentation therapy on the loss of lung tissue: an integrated analysis of 2 randomised clinical trials using computed tomography densitometry. Respir Res 2010; 11:136.
- U.S. Food and Drug Administration (FDA). Clinical and surrogate endpoints for evaluating efficacy of alpha1-proteinase inhibitor (human) augmentation therapy. Blood Products Advisory Committee 95th Meeting July 20–21, 2009. Available from: http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmater-ials/bloodvaccinesandotherbiologics/bloodproductsadvisory-committee/ucm171091.pdf (accessed April15 2016).
- Craig TJ. Suspecting and testing for Alpha-1 antitrypsin deficiency—an allergist's and/or immunologist's perspective. J Allergy Clin Immunol Pract 2015; 3(4):506–511.
- Wielputz MO, Bardarova D, Weinheimer O, et al. Variation of densitometry on computed tomography in COPD—influence of different software tools. PLoS One 2014; 9(11):e112898.
- Dawkins PA, Dowson LJ, Guest PJ, Stockley RA. Predictors of mortality in alpha1-antitrypsin deficiency. Thorax 2003; 58(12):1020–1026.
- Kohnlein T, Janciauskiene S, Welte T. Diagnostic delay and clinical modifiers in alpha-1 antitrypsin deficiency. Ther Adv Respir Dis 2010; 4(5):279–287.
- Wozniak J, Wandtke T, Kopinski P, Chorostowska-Wynimko J. Challenges and prospects for alpha-1 antitrypsin deficiency gene therapy. Hum Gene Ther 2015; 26(11):709–718.
- Muñiz-Hernández S, Hernández-Pedro N, Macedo-Pérez OE, Arrieta O. Alterations in retinoic acid receptors in non-small cell lung cancer and their clinical implications. J Cancer Ther 2015; 6:648–664.
- Stolk J, Stockley RA, Stoel BC, et al. Randomised controlled trial for emphysema with a selective agonist of the gamma-type retinoic acid receptor. Eur Respir J 2012; 40(2):306–312.
- Belloni PN, Garvin L, Mao CP, et al. Effects of all-trans-retinoic acid in promoting alveolar repair. Chest 2000; 117(5 Suppl 1):235S–241S.
- Massaro GD, Massaro D. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nat Med 1997; 3(6):675–677.
- Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 2010; 329(5988):229–232.
- Clinicaltrials.gov. Carbamazepine in Severe liver disease due to alpha-1 antitrypsin deficiency. Available from: https://clinicaltrials.gov/ct2/show/NCT01379469 (published February 2016, accessed April 2016).
- Guo S, Booten SL, Aghajan M, et al. Antisense oligonucleotide treatment ameliorates alpha-1 antitrypsin-related liver disease in mice. J Clin Invest 2014; 124(1):251–261.
- Clinicaltrials.gov. A study of ARC-AAT in healthy volunteer subjects and patients with alpha-1 antitrypsin deficiency (AATD). Available from: https://clinicaltrials.gov/ct2/show/NCT02363946 (published July 2015, accessed April 2016).
- Mahler DA, Criner GJ. Assessment tools for chronic obstructive pulmonary disease: do newer metrics allow for disease modification? Proc Am Thorac Soc 2007; 4(7):507–511.